1. Introduction
Interleukin (IL)-36 is a group of cytokines that
contains three receptor agonists IL-36α, IL-36β IL-36γ and an
antagonist IL-36 receptor a (IL-36Ra). The IL-36 genome, located at
human genome 2Q14, was first discovered by genome screening around
the year 2000 (1,2). It is thought to be a homologue of
IL-1 cytokines because of its similarity to IL-1 in conserved
sequence and predicted structure (3). Thus, the IL-36 cytokine family was
originally named as IL-1F6, IL-1F8, IL-1F9 and IL-1F5, before being
formally unified in 2011 (4,5).
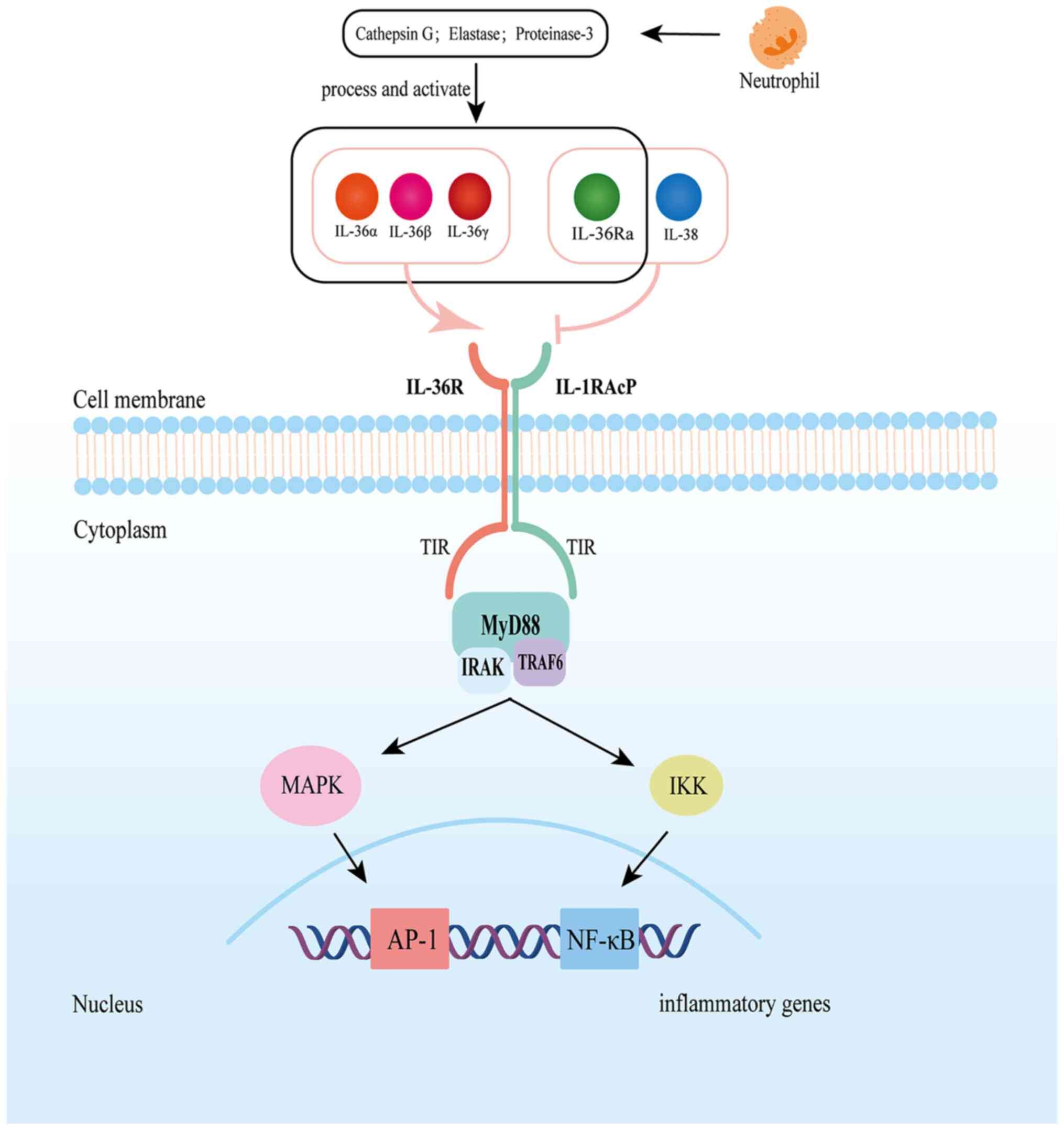
IL-36α, IL-36β and IL-36γ are agonistic ligands of IL-36R, while
IL-36Ra is an inhibitory ligand. All of them require protease
shearing to be active (6,7). Upon binding to the IL-36 functional
receptor complex, which consists of the IL-36R and the IL-1
receptor accessory protein (IL-1RAcP), IL-36 agonists activate the
mitogen-activated protein kinase (MAPK) and nuclear factor-κB
(NF-κB) signaling pathways and then regulate the downstream immune
and inflammatory responses (8-10).
IL-36Ra, as a receptor antagonist, hinders dimerization of the
IL-36R/IL-1RAcP complex by inhibiting IL-1RAcP recruitment, hence
suppressing downstream inflammatory signaling (11). IL-36 ligands and IL-36R have been
shown to be broadly expressed in human tissues, especially in
mucosal barrier regions such as the skin, lung and intestine
(8). Its role in the etiology of
skin inflammation, in particular, has been successfully developed
as a therapeutic target and has been approved by the Food and Drug
Administration (FDA) for the treatment of generalized pustular
psoriasis after phase III clinical trials (12).
The role of IL-36 in the gut is complicated. Several
previous studies in inflammatory diseases have shown a key role for
IL-36 in regulating inflammation, particularly in chronic
inflammation as a pro-inflammatory factor that exacerbates
inflammation (13,14). In chronic intestinal inflammation,
IL-36 is not only involved in intestinal inflammation but also
promotes the development of intestinal fibrosis (15). However, it also has a protective
role and may exert a protective function in acute intestinal
inflammation by promoting healing after mucosal injury (16). Cancer is closely associated with
inflammation and the mechanism of action of IL-36 has been
identified in many types of cancer (17,18).
In colorectal cancer (CRC), IL-36α and IL-36γ have been more
extensively studied, and their expression levels appear to be used
as biomarkers for predicting colorectal cancer prognosis (19). Available evidence suggests that
IL-36α has antitumor effects, whereas IL-36γ has a more complex
role, both in promoting antitumor immune responses, such as
modulating tumor-infiltrating immune cells, and inducing genes
involved in the IL-17/IL-23 axis to promote colorectal cancer cell
proliferation (20,21). The present review briefly
summarizes the regulation of IL-36 expression and function,
discusses the role of IL-36 in intestinal inflammation and
colorectal cancer and the latest research progress in the
development of targeted IL-36 signaling therapy.
2. Expression and function of IL-36
IL-36 is a novel cytokine subfamily of the IL-1
superfamily. Based on the gene sequences and binding receptors,
this family also contains IL-1, IL-18 and IL-33 and has been shown
to have a significant role in the regulation of immunity and
inflammation (22). Because of its
near proximity and similar conserved sequence to the IL-1 genome,
IL-36 has been extensively researched in numerous immunological
disorders and inflammatory diseases (1,23-26).
IL-36 has been found in a variety of human tissues, such as the
skin, lung, intestine and synovium, and can be expressed by a
variety of cells, including epithelial cells such as keratinocyte,
bronchial and intestinal epithelial cells, as well as immune cells
such as neutrophils, monocyte macrophages and T cells (27,28).
Under physiological conditions, the production of IL-36 in human
tissues is low; however, under inflammatory conditions, the
expression of IL-36 is greatly elevated after proinflammatory
cytokine stimulation or signal activation by Toll-like receptors,
particularly in the epithelial cells of the barrier tissues of the
skin, respiratory tract and digestive tract, where they are in
close contact with external pathogens (29-31).
