Introduction
Ankylosing spondylitis (AS) is a chronic autoimmune
inflammatory disease that primarily invades the spine and involves
the cuboid iliac joint (1). The
disability rate of patients with AS is high and AS has a serious
impact on quality of life (2,3).
Studies have shown that AS is associated with genetics, infection,
immunity, inflammatory cell infiltration, cytokine imbalance and
other factors (4,5).
In the ligaments of patients with early AS,
increased chondrocyte differentiation promotes cartilage formation,
which is accompanied by cartilage calcification Chen et al
(4) found that long non-coding RNA
metastasis-associated lung adenocarcinoma transcript 1 and
gasdermin-D expression is increased in AS cartilage tissue and
chondrocytes, whereas microRNA-558 expression is decreased in AS
chondrocytes, thereby promoting chondrocyte pyroptosis and
activating the inflammatory response in vivo (4,5).
Moreover, one of the hallmarks of AS joint remodeling is cartilage
degeneration and increased chondrocyte apoptosis (6). The aforementioned studies suggest
that chondrocyte death accelerates cartilage degeneration and
promotes the disease course of AS.
Ferroptosis is a cell death pathway that is distinct
from apoptosis, necrosis and autophagy at the biochemical,
morphological and genetic levels (7). Several studies confirmed that
excessive autophagy could promote ferroptosis (8-10).
In immortalized mouse embryonic fibroblasts, PANC1 and PANC2.03
human pancreatic cancer cell lines as well as human fibrosarcoma
cell line HT-1080, knockdown of autophagy-related 5 and
autophagy-related 7 inhibits autophagy and levels of intracellular
free iron levels and lipid peroxidation end products [such as
malondialdehyde (MDA)] decrease significantly (9,11).
Moreover, expression of ferritin heavy chain 1 (FTH1), an
intracellular ferritin marker, is significantly increased (9,11).
In addition, autophagy is accompanied by a decrease in glutathione
under conditions such as starvation of fetal bovine serum (FBS) and
oxidative stress (12). By
contrast, glutathione peroxidase 4 (GPX4) overexpression inhibits
reactive oxygen species (ROS)-mediated autophagy (13). Other ferroptosis regulators such as
solute carrier family 7 member 11 (SLC7A11) have also been shown to
be potential regulators of autophagy (14). Through weighted gene co-expression
network analysis, Rong et al (13) demonstrated that
ferroptosis-associated gene expression is abnormal, e.g., that of
small nucleolar RNA host gene 16 and mitogen-activated protein
kinase 1, in patients with AS, leading to the speculation that
ferroptosis is involved in the pathological development of AS.
Acrylamide (ACR) is a synthetic compound utilized
commercially and is also a cytotoxic and genotoxic compound that
forms during heat treatment of foodstuffs, such as potatoes, bakery
products and plant derivatives, as a β-unsaturated (conjugated)
reactive molecule (15).
Increasing studies have suggested ACR as a probable malignant
neoplastic disease agent in all organs and tissues of the human
body (16,17). For example, ACR induces human
chondrocyte death by mitochondrial dysfunction (15). However, to the best of our
knowledge, if ACR contributes to AS pathogenesis via ferroptosis
remains unclear.
Materials and methods
Cell culture
Human chondrocytes were purchased from Procell Life
Science & Technology Co., Ltd. (cat. no. CP-H107). Chondrocytes
were cultured in human chondrocyte complete culture medium (cat.
no. CM-H107; Procell Life Science & Technology Co., Ltd.)
supplemented with 10% FBS (Invitrogen; Thermo Fisher Scientific,
Inc.), streptomycin (100 mg/ml) and penicillin (100 U/ml) at 37˚C
in a humidified atmosphere with 5% CO2. Human
chondrocytes were treated with ACR (ACR group; cat. no. V900845;
Sigma-Aldrich; Merck KGaA) or an equal volume of sterilized water
(Con group) for 24 h at 37˚C.
Cell counting kit (CCK)-8 assay
Human chondrocytes were seeded into a 96-well plate
at a density of 5.0x103 cells/well. Following exposure
to 0.1, 0.2, 0.4, 0.8, 1.6, 3.2 or 6.4 µg/ml ACR for 24 h at 37˚C,
chondrocyte viability was determined using a CCK-8 assay kit
(Beijing Solarbio Science & Technology Co., Ltd.) according to
the manufacturer's instructions. In brief, human chondrocytes were
incubated with 10 µl CCK-8 regent at 37˚C for 1 h. Next,
cell viability was calculated from the optical density measured at
450 nm using a microplate reader and PBS without any cells was used
as a blank control (Thermo Fisher Scientific, Inc.). The
half-maximal inhibitory concentration (IC50) values were
calculated via the Reed-Muench method (18).
Human chondrocytes were also seeded in a 96-well
plate at a density of 5,000 cells/well. To validate which type of
cell death could be induced by ACR, human chondrocytes were
preincubated with ferrostatin-1 (Fer-1) (1 µM; a ferroptosis
inhibitor), benzyloxycarbonylvalyl-alanyl-aspartyl fluoromethyl
ketone (Z-VAD-FMK) (20 µM; a caspase-3 inhibitor), necrostatin-1
(Nec-1) (20 µM; a necroptosis inhibitor) and 3-MA (20 µM; an
autophagy inhibitor) for 1 h. The human chondrocytes were then
further treated with 0.35 µg/ml ACR at 37˚C for 24 h.
Chondrocyte viability was determined using a CCK-8 assay kit
(Beijing Solarbio Science & Technology Co., Ltd.) according to
the manufacturer's instructions. The absorbance values were
determined at OD450 nm using a microplate reader (Thermo Fisher
Scientific, Inc.). Dead cells (%)=100-OD experimental group/OD
control group x100.
EdU staining
Human chondrocytes were seeded in a 96-well plate at
a density of 5.0x103 cells/well and treated with 0.35
µg/ml ACR for 24 h at 37˚C. EdU-positive cells were stained with a
Cell-Light™ EdU Apollo In Vitro kit (Guangzhou
RiboBio Co., Ltd.) according to the manufacturer's instructions. A
total of 100 µl of 50 µM EdU medium was added to each well and
incubated at 37˚C for 2 h. The medium was then
discarded. Afterwards, the cells were washed twice with PBS for 5
min each time. Next, 100 µl 4% paraformaldehyde (Beijing Solarbio
Science & Technology Co., Ltd.) was added to each well and
incubated for 30 min at room temperature, and 100 µl of 1X DAPI
(cat. no. LI-9557; OriGene Technologies, Inc.) was added to each
well and incubated for 30 min at room temperature, avoiding light
and then discarded. Once the staining reaction solution was
discarded, 100 µl PBS was added to each well. After washing three
times, the slides were observed under a fluorescence microscope
(Keyence Corporation).
EdU is a thymidine nucleoside analogue that is
incorporated into replicating DNA molecules instead of thymine
during cell proliferation and rapidly labels the replication
activity of DNA in cells by the specific reaction of EdU with
fluorescent dyes (19).
EdU-positive cells indicate active replication.
Senescence-associated β-galactosidase
staining
Human chondrocytes were seeded into a six-well plate
at a density of 1x106 cells/well and treated with 0.35
µg/ml ACR for 24 h at 37˚C. Subsequently, chondrocytes were stained
with Senescence β-Galactosidase Staining kit (cat. no. C0602;
Beyotime Institute of Biotechnology) according to the
manufacturer's instructions. In brief, cells were fixed with 4%
paraformaldehyde (Beyotime Institute of Biotechnology) for 10 min
at room temperature. The cells were washed three times with PBS (5
min/time). After that, the PBS was discarded, and 1 ml β-gal
staining solution (cat. no. C0602; Beyotime Institute of
Biotechnology) was added to each well and incubated at 37˚C for 1 h
followed by three washes with PBS (5 min/time). Aging cells were
observed under a light microscope and determined by counting the
number of blue-stained cells. The cytoplasm of
β-galactosidase-containing cells was blue or dark blue. A total of
five fields of view were randomly selected from each section, the
number of blue or dark blue cells was counted manually, and the
average was taken for statistical analysis.
