Introduction
Esophageal carcinoma (ESCA) is one of the most
common cancers in the world. According to statistics, there were
over 600,000 new cases of esophageal cancer worldwide and over
540,000 related deaths in 2020; it ranked 7th in incidence and 6th
in mortality among all cancers (1). The tissue subtypes of esophageal
cancer are divided into esophageal squamous cell carcinoma (ESCC)
and esophageal adenocarcinoma (EAC), of which ESCC is the main
subtype. The incidence and distribution of histological types vary
according to geographic location. ESCC accounts for more than 90%
of esophageal cancer cases in developing countries, and
approximately 50% of new cases occur in China; its incidence is
increasing each year (2,3). With the rapid development of medical
technology, many methods are being used to treat ESCC, and these
methods include surgery, chemotherapy, radiotherapy, and immune
checkpoint inhibitor therapy.
However, the early symptoms of ESCC patients are not
obvious, and the disease is typically in an advanced stage at
diagnosis, which leads to a poor prognosis in ESCC patients; ESCC
has a 5-year survival rate of only 14~22% (4-6).
Esophageal cancer has become one of the deadliest cancers, mainly
due to its high aggressiveness and low survival rate. Therefore,
novel biomarkers of esophageal cancer are urgently required to
facilitate early diagnosis, tumor-targeted drug development and
ESCA prognosis prediction.
Necroptosis is a form of programmed inflammatory
cell death that was first reported by Degterev et al
(7). Necroptosis involves a
phosphorylation signal mediated by serine/threonine protein kinase
1/3 (RIPK1/RIPK3). Necrosis shares a common pathway with apoptosis:
the mixed lineage kinase domain protein (MLKL/pMLKL) pathway
(8,9). This pattern involves necrotic cell
death; the related morphological features include lysosomal
membrane degradation, cytoplasmic vacuolation, plasma membrane
disintegration, and finally, explosive cell rupture (10,11).
In recent years, increasing evidence has shown that necroptosis
plays an important role in tumorigenesis, metastasis, and the tumor
immune response. It has been reported that activation of the
necrosis-related genes RIPK1 and RIPK3 or necrosis-related
signaling pathways is involved in the regulation of tumor cell
metabolic biological processes and the tumor microenvironment
(12-15).
Studies have shown that necroptosis can not only promote the
occurrence and development of cancer but also inhibit the
occurrence and development of cancer, and its specific role depends
on the type of tumor and its developmental stage. Low expression of
RIPK3 facilitates resistance to necroptosis in some tumors, such as
acute myeloid leukemia (16),
chronic lymphocytic leukemia (17)
and breast adenocarcinoma (18),
and thus can readily promote tumor growth. However, high levels of
phosphorylated MLKL have been shown to be associated with a poor
prognosis in patients with colon cancer (19) and esophageal cancer (20). Necroptosis may play an important
role in eliciting immunogenicity and promoting natural anticancer
immune surveillance. Studies have shown that tumor cells undergo
necroptosis to release IL-lα to activate dendritic cells (DCs).
Activated DCs induce antitumor immune responses by producing the
cytotoxic cytokine IL-12 and activating CD8+ T cells to eliminate
cancer cells (21,22). Similarly, damage-associated
molecular patterns (DAMPs) from necrotic tumor cells strongly
induce the activation of antitumor CD8+ T cells (23). It has also been shown that NKT
cells are involved in RIPK3-mediated antitumor immune responses, as
the loss of RIPK3 impairs the activation of NKT cells to prevent
tumor killing (24). All these
findings suggest that necroptosis inhibits tumor initiation and
progression. On the other hand, tumor cell metastasis is the main
cause of death in cancer patients. Metastasis refers to the
dissemination of individual tumor cells through the circulatory
system to other distant organs for further growth. Necroptosis can
promote tumor development and metastasis through a variety of
mechanisms. Extravasation of tumor cells through the endothelium is
an important step in metastatic spread. Tumor cells induce the
necroptosis of endothelial cells by activating death receptor 6
(DR6 encoded by TNFRSF21), another member of the death receptor
family, thereby promoting tumor cell extravasation and metastasis.
In mice treated with Nec-1 or mice with endothelial cell-specific
knockout of RIPK3 or MLKL, we observed a reduction in tumor
cell-induced necroptosis of endothelial cells, which thereby
reduced tumor cell extravasation and metastasis (25). In addition, metastasis also
involves a complex interaction between cancer cells and the tumor
microenvironment, which involves factors including immune cell
infiltration and cytokine secretion. Necroptosis is a type of
proinflammatory cell death, and the inflammatory response caused by
necroptosis may lead to tumor metastasis. In pancreatic ductal
adenocarcinoma (PDAC), RIPK3 knockout tumor cells undergo
necroptosis and release soluble cytokines that bind to receptors on
inflammatory cells; for example, cytokine SAP130 is released and
binds its cognate receptor Mincle, thereby initiating an
immunosuppressive tumor microenvironment and promoting the
progression of PDAC (26).
Therefore, studying necroptosis-related genes in tumor cells and
their regulatory mechanisms is expected to reveal a target for ESCA
therapy.
Long noncoding RNAs (lncRNAs) refer to noncoding
RNAs with a length greater than 200 nucleotides that lack
protein-coding ability and can play a variety of important
biological roles in cells (27).
It has been reported that lncRNAs participate in and exacerbate
tumor inflammation and tumor microenvironment disorder and prevent
tumor immune escape (28).
LINC00680 promotes esophageal squamous cell carcinoma progression
through the miR-423-5p/PAK6 axis. Knockdown of LINC00680 was found
to inhibit ESCC cell proliferation, colony formation, migration and
invasion in vitro and tumor growth in vivo (29). LncRNA ZEB2-AS1 is upregulated in
ESCC tissues and cells vs. corresponding controls. It has been
verified to promote the proliferation, migration and invasion of
esophageal squamous cell carcinoma cells by modulating the
miR-574-3p/HMGA2 axis (30). In
addition, lncRNA necrosis-related factor (NRF) regulates
cardiomyocyte necroptosis by targeting miR-873 and
RIPK1/RIPK3(31). Previous studies
have reported that lncRNAs associated with necrosis can be used to
predict the features of many tumors (including head and neck
squamous cancer, gastric cancer, colon cancer and breast cancer)
and their immune environment (32-35).
However, the relationship between lncRNAs and necrosis in ESCA
remains unclear and needs further investigation. Therefore, a
prognostic risk model of necrosis-related lncRNAs (nrlncRNAs) was
constructed in this study to predict the prognosis of ESCA patients
and provide guidance for clinical diagnosis and treatment.
Materials and methods
Acquisition of transcriptome-level
gene expression data and clinical data for patients with esophageal
cancer
RNA sequencing-seq (RNA-seq) transcriptome data for
patients with ESCA were downloaded from the TCGA-ESCA dataset
(https://portal.gdc.cancer.gov/); the
related samples included 11 normal samples and 161 ESCA tumor
samples (last accessed: 10 May 2022). To reduce potential
statistical bias in the sample analysis, patients with no OS data
or short OS time (<30 days) were excluded, and patients in the
TCGA-ESCA cohort were randomly divided into the training risk group
and the test risk group with Strawberry Perl at a ratio of 1:1.
Identification of nrlncRNAs
According to previous studies, we downloaded the
necroptosis gene set M24779.gmt from the Gene Set Enrichment
Analysis (GSEA) website (http://www.gsea-msigdb.org/gsea/index.jsp) and further
extracted 8 necroptosis-related genes and 59 other genes (36). Then, correlation analysis was
performed for 67 necroptosis-related genes and differentially
expressed lncRNAs in the combined matrices via Strawberry Perl and
R software. Then, nrlncRNAs were identified with the criteria of
|Pearson R|>0.4 and P<0.001.