It is therefore hypothesized that IL-36 is specialized in the
regulation of mucosal innate and adaptive immunity. When IL-36
signaling is activated in cells, it can activate a variety of
immune cells, epithelial cells, fibroblasts and keratinocytes in an
autocrine or paracrine manner and induce the expression of various
cytokines and chemokines, thus promoting the restoration of mucosal
homeostasis and epithelial repair, as well as inducing the
development of inflammatory responses and even promoting the
progression of fibrosis and malignant tumors (21,22,27,32).
In addition, as an inflammatory regulator, IL-36 also participates
in the host immune response to bacterial and viral infections
(33-36).
IL-36 cytokines must be processed with
neutrophil-derived proteases before they are activated. After
truncating the N-terminal with different enzymes, the affinity of
IL-36 ligand to IL-36R can be magnified by several hundred times
(6,10). Cathepsin G and elastase can
activate IL-36α, whereas IL-36β and IL-36γ are activated by
cathepsin G and elastase respectively (6). Similarly, in order to have an active
antagonistic form, IL-36Ra must be cleaved by elastase (7). IL-36R is a 575-amino-acid
multi-domain transmembrane receptor with high sequence homology
with IL-1R that is comprised of three immunoglobulin-like
extracellular domains and an intracellular Toll-IL-1-receptor
domain (36). The helper protein,
IL-1RAcP, is structurally similar to IL-36R and is a critical
co-receptor shared by family members of IL-1 that is significantly
engaged in cell signaling transmission (36,37).
IL-36Ra is a natural IL-36R antagonist encoded by the IL-36RN gene.
The fact that IL-36Ra binds to IL-36R with greater affinity and
slower off-rate compared with the IL-36 agonist reveals that
activation of the IL-36 signaling pathway has a strong
self-negative regulation (38).
Thus, IL-36 activation will be out of control and worsen the
inflammatory response when IL-36Ra is mutated or deleted, as has
been shown in patients with GPP (39). This is a severe and potentially
life-threatening pustular skin condition caused by congenital
deletion of the IL-36RN gene (40). Furthermore, IL-38 has also been
revealed to be an antagonist of IL-36, exerting comparable
biological activity to IL-36Ra by binding to IL-36R (41). This may be associated with its
chromosomal location and gene sequence structure that is similar to
that of IL-36Ra. This is why numerous studies often classify IL-38
as part of the IL-36 family for discussion (23,42-44).
IL-36 activators bind to the receptor complex formed
by IL-36R and IL-1RAcP and then recruit the intracellular signaling
molecules myeloid differentiation factor 88 (Myd88), IL-1R-related
kinase and tumor necrosis factor receptor-related factor 6 to
activate the NF-κB and MAPK signaling pathways (10). This, in turn, promotes the nuclear
translocation and activation of activator protein 1 and NF-κB,
thereby regulating the transcription and expression of downstream
pro-inflammatory genes (8-10,45).
MyD88 is essential for IL-36 signaling. A gene expression
sequencing analysis of primary keratinocytes has revealed that
silencing MyD88 via CRISPS/Cas9 inhibits IL-36 responsiveness in
epithelial cells (Fig. 1)
(46).
3. Role of IL-36 in chronic intestinal
inflammation
Inflammatory bowel disease (IBD) is a chronic
non-specific inflammatory disorder of the gastrointestinal tract
that is frequently divided into Crohn's disease (CD) and Ulcerative
colitis (UC) based on disease features. The pathophysiology of IBD
is not yet fully determined, and the available evidence attributes
its etiology to a variety of factors, including genetic,
environmental, intestinal flora and immune (35). Immune attributes have been
thoroughly researched as therapeutic targets for IBD and have been
successfully applied in clinical therapy with remarkable efficacy
(36). IL-36, an emerging
inflammatory factor, is significantly dysregulated in patients with
IBD and mice with colitis and is engaged in intestinal homeostasis
and inflammation (43).
In the inflamed colon of patients with active IBD,
mRNA expression of IL-36 cytokines is elevated compared with
non-inflamed and normal mucosal tissues, especially in the colonic
mucosa of patients with active UC, where IL-36α and IL-36γ
expression is significantly upregulated and correlated with the
degree of inflammation (14,47).
The expression of IL-36β has been observed to be higher in the
plasma membrane, muscle and submucosa of active CD (48). Since IL-36α and IL-36γ have a more
comparable gene sequence compared with IL-36β, they are also more
functionally similar and hence more consistent in intestinal
inflammation (3,14). Mucosal tissue biopsies from
patients with IBD have demonstrated that both non-immune and immune
cells, such as intestinal epithelial cells, macrophages, monocytes
and T cells, can produce IL-36α, IL-36β, IL-36γ and IL-36Ra
(48). According to Scheibe et
al, IL-36α is mostly expressed in CD14 inflammatory
macrophages, whereas intestinal epithelial cells primarily express
IL-36γ (49). Overall, IL-36 is
broadly expressed in inflammatory tissues of the intestine, and its
degree of expression corresponds with tissue inflammation.
The complex role of IL-36 in IBD has been further
demonstrated by several studies in experimental models of colitis
(15,47,50).
It has been revealed that dextran sodium sulfate (DSS)-induced
colitis is ameliorated in IL-36R-/- mice (IL-36R knockout),
accompanied by a decreased infiltration of innate inflammatory
cells in the colonic lamina propria (50). This has also been observed in the
Citrobacter rodentium-infected IL36R-/- mouse model. Unlike
the DSS model, which often induces only an innate immune response,
the Citrobacter rodentium-infected colitis model also
induces adaptive immunity, resulting in an altered T helper cell
response, manifested by an enhanced Th17 response and a diminished
Th1 response (50). These findings
suggest that IL-36R signaling can modulate both innate and T-cell
immunity in the intestinal mucosa. Thus, in an oxazolone-induced T
cell-dependent colitis model, IL-36 or IL-36R absence reduces
colonic inflammation (51). This
is associated with IL-36γ inhibiting regulatory T cell (Treg)
differentiation through MyD88 and NFκBp50-dependent signaling and
promoting IL-9-producing pathogenic CD4 helper T cell (TH9)
polarization via an IL-2-signal transducer and activator of
transcription 5 (STAT5) and IL-4-STAT6-dependent manner. Therefore,
mice blocking IL-36γ/IL-36R signaling are protected from effector T
cell-driven intestinal inflammation and colonic immune cells
exhibit elevated Treg cells and decreased Th9 cells (51). Kanda et al have also
demonstrated that IL-36α and IL-36γ promotes intestinal
inflammation through the induction of pro-inflammatory mediators,
such as IL-6 and chemokine C-X-C ligand (CXCL)1, CXCL2 and CXCL8,
by colonic subepithelial myofibroblasts (52). This process can be synergized by
the pro-inflammatory cytokines IL-17A and TNF-α (52). Another report also revealed the
pathogenic role of IL-36β. The research revealed an exacerbation of
colitis compared with the control group by intraperitoneal
injection of IL-36β in a DSS mouse model (53). It was hypothesized that this result
is associated with enhanced Th2 response and inhibition of Treg
response by IL-36β (53). In
conclusion, all three IL-36 agonists demonstrate pro-inflammatory
effects in the experimental model. Hence, in both DSS and
2,4,6-trinitro benzene sulfonic acid-induced colitis mice models,
inhibiting IL-36R activation through gene deletion or neutralizing
antibodies decreases colonic mucosal inflammation and
histopathology, the degree of colonic mucosal inflammation and
histopathology (15). Moreover,
IL-38, another natural antagonist of IL-36, is highly expressed in
IBD-inflamed tissues, and blocking IL-36 signaling with IL-38
improves DSS-induced colitis and inhibits the expression of
macrophage-associated pro-inflammatory molecules, such as IL-1β and
TNF-α (54). All of these findings
indicate that IL-36 has a pro-inflammatory pathogenic property in
chronic intestinal inflammation.