Immunofluorescence (IF) staining
Human chondrocytes (1x106 cells/well)
were seeded on coverslips, fixed with 4% paraformaldehyde (Beyotime
Institute of Biotechnology) at room temperature for 10 min and
permeabilized with 1% Triton X-100 (Beyotime Institute of
Biotechnology) for 15 min at room temperature. Slides were blocked
with 8% BSA (Beijing Solarbio Science & Technology Co., Ltd.)
at room temperature for 2 h and then incubated with γ-H2A histone
family member X primary antibody (γ-H2AX; cat. no. C2035S; 1;100;
DNA Damage Assay kit; Beyotime Institute of Biotechnology) or
anti-AMP-activated protein kinase primary antibody (cat. no. 5832,
anti-AMPK; 1:50; CST Biological Reagents Co., Ltd.) at 4˚C
overnight. Then, slides were incubated with FITC-conjugated
secondary antibody (cat. no. ZF-0314; OriGene Technologies, Inc.)
at room temperature for 2 h in the dark and mounted with
fluorescent mounting medium with DAPI (cat. no. LI-9557; OriGene
Technologies, Inc.). The slides were observed under a light
microscope (Keyence Corporation). The density of relative
fluorescence intensity was analyzed using ImageJ 1.43b software
(National Institutes of Health).
Western blotting
Human chondrocytes (2x106 cells in total
for each sample) were lysed with RIPA buffer (cat. no. R0010,
Beijing Solarbio Science & Technology Co., Ltd.) containing
protease and phosphatase inhibitors (Sigma-Aldrich; Merck KGaA) for
30 min on ice. The lysates were centrifuged at 11,000 x g for 30
min at 4˚C and protein concentration was determined using a BCA
Protein Assay kit (Pierce; Thermo Fisher Scientific, Inc.). Equal
amounts of proteins (20 µg/lane) were isolated by SDS-PAGE on a 12%
gel and transferred onto polyvinylidene fluoride membranes
(MilliporeSigma). The membranes were blocked with 6% non-fat milk
(MilliporeSigma) at room temperature for 2 h and incubated with
primary antibodies as follows: p53 (cat. no. 2527; 1:1,000),
cyclin-dependent kinase inhibitor 1 (p21; cat. no. 2947; 1:1,000),
cyclin-dependent kinase inhibitor protein (p16; cat. no. 80772;
1:1,000), light chain (LC) 3I/II (cat. no. 3868; 1:1,000),
sequestosome 1 (p62; cat. no. 88588; 1:1,000), GPX4 (cat. no.
59735, 1:1,000), SLC7A11 (cat. no. 12691; 1:1,000; SLC7A11);
transferrin receptor protein 1 (TfR1; cat. no. 13113; 1:1,000;
SLC7A11), FTH1 (cat. no. 4393, 1:1,000), AMPK (cat. no. 5832,
1:1,000), phosphorylated (p)-AMPK (cat. no. 2537, 1:1,000),
serine/threonine-protein kinase ULK1 (cat. no. 8054, 1:1,000),
p-ULK1 (cat. no. 14202, 1:1,000), p-mTOR (cat. no. 5536, 1:1,000);
mTOR (cat. no. 2983, 1:1,000) and GAPDH (cat. no. 5174, 1:1,000)
(all CST Biological Reagents Co., Ltd.) at 4˚C overnight. After
washing with PBST (0.1% Tween-20 in PBS; Beijing Solarbio Science
& Technology Co., Ltd.) three times, membranes were incubated
with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG
(cat. no. ZB-2301; 1:5,000; OriGene Technologies, Inc.) for 2 h at
room temperature. The protein signal was determined using Immobilon
Western Chemiluminescent HRP Substrate (cat. no. WBKLS0500;
MilliporeSigma). Relative protein expression was normalized to that
of GAPDH. The density of each band was analyzed using ImageJ 1.43b
software (National Institutes of Health).
Knockdown of AMPK
Specific small interfering (si)RNA targeting AMPK
(siRNA-AMPK; 5'-AAAGTGAAGGTTGGCAAACATGA-3') or negative control
(NC; 5'-CCGAUAGGUUUACUGCCAATT-3') was purchased from Guangzhou
RiboBio Co., Ltd. and transfected using HiperFect Transfection
Reagent (Qiagen GmbH) according to the manufacturer's instructions,
and subsequent experiment was performed after 48 h of transfection.
In brief, human chondrocytes were seeded into a six-well plate at a
density of 1x106 cells/well for 24 h and then 10 µl
HiperFect Transfection Reagent was mixed with 100 µl serum-free
culture medium. Subsequently, 4 µl siRNA-AMPK (10 µM) or NC (10 µM)
was added and it was incubated at room temperature for 10 min.
Subsequently, the mixture was added to each well of a six-well
plate at a final concentration of 20 nM and incubated at 37˚C for
24 h before cells were collected for further study.
Determination of lipid ROS
Human chondrocytes were seeded into a 6-well plate
at a density of 1x106 cells/well and lipid ROS levels
were determined following incubation with 5 µM C11-BODIPY581/591
fluorescent probe (MedChemExpress) for 20 min at 37˚C in the dark.
Then, fluorescence was observed under a light microscope (Keyence
Corporation).
Mitochondrial membrane potential (MMP)
evaluation
MMP was determined using the JC-1 MMP Assay kit
(cat. no. ab113850; Abcam) according to the manufacturer's
instructions. Human chondrocytes were seeded into a six-well plate
at a density of 1x106 cells/well and treated with 0.35
µg/ml ACR for 24 h at 37˚C. Subsequently, cells were incubated with
1X JC-1 staining solution for 20 min in the dark at 37˚C and
observed under a fluorescence microscope (Keyence Corporation).
Normal MMP was shown in red with JC-1 dimers whereas depolarized
membrane potential was shown in green with JC-1 monomers.
Determination of intracellular
Fe2+ levels
Human chondrocytes were stained with 1 µM
FerroOrange (Dojindo Laboratories, Inc.) in Hanks' balanced salt
solution (cat. no. H1045; Beijing Solarbio Science & Technology
Co., Ltd.) at 37˚C for 30 min in the dark and immediately observed
under a fluorescence microscope (Keyence Corporation).
Determination of ROS and MDA
levels
ROS and MDA content were measured using a ROS Assay
kit (cat. no. S0033S; Beyotime Institute of Biotechnology) and
Lipid Peroxidation MDA Assay kit (cat. no. S0131S; Beyotime
Institute of Biotechnology), respectively, according to the
manufacturer's instructions. Human chondrocytes were seeded into a
6-well plate at a density of 1x106 cells/well and were
treated with 0.35 µg/ml ACR for 24 h at 37˚C. After that, the cells
were collected for further determination of intracellular ROS and
MDA contents.
For ROS quantification, human chondrocytes were
incubated with 10 µM DCFH-DA for 20 min at 37˚C in the
dark and immediately observed under a fluorescence microscope
(Keyence Corporation). The density of relative fluorescence
intensity was analyzed using ImageJ 1.43b software (National
Institutes of Health).
For MDA quantification, 0.1 ml of sample or PBS
(blank control) was added to the centrifuge tube, followed by 0.2
ml of MDA assay working solution. The mixture was heated at 100˚C
for 15 min. The water bath was cooled to room temperature and
centrifuged at 1,000 x g for 10 min at room temperature. After
that, 200 µl of supernatant was added to a 96-well plate, followed
by determination of absorbance at 532 nm using a microplate reader
(Thermo Fisher Scientific, Inc.). The relative MDA content in the
sample solution was calculated from the standard curve.
Transmission electron microscopy
Human chondrocytes were fixed in 2.5% glutaraldehyde
solution at room temperature for 2 h, post-fixed in 1% aqueous
osmium tetroxide at room temperature for 2 h and dehydrated in an
ethanol gradient (50, 70 and 90%) at room temperature for 15 min
separately and 100% acetone at room temperature for 15 min. The
samples were prepared by gradient infiltration of anhydrous acetone
and epoxy resin overnight, embedded in resin and polymerized at
60˚C for 48 h. The samples were cut into 50 nm ultrathin sections
using an ultramicrotome (Leica Microsystems GmbH), stained with 3%
uranium acetate-lead citrate (Beijing Zhongxing Bairui Technology
Co., Ltd.) at room temperature for 15 min and examined by 120 keV
transmission electron microscopy (Tecnai G2 20 TWIN; Thermo Fisher
Scientific, Inc.).
Flow cytometry analysis
Human chondrocytes were seeded into a six-well plate
at a density of 1x106 cells/well and treated with 0.35
µg/ml ACR for 24 h at 37˚C. Subsequently, the cells were collected
for cell death and cell cycle analysis using an Annexin V-PE/7-AAD
Apoptosis Detection (cat. no. 40310ES50; Shanghai Yeasen
Biotechnology Co., Ltd.) and Cell Cycle Assay Kit (Red
Fluorescence; E-CK-A351; Elabscience Biotechnology Co., Ltd.)
according to the manufacturer's instructions.
The cells were centrifuged at 300 x g at 4˚C for 5
min to collect the cells. The cells were washed twice with PBS
precooled at 4˚C and centrifuged at 300 x g at 4˚C for 5 min.