Establishment and validation of the
risk signature according to nrlncRNAs in ESCA
Univariate Cox (Uni-Cox) regression was used to
screen lncRNAs associated with prognosis with P<0.05. Then,
LASSO Cox analysis was performed to screen nrlncRNAs by 10-fold
cross-validation, and the risk model P-value was 0.05. The number
of resamples was increased to more than 1,000 to prevent
overfitting error. Finally, the previously defined formula Risk
score=∑(nrlncRNAi) coefficient x (nrlncRNAi)
expression was used to establish the prognostic risk profiles with
eligible nrlncRNAs. ESCA patients in the TCGA cohort were divided
into a low-risk group and a high-risk group in accordance with the
median risk score. Kaplan-Meier (KM) survival analysis and log-rank
test were performed using the survival R package to analyze whether
there were differences in OS in the low-risk group of ESCA
patients. That survival curves cross over may affect the results of
log-rank analysis. When survival curves cross over, we reanalyze
this dataset by the two-stage test (R package: https://cran.r-project.org/web/packages/TSHRC/TSHRC.pdf).
The chi-square test was used to analyze the relationship between
the model and clinical features to assess the prognostic value of
the established risk features. Receiver operating characteristic
(ROC) curves (1-year, 3-year, and 5-year survival rates) and the
area under the ROC curve (AUC) values were evaluated to demonstrate
the validity of the prediction model and compare its performance to
that of models based on other clinical characteristics. Nomogram
analysis was carried out via Uni-Cox and Multi-Cox analyses to
evaluate survival predictions.
Enrichment analysis of genes
The related gene set (KEGG.v7.4.symbols. GMT) was
analyzed by GSEA software version 4.2.3. The top 5 pathways in the
low- and high-risk groups were selected based on the criterion of
P<0.05.
Assessment of immune factors and the
tumor microenvironment
We downloaded all the data on TCGA tumor invasion
estimates from the website http://timer.cistrome.org/. Then, a variety of
methods, including the Wilcoxon signed-rank test and analyses with
the limma, scales, ggplot2, ggtext, tidyverse and ggpubr R
packages, were employed, consistent with a previous report
(37). Moreover, the stromal
score, immune score, and ESTIMATE score (stromal score + immune
score) of each patient were calculated. Then, single-sample GSEA
(ssGSEA) was used to score the infiltrating immune cells in ESCA,
and their relative content was quantified by the ‘GSVA’ package.
The immune cell scores and pathways scores for various groups are
shown on multiple boxplots. Finally, we analyzed and explored the
differences in immune functions, immune cell infiltration and
checkpoint expression levels between the low- and high-risk groups
via the ggpubr R package.
Association between the risk model and
clinical treatment
According to half-maximal inhibitory concentration
(IC50) data for ESCA from the Genomics of Drug Sensitivity in
Cancer (GDSC) database (https://www.cancerrxgene.org/), we used the
‘pRRophetic’ package to compare the therapeutic response between
the low-risk and high-risk groups (38). This study focused on the analysis
and comparison of the response to 48 commonly used chemotherapy
drugs. When the P-value was less than 0.05, the IC50 values of the
low-risk group and high-risk group were considered to be
significantly different according to the Wilcoxon rank sum
test.
Cluster analysis of prognostic
nrlncRNAs
We evaluated the expression of nrlncRNAs related to
prognosis using the ‘ConsensusClusterPlus’ package and assessed
their ability to predict the response of ESCA patients to
immunotherapy (39). Subsequently,
K-M survival analysis, principal component analysis (PCA), and
t-distributed stochastic neighbor embedding (t-SNE) were carried
out with the Rtsne R package. Additionally, analyses of immune
features and drug susceptibility were performed were performed with
the GSVA and the pRRophetic R packages.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The esophageal epithelial cell line HET-1A and human
esophageal cancer cell lines KYSE150 and TE1 were provided by the
Research Center of the Fourth Hospital of Hebei Medical University.
Total RNA of cells was extracted and purified using the TransZol Up
Plus RNA Kit (TransGen Biotech, Beijing, China) according to the
manufacturer's instructions. Total RNA was used to synthesize cDNA
via the lnRcute lncRNA First-Strand cDNA kit (TIANGER, China).
BlasTaq 2X qPCR Master Mix (Ab, China) was used for the PCR
amplification process. All experimental procedures were carried out
according to the commercial instructions. Because the AC090912.2,
AC244197.2, AL158166.1, AC079684.1, AP003696.1, and AC026741.1
products may be artifacts or may not correspond to gene
annotations, six gene primer pairs were designed for verification.
Primers for LINC02811 and LINC00299 were designed and validated by
PCR in three types of esophageal cells. The primer sequences for
PCR amplification were as follows: LINC02811, forward:
5'-TTGGCACACTTAGCAAGGACTGAC-3', reverse:
5'-CTTCTGCCTCATTTCTGTCCTCCAC-3'; and LINC00299, forward:
5'-TCTGAAGTCACCTGCCCTATCTGG-3', reverse:
5'-TCCACTTGCCACTGCTTGCTTATC-3'. Each condition was replicated three
times, and the quantitative analysis of gene expression was
calculated by the 2-ΔΔCq method. GAPDH was used as the
internal control. The differences in LINC02811 and LINC00299
expression among HET-1A, KYSE150 and TE1 were assessed by a one-way
ANOVA and Bonferroni correction post hoc test. GraphPad Prism
(version 8.0.2) was applied to create the graphs.
Statistical analysis
All statistical evaluations utilized R (version
4.2.0). For each analysis, P<0.05 confirmed statistical
significance. Univariate/multivariate Cox proportional hazard
regression analyses were used for constructing a
necroptosis-associated lncRNA model used as a predictive risk
model. Chi-squared test was used to assess the medical features
between different study groups. The log-rank test and Kaplan-Meier
(KM) survival analysis was used to compare the differences in OS
between low- and high-risk groups. The survival receiver operating
characteristic (ROC) curves and their areas under the curves (AUCs)
were used to assess the efficacy of prediction, comparing with
other clinical characteristics. Kruskal-Wallis test followed by
Dunn's post hoc test was used to analyze the association between
clusters, immune factors and the TME, and to compare drug
susceptibility. Wilcoxon rank sum test was used to analyze the
difference in IC50 values between the low-risk and
high-risk groups. The relative expression of each gene was
calculated and compared using the 2-ΔΔCq method, and was
analyzed by one-way ANOVA and Bonferroni correction. GraphPad Prism
(version 8.0.2) was applied to create the graphs
(*P<0.01, **P<0.001 and
***P<0.0001). Experiments were repeated three
times.