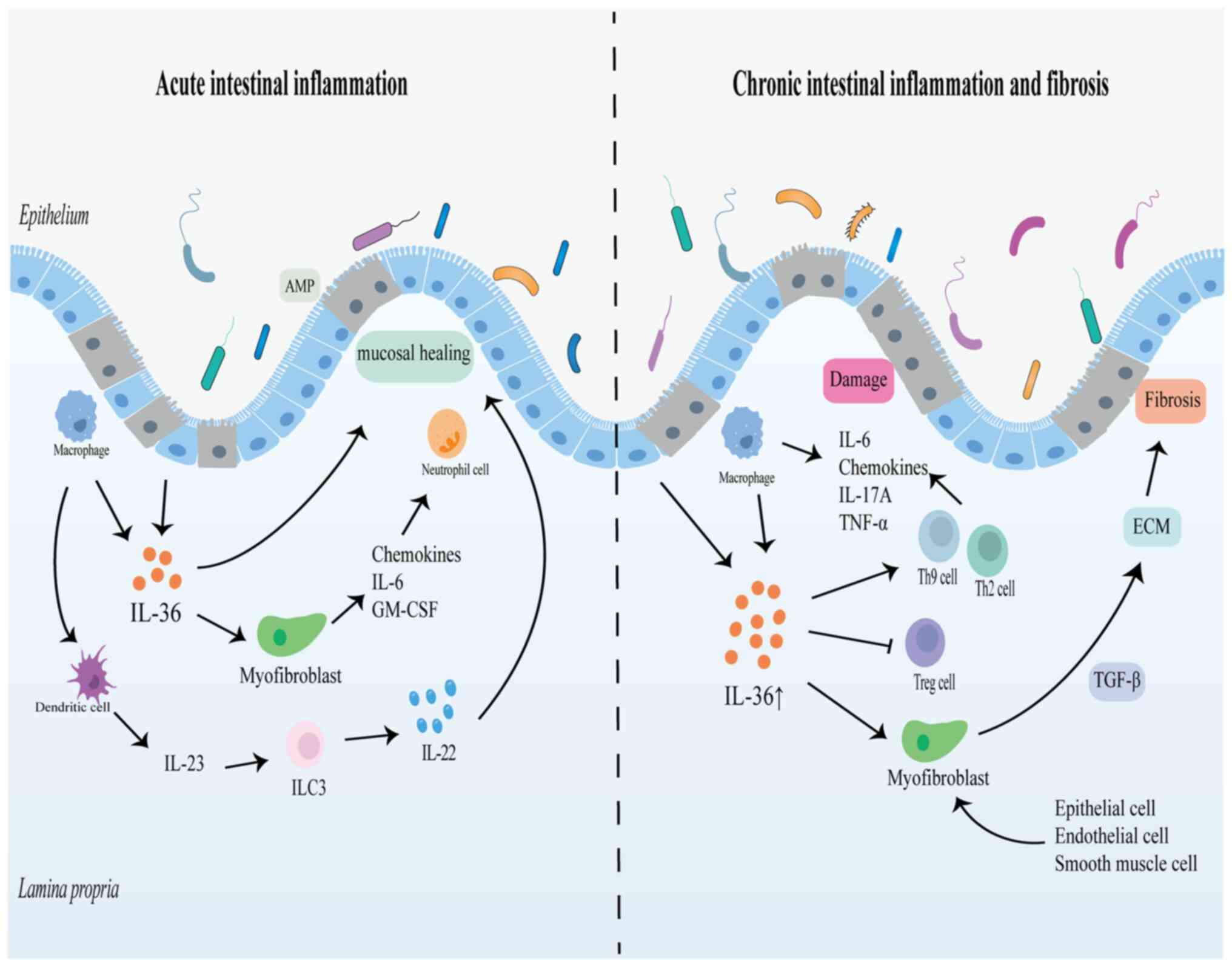
However, the detrimental role of IL-36 in the gut is
somewhat controversial. Numerous studies have discovered that IL-36
may act as a protective factor in intestinal mucosal repair,
particularly in acute inflammation, which appears to be associated
with the role of IL-22 in stimulating intestinal epithelial cell
proliferation, promoting mucosal healing and maintaining intestinal
epithelial barrier integrity (16,49,55).
IL-22 is currently found to be mainly derived from group 3 innate
lymphocytes, which are key sentinels of the intestinal innate
immune response, after sensing cytokine signals such as IL-1 and
IL-23 from mononuclear phagocytes activate rapidly and secrete
IL-22 effector molecules to participate in mucosal immunity
(56). In the acute DSS-induced
colitis model, mice with IL-36R deficiency are unable to recover
from acute colitis and have increased disease activity and
decreased survival, accompanied by a significant decrease in IL-22
(16,49,57).
Meanwhile, the impairment of intestinal mucosal repair capacity
caused by IL-36R deficiency is restored by exogenous administration
of IL-23(16). In addition, IL-36γ
can stimulate the increased expression of IL-22 and IL-23 in an
in vitro experiment with colonic explants, suggesting a role
for the cytokine network between IL-36 and IL-23/IL-22 in
intestinal mucosal barrier repair and innate immune response
(16).
In addition, IL-36 potentially affects intestinal
mucosal healing by influencing the proliferation and activation of
intestinal epithelial cells and fibroblasts. Analysis of intestinal
epithelial cell proliferation marker Ki-67 and antimicrobial
protein lipocalin-2 (LCN2) expression by immunofluorescence and
quantitative PCR after injection of IL-36R ligand into the
peritoneal cavity of mice has revealed that Ki-67 and LCN2
expression levels are reduced in IL-36R-/- knockout mice compared
with wild-type mice (49). The
IL-36R agonists IL-36α and IL-36γ also promote the expression of
colonic fibroblasts and promote mucosal repair by inducing
chemokines, granulocyte-macrophage colony stimulating factor and
IL-6 to recruit leukocytes (49,58).
Although inflammatory factors adversely affect the intestinal
mucosal barrier, they also have a host protective role during the
acute inflammatory phase (59).
The release of large amounts of inflammatory mediators by cells
into the tissue microenvironment also participates in mucosal
immune defense, recruiting immune cells to remove pathogens and
necrotic material and stimulating epithelial repair responses and
the synthesis of mediators that contribute to the restoration of
mucosal homeostasis (59). The
intestinal epithelium, the interface between the luminal
microenvironmental and the mucosal immune system, is exposed to
additional challenges and undertakes more complex and demanding
tasks, the integrity of which is fundamental to the maintenance of
intestinal homeostasis (60).
Therefore, the impact of IL-36, which is widely present in a
variety of cells and tissues, in intestinal inflammation and
mucosal repair is not destined to be singular, and its main role as
pathogenic or protective needs to be seen in context (Fig. 2) (17,43).
4. Function of IL-36 in intestinal
fibrosis
The essence of fibrosis is the healing and repair
response to tissue injury. The recruitment of immune cells and the
activation of the inflammatory response are the first steps in this
process. Following the activation of immune cells such as
macrophages, eosinophils, basophils, innate lymphocytes and T
cells, a series of soluble pro-fibrotic cytokines are produced to
stimulate myofibroblast proliferation and activation and to
generate extracellular matrix (ECM) (61). Transforming growth factor-β is
considered to be the most effective fiber-activating cytokine,
which is not only derived from fibroblasts, but also expressed by
macrophages and eosinophils (62).
Additionally, peripheral non-immune cells such epithelial,
endothelial and smooth muscle cells can also transform to
myofibroblast-like phenotypes during fibrosis, generating soluble
cytokines that hasten the progression of fibrosis. Therefore, the
most notable feature of fibrosis is the excessive synthesis and
aberrant deposition of collagen-rich ECM, which is mainly derived
from mesenchymal cells including fibroblasts and myofibroblasts
(61,63). Owing to its ability to stimulate
fibroblasts and promote the secretion of pro-fibrotic soluble
mediators, IL-36 is implied to play a role in fibrotic diseases
(64). Several studies have
revealed the function of IL-36 in fibrotic diseases of the kidney,
lung, intestine and pancreas, and it has been hypothesized that
IL-36 may mediate the fibrotic process by regulating immune cells,
such as macrophages, fibrotic cytokines such as IL-17 and collagen
remodeling (65-67).