Subsequently, 250 µl 1X binding buffer was added to resuspend the
cells and the concentration was adjusted to 1x106
cells/ml. Then, 100 µl cell suspension was transferred into a 5 ml
flow tube and incubated with 5 µl annexin V/PE and 10 µl 7-AAD in
the dark at room temperature for 15 min. The cells were tested
within 1 h using a flow cytometer (BD LSRFortessa™; BD
Biosciences). The data was analyzed using FlowJo 10.8.1 software
(FlowJo LLC).
Approximately 5x105 human chondrocytes
were collected. The cells were centrifuged at 300 x g for 5 min and
the supernatant was discarded. Next, 0.3 ml PBS was added to
resuspend the cells. Afterwards, 1.2 ml of-20˚C anhydrous ethanol
was added, mixed thoroughly and the samples were placed at -20˚C
for 1 h. Next, the samples were centrifuged at 300 x g for 5 min,
the supernatant was discarded and 1 ml PBS was added to resuspend
the cells for 15 min at room temperature. Afterwards, the mixture
was centrifuged at 300 x g for 5 min, the supernatant was discarded
and 100 µl of RNase A was added. Finally, 400 µl PI Reagent (50
µg/ml) was added and mixed through thoroughly, and the cells were
incubated for 30 min at 4˚C under low light. The red fluorescence
at 488 nm was recorded immediately on a BD LSRFortessa flow
cytometer (BD Biosciences). The data was analyzed using FlowJo
10.8.1 software (FlowJo LLC).
Determination of autophagic flux
Human chondrocytes were seeded into a six-well plate
at a density of 1x106 cells/well and were transfected
with GFP-mCherry-LC3 adenovirus at 100 multiplicity of infection
(MOI) for 24 h at 37˚C. Following treatment with 0.35 µg/ml ACR for
24 h at 37˚C, cells were fixed with 4% paraformaldehyde (Beyotime
Institute of Biotechnology) at room temperature for 10 min and
immediately observed under a microscope (Keyence Corporation). GFP
degraded in the acidic environment. After the red-green
fluorescence was merged, the yellow spots that appeared after the
merge were only autophagosomes. The red spots indicated
autolysosomes. If the autophagic flow was unblocked, the red
fluorescence was more than the yellow fluorescence. If the
autophagic flow was blocked, it was mainly yellow fluorescence.
Autophagic flux was determined by counting the cells with
GFP+/mCherry+ (yellow) or GFP-/mCherry+ (red) puncta manually using
a fluorescence microscope (Keyence Corporation).
Statistical analysis
The data are expressed as the mean ± standard
deviation of three independent experimental repeats. Unpaired
Student's t test was used for comparisons between two groups.
Comparisons between three or more groups were performed via one-way
ANOVA followed by Bonferroni's post hoc test. All analyses were
performed using GraphPad Prism 8 statistical software (GraphPad
Software, Inc.). P<0.05 was considered to indicate a
statistically significant difference.
Results
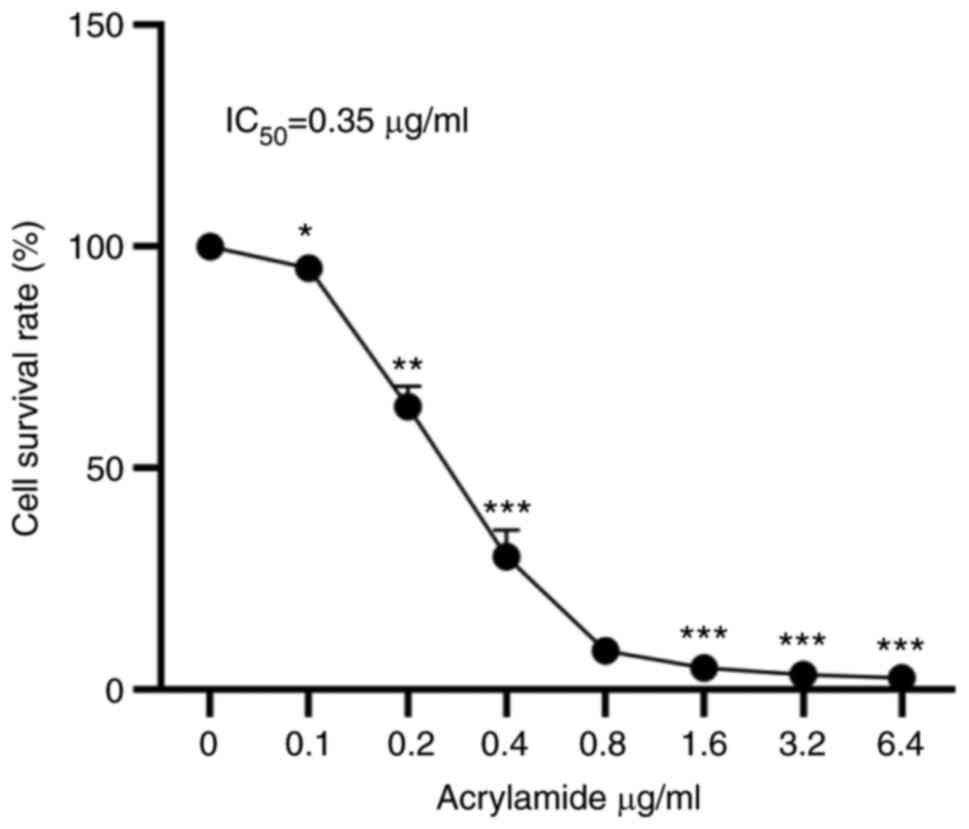
ACR decreases human chondrocyte
survival rate in a dose-dependent manner
To analyze the effects of ACR on human chondrocyte
viability, cells were treated with ACR at 0.1, 0.2, 0.4, 0.8, 1.6,
3.2 and 6.4 µg/ml ACR for 24 h. Compared with the control, the cell
survival rate of human chondrocytes decreased significantly in a
dose-dependent manner (P<0.05; Fig.
1). The IC50 of ACR in human chondrocytes was 0.35
µg/ml.
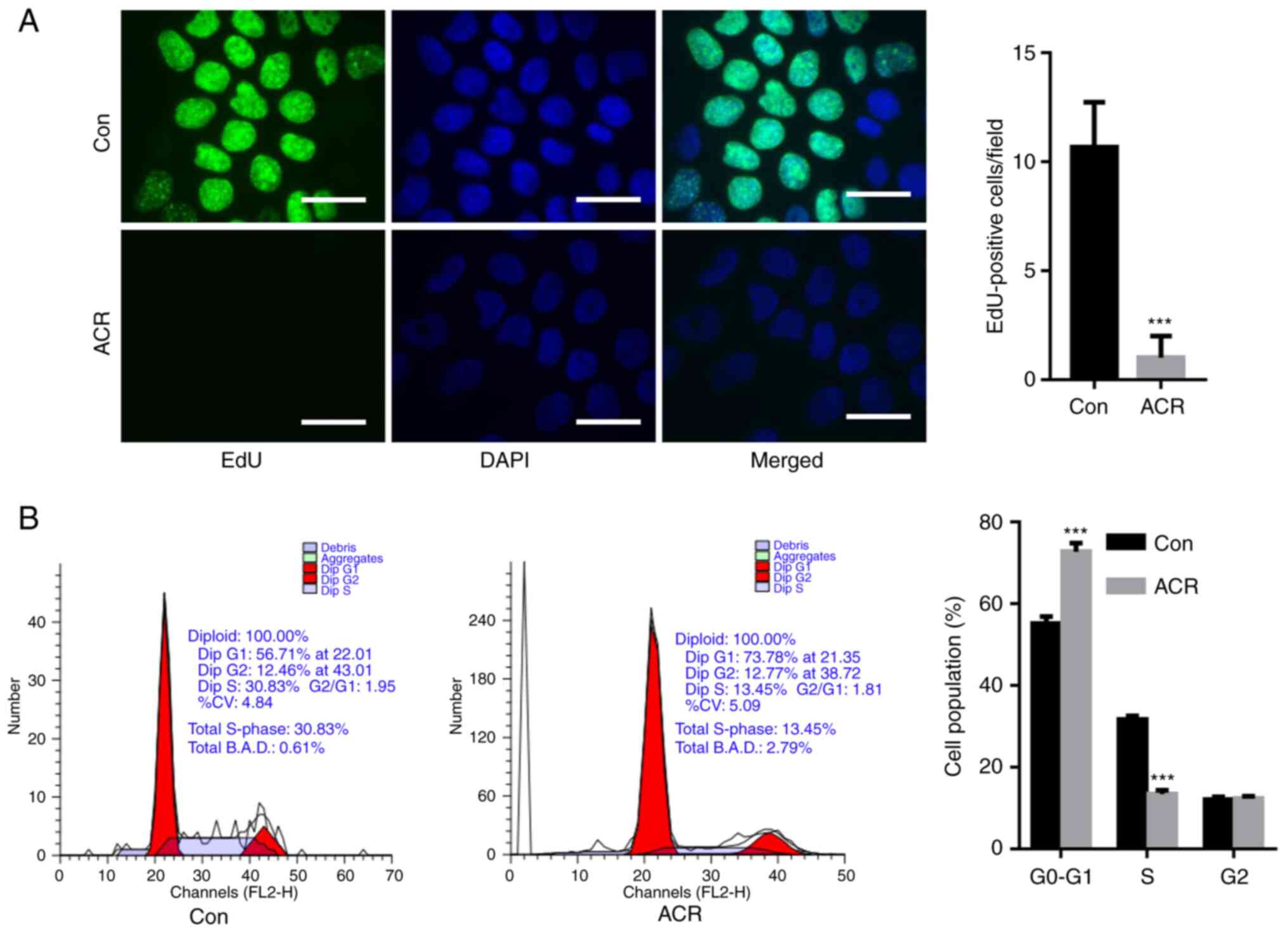
ACR suppresses chondrocyte
proliferation
To determine the effect of ACR on cell
proliferation, human chondrocytes were incubated with 0.35 µg/ml
ACR at 37˚C for 24 h and EdU staining was performed. EdU-positive
cells/field were significantly decreased in chondrocytes treated
with ACR compared with Con chondrocytes (P<0.001; Fig. 2A). In addition, cell cycle arrest
was observed in human chondrocytes treated with ACR, as evidenced
by elevation in the G0-G1 phase cell
population and downregulation of the S phase cell population
compared with Con (P<0.001; Fig.