Results
Analysis of nrlncRNAs in ESCA
patients
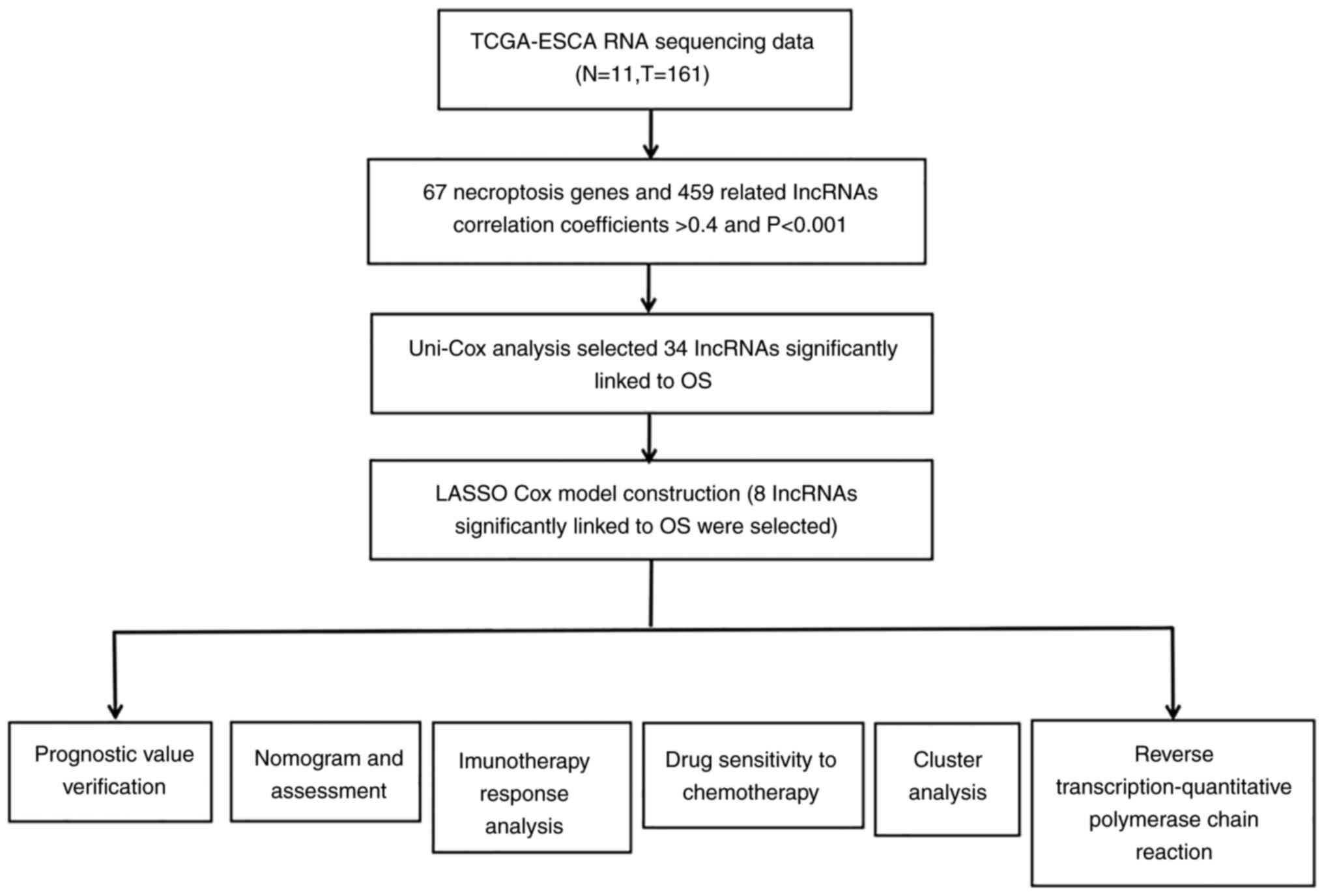
The detailed flow diagram of the study design is
shown in Fig. 1. The transcriptome
data of ESCA downloaded from the TCGA included data for 11 normal
samples and 161 ESCA tumor samples. According to the GTF file, the
mRNAs and lncRNAs were distinguished in the transcriptome data. The
data showed that 67 necrosis-associated genes were differentially
expressed in normal and tumor samples. In addition, we finally
obtained 459 nrlncRNAs, of which 103 were downregulated and 356
were upregulated based on the criteria of |log2-fold fold change
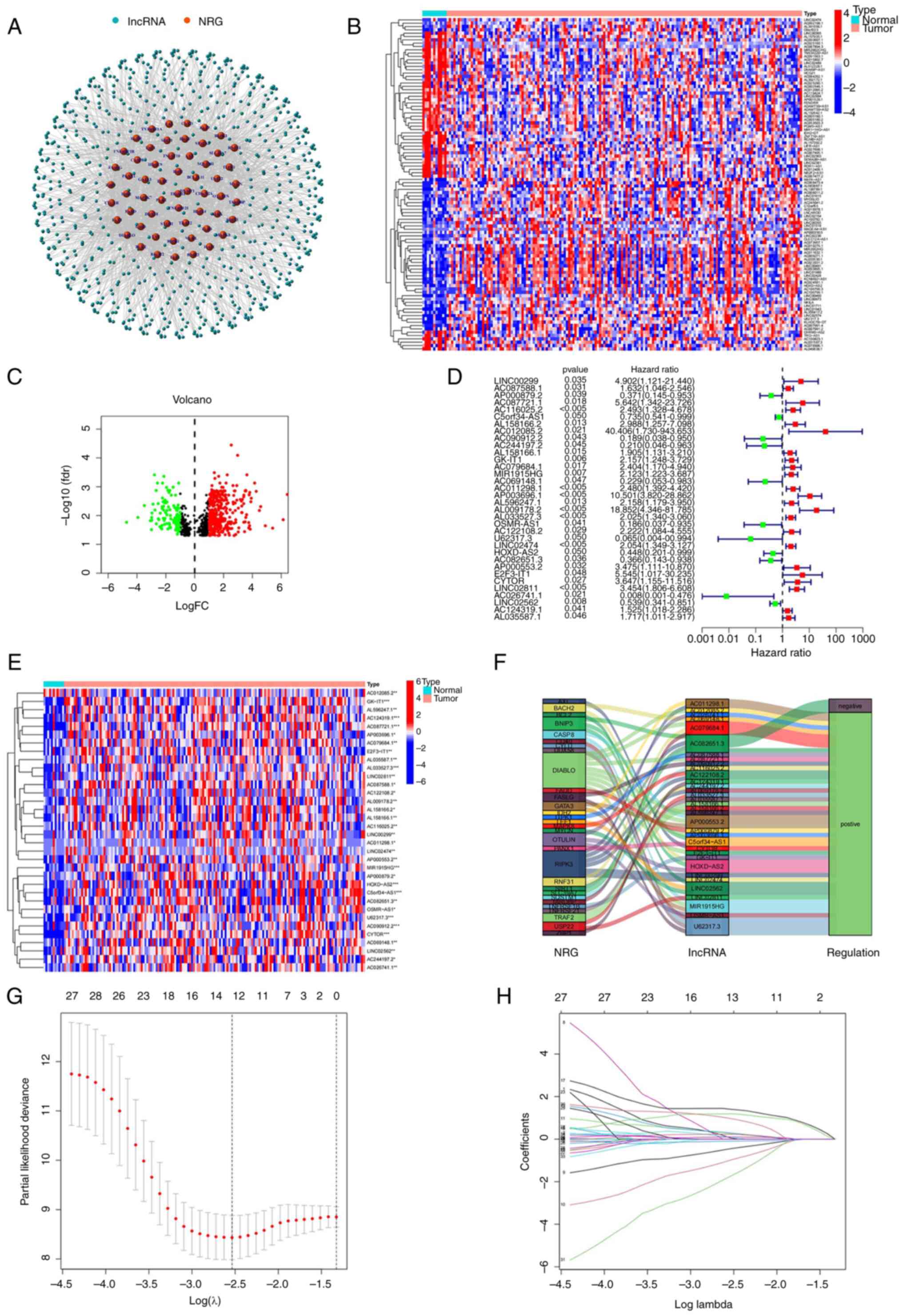
(FC)|>1 and P<0.05. As shown in Fig. 2A, the analysis revealed the
interaction relationship between genes and lncRNAs via a network
diagram. A heatmap (Fig. 2B) and
volcano plot (Fig. 2C) were used
to reflect the differential expression of nrlncRNAs between normal
and tumor samples.