Intestinal fibrosis is a serious complication of IBD that is
frequently encountered in patients with CD of the small intestine
(68). Under the long-term
stimulation of chronic inflammatory agents, intestinal fibrosis
remodeling in patients with IBD proceeds constantly, resulting in a
thickening of the intestinal wall and structural alterations of the
intestinal canal, eventually leading to intestinal stenosis and
even serious consequences such as intestinal obstruction requiring
surgical treatment (69).
Scheibe et al revealed elevated IL-36α
expression in the tissues of patients with fibrous stenosis CD, and
macrophages were its main source (15). Subsequently, it was revealed in a
mouse model that activation of IL-36R consistently induces
fibroblast activation and type VI collagen production, while the
intestinal fibrosis process is significantly reduced in IL-36R-/-
mice or in mice with antibodies that inhibit IL-36R activity
(15). These results supported the
hypothesis that the IL-36R signaling pathway is associated with
intestinal fibrosis. Previous studies have reported that IL-36 can
be derived from a variety of cells, such as fibroblasts, epithelial
cells and macrophages, and that IL-36R-expressing epithelial cells
and fibroblasts are also regulated by IL-36 cytokines (49,52,66).
Furthermore, human colonic myofibroblasts can produce large amounts
of IL-36γ when stimulated with IL-1β or IL-1β plus TNF-α in
vitro (70). RNA-sequencing
and gene enrichment analyses of human and mice colonic fibroblasts
have indicated that IL-36R signaling upregulates the inflammatory
factor IL-6 as well as fibrosis-related genes, such as CXCL1,
CXCL5, matrix metalloproteinase (MMP)1, MMP3, MMP10 and
MMP13(15). Therefore, it is
postulated that the IL-36 in intestinal fibrosis may predominantly
be IL-36α, which boosts fibroblast activation and collagen
generation by modulating cytokines involved in fibrosis and tissue
remodeling (15). This procedure
is dependent on MyD88 signaling, which is consistent with earlier
research (15,46). In addition, the IL-36R antibody has
also showed positive effects on reversing chronic intestinal
fibrosis in established mouse models, as evidenced by a decrease in
tissue fibrosis scores, α-smooth muscle fibroblasts and type VI
collagen (15). Notably, this
research demonstrates the potential value of IL-36R inhibition in
halting and reversing the fibrosis process, as well as the
potential of IL-36 for the treatment of intestinal fibrosis and
stricture in IBD.
In summary, IL-36 cytokine regulates the intestinal
immune response and promotes intestinal inflammation and fibrosis,
therefore targeting IL-36 may be one of the strategies to treat IBD
and inhibit the progression of IBD fibrosis. However, its practical
application to human intestinal fibrosis requires further
investigation in more animal models and clinical trials.
5. Role of IL-36 in colorectal cancer
To the best of our knowledge, although evidence on
IL-36 in cancer are few, it is reasonable to infer that IL-36 has a
role in cancer response since it is an important modulator of
intestinal inflammation, and inflammation has been linked to
cancer. Several experimental investigations have demonstrated that
IL-36 has an anticancer impact in a variety of cancers, including
ovarian cancer, breast cancer, hepatocellular carcinoma and
melanoma (17,21). CRC, as the most dominant malignant
tumor of the intestine, has become the third most prevalent cancer
in humans and the second leading cause of cancer-related deaths
worldwide based on cancer statistics analysis in 2020(71). Evidence has indicated that it can
develop from IBD through the inflammatory pathway such as NF-κB,
IL-6/STAT3 and IL-23/IL-17 pathways (72,73).
Therefore, the search for sensitive early screening and diagnostic
markers remains one of the aims of CRC research. Current research
evidence of IL-36 in CRC seems to indicate its diagnostic and
therapeutic value (Table I). Wang
et al in 2014 detected a negative correlation between high
IL-36α expression in tumor tissues and corresponding
clinicopathological parameters such as tumor size and TNM stage
(74). Kaplan-Meier analysis and
Cox regression analysis have demonstrated that low IL-36α
expression levels predict poorer prognosis and decreased survival
in CRC. However, this finding needs to be further validated because
it was not compared with non-cancerous tissues (74). Following that, Chen et al
revealed that colonic IL-36α, IL-36β and IL-36γ are significantly
lower in CRC (19). Moreover, high
IL-36α expression and low IL-36γ expression are associated with
higher survival in patients with CRC through immunohistochemical
and histopathological analysis of 185 tissue arrays from patients
undergoing colorectal cancer surgery and non-cancerous tissues.
This indicates that IL-36α and IL-36γ levels in CRC tissues might
be used as biomarkers to predict CRC prognosis. This also implies
that IL-36α and IL-36γ play distinct roles in tumor growth, despite
both having pro-inflammatory effects in reports of chronic
intestinal inflammation (19).
Although there was no statistically significant difference in
survival, IL-36β has been demonstrated to be reduced by 80% in CRC
colon tissues, suggesting that IL-36β may contribute to colorectal
cancer development (19). An
experiment has shown that IL-36β can boost CD8+ T cell
activation and then enhance anti-tumor immune responses by
activating phosphatidylinositol 3 kinase (PI3K)/protein kinase B
(Akt), IκB kinase and MyD88 dependent mammalian target of rapamycin
complex1 pathways (75).
 | Table IRole of IL-36 in colorectal
cancer. |
Table I
Role of IL-36 in colorectal
cancer.
| Cytokine | Function | Mechanism | (Refs.) |
|---|
| IL-36α | Predict
prognosis | - | (19,74) |
| | Suppress tumor | Enhance the
function of CD8 T lymphocytes | (78) |
| IL-36β | Promote tumor | Regulate p42/44
MAPK and PI3K/AKT pathway | (88) |
| | Suppress tumor | Enhance CD8 T cells
proliferation and activation | (75) |
| IL-36γ | Predict
prognosis | - | (19) |
| | Promote tumor | Regulate p42/44
MAPK and PI3K/AKT pathway | (88) |
| | Promote tumor | Induce cell-matrix
adhesion molecules expression and Wnt signaling | (87) |
| | Promote tumor | Synergize with
IL-17/IL-23 axis | (32) |
| | Suppress tumor | Recruit immune cell
infiltration and promote the formation and maintenance of TLS | (81,84) |
| | Suppress tumor | Combination with
OX40L and IL-23 increases tumor sensitivity to immune checkpoint
blockade therapy | (92) |
| IL-36Ra | Suppress tumor | - | (87,88) |
Considering IL-36α drives antitumor immune responses
in hepatocellular carcinoma cells by recruiting CD3 and CD8 T
lymphocytes to activate adaptive immune cells, it hypothesized that
the anticancer impact of IL-36α in CRC may potentially be connected
with T lymphocyte activation (76). This speculation was confirmed in a
study by Wei et al (77).
This revealed that IL-36α inhibits the growth and metastasis of
colorectal cancer by constructing CT26-IL-36α and HT29-IL-36α cell
lines overexpressing IL-36α, which enhances the infiltration and
activity of CD8 T lymphocytes by upregulating the expression of the
chemokines CXCL10 and CXCL11(77).
By contrast, the role of IL-36γ in CRC is more complicated. In
vitro experiments, IL-36γ stimulates the proliferation and
secretion of IFN-γ by type 1 lymphocytes, including CD8+
T cells, natural killer cells and γδ T cells, which are significant
antitumor immune effector cells (78-80).
Wang et al then demonstrated in melanoma and breast cancer
models that IL-36γ/IL-36R signaling promotes the differentiation of
type 1 lymphocytes in the tumor microenvironment and inhibits the
tumor growth, implying that IL-36 has an important tumor
immunomodulatory role by regulating the tumor microenvironment and
tumor infiltrating cells (78).