2B).
ACR induces human chondrocyte
senescence
Compared with Con, 0.35 µg/ml ACR significantly
increased the number of SA-β-gal-positive cells after 24 h
(P<0.001; Fig. 3A). In
addition, relative fluorescence of γ-H2AX (a biomarker of DNA
damage) was increased in human chondrocytes treated with ACR
compared with Con (Fig. 3B).
Moreover, flow cytometry analysis showed that ACR increased the
number of dead cells to ~28% compared with that of Con (11%;
P<0.001; Fig. 3C). Western
blotting showed that the expression levels of cell
arrest-associated proteins, including p53, p21 and p16, were
significantly enhanced in human chondrocytes treated with ACR
compared with those in Con chondrocytes (P<0.05; Fig. 3D). These observations indicated
that ACR contributed to the senescence of human chondrocytes.
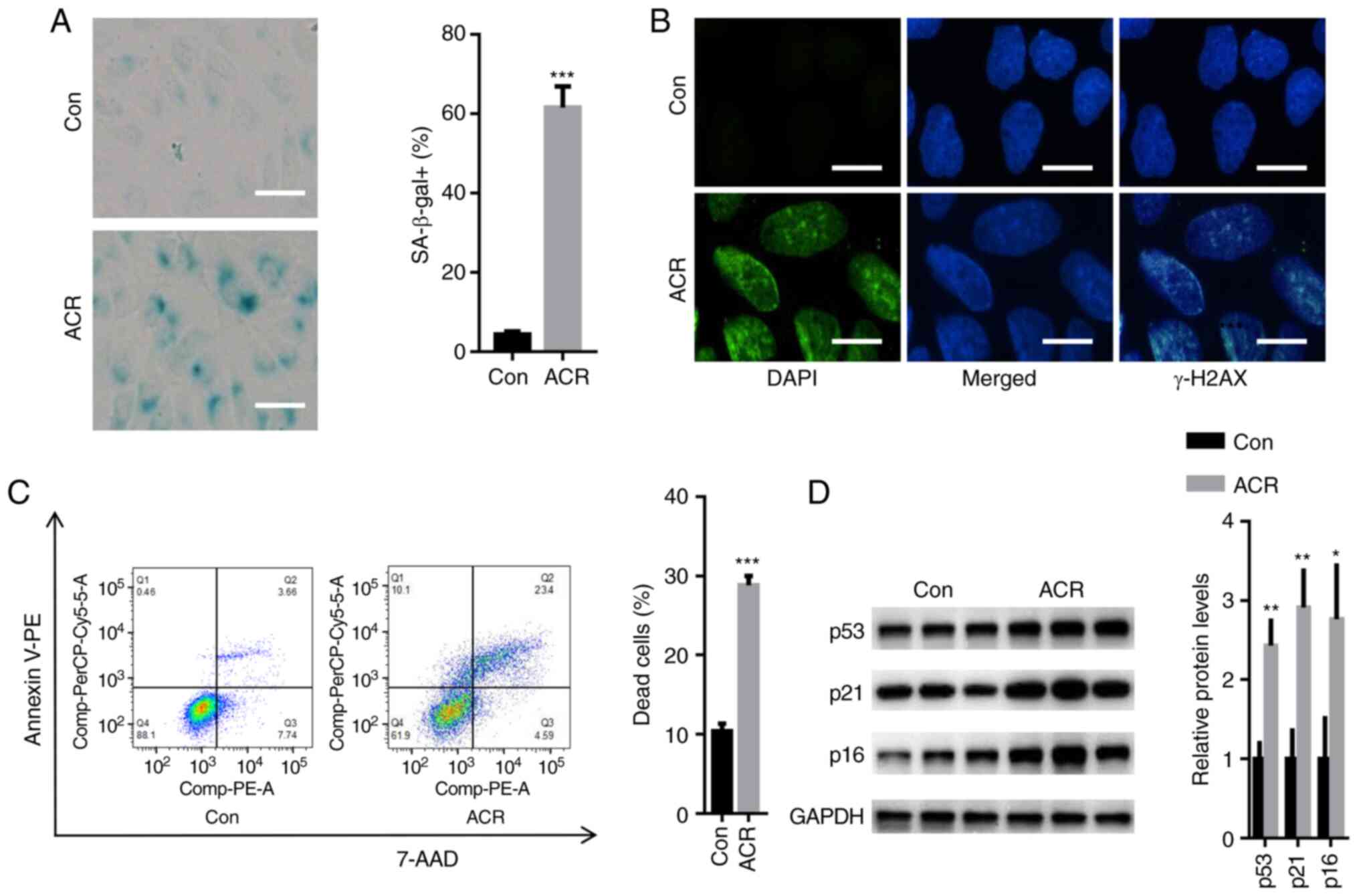 | Figure 3ACR induces human chondrocyte
senescence. (A) SA-β-gal staining showed that 0.35 µg/ml ACR
significantly increased the number of SA-β-gal-positive cells
(magnification, x20; scale bar, 20 µm). (B) Immunofluorescence
staining showed that the relative fluorescence of γ-H2A histone
family member X was increased in human chondrocytes treated with
ACR (magnification, x40; scale bar, 10 µm). (C) Flow cytometry
showed that ACR increased the number of dead cells. (D) Western
blot assay showed that expression levels of p53, cyclin-dependent
kinase inhibitor 1 and cyclin-dependent kinase inhibitor protein
were significantly increased in human chondrocytes treated with
ACR. *P<0.05, **P<0.01 and
***P<0.001 vs. Con. ACR, acrylamide; Con, control;
SA-β-gal, senescence-associated β-galactosidase; γ-H2AX, γ-H2A
histone family member X. |
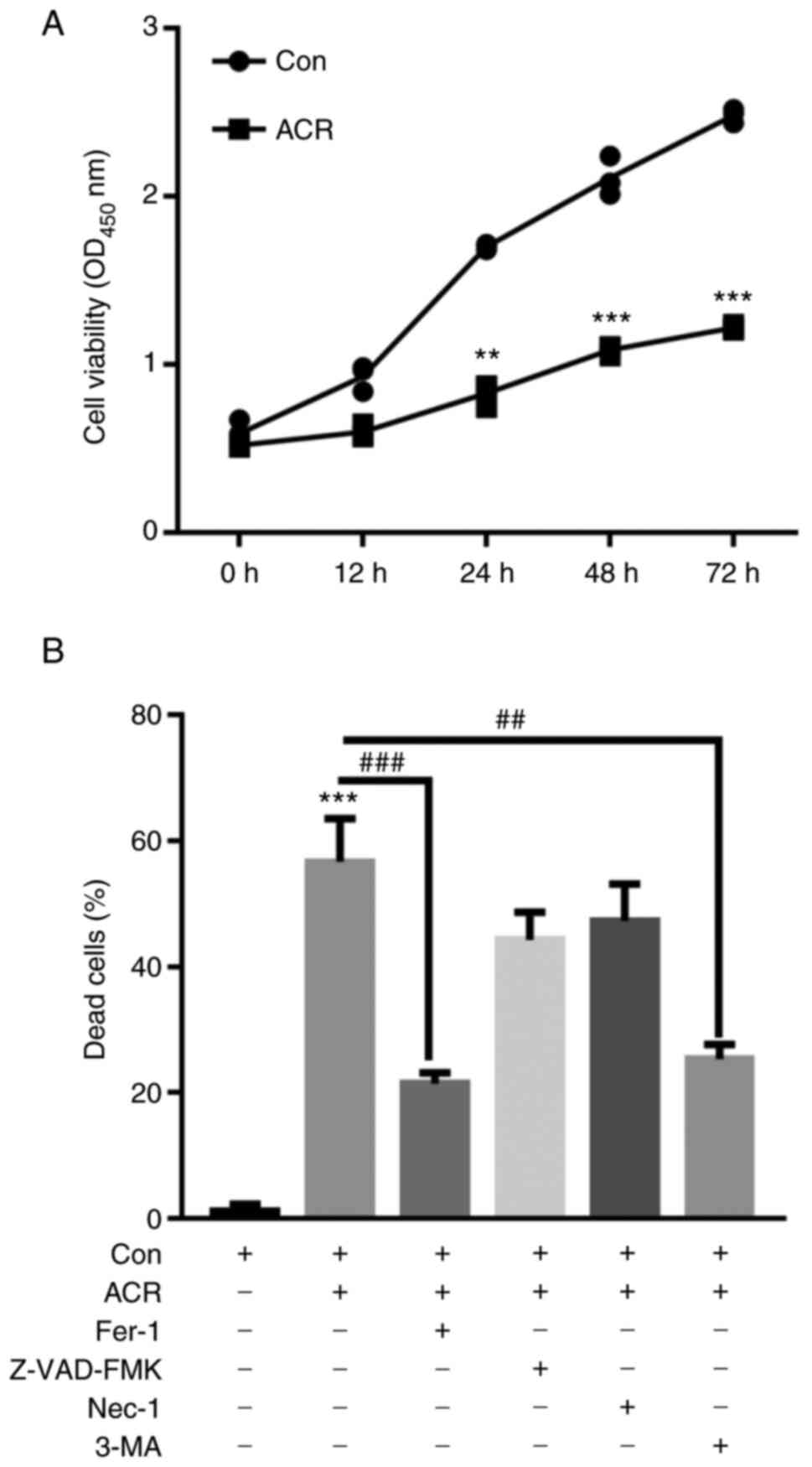
Ferrostatin-1 (Fer-1) and
3-methyladenine (3-MA) reverse ACR-induced cell death in human
chondrocytes
It was investigated whether 0.35 µg/ml ACR decreased
human chondrocyte cell death in a time-dependent manner. CCK-8
assay showed that cell viability was significantly decreased in
human chondrocytes treated with 0.35 µg/ml ACR at 24, 48 and 72 h
(P<0.01; Fig. 4A). To validate
which type of cell death was induced by ACR, human chondrocytes
were preincubated with Fer-1, Z-VAD-FMK, Nec-1 and 3-MA for 1 h.
Human chondrocytes were further treated with 0.35 µg/ml ACR at 37˚C
for 24 h. Fer-1 and 3-MA significantly reversed ACR-induced cell
death (P<0.01; Fig. 4B). These
findings demonstrated that ACR promoted chondrocyte cell death by
inducing ferroptosis and autophagy.
ACR induces autophagy in human
chondrocytes
Ultrastructural analysis indicated a higher level of