Construction and verification of the
nrlncRNA risk model
Uni-Cox regression analysis showed that 34 nrlncRNAs
were associated with overall survival (OS) (all P<0.05), as
shown in Fig. 2D (Uni-Cox forest
map) and Fig. 2E (heatmap). For
ESCA patients, 23 nrlncRNAs were identified as indicators of a poor
prognosis (hazard ratio, HR>1); however, the remaining nrlncRNAs
were favorable prognostic factors.
To avoid overfitting the prognostic model, the 34
nrlncRNAs were analyzed by LASSO regression analysis to establish
prognostic variables. As shown in Fig.
2G and H, 8 nrlncRNAs were
selected to develop the prognostic model according to nrlncRNA
expression and the Multi-Cox regression analysis results.
Additionally, the Sankey diagram analysis, as shown in Fig. 2F, revealed that all 34 nrlncRNAs
were upregulated in ESCA patients.
The risk score formula was as follows: Risk
score=(2.79718675565941 x LINC00299 expression)-(1.73124557102057 x
AC090912.2 expression)-(2.81846794900141 x AC244197.2 expression)+
(0.810897179352003 x AL158166.1 expression)+ (0.90494089310592 x
AC079684.1 expression)+ (2.58956796584768 x AP003696.1expression)+
(1.52401713479381 x LINC02811 expression)- (5.43761185912726 x
AC026741.1 expression).
Based on the median risk score, the ESCA patients
were divided high and low risk groups and then divided into
training and testing groups. A total of 155 ESCA patients were
enrolled, including 77 with high risk scores and 78 with low risk
scores. In the training group, there were 39 patients each in the
high risk and low risk subgroups. In the test group, there were 38
patients with high risk scores and 39 patients with low risk scores
(Fig. 3A-C). Furthermore,
comparison of the survival status, survival time and related lncRNA
expression levels among these groups (Fig. 3D-L) suggested that the high-risk
group had a poor prognosis.
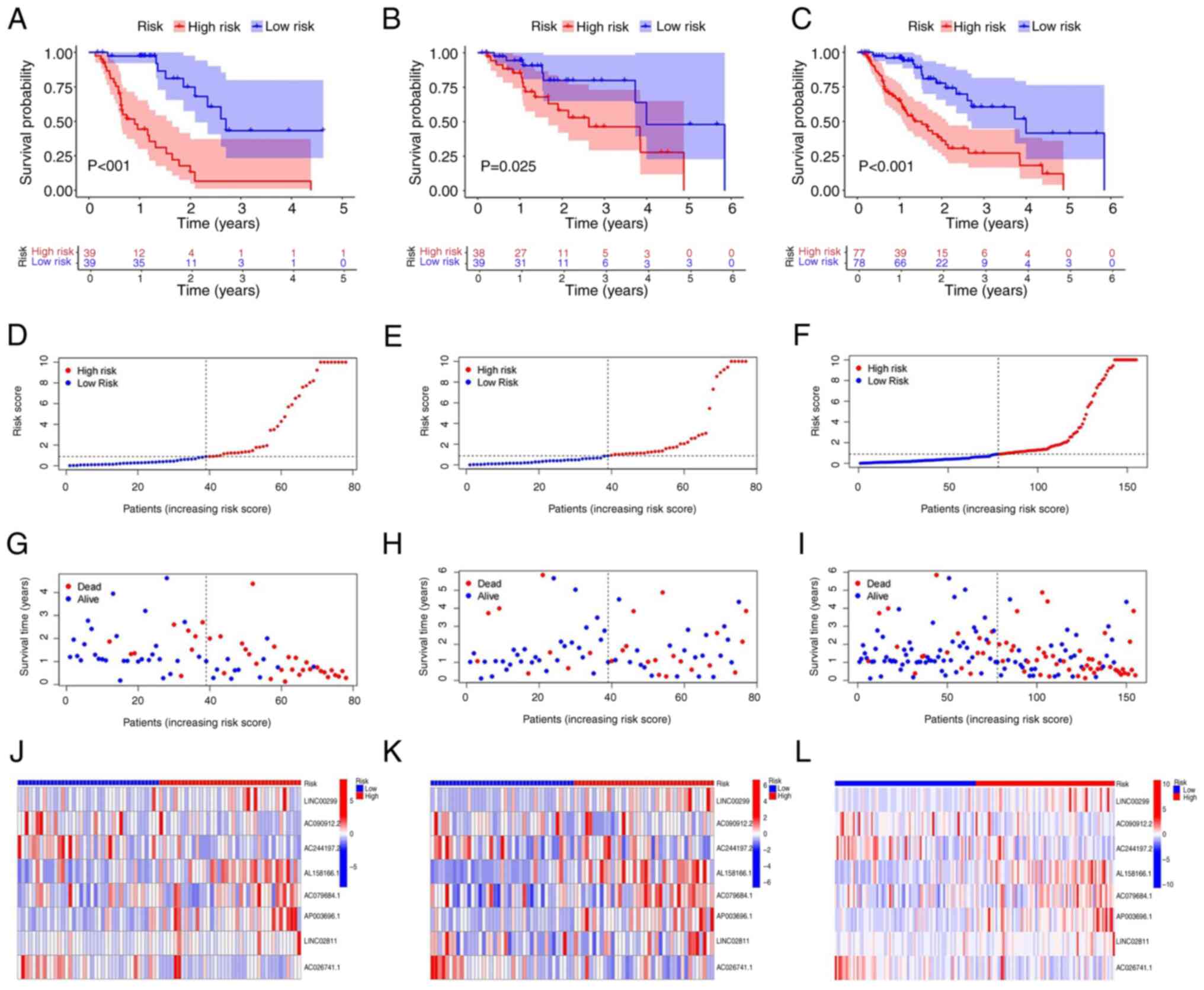 | Figure 3Prognostic value of the 8 signature
nrlncRNAs in the training, testing, and entire datasets. (A-C) K-M