Furthermore, IL-36γ expression has been indicated to be adversely
linked with the advancement of human melanoma and lung cancer,
supporting the anticancer potential of IL-36γ (78). In the MC38 mice model of CRC,
IL-36γ delivered by dendritic cells (DC) has previously been shown
to slow tumor development with recruitment of T cells into the
tumor microenvironment and the formation of tumor-associated
tertiary lymphoid structures (TLS) (81). T lymphocytes are the most common
lymphocytes seen in tumors and are linked to tumor immune response
and prognosis (82). The presence
of TLS has also been linked to an improved prognosis in a variety
of malignancies including CRC, breast cancer, lung cancer and head
neck squamous cell carcinoma (83). Some studies reported that IL-36γ
can be generated by M1 macrophages, vascular endothelial cells and
smooth muscle cells in the CRC tumor microenvironment and
contributes to the activation of anti-tumor response by
upregulating vascular cell adhesion molecule, intercellular
adhesion molecule-1 and the chemokines CCL2 and CCL20 to recruit
immune cell populations and promote CD4 central memory T cell
infiltration, as well as the maintenance and formation of TLS
(78,84-86).
However, several reports have proposed the opposite
view that IL-36γ promotes the development of CRC (32,87,88).
In the intestinal cancer model, injection of anti-IL-36γ
significantly inhibits the tumorigenesis and tumor development in
the colon and the small intestine along with the expression of
cell-matrix adhesion molecules and Wnt downstream genes in the
colon tumors (87). Therefore,
IL-36γ may contribute to tumorigenesis by indirectly regulating Wnt
signaling through inducing the expression of cell-matrix adhesion
molecules (87). Another study has
reported that IL-36R agonists induce pro-tumor gene expression and
promote tumor cell proliferation, migration and invasion in
vitro colon cancer cell line. Among them, IL-36β and IL-36γ
were more prominent in tumor invasion compared with IL-36α, which
were possibly mediated through the p42/44 MAPK and PI3K/AKT
pathways (75). Subsequently,
inhibition of the IL-36R signaling pathway has been shown to reduce
tumor cell proliferation and tumor burden in a CT26 mouse colon
cancer model by administration of IL-36Ra or knockdown of IL-36R
gene (88). The reduced expression
of IL-36R in tumors has also been shown to be associated with an
improved prognosis for patients (32). IL-36Ra administration more strongly
inhibits tumor growth in mice, which is consistent with previous
findings of increased incidence of colon tumorigenesis in the
absence of IL-36Ra, both supporting IL-36Ra as a potentially potent
target for the treatment of CRC and the anticancer potential of
inhibiting the IL-36R (87,88).
IL-36 also synergizes with inflammatory factors to promote tumors.
Differential gene expression analysis of patient samples and cell
lines has revealed the role of the IL-36/IL-17/IL-23 axis in colon
tumorigenesis, showing that IL-36γ synergistically induces various
genes involved in the IL-17/IL-23 axis in CRC cells, thereby
inducing cancer cell proliferation (32).
The discovery of immune checkpoints such as
programmed cell death (PD)-1/PD ligand 1 and cytotoxic T-lymphocyte
antigen-4 has revolutionized the course of tumor immunotherapy
(89,90). However, the responsiveness to
immune checkpoint inhibitors varies by cancer type and by mutation
type in the same cancer. Therefore, new research approaches are
exploring how to improve the therapeutic sensitivity of immune
checkpoint inhibitors, such as combination with co-stimulatory
signals and pro-inflammatory factors to modulate the recruitment
and activation of tumor-infiltrating lymphocytes, thus enhancing
the anti-tumor immune response (91). As IL-36γ is involved in reducing
the expression of immune checkpoint molecules and coordinately
activating DCs and T cells in the TME in support of strong
anti-tumor CD8+ T cell responses, it has been explored
as a direction to modulate antitumor immunity and immune checkpoint
inhibitor therapy (78).
Treatments with IL-23/IL-36γ/OX40L triplet mRNA mixture facilitates
infiltration of immune cells, including cross-presenting DCs and
cytotoxic CD8+ T cells, into tumor tissue and
effectively destroys tumor cells in HT22 hepatoma, MC38 colon
carcinoma and B16 melanoma (92).
Notably, the combination therapy of IL-36γ with OX40L and IL-23
significantly increases tumor susceptibility to immune checkpoint
blockade therapy in a murine colon cancer model (92). Collectively, these findings reveal
the complexity of the IL-36 family function in colorectal cancer,
but also provide lateral evidence of the potential value of the
IL-36 family in the treatment of CRC.
6. Clinical trials of IL-36 antibody in
IBD
Targeting the IL-36 signaling pathway in
inflammatory diseases is currently dominated by targeting IL-36R.
Several clinical trials are currently investigating the
neutralization of antibodies targeting IL-36R in GPP, palmoplantar
pustulosis (PPP), atopic dermatitis (AD) and IBD to evaluate the
safety and efficacy of clinical treatment. Spesolimab (BI 65513), a
novel humanized IL-36R monoclonal immunoglobulin G1 antibody
developed by Boehringer Ingelheim, has demonstrated a good safety
profile and significant efficacy in phase I and II clinical trials
for the treatment of GPP and was approved by the FDA as a treatment
option for GPP flares in adults on September of this year (12,93).
In the three clinical trials of patients with moderate to severe
active UC, although the efficacy endpoints were not reached, the
spesolimab shown to be generally well tolerated in patients with UC
with no unexpected safety concerns or clinically relevant
hypersensitivity or opportunistic infections (NCT03482635;
NCT03123120; NCT03100864) (94).
Another trial on patients who have moderate to severely active UC
and have completed a previous treatment trial is evaluating the
long-term safety of spesolimab (NCT03648541). Moreover, IL-36R
targeted antibody are also undergoing phase II clinical trial in
patients with CD specifically targeting structuring and fistulizing
CD. The causes of fistula development in CD and the effect of
spesolimab on the treatment of patients with fistulizing CD have
been studied in a phase II trial (NCT03752970), while another study
on whether spesolimab administration improves intestinal stricture
caused by CD was terminated in 2022 (NCT05013385). In addition, the
long-term safety and efficacy of spesolimab is currently under
evaluation in patients with perianal fistulizing CD who had
completed previous treatments (NCT04362254). In conclusion, the
safety of IL-36 in patients with IBD has been demonstrated, but
clinical efficacy remains to be determined by additional clinical
trials and data.
7. Conclusion
Inflammatory bowel disease and colorectal cancer,
two of the most predominant heterogeneous diseases of the
intestine, are now becoming increasingly common worldwide. The
search for markers and targets for the diagnosis and treatment of
both diseases still remains an important research direction. The
IL-36 cytokine, an important member of the IL-1 superfamily, has
been identified to have role in regulating innate and adaptive
immunity in a variety of tissues, including the intestine, lung,
joints and kidney, and from experimental models and a growing body
of evidence from clinical samples supports the function of the
IL-36 signaling pathway in promoting inflammation and fibrosis in
the context of chronic intestinal inflammation. Some of the agents
that target IL-36 signaling have been or are being evaluated in
phase II clinical trials in patients with CD and UC and have shown
a favorable safety profile. Although investigations of IL-36 in
cancer are still scarce, the existing data suggests that IL-36 has
an anticancer effect in the tumor microenvironment by regulating
tumor-infiltrating cells and tertiary lymphoid bodies, although
this conclusion is being challenged by numerous studies. In
conclusion, all of these findings revealed that the IL-36 signaling
pathway has therapeutic potential in IBD and CRC and more
exploration is required in the future to investigate more precise
mechanisms of IL-36 function.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
Not applicable.
Authors' contributions
JZ and ML conceived and designed the article. ML,
WJ, ZW, YL and JZ were involved in drafting of the manuscript and
in revising it critically for important intellectual content. All
authors have read and approved the final manuscript. Data
authentication is not applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Taylor SL, Renshaw BR, Garka KE, Smith DE
and Sims JE: Genomic organization of the interleukin-1 locus.
Genomics. 79:726–733. 2002.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Smith DE, Renshaw BR, Ketchem RR, Kubin M,
Garka KE and Sims JE: Four new members expand the interleukin-1
superfamily. J Biol Chem. 275:1169–1175. 2000.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Dunn E, Sims JE, Nicklin MJ and O'Neill
LA: Annotating genes with potential roles in the immune system: Six
new members of the IL-1 family. Trends Immunol. 22:533–536.
2001.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Dinarello C, Arend W, Sims J, Smith D,
Blumberg H, O'Neill L, Goldbach-Mansky R, Pizarro T, Hoffman H,
Bufler P, et al: IL-1 family nomenclature. Nat Immunol.