outer mitochondrial membrane rupture in chondrocytes treated with
0.35 µg/ml ACR at 37˚C for 24 h compared with that in Con
chondrocytes (Fig. 5A). Meanwhile,
the mitochondrial ridge was decreased or disappeared in human
chondrocytes treated with ACR (Fig.
5A). By contrast, preincubation with 3-MA at 37˚C for 1 h
reversed these effects (Fig. 5A).
Autophagic flux was activated in human chondrocytes treated with
ACR, as evidenced by the elevation in red puncta, whereas 3-MA
preincubation significantly decreased the red puncta in human
chondrocytes (P<0.001; Fig.
5B). Western blotting of autophagic flux showed that the
LC3II/LC3I ratio was increased, whereas expression of p62 was
decreased in human chondrocytes treated with ACR (P<0.01;
Fig. 5C). By contrast, 3-MA
preincubation decreased the LC3II/LC3I ratio and increased the
protein expression of p62 compared with the ACR treatment alone
(P<0.01; Fig. 5C). These
results indicate that ACR activated autophagic flux in human
chondrocytes.
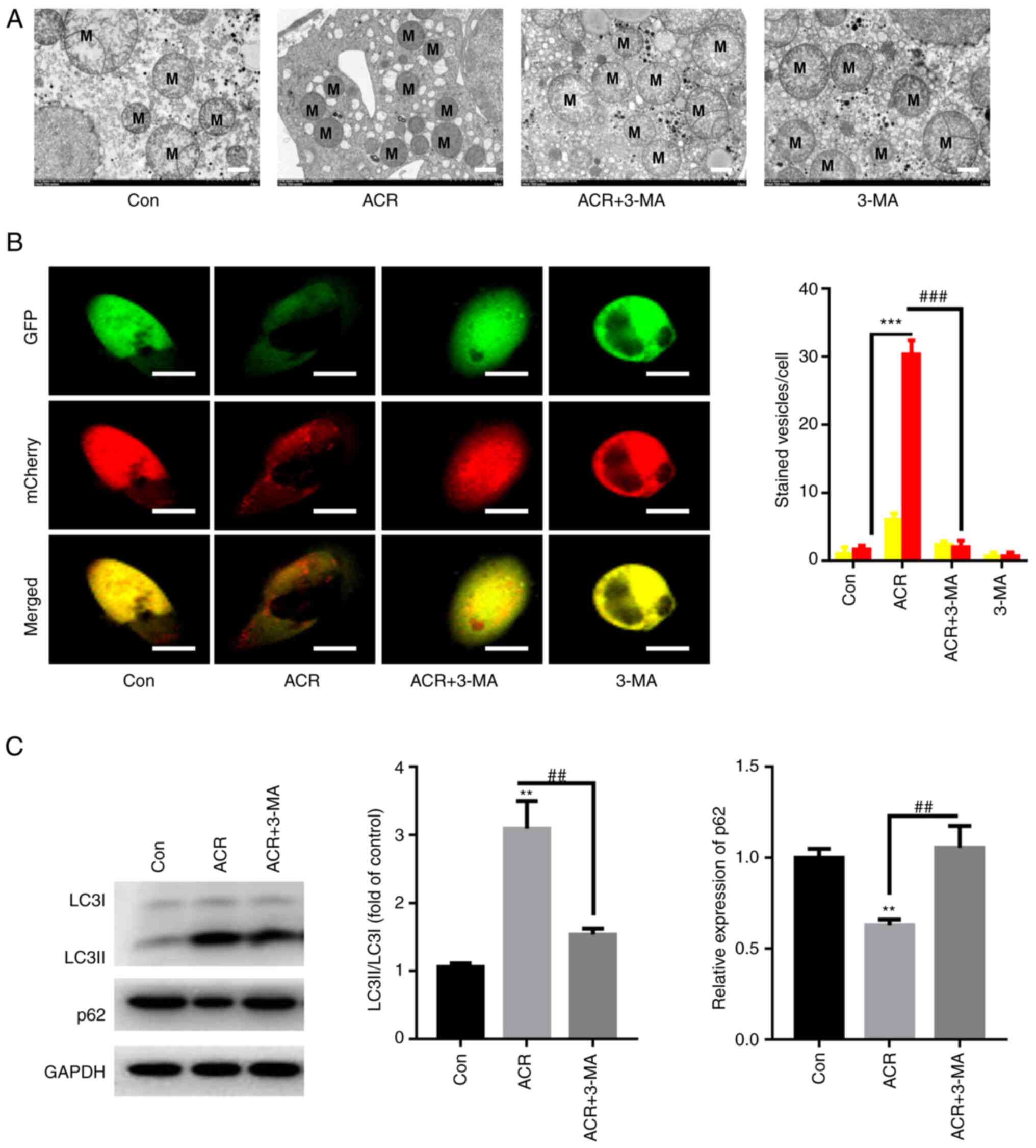 | Figure 5ACR induces autophagy in human
chondrocytes. (A) Transmission electron microscopy demonstrated
that ACR induced rupture of the mitochondrial membrane in human
chondrocytes (magnification, x400; scale bar, 2 µm). (B) ACR
increased the number of red puncta in human chondrocytes whereas
preincubation with 3-MA reversed these effects (magnification, x40;
scale bar, 10 µm). (C) Western blot analysis showed that ACR
increased LC3II/LC3I ratio whereas it decreased sequestosome 1
expression in human chondrocytes. **P<0.01 and
***P<0.001 vs. Con. ##P<0.01 and
###P<0.001 vs. ACR. ACR, acrylamide; Con, control;
p62, sequestosome 1; 3-MA, 3-methyladenine; LC, light chain; M,
mitochondria. |
ACR promotes ferroptosis in human
chondrocytes
Changes in mitochondrial function are a hallmark of
ferroptosis (20). Hence, cellular
MMP was evaluated in human chondrocytes treated with 0.35 µg/ml ACR
at 37˚C for 24 h. When human chondrocytes were treated with ACR,
poly JC-1 molecules were dissociated into mono JC-1 molecules,
suggesting a reduction or loss of MMP (Fig. 6A). In the Fer-1 group, poly JC-1
was enhanced whereas mono JC-1 was decreased in human chondrocytes
(Fig. 6A). FerroOrange staining
showed an increase in red fluorescence density after ACR treatment
whereas Fer-1 preincubation decreased the red fluorescence density
in chondrocytes (Fig. 6B). A C11
BODIPY fluorescent probe was used to detect intracellular lipid ROS
content. The data showed that ACR increased lipid-ROS accumulation
in chondrocytes whereas preincubation with Fer-1 decreased
intracellular lipid-ROS levels (Fig.