survival curves of the OS probability of patients between low- and
high-risk groups in (A) training group, (B) testing group and (C)
entire group, respectively. (D-F) Demonstration of the nrlncRNA
model based on the risk score in (D) training group, (E) testing
group and (F) entire group, respectively. (G-I) Survival time and
survival status between the low- and high-risk groups in (G)
training group, (H) testing group and (I) entire group,
respectively. (J-L) Heatmap of the expression of 8 lncRNAs in (J)
training group, (K) testing group and (L) entire group,
respectively. K-M, Kaplan-Meier; nrlncRNAs, necrosis-related
lncRNAs. |
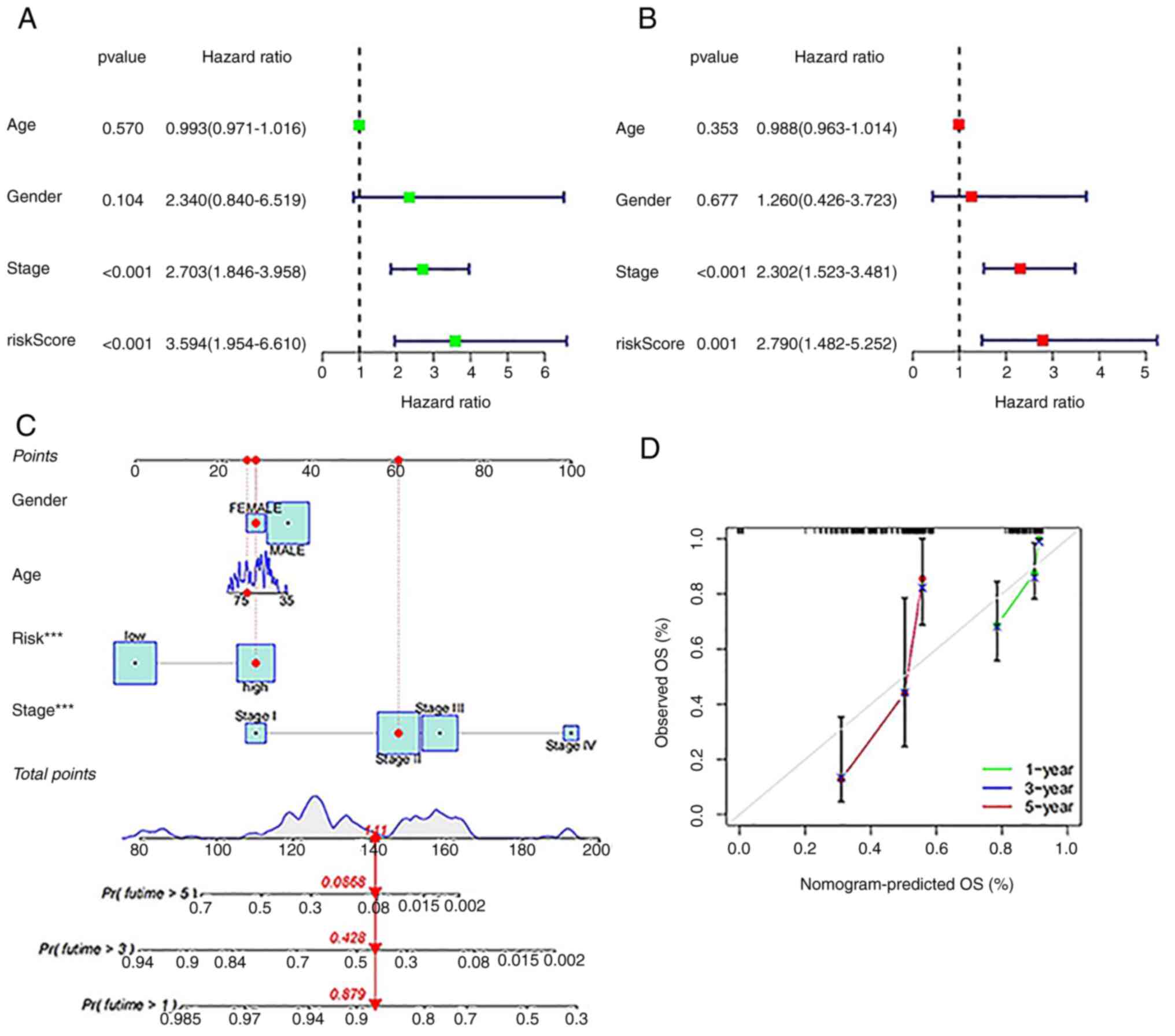
Additionally, this risk score had high predictive
efficiency in patients grouped by for age (Fig. 4A and B), sex (Fig.
4C and D), stage I-II
(Fig. 4E), T stage (Fig. 4F and G), N stage (Fig. 4H and I) and M stage (Fig. 4J). For the different subgroups, the
OS of the ESCA patients in the low-risk group was obviously longer
than that of the patients in the high-risk group. These results
suggest that the predictive signature can also predict the
prognosis of ESCA patients in all subgroups based on age, sex, T
stage and N stage and in the stage I-II and M0 stage subgroups. The
area under the ROC curve (AUC) value of the risk model was 0.746,
as shown in Fig. 4K, and this
value was obviously higher than that of the model based on clinical
characteristics, including age (0.545), sex (0.492), and stage
(0.665; Fig. 4L).
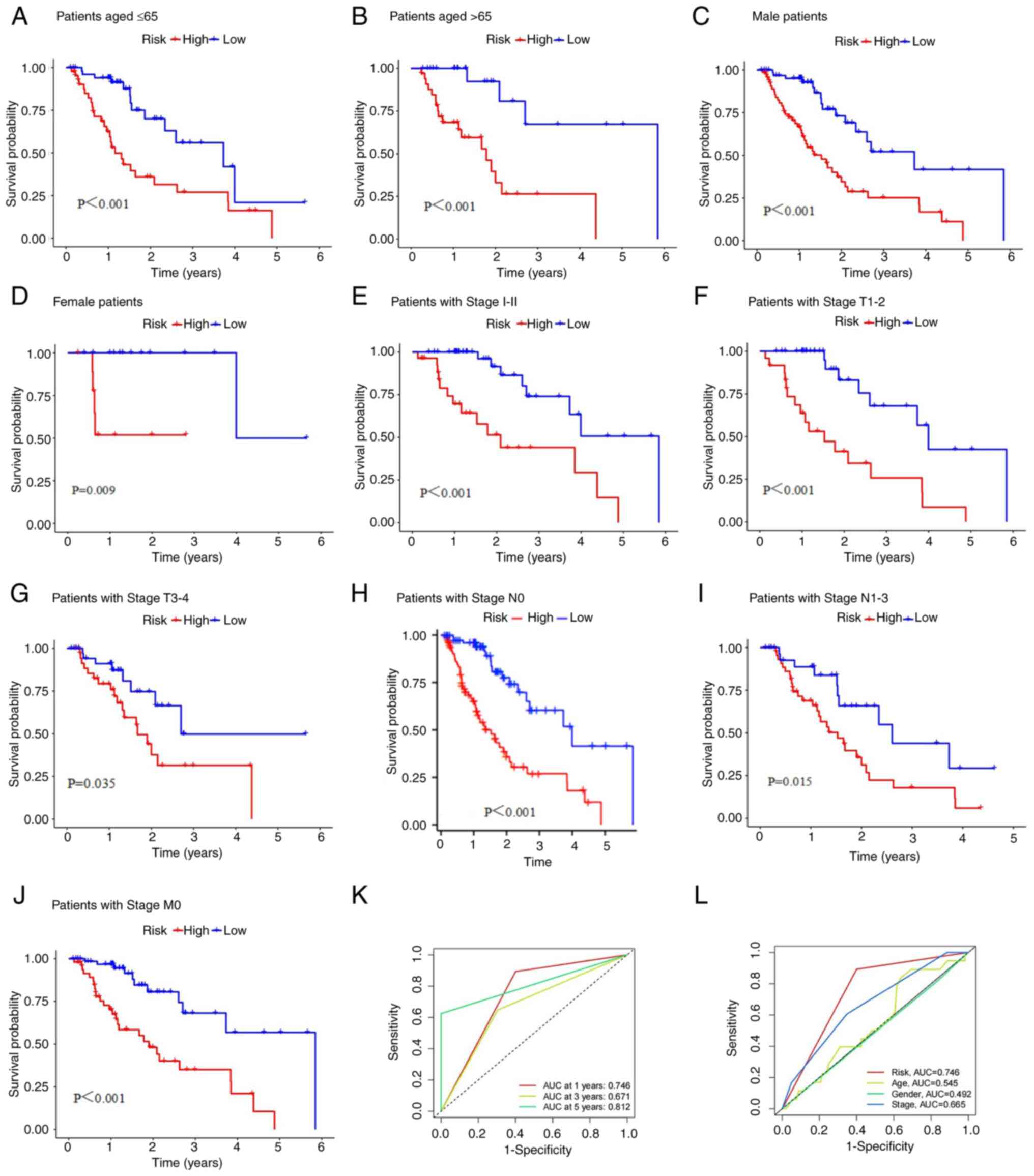 | Figure 4K-M survival curves of low- and
high-risk groups in the entire dataset. (A) Patients aged ≤65, (B)
patients aged >65, (C) male patients, (D) female patients, (E)
patients with Stage I-II, (F) patients with Stage T1-2, (G)
patients with Stage T3-4, (H) patients with Stage N0, (I) patients
with Stage N1-3 and (J) patients with Stage M0. (K) 1-, 3-, and
5-year ROC curves of the entire dataset. (L) ROC curves of the risk
score and clinicopathologic features such as age, sex, and stage.