11(973)2010.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Sims JE, Nicklin MJ, Bazan JF, Barton JL,
Busfield SJ, Ford JE, Kastelein RA, Kumar S, Lin H, Mulero JJ, et
al: A new nomenclature for IL-1-family genes. Trends Immunol.
22:536–537. 2001.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Henry CM, Sullivan GP, Clancy DM, Afonina
IS, Kulms D and Martin SJ: Neutrophil-derived proteases escalate
inflammation through activation of IL-36 family cytokines. Cell
Rep. 14:708–722. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Macleod T, Doble R, McGonagle D, Wasson
CW, Alase A, Stacey M and Wittmann M: Neutrophil elastase-mediated
proteolysis activates the anti-inflammatory cytokine IL-36 receptor
antagonist. Sci Rep. 6(24880)2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Bassoy EY, Towne JE and Gabay C:
Regulation and function of interleukin-36 cytokines. Immunol Rev.
281:169–178. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Gabay C and Towne JE: Regulation and
function of interleukin-36 cytokines in homeostasis and
pathological conditions. J Leukocyte Biol. 97:645–652.
2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Towne JE, Garka KE, Renshaw BR, Virca GD
and Sims JE: Interleukin (IL)-1F6, IL-1F8, and IL-1F9 signal
through IL-1Rrp2 and IL-1RAcP to activate the pathway leading to
NF-kappaB and MAPKs. J Biol Chem. 279:13677–13688. 2004.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Towne JE, Renshaw BR, Douangpanya J,
Lipsky BP, Shen M, Gabel CA and Sims JE: Interleukin-36 (IL-36)
ligands require processing for full agonist (IL-36α, IL-36β, and
IL-36γ) or antagonist (IL-36Ra) activity. J Biol Chem.
286:42594–42602. 2011.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Mullard A: FDA approves first anti-IL-36
receptor antibody for rare skin disease. Nat Rev Drug Discov.
21(786)2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ding L, Wang X, Hong X, Lu L and Liu D:
IL-36 cytokines in autoimmunity and inflammatory disease.
Oncotarget. 9:2895–2901. 2017.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Boutet MA, Bart G, Penhoat M, Amiaud J,
Brulin B, Charrier C, Morel F, Lecron JC, Rolli-Derkinderen M,
Bourreille A, et al: Distinct expression of interleukin (IL)-36α, β
and γ, their antagonist IL-36Ra and IL-38 in psoriasis, rheumatoid
arthritis and Crohn's disease. Clin Exp Immunol. 184:159–173.
2016.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Scheibe K, Kersten C, Schmied A, Vieth M,
Primbs T, Carlé B, Knieling F, Claussen J, Klimowicz AC, Zheng J,
et al: Inhibiting interleukin 36 receptor signaling reduces
fibrosis in mice with chronic intestinal inflammation.
Gastroenterology. 156:1082–1097.e11. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ngo VL, Abo H, Maxim E, Harusato A, Geem
D, Medina-Contreras O, Merlin D, Gewirtz AT, Nusrat A and Denning
TL: A cytokine network involving IL-36γ, IL-23, and IL-22 promotes
antimicrobial defense and recovery from intestinal barrier damage.
Proc Natl Acad Sci USA. 115:E5076–E5085. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Queen D, Ediriweera C and Liu L: Function
and regulation of IL-36 signaling in inflammatory diseases and
cancer development. Front Cell Dev Biol. 7(317)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Xu P, Guan H, Xiao W and Sun L: The role
of IL-36 subfamily in intestinal disease. Biochem Soc Trans.
50:223–230. 2022.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Chen F, Qu M, Zhang F, Tan Z, Xia Q,
Hambly BD, Bao S and Tao K: IL-36 s in the colorectal cancer: Is
interleukin 36 good or bad for the development of colorectal
cancer? Bmc Cancer. 20(92)2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Bao S, Hu R and Hambly BD: IL-34, IL-36
and IL-38 in colorectal cancer-key immunoregulators of
carcinogenesis. Biophys Rev. 12:925–930. 2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Byrne J, Baker K, Houston A and Brint E:
IL-36 cytokines in inflammatory and malignant diseases: Not the new
kid on the block anymore. Cell Mol Life Sci. 78:6215–6227.
2021.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhou L and Todorovic V: Interleukin-36:
Structure, signaling and function. Adv Exp Med Biol. 21:191–210.
2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Namba T, Ichii O, Nakamura T, Masum MA,
Otani Y, Hosotani M, Elewa YHA and Kon Y: Compartmentalization of
interleukin 36 subfamily according to inducible and constitutive
expression in the kidneys of a murine autoimmune nephritis model.
Cell Tissue Res. 386:59–77. 2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Buhl AL and Wenzel J: Interleukin-36 in
infectious and inflammatory skin diseases. Front Immunol.
10(1162)2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Mai SZ, Li CJ, Xie XY, Xiong H, Xu M, Zeng
FQ, Guo Q and Han YF: Increased serum IL-36α and IL-36γ levels in
patients with systemic lupus erythematosus: Association with
disease activity and arthritis. Int Immunopharmacol. 58:103–108.
2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Chen WJ, Yu X, Yuan XR, Chen BJ, Cai N,
Zeng S, Sun YS and Li HW: The role of IL-36 in the
pathophysiological processes of autoimmune diseases. Front
Pharmacol. 12(727956)2021.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Dinarello CA: The IL-1 family of cytokines
and receptors in rheumatic diseases. Nat Rev Rheumatol. 15:612–632.
2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Vigne S, Palmer G, Lamacchia C, Martin P,
Talabot-Ayer D, Rodriguez E, Ronchi F, Sallusto F, Dinh H, Sims JE
and Gabay C: IL-36R ligands are potent regulators of dendritic and
T cells. Blood. 118:5813–5823. 2011.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Foster AM, Baliwag J, Chen CS, Guzman AM,
Stoll SW, Gudjonsson JE, Ward NL and Johnston A: IL-36 promotes
myeloid cell infiltration, activation, and inflammatory activity in
skin. J Immunol. 192:6053–6061. 2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Carrier Y, Ma HL, Ramon HE, Napierata L,
Small C, O'Toole M, Young DA, Fouser LA, Nickerson-Nutter C,
Collins M, et al: Inter-regulation of Th17 cytokines and the IL-36
cytokines in vitro and in vivo: implications in psoriasis
pathogenesis. J Invest Dermatol. 131:2428–2437. 2011.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Catapano M, Vergnano M, Romano M, Mahil
SK, Choon SE, Burden AD, Young HS, Carr IM, Lachmann HJ, Lombardi
G, et al: IL-36 promotes systemic IFN-I responses in severe forms
of psoriasis. J Invest Dermatol. 140:816–826. 2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Baker KJ, Brint E and Houston A:
Transcriptomic and functional analyses reveal a tumour-promoting
role for the IL-36 receptor in colon cancer and crosstalk between
IL-36 signalling and the IL-17/ IL-23 axis. Brit J Cancer.
128:735–747. 2023.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Aoyagi T, Newstead MW, Zeng X, Nanjo Y,
Peters-Golden M, Kaku M and Standiford TJ: Interleukin-36γ and
IL-36 receptor signaling mediate impaired host immunity and lung
injury in cytotoxic Pseudomonas aeruginosa pulmonary infection:
Role of prostaglandin E2. PLoS Pathog. 13(e1006737)2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Gao Y, Wen Q, Hu S, Zhou X, Xiong W, Du X,
Zhang L, Fu Y, Yang J, Zhou C, et al: IL-36γ promotes killing of
mycobacterium tuberculosis by macrophages via WNT5A-induced
noncanonical WNT signaling. J Immunol. 203:922–935. 2019.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Kovach MA, Singer B, Martinez-Colon G,
Newstead MW, Zeng X, Mancuso P, Moore TA, Kunkel SL, Peters-Golden
M, Moore BB and Standiford TJ: IL-36γ is a crucial proximal
component of protective type-1-mediated lung mucosal immunity in
Gram-positive and -negative bacterial pneumonia. Mucosal Immunol.