6C). Intracellular ROS and MDA levels were investigated. Fer-1
attenuated ACR-induced upregulation of ROS and MDA contents
(P<0.01; Fig. 6D). Western
blotting of ferroptosis-associated proteins demonstrated that ACR
reduced expression of GPX4, SLC7A11, TfR1 and FTH1 in chondrocytes
whereas Fer-1 abolished these effects (P<0.05; Fig. 6E). These observations indicated
that ACR induced chondrocyte ferroptosis.
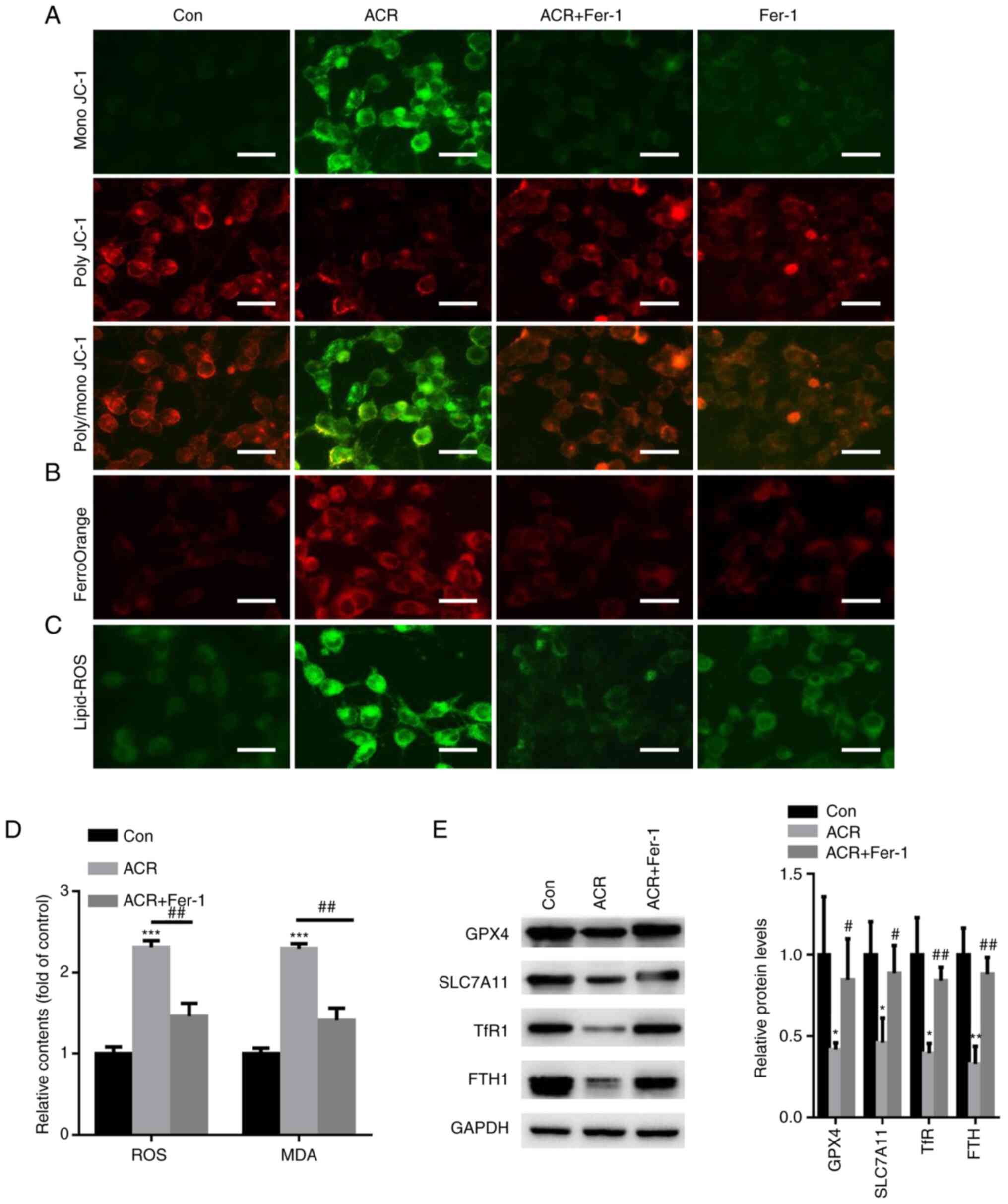 | Figure 6ACR promotes ferroptosis in human
chondrocytes. (A) Cell mitochondrial membrane potential was
detected in human chondrocytes treated with ACR (magnification,
x20; scale bar, 20 µm). (B) FerroOrange staining showed that ACR
increased intracellular Fe2+ levels in human
chondrocytes (magnification, x20; scale bar, 20 µm). (C) C11 BODIPY
fluorescent staining showed that ACR increased lipid-ROS
accumulation (magnification, x20; scale bar, 20 µm). (D) Fer-1
attenuates ACR-induced upregulation of ROS and malondialdehyde
content in human chondrocytes. (E) Western blot analysis
demonstrated that ACR decreased expression of GPX4, SLC7A11, TfR1
and FTH1 in chondrocytes. *P<0.05 vs. Con,
**P<0.01 and ***P<0.001;
#P<0.05, ##P<0.01 vs. ACR. ACR,
acrylamide; Con, control; ROS, reactive oxygen species; MDA,
malondialdehyde; GPX4, glutathione peroxidase 4; TfR1, transferrin
receptor protein 1; SLC7A11, solute carrier family 7 member 11;
FTH1, ferritin heavy chain 1; Fer-1, ferrostatin-1. |
ACR activates AMPK/ULK1 signaling in
human chondrocytes
Some studies have demonstrated an important role of
AMPK/ULK1 in autophagy and ferroptosis (21,22);
thus, the present study investigated the effects of ACR on AMPK
signaling. ACR increased the phosphorylation of AMPK and ULK1 but
suppressed activation of mTOR in human chondrocytes at 24 and 48 h
(P<0.05: Fig. 7A). To elucidate
if ACR induced autophagy and ferroptosis by activating AMPK
signaling, a specific siRNA targeting AMPK was used. Western
blotting showed that transfection with siRNA AMPK significantly
reduced expression of AMPK compared with NC (Fig. 7B). IF staining showed that siRNA
AMPK notably suppressed the fluorescence density of AMPK in human
chondrocytes even in the presence of ACR (Fig. 7C). Furthermore, ACR-induced
mitochondrial membrane rupture was reversed by silencing AMPK in
human chondrocytes (Fig. 7D).
Moreover, the upregulated levels of lipid ROS and Fe2+
were blocked by transfection with siRNA-AMPK (Fig. 7E and F). These data showed that ACR-induced
ferroptosis and autophagy were achieved by activating AMPK/ULK1
signaling in human chondrocytes.