K-M, Kaplan-Meier; ROC, receiver operating characteristic. |
The ability of the model to predict prognosis was
evaluated independent of clinical characteristics such as age, sex
and stage. As shown in Fig. 5A and
B, Uni-Cox regression and
multi-Cox regression showed that the HR values of the risk score
were 3.594 (95% CI: 1.954 to 6.610) and 2.790 (95% CI: 1.482 to
5.252), respectively. Moreover, the HR values for disease stage
were 2.703 (95% CI: 1.846 to 3.958) in the Uni-Cox regression
analysis and 2.302 (95% CI: 1.523 to 3.481) in the Multi-Cox
regression analysis, indicating that stage, as an independent
prognostic parameter, seems to be the main factor affecting
prognosis.
Based on the risk score and independent clinical
factors, a nomogram was constructed to predict the 1-, 3-, and
5-year survival rates of ESCA patients (Fig. 5C). Calibration plots confirmed the
nomogram predictions had excellent concordance with the actual
observations (Fig. 5D).
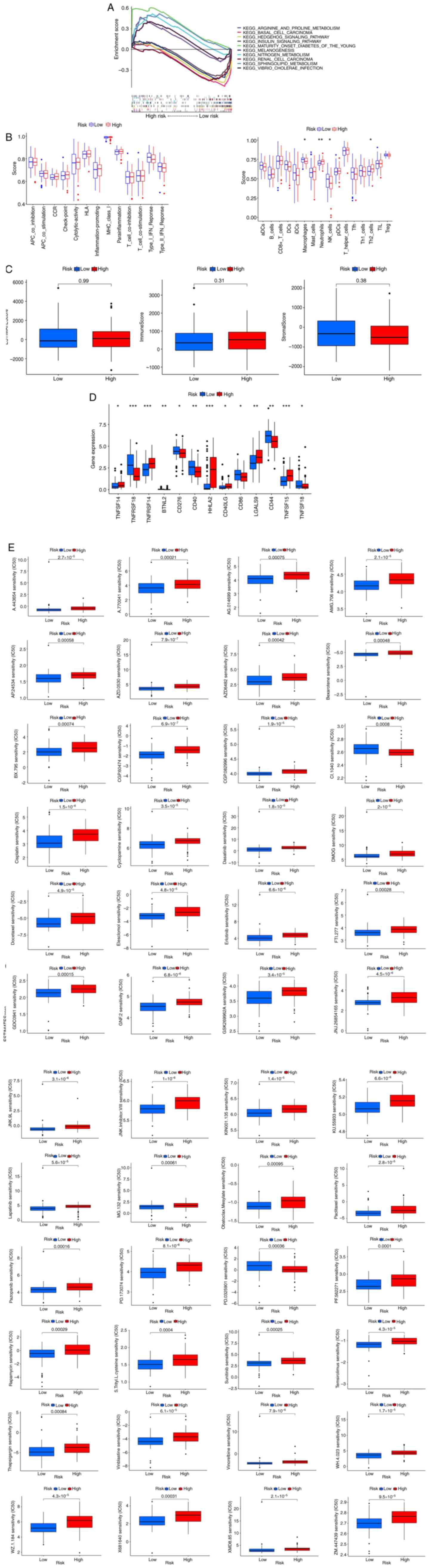
Gene enrichment analysis
To investigate the function of lncRNAs
differentially expressed between the low- and high-risk groups in
the whole set, As shown in Fig. 6A
we explored the Kyoto Encyclopedia of Genes and Genomes (KEGG)
pathways enriched in genes upregulated or downregulated in the
high-risk group. The top 5 KEGG pathways enriched in upregulated
and downregulated genes were highly related to tumor infiltration
and immune escape. The analysis of these 10 pathways showed that
regardless of the FDR value, the P-value was less than 0.05.
Correlations of the risk score with
tumor immunity and microenvironment features
The relationship between the risk score and immune
cell infiltration or functions was further investigated according
to the enrichment score, and we quantified the enrichment scores of
single-sample GSEA for different immune cell subsets, related
functions or pathways. The boxplot suggested that the high-risk
group had more neutrophils and Th2 cells, while the low-risk group
had more macrophages and NK cells. There was no statistically
significant difference in related immune functions between the two
groups (Fig. 6B). We also found no
significant differences in matrix scores, immune scores and
ESTIMATE scores between the two groups (Fig. 6C).
Furthermore, as shown in Fig. 6D, we found that most of the immune
checkpoints (TNFRSF18, BTNL2, CD276, CD40, CD86, CD44, and TNFSF18)
showed greater activation in the low-risk group than in the other
risk groups.
Clinical treatment response of the
risk groups
The potential effective therapeutic drugs in the
high-risk group were predicted by comparing the IC50 values of
drugs in the low- and high-risk groups according to the pRRophetic
method. As shown in Fig. 6E, we
found that 48 chemotherapies or targeted drugs relevant to ESCA
therapy showed lower IC50 values in the high-risk group.
Cluster analysis of prognostic
nrlncRNAs
To further analyze the immune microenvironment and
response of different tumor subtypes, the clusters of ESCA patients
were regrouped. As shown in Fig.
7A, we ultimately used the ‘ConsensusClusterPlus’ package to
divide patients into 3 clusters based on the 8 nrlncRNAs that
constituted the risk model. Moreover, as shown in the Sankey
diagram (Fig. 7B), most low-risk
group patients (blue) were classified into cluster 1. On the other
hand, high-risk group patients were mainly concentrated in cluster
2 and cluster 3. Among them, cluster 1 had a better OS (P=0.016)
than cluster 2 and cluster 3 (Fig.
7C). As shown in Fig. 7D, the
PCA results showed that PCs in the risk group and cluster were
different, and clusters could be clearly distinguished through
t-SNE verification (Fig. 7E). We
also analyzed the correlation between cluster, immune factors and
the TME by the Kruskal-Wallis test followed by Dunn's post hoc
test. However, from the results of the boxplot, there were no
significant differences in the stromal score, immune score and
ESTIMATE score among the three groups (Fig. 7F). As shown in Fig. 7G, the heatmap showed that the
differences in immune cell infiltration in the cluster were in
accordance with the analysis of immune infiltration by various
platforms. Moreover, immune checkpoints, such as CD28, TNFSF14,
TNFRSF14, TNFRSF14, TNFRSF9, LAIR1, HHLA2, LGALS9 and TNFSF15, were
highly expressed in Cluster 3 (Fig.
7H). Finally, in the drug susceptibility comparison, we found
49 immunotherapeutic drugs related to systemic treatment of ESCA
(Fig. 7I) by the Kruskal-Wallis
test followed by Dunn's post hoc test. However, only 6 showed a
significant IC50 difference between the groups.