10:1320–1334. 2017.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Yi G, Ybe JA, Saha SS, Caviness G, Raymond
E, Ganesan R, Mbow ML and Kao CC: Structural and functional
attributes of the interleukin-36 receptor. J Biol Chem.
291:16597–16609. 2016.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Zarezadeh Mehrabadi A, Aghamohamadi N,
Khoshmirsafa M, Aghamajidi A, Pilehforoshha M, Massoumi R and Falak
R: The roles of interleukin-1 receptor accessory protein in certain
inflammatory conditions. Immunology. 166:38–46. 2022.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zhou L, Todorovic V, Kakavas S, Sielaff B,
Medina L, Wang L, Sadhukhan R, Stockmann H, Richardson PL,
DiGiammarino E, et al: Quantitative ligand and receptor binding
studies reveal the mechanism of interleukin-36 (IL-36) pathway
activation. J Biol Chem. 293:403–411. 2018.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Ganesan R, Raymond EL, Mennerich D, Woska
JR Jr, Caviness G, Grimaldi C, Ahlberg J, Perez R, Roberts S, Yang
D, et al: Generation and functional characterization of anti-human
and anti-mouse IL-36R antagonist monoclonal antibodies. MAbs.
9:1143–1154. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Onoufriadis A, Simpson MA, Pink AE, Di
Meglio P, Smith CH, Pullabhatla V, Knight J, Spain SL, Nestle FO,
Burden AD, et al: Mutations in IL36RN/IL1F5 are associated with the
severe episodic inflammatory skin disease known as generalized
pustular psoriasis. Am J Hum Genet. 89:432–437. 2011.PubMed/NCBI View Article : Google Scholar
|
|
41
|
van de Veerdonk FL, Stoeckman AK, Wu G,
Boeckermann AN, Azam T, Netea MG, Joosten LA, van der Meer JW, Hao
R, Kalabokis V and Dinarello CA: IL-38 binds to the IL-36 receptor
and has biological effects on immune cells similar to IL-36
receptor antagonist. Proc Natl Acad Sci USA. 109:3001–3005.
2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Li JM, Lu R, Zhang Y, Lin J, Hua X,
Pflugfelder SC and Li DQ: IL-36α/IL-36RA/IL-38 signaling mediates
inflammation and barrier disruption in human corneal epithelial
cells under hyperosmotic stress. Ocul Surf. 22:163–171.
2021.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Ngo VL, Kuczma M, Maxim E and Denning TL:
IL-36 cytokines and gut immunity. Immunology. 163:145–154.
2021.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Han Y, Huard A, Mora J, da Silva P, Brüne
B and Weigert A: IL-36 family cytokines in protective versus
destructive inflammation. Cell Signal. 75(109773)2020.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Verstak B, Nagpal K, Bottomley SP,
Golenbock DT, Hertzog PJ and Mansell A: MyD88 adapter-like
(Mal)/TIRAP interaction with TRAF6 is critical for TLR2- and
TLR4-mediated NF-kappaB proinflammatory responses. J Biol Chem.
284:24192–24203. 2009.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Swindell WR, Beamer MA, Sarkar MK, Loftus
S, Fullmer J, Xing X, Ward NL, Tsoi LC, Kahlenberg MJ, Liang Y and
Gudjonsson JE: RNA-Seq analysis of IL-1B and IL-36 responses in
epidermal keratinocytes identifies a shared MyD88-dependent gene
signature. Front Immunol. 9(80)2018.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Nishida A, Hidaka K, Kanda T, Imaeda H,
Shioya M, Inatomi O, Bamba S, Kitoh K, Sugimoto M and Andoh A:
Increased expression of interleukin-36, a member of the
interleukin-1 cytokine family, in inflammatory bowel disease.
Inflamm Bowel Dis. 22:303–314. 2016.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Fonseca-Camarillo G, Furuzawa-Carballeda
J, Iturriaga-Goyon E and Yamamoto-Furusho JK: Differential
expression of IL-36 family members and IL-38 by immune and
nonimmune cells in patients with active inflammatory bowel disease.
Biomed Res Int. 2018(5140691)2018.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Scheibe K, Backert I, Wirtz S, Hueber A,
Schett G, Vieth M, Probst HC, Bopp T, Neurath MF and Neufert C:
IL-36R signalling activates intestinal epithelial cells and
fibroblasts and promotes mucosal healing in vivo. Gut. 66:823–838.
2017.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Russell SE, Horan RM, Stefanska AM, Carey
A, Leon G, Aguilera M, Statovci D, Moran T, Fallon PG, Shanahan F,
et al: IL-36α expression is elevated in ulcerative colitis and
promotes colonic inflammation. Mucosal Immunol. 9:1193–1204.
2016.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Harusato A, Abo H, Ngo VL, Yi SW,
Mitsutake K, Osuka S, Kohlmeier JE, Li JD, Gewirtz AT, Nusrat A and
Denning TL: IL-36γ signaling controls the induced regulatory T
cell-Th9 cell balance via NFκB activation and STAT transcription
factors. Mucosal Immunol. 10:1455–1467. 2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Kanda T, Nishida A, Takahashi K, Hidaka K,
Imaeda H, Inatomi O, Bamba S, Sugimoto M and Andoh A:
Interleukin(IL)-36α and IL-36γ induce proinflammatory mediators
from human colonic subepithelial myofibroblasts. Front Med
(Lausanne). 2(69)2015.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Zhu J, Xu Y, Li Z, Liu S, Fu W and Wei Y:
Interleukin-36β exacerbates DSS-induce acute colitis via inhibiting
Foxp3+ regulatory T cell response and increasing Th2
cell response. Int Immunopharmacol. 108(108762)2022.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Xie C, Yan W, Quan R, Chen C, Tu L, Hou X
and Fu Y: Interleukin-38 is elevated in inflammatory bowel diseases
and suppresses intestinal inflammation. Cytokine.
127(154963)2020.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Sonnenberg GF, Fouser LA and Artis D:
Border patrol: Regulation of immunity, inflammation and tissue
homeostasis at barrier surfaces by IL-22. Nat Immunol. 12:383–390.
2011.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Longman RS, Diehl GE, Victorio DA, Huh JR,
Galan C, Miraldi ER, Swaminath A, Bonneau R, Scherl EJ and Littman
DR: CX3CR1+ mononuclear phagocytes support
colitis-associated innate lymphoid cell production of IL-22. J Exp
Med. 211:1571–1583. 2014.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Medina-Contreras O, Harusato A, Nishio H,
Flannigan KL, Ngo V, Leoni G, Neumann PA, Geem D, Lili LN, Ramadas
RA, et al: Cutting edge: IL-36 receptor promotes resolution of
intestinal damage. J Immunol. 196:34–38. 2016.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Parkos CA: Neutrophil-Epithelial
Interactions: A Double-Edged Sword. Am J Pathol. 186:1404–1416.
2016.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Luissint AC, Parkos CA and Nusrat A:
Inflammation and the intestinal barrier: Leukocyte-epithelial cell
interactions, cell junction remodeling, and mucosal repair.
Gastroenterology. 151:616–632. 2016.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Peterson LW and Artis D: Intestinal
epithelial cells: Regulators of barrier function and immune
homeostasis. Nat Rev Immunol. 14:141–153. 2014.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Melton E and Qiu H: Interleukin-36
cytokine/receptor signaling: A new target for tissue fibrosis. Int
J Mol Sci. 21(6458)2020.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Yun SM, Kim SH and Kim EH: The molecular
mechanism of transforming growth factor-β signaling for intestinal
fibrosis: A mini-review. Front Pharmacol. 10(162)2019.PubMed/NCBI View Article : Google Scholar
|
|
63
|
D'Alessio S, Ungaro F, Noviello D, Lovisa
S, Peyrin-Biroulet L and Danese S: Revisiting fibrosis in
inflammatory bowel disease: The gut thickens. Nat Rev Gastroenterol
Hepatol. 19:169–184. 2022.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Elias M, Zhao S, Le HT, Wang J, Neurath
MF, Neufert C, Fiocchi C and Rieder F: IL-36 in chronic
inflammation and fibrosis-bridging the gap? J Clin Invest.