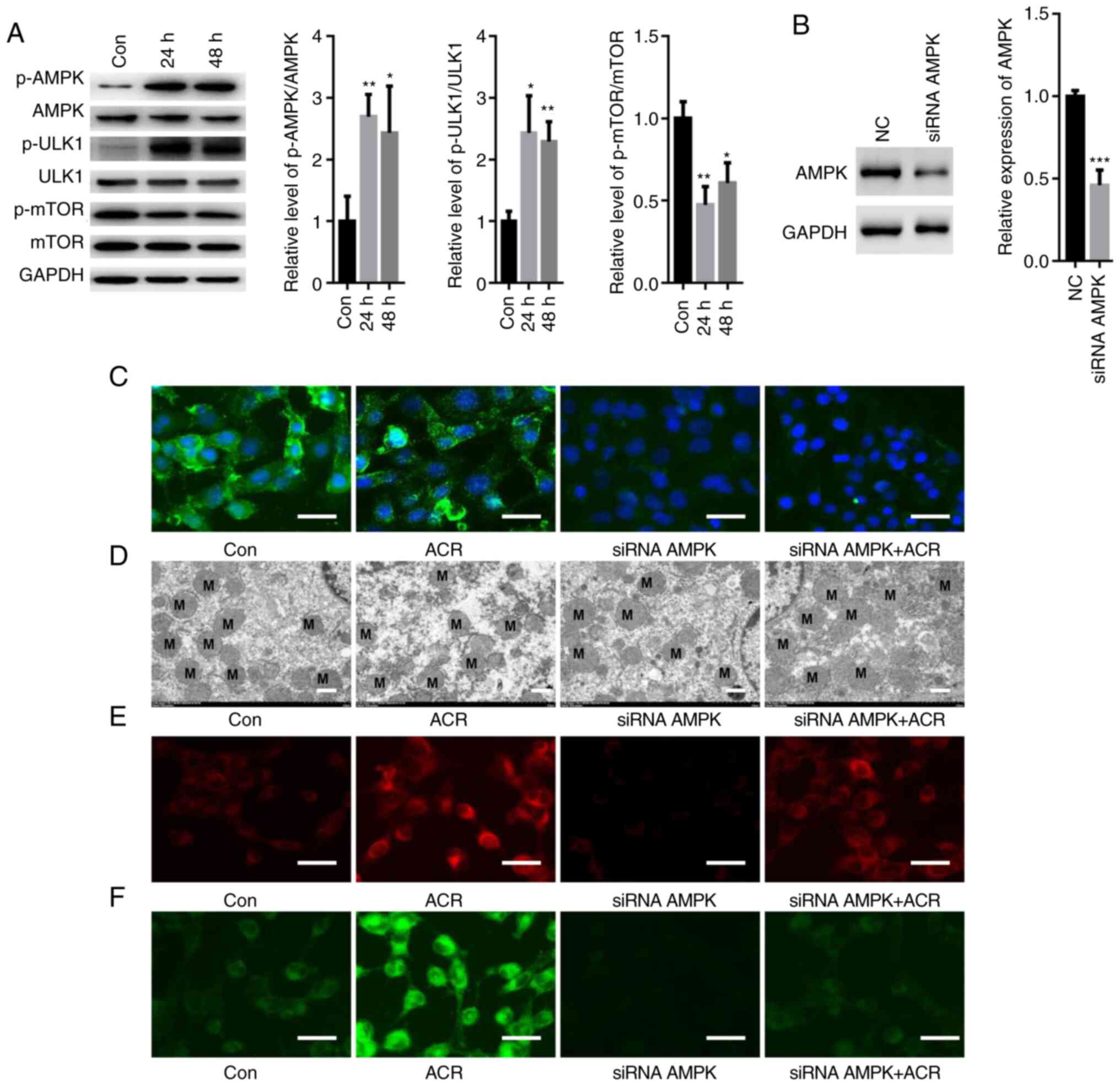 | Figure 7ACR activates AMPK/ULK1 signaling in
human chondrocytes. (A) Western blot analysis showed that ACR
increased phosphorylation of AMPK and ULK1 while suppressing the
activation of mTOR in human chondrocytes at 24 and 48 h. (B)
Western blot assay showed that transfection with siRNA-AMPK
significantly suppressed expression of AMPK. (C) Immunofluorescence
staining showed that siRNA AMPK significantly suppressed
fluorescence density of AMPK in human chondrocytes even in the
presence of ACR (magnification, x20; scale bar, 20 µm). (D)
Transmission electron microscopy showed that ACR-induced
mitochondrial membrane rupture was notably reversed by silencing
AMPK in human chondrocytes (magnification, x400; scale bar, 2 µm).
Upregulation of (E) lipid ROS and (F) Fe2+ was blocked
by transfection with siRNA-AMPK (magnification, x20; scale bar, 20
µm). *P<0.05, **P<0.01 and
***P<0.001 vs. Con. AMPK, AMP-activated protein
kinase; ACR, acrylamide; Con, control; NC, negative control; p-,
phosphorylated; si, small interfering. |
Discussion
In 2002, Swedish researchers demonstrated notable
levels of ACR in many heat-treated carbohydrate-rich foods
(23). ACR is extensively found in
deep-fried sugar-rich foods, such as potato chips, bread and
breakfast cereals (24). Exposure
to ACR has become a public issue due to its carcinogenicity and
neurotoxicity in humans (15). To
the best of our knowledge, however, whether ACR is involved in the
pathology of AS has not been explored.
To the best of our knowledge, the present study
showed for the first time that ACR suppressed human chondrocyte
proliferation in a dose-dependent manner. Moreover, ACR
significantly promoted chondrocyte senescence and elevated
expression of cell cycle arrest-associated proteins, including p53,
p21 and p16, in human chondrocytes. Similarly, DNA damage was also
enhanced following ACR treatment in chondrocytes since γ-H2AX
positive cells were increased. These observations indicated a
pathogenic role of ACR in human chondrocytes.
Previous studies have shown that autophagic cell
death, apoptosis and necrosis are involved in the development of AS
(25,26). Ferroptosis is an iron-dependent
cell death pathway that is associated with pathological conditions,
including osteoarthritis (27).
However, whether ACR induces ferroptosis in the progression of AS
remains unclear. The present study aimed to define which type of
cell death could be induced by ACR in human chondrocytes and found
that the ferroptosis-specific inhibitor Fer-1 and the autophagy
inhibitor 3-MA abolished ACR-induced cell death in chondrocytes.
Thus, it was hypothesized that ACR induced autophagic cell death
and ferroptosis in human chondrocytes.
The pathological process of AS remains unclear and
current treatments (e.g., sarilumab and tofacitinib) have had
limited success (28,29). Recently, autophagy was described as
a potential player in the pathogenesis of AS (28). Autophagy is an extremely preserved
mechanism, from yeast to mammals (30). Under nutrient depletion conditions,
autophagy recycles intracellular energy resources by degrading
cytotoxic proteins and damaged organelles (30). However, excessive autophagy leads
to aberrant cell death (30). The
present study found that the levels of LC3II were increased and p62
expression was decreased by ACR treatment in human chondrocytes.
ACR-induced cell autophagy was reversed by the autophagy inhibitor
3-MA. It was hypothesized speculate that ACR induced autophagy in
human chondrocytes.
Ferroptosis is an autophagic cell death mode because
inhibition of autophagy or knockout of autophagy-related 5 protein
decreases the cytosolic labile iron pool and intracellular
peroxidation (31). The present
study showed that ACR induced ferroptosis in human chondrocytes via
ferritinophagy. Ferritin, which contains FTH1 and ferritin light
chain, acts as a key storage molecule of excess iron to maintain
iron homeostasis (9). Autophagy
activation degrades proteins to elevate intracellular iron levels
and leads to oxidative injury via the Fenton reaction (32). Nuclear receptor coactivator 4
(NCOA4) is a selective cargo receptor for turnover of autophagic
proteins in lysosomes (33).
NCOA4-mediated degradation of FTH1 triggers ferroptosis (33). The present study found that
expression of FTH1 was significantly suppressed by ACR in
chondrocytes, indicating that iron equilibrium was disrupted by
ACR, resulting in ferroptosis. In addition, ACR also decreased the
protein expression of SLC7A11, a key component of the Xc-system
that provides adequate concentrations of cysteine for the synthesis
of glutathione, resulting in increased lipid peroxidation (34). These findings suggested induction
of ferroptosis via ACR in human chondrocytes.
AMPK phosphorylation is known to phosphorylate ULK1
and inhibit mTOR phosphorylation, thereby activating autophagy
(35). The present data showed
that ACR significantly elevated the phosphorylation levels of AMPK
and ULK1 in human chondrocytes. Notably, the effect of ACR was
diminished by siRNA AMPK treatment, as evidenced by the decreased
lipid ROS accumulation, Fe2+ levels and cell autophagy.
Hence, it was hypothesized that ACR activated autophagy-dependent
ferroptosis by activating AMPK signaling.
However, there are limitations in the present study.
First, ACR groups with different treatment doses were not
considered. Secondly, a positive control group was not implemented.
In the future, a positive group, such as interleukin-1β, should be
included.