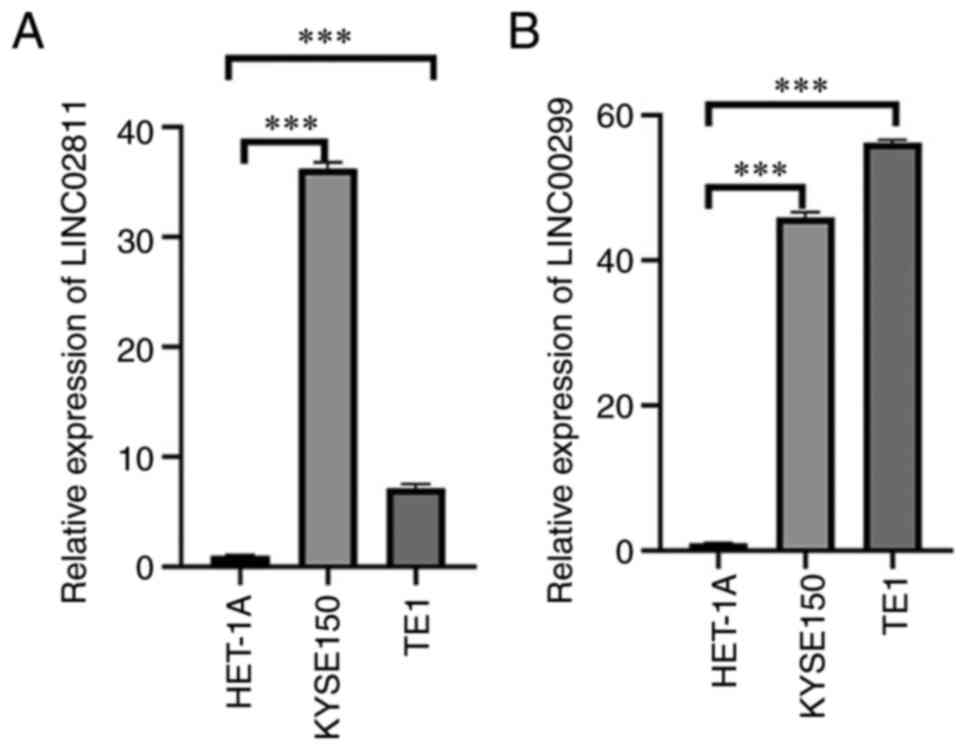
RT-qPCR analysis
Two nrlncRNAs (LINC02811 and LINC00299) were
selected and tested in various cell lines (HET-1A, KYSE150 and
TE1). As shown in Fig. 8, there
were significant differences in the expression levels of these
lncRNAs between tumor and normal cells. All the results showed that
the expression of these factors was different, which further
confirmed the reliability of the risk model.
Discussion
In China, due to the lack of typical symptoms, most
esophageal cancer cases are diagnosed in the middle and late
stages. In recent years, its morbidity and mortality rates have
been increasing. It remains a huge threat to human health (1). Multiple studies have confirmed that
lncRNAs play an important role in esophageal cancer, and their
functions and molecular mechanisms have been explored and
emphasized. Furthermore, studies have confirmed that lncRNAs play
an important indicative role in the diagnosis, treatment and
prognosis evaluation of esophageal cancer, indicating that lncRNAs
could be used as biomarkers and potential targets for esophageal
cancer (40).
LncRNAs play an important biological role in the
occurrence and development of cancer. LncRNAs compete with miRNA
via the competing endogenous RNA (ceRNA) mechanism or directly
regulate downstream proteins. LncRNAs have been found to play these
roles in esophageal cancer (41).
Tang et al performed RNA-Seq experiments on preimmortalized
human embryonic esophageal epithelial SHEE cells at passages 26 and
79 and found that LINC00885 could play a positive regulatory role
in esophageal cancer (42). It is
well known that lncRNAs affect not only the tumorigenesis and
progression of ESCA but also the sensitivity of cancer cells to
chemoradiotherapy. Therefore, reducing resistance to
chemoradiotherapy by regulating lncRNA expression in ESCA patients
could be regarded as a new way to enhance the therapeutic effect
and prevent the development of tumors. Li et al (43) transplanted MALAT1-overexpressing
cells into nude mice to induce tumor formation and then treated the
tumors with radiotherapy. The results showed that MALAT1
overexpression could suppress radiation-induced apoptosis of tumor
cells and enhance the resistance of tumor cells to radiotherapy,
whereas another study reported that MALAT1 silencing could enhance
the sensitivity of tumor cells to radiotherapy (44). In recent years, more studies have
emphasized the key role of lncRNAs in ESCA, but the relationship
between lncRNAs and ESCA has not been fully clarified and is a
research hotspot.
Currently, it has been reported that the
tumorigenesis and tumor progression are closely related to tumor
immune function, and necroptosis has been confirmed to play an
important role in this process (45,46).
McComb et al demonstrated that necrosis-related molecules
(CIAPs) effectively reduce macrophage necrosis and enhance the
body's response to pathogen invasion (47). Although an increasing number of
studies have shown that necroptosis is closely involved in
tumorigenesis, its mechanism has not been fully elucidated. In the
present study, we found that necrosis-associated lncRNAs could be
used to divide patients into different subgroups, illustrating for
the first time their usefulness as prognostic markers. We also
systematically investigated the correlation between the tumor
microenvironment, immune cell infiltration and immune checkpoints,
which is expected to be applied to clinical diagnosis and treatment
in the future. We also systematically investigated the associations
between the expression of these lncRNAs and tumor microenvironment
features and immune checkpoint expression to assess their potential
application in clinical diagnosis and treatment in the future.
Our results showed that 34 nrlncRNAs affected the
survival of HNSCC patients, and 8 lncRNAs, including LINC00299,
AC090912.2, AC244197.2, AL158166.1, AC079684.1, AP003696.1,
LINC02811 and AC026741.1, could be used as prognostic markers. Liu
et al identified that Linc00299 regulates the migration and
proliferation of endothelial cells and vascular smooth muscle cells
(VSMCs) during atherosclerosis by regulating miR-490-3p, which
targets Aurora kinase A (AURKA) (48). Chang et al studied the role
of the LINC00299/miR-135a-5p/XBP1 axis in the regulation of
atherosclerosis progression, ox-LDL-induced oxidative damage, and
human aortic vascular smooth muscle cell migration and invasion
(49). Moreover, Manoochehri et
al found that the occurrence of TNBC was often accompanied by
hypermethylation of LINC00299 in young women, indicating a
potential association between them (50). Talkowski et al found a
functional role for these specific noncoding RNAs in brain
development in subjects with LINC00299 mutations and raised the
possibility that lncRNAs, as a class of abnormalities, might play
an important role in human developmental disorders (51). Li et al suggested that
LINC00299 may be an important regulator in human hypertrophic scars
(HSs) that exerts its effects via its coexpressed genes (52). The other seven genes have not been
further reported in the literature.
The 1-, 3-, and 5-year AUC values for the risk score
model were significantly higher than those for models based on
other clinical characteristics (age, sex and stage), suggesting
that risk score model performs better in predicting the prognosis
of ESCA patients. Cox regression analysis showed that the risk
score of ESCA patients was an independent risk indicator and
negatively correlated with OS. Furthermore, the correlation of a
high-risk score with older age, later T and N stages and worse
prognosis suggested that the risk score plays a crucial role in the
classification of patient survival status. In addition, four
independent factors, including risk score, age, sex and stage, were
used to establish a nomogram prediction model for OS, and the
calibration plots of 1-, 3-, and 5-year OS showed high consistency
between the predictions and actual observations. Hence, all results
indicated that the risk model is robust and an effective indicator
of outcomes in ESCA patients.
In recent years, an increasing number of scholars
have focused on the function and application of immune checkpoint
inhibitors. Clinical trials have shown that immunotherapy
strategies in ESCA patients may shift from metastatic therapy to
neoadjuvant/adjuvant therapy. The addition of immunotherapy brings
a good survival benefit to ESCA patients (53). Previous studies have shown that the
tumor microenvironment has a significant influence on immunotherap
(54,55). Immune infiltration analysis showed
that CD4+ T cells and other infiltrating immune cells were more
abundant in the low-risk group. Moreover, regulatory T cells
(Tregs) are immunosuppressive subsets of CD4+ T cells that play an
important role in maintaining self-tolerance and immune
homeostasis. In tumor immunity, Tregs disrupt the immune
surveillance of cancer in healthy individuals and impair the
antitumor immune response of tumor-bearing hosts. Thus, this
evidence suggests that Tregs accelerate the immune escape of tumor
cells and further lead to various types of tumor development and
progression (56). It has been
demonstrated that tumor markers play an important role in ESCA by
analyzing tumor immunity and immune cell levels. All results
suggest that necrosis-associated lncRNAs may be associated with
tumor immune infiltration. Our study predicted compounds that may
be useful for the clinical treatment of diseases, which could
provide a meaningful reference for future exploration of new
therapeutic targets.
We divided the ESCA patients into 3 clusters, and
the survival analysis showed that cluster 1 had better survival
than the other groups (P=0.016). Moreover, immune checkpoints, such
as CD28, TNFSF14, TNFRSF14, TNFRSF14, TNFRSF9, LAIR1, HHLA2, LGALS9
and TNFSF15, displayed higher expression in cluster 3. These
analysis results suggest that these immune checkpoints are closely
related to both patient prognosis and the immune microenvironment,
suggesting that immunotherapy can be used as a new treatment for
ESCA patients. In addition, drug susceptibility analysis results
suggested that risk models and tumor subtypes could be used to
guide ESCA treatment decision making.
We performed PCR verification of the selected
nrlncRNAs and concluded that LINC02811 and LINC00299 were
significantly differentially expressed between tumor and normal
cells. Thus, they may be markers of prognosis and immune invasion
in esophageal cancer. Further mechanistic research is still
needed.
These studies provide meaningful references for the
identification of new therapeutic targets in the future. However,
there are several limitations to our study. First, all of our data
came from the TCGA. Although risk profiles are valuable in
assessing prognosis, there is a lack of lncRNA data from other
databases and clinical information for external cohort. However, we
might obtain different results by combining data from other
databases. Second, we verified nrlncRNAs by cell RT-qPCR analysis,
but PCR assessment of clinical samples in a future study is needed.
the functions and mechanisms of these RNAs need further study.
Finally, there is a lack of clinical follow-up data to confirm the
value of our prognostic model. In addition, extensive clinical
trials are needed to confirm the relationship between risk score
and the response to immunotherapy and chemotherapy. Nevertheless,
immune cell correlation analysis based on various experimental
platforms can be regarded as external validation in some sense.
Thus, all evidence indicates that the nrlncRNA risk model can be
considered reliable and acceptable. In the future, large-scale
studies should further focus on immunotherapy and chemotherapy
based on bioinformatics analysis.
In this paper, we systematically evaluated the value
and function of lncRNAs associated with necrosis in predicting
survival, tumor microenvironment features and immune cell
infiltration. We also analyzed the potential regulatory mechanisms
of lncRNAs associated with necrosis, as well as the anticipated
response to immunotherapy and chemotherapy agents for ESCA. Eight
nrlncRNAs could predict the survival of patients with ESCA and may
promote the development of personalized and precision therapies for
ESCA patients. According to cell RT-qPCR analysis, LINC02811 and
LINC00299 may be markers of esophageal cancer prognosis and immune
invasion.
Acknowledgements
Not applicable.
Funding
Funding: This work was supported by the Medical Science Research
Project of Hebei Province (grant no. 20230971).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request. The RNA sequencing transcriptome data of patients with
ESCA were downloaded from The Cancer Genome Atlas-ESCA dataset
(https://portal.gdc.cancer.gov/).
Authors' contributions
XD, HD and JS conceived and designed the study. XD
and JS confirm the authenticity of all raw data. XD, HD, MY, LL and
RL downloaded and analyzed the data. XD and HD performed
experiments and wrote the manuscript. XD, HD, MY, LL and RL
organized the figures and tables. JS reviewed and revised the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Sung H, Ferlay J, Siegel RL, Laversanne M,
Soerjomataram I, Jemal A and Bray F: Global Cancer Statistics 2020:
GLOBOCAN estimates of incidence and mortality worldwide for 36
cancers in 185 countries. CA Cancer J Clin. 71:209–249.
2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Abnet CC, Arnold M and Wei WQ:
Epidemiology of esophageal squamous cell carcinoma.
Gastroenterology. 154:360–373. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Long Z, Liu W, Lin L, et al: Disease
burden analysis of esophageal cancer in China from 1990 to 2017.
Chin J Chronic Disease Prevention Control. 29:571–575, 581.
2021.
|
|
4
|
Li MX, Cheng WG, Chen P, et al:
Identification and prognostic analysis of key genes in lymph node
metastasis of esophageal squamous cell carcinoma. J Natural Sci Hun
Normal Univ. 44:92–100. 201.
|
|
5
|
Short MW, Burgers KG and Fry VT:
Esophageal cancer. Am Fam Physician. 95:22–28. 2017.PubMed/NCBI
|
|
6
|
Kyle JN, Mary S and Subhasis M: Esophageal
cancer: A review of epidemiology, pathogenesis, staging workup and
treatment modalities. World J Gastrointest Oncol. 6:112–120.
2014.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Degterev A, Huang Z, Boyce M, Li Y, Jagtap
P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA and Yuan J:
Chemical inhibitor of nonapoptotic cell death with therapeutic
potential for ischemic brain injury. Nat Chem Biol. 1:112–119.
2005.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Marshall KD and Baines CP: Necroptosis: Is
there a role for mitochondria? Front Physiol. 5:323–328.
2014.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Declercq W, Vanden BT and Vandenabeele P:
RIP kinases at the crossroads of cell death and survival. Cell.
138:229–232. 2009.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Krysko O, Aaes TL, Kagan VE, D'Herde K,
Bachert C, Leybaert L, Vandenabeele P and Krysko DV: Necroptotic
cell death in anti-cancer therapy. Immunol Rev. 280:207–219.
2017.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Jiao D, Cai Z, Choksi S, Ma D, Choe M,
Kwon HJ, Baik JY, Rowan BG, Liu C and Liu ZG: Necroptosis of tumor
cells leads to tumor necrosis and promotes tumor metastasis. Cell
Res. 28:868–870. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Gong Y, Fan Z, Luo G, Yang C, Huang Q, Fan
K, Cheng H, Jin K, Ni Q, Yu X and Liu C: The role of necroptosis in
cancer biology and therapy. Mol Cancer. 18(100)2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Snyder AG, Hubbard NW, Messmer MN, Kofman
SB, Hagan CE, Orozco SL, Chiang K, Daniels BP, Baker D and Oberst
A: Intratumoral acti-vation of the necroptotic pathway components
RIPK1 and RIPK3potentiates anti tumor immunity. Sci Immunol.
4(eaaw