131(e144336)2021.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Chi HH, Hua KF, Lin YC, Chu CL, Hsieh CY,
Hsu YJ, Ka SM, Tsai YL, Liu FC and Chen A: IL-36 signaling
facilitates activation of the NLRP3 inflammasome and IL-23/IL-17
axis in renal inflammation and fibrosis. J Am Soc Nephrol.
28:2022–2037. 2017.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Sommerfeld SD, Cherry C, Schwab RM, Chung
L, Maestas DR Jr, Laffont P, Stein JE, Tam A, Ganguly S, Housseau
F, et al: Interleukin-36γ-producing macrophages drive
IL-17-mediated fibrosis. Sci Immunol. 4(eaax4783)2019.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Nishida A, Inatomi O, Fujimoto T, Imaeda
H, Tani M and Andoh A: Interleukin-36α induces inflammatory
mediators from human pancreatic myofibroblasts via a MyD88
dependent pathway. Pancreas. 46:539–548. 2017.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Santacroce G, Lenti MV and Di Sabatino A:
Therapeutic targeting of intestinal fibrosis in Crohn's disease.
Cells. 11(429)2022.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Wang J, Lin S, Brown JM, van Wagoner D,
Fiocchi C and Rieder F: Novel mechanisms and clinical trial
endpoints in intestinal fibrosis. Immunol Rev. 302:211–227.
2021.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Takahashi K, Nishida A, Shioya M, Imaeda
H, Bamba S, Inatomi O, Shimizu T, Kitoh K and Andoh A: Interleukin
(IL)-1β is a strong inducer of IL-36γ expression in human colonic
myofibroblasts. PLoS One. 10(e138423)2015.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global cancer statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Shah SC and Itzkowitz SH: Colorectal
cancer in inflammatory bowel disease: Mechanisms and management.
Gastroenterology. 162:715–730.e3. 2022.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Hirano T, Hirayama D, Wagatsuma K,
Yamakawa T, Yokoyama Y and Nakase H: Immunological mechanisms in
inflammation-associated colon carcinogenesis. Int J Mol Sci.
21(3062)2020.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Wang ZS, Cong ZJ, Luo Y, Mu YF, Qin SL,
Zhong M and Chen JJ: Decreased expression of interleukin-36α
predicts poor prognosis in colorectal cancer patients. Int J Clin
Exp Patho. 7:8077–8081. 2014.PubMed/NCBI
|
|
75
|
Zhao X, Chen X, Shen X, Tang P, Chen C,
Zhu Q, Li M, Xia R, Yang X, Feng C, et al: IL-36β promotes
CD8+ T cell activation and antitumor immune responses by
activating mTORC1. Front Immunol. 10(1803)2019.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Pan QZ, Pan K, Zhao JJ, Chen JG, Li JJ, Lv
L, Wang DD, Zheng HX, Jiang SS, Zhang XF and Xia JC: Decreased
expression of interleukin-36α correlates with poor prognosis in
hepatocellular carcinoma. Cancer Immunology, Immunotherapy.
62:1675–1685. 2013.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Wei X, Yao Y, Wang X, Sun J, Zhao W, Qiu
L, Zhai W, Qi Y, Gao Y and Wu Y: Interleukin-36α inhibits
colorectal cancer metastasis by enhancing the infiltration and
activity of CD8+ T lymphocytes. Int Immunopharmacol.
100(108152)2021.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Wang X, Zhao X, Feng C, Weinstein A, Xia
R, Wen W, Lv Q, Zuo S, Tang P, Yang X, et al: IL-36γ transforms the
tumor microenvironment and promotes type 1 lymphocyte-mediated
antitumor immune responses. Cancer Cell. 28:296–306.
2015.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Stolk D, van der Vliet HJ, de Gruijl TD,
van Kooyk Y and Exley MA: Positive & negative roles of innate
effector cells in controlling cancer progression. Front Immunol.
9(1990)2018.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Uzhachenko RV and Shanker A:
CD8+ T lymphocyte and NK cell network: Circuitry in the
cytotoxic domain of immunity. Front Immunol.
10(1906)2019.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Weinstein AM, Chen L, Brzana EA, Patil PR,
Taylor JL, Fabian KL, Wallace CT, Jones SD, Watkins SC, Lu B, et
al: Tbet and IL-36γ cooperate in therapeutic DC-mediated promotion
of ectopic lymphoid organogenesis in the tumor microenvironment.
Oncoimmunology. 6(e1322238)2017.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Lei X, Lei Y, Li JK, Du WX, Li RG, Yang J,
Li J, Li F and Tan HB: Immune cells within the tumor
microenvironment: Biological functions and roles in cancer
immunotherapy. Cancer Lett. 470:126–133. 2020.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Schumacher TN and Thommen DS: Tertiary
lymphoid structures in cancer. Science.
375(eabf9419)2022.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Weinstein AM, Giraldo NA, Petitprez F,
Julie C, Lacroix L, Peschaud F, Emile JF, Marisa L, Fridman WH,
Storkus WJ and Sautès-Fridman C: Association of IL-36γ with
tertiary lymphoid structures and inflammatory immune infiltrates in
human colorectal cancer. Cancer Immunol Immunother. 68:109–120.
2019.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Yang M, Giehl E, Feng C, Feist M, Chen H,
Dai E, Liu Z, Ma C, Ravindranathan R, Bartlett DL, et al:
IL-36γ-armed oncolytic virus exerts superior efficacy through
induction of potent adaptive antitumor immunity. Cancer Immunol
Immunother. 70:2467–2481. 2021.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Weinstein AM and Storkus WJ: Therapeutic
lymphoid organogenesis in the tumor microenvironment. Adv Cancer
Res. 128:197–233. 2015.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Yang W, Dong HP, Wang P, Xu ZG, Xian J,
Chen J, Wu H, Lou Y, Lin D and Zhong B: IL-36γ and IL-36Ra
reciprocally regulate colon inflammation and tumorigenesis by
modulating the cell-matrix adhesion network and Wnt signaling. Adv
Sci (Weinh). 9(e2103035)2022.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Baker K, O'Donnell C, Bendix M, Keogh S,
Byrne J, O'Riordain M, Neary P, Houston A and Brint E: IL-36
signalling enhances a pro-tumorigenic phenotype in colon cancer
cells with cancer cell growth restricted by administration of the
IL-36R antagonist. Oncogene. 41:2672–2684. 2022.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Kaushik I, Ramachandran S, Zabel C,
Gaikwad S and Srivastava SK: The evolutionary legacy of immune
checkpoint inhibitors. Semin Cancer Biol. 86:491–498.
2022.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Wei SC, Duffy CR and Allison JP:
Fundamental mechanisms of immune checkpoint blockade therapy.
Cancer Discov. 8:1069–1086. 2018.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Peña-Asensio J, Calvo H, Torralba M,
Miquel J, Sanz-de-Villalobos E and Larrubia JR: Anti-PD-1/PD-L1
based combination immunotherapy to boost antigen-specific
CD8+ T cell response in hepatocellular carcinoma.
Cancers (Basel). 13(1922)2021.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Hewitt SL, Bai A, Bailey D, Ichikawa K,
Zielinski J, Karp R, Apte A, Arnold K, Zacharek SJ, Iliou MS, et
al: Durable anticancer immunity from intratumoral administration of
IL-23, IL-36γ, and OX40L mRNAs. Sci Transl Med.
11(eaat9143)2019.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Bachelez H, Choon SE, Marrakchi S, Burden
AD, Tsai TF, Morita A, Navarini AA, Zheng M, Xu J, Turki H, et al:
Trial of spesolimab for generalized pustular psoriasis. New Engl J
Med. 385:2431–2440. 2021.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Ferrante M, Irving PM, Selinger CP,
D'Haens G, Kuehbacher T, Seidler U, Gropper S, Haeufel T, Forgia S,
Danese S, et al: Safety and tolerability of spesolimab in patients
with ulcerative colitis. Expert Opin Drug Saf. 22:141–152.
2023.PubMed/NCBI View Article : Google Scholar
|