The present findings indicated that ACR inhibited
cell proliferation and contributed to cell death by inducing
autophagy-dependent ferroptosis and that ACR promoted autophagy by
activating AMPK/ULK1/mTOR signaling in human chondrocytes. It was
hypothesized that the presence of ACR in foodstuffs may increase
the risk of AS and that decreasing ACR in food products is of
importance.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by the Natural Science
Foundation of Shandong Province (grant no. 83562185).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
HW performed the experiments. ZT, SL and KX
performed the western blot experiments. HW and HZ designed the
experiments and analyzed the data. All authors have read and
approved the final manuscript. HW and HZ confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
Ethics approval for commercially purchased primary
human chondrocytes was obtained from the Ethics Review Committee of
Zaozhuang Municipal Hospital (approval no. ZMH-2021aj32).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Qian H, Chen R, Wang B, Yuan X, Chen S,
Liu Y and Shi G: Associations of platelet count with inflammation
and response to anti-TNF-α therapy in patients with ankylosing
spondylitis. Front Pharmacol. 11(559593)2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Song GG and Lee YH: Red cell distribution
width, platelet-to-lymphocyte ratio, and mean platelet volume in
ankylosing spondylitis and their correlations with inflammation: A
meta-analysis. Mod Rheumatol. 30:894–899. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Steiner M, Del Mar Esteban-Ortega M,
Thuissard-Vasallo I, García-Lozano I, Moriche-Carretero M,
García-González AJ, Pérez-Blázquez E, Sambricio J, García-Aparicio
Á, Casco-Silva BF, et al: Measuring choroid thickness as a marker
of systemic inflammation in patients with ankylosing spondylitis. J
Clin Rheumatol. 27:e307–e311. 2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Chen W, Wang F, Wang J, Chen F and Chen T:
The molecular mechanism of long non-coding RNA MALAT1-mediated
regulation of chondrocyte pyroptosis in ankylosing spondylitis. Mol
Cells. 45:365–375. 2022.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Yu T, Zhang J, Zhu W, Wang X, Bai Y, Feng
B, Zhuang Q, Han C, Wang S, Hu Q, et al: Chondrogenesis mediates
progression of ankylosing spondylitis through heterotopic
ossification. Bone Res. 9(19)2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Bleil J, Maier R, Hempfing A, Schlichting
U, Appel H, Sieper J and Syrbe U: Histomorphologic and
histomorphometric characteristics of zygapophyseal joint remodeling
in ankylosing spondylitis. Arthritis Rheumatol. 66:1745–1754.
2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kang R and Tang D: Autophagy and
ferroptosis-what's the connection? Curr Pathobiol Rep. 5:153–159.
2017.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Park E and Chung SW: ROS-mediated
autophagy increases intracellular iron levels and ferroptosis by
ferritin and transferrin receptor regulation. Cell Death Dis.
10(822)2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh
HJ III, Kang R and Tang D: Autophagy promotes ferroptosis by
degradation of ferritin. Autophagy. 12:1425–1428. 2016.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Li J, Liu J, Xu Y, Wu R, Chen X, Song X,
Zeh H, Kang R, Klionsky DJ, Wang X and Tang D: Tumor heterogeneity
in autophagy-dependent ferroptosis. Autophagy. 17:3361–3374.
2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zhou Q, Fu X, Wang X, Wu Q, Lu Y, Shi J,
Klaunig JE and Zhou S: Autophagy plays a protective role in
Mn-induced toxicity in PC12 cells. Toxicology. 394:45–53.
2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Garg AD, Dudek AM, Ferreira GB, Verfaillie
T, Vandenabeele P, Krysko DV, Mathieu C and Agostinis P:
ROS-induced autophagy in cancer cells assists in evasion from
determinants of immunogenic cell death. Autophagy. 9:1292–1307.
2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Rong T, Jia N, Wu B, Sang D and Liu B: New
insights into the regulatory role of ferroptosis in ankylosing
spondylitis via consensus clustering of ferroptosis-related genes
and weighted gene co-expression network analysis. Genes (Basel).
13(1373)2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Mukhopadhyay S, Biancur DE, Parker SJ,
Yamamoto K, Banh RS, Paulo JA, Mancias JD and Kimmelman AC:
Autophagy is required for proper cysteine homeostasis in pancreatic
cancer through regulation of SLC7A11. Proc Natl Acad Sci USA.
118(e2021475118)2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Koszucka A, Nowak A, Nowak I and Motyl I:
Acrylamide in human diet, its metabolism, toxicity, inactivation
and the associated European Union legal regulations in food
industry. Crit Rev Food Sci Nutr. 60:1677–1692. 2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Rifai L and Saleh FA: A review on
acrylamide in food: Occurrence, toxicity, and mitigation
strategies. Int J Toxicol. 39:93–102. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Michalak J, Czarnowska-Kujawska M and
Gujska E: Acrylamide and thermal-processing indexes in
market-purchased food. Int J Environ Res Public Health.
16(4724)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Welkos S and O'Brien A: Determination of
median lethal and infectious doses in animal model systems. Methods
Enzymol. 235:29–39. 1994.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zeng C, Pan F, Jones LA, Lim MM, Griffin
EA, Sheline YI, Mintun MA, Holtzman DM and Mach RH: Evaluation of
5-ethynyl-2'-deoxyuridine staining as a sensitive and reliable
method for studying cell proliferation in the adult nervous system.
Brain Res. 1319:21–32. 2010.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Miao Y, Chen Y, Xue F, Liu K, Zhu B, Gao
J, Yin J, Zhang C and Li G: Contribution of ferroptosis and GPX4's
dual functions to osteoarthritis progression. EBioMedicine.
76(103847)2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
22
|
Lee H, Zandkarimi F, Zhang Y, Meena JK,
Kim J, Zhuang L, Tyagi S, Ma L, Westbrook TF, Steinberg GR, et al:
Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat
Cell Biol. 22:225–234. 2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Tareke E, Rydberg P, Karlsson P, Eriksson
S and Törnqvist M: Analysis of acrylamide, a carcinogen formed in
heated foodstuffs. J Agric Food Chem. 50:4998–5006. 2002.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Dodić J, Pejin D, Dodić S, Popov S,
Mastilović J, Popov-Raljić J and Zivanovic S: Effects of
hydrophilic hydrocolloids on dough and bread performance of samples
made from frozen doughs. J Food Sci. 72:S235–S241. 2007.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Ni WJ and Leng XM: Down-regulated miR-495
can target programmed cell death 10 in ankylosing spondylitis. Mol
Med. 26(50)2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Ma C, Wen B, Zhang Q, Shao PP, Gu W, Qu K,
Shi Y and Wang B: Emodin induces apoptosis and autophagy of
fibroblasts obtained from patient with ankylosing spondylitis. Drug
Des Devel Ther. 13:601–609. 2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Yao X, Sun K, Yu S, Luo J, Guo J, Lin J,
Wang G, Guo Z, Ye Y and Guo F: Chondrocyte ferroptosis contribute
to the progression of osteoarthritis. J Orthop Translat. 27:33–43.
2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Tan M, Zhang QB, Liu TH, Yang YY, Zheng
JX, Zhou WJ, Xiong Q and Qing YF: Autophagy dysfunction may be
involved in the pathogenesis of ankylosing spondylitis. Exp Ther
Med. 20:3578–3586. 2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Garcia-Montoya L, Gul H and Emery P:
Recent advances in ankylosing spondylitis: Understanding the
disease and management. F1000Res. 7:F1000 Faculty Rev-1512.
2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wang Y, Luo J, Wang X, Yang B and Cui L:
MicroRNA-199a-5p induced autophagy and inhibits the pathogenesis of
ankylosing spondylitis by modulating the mTOR signaling via
directly targeting ras homolog enriched in brain (Rheb). Cell
Physiol Biochem. 42:2481–2491. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Qin X, Zhang J, Wang B, Xu G, Yang X, Zou
Z and Yu C: Ferritinophagy is involved in the zinc oxide
nanoparticles-induced ferroptosis of vascular endothelial cells.
Autophagy. 17:4266–4285. 2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Liu J, Kuang F, Kroemer G, Klionsky DJ,
Kang R and Tang D: Autophagy-dependent ferroptosis: Machinery and
regulation. Cell Chem Biol. 27:420–435. 2020.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Zhang Y, Kong Y, Ma Y, Ni S, Wikerholmen
T, Xi K, Zhao F, Zhao Z, Wang J, Huang B, et al: Loss of COPZ1
induces NCOA4 mediated autophagy and ferroptosis in glioblastoma
cell lines. Oncogene. 40:1425–1439. 2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Koppula P, Zhuang L and Gan B: Cystine
transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient
dependency, and cancer therapy. Protein Cell. 12:599–620.
2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Tang X, Ding H, Liang M, Chen X, Yan Y,
Wan N, Chen Q, Zhang J and Cao J: Curcumin induces ferroptosis in
non-small-cell lung cancer via activating autophagy. Thorac Cancer.
12:1219–1230. 2021.PubMed/NCBI View Article : Google Scholar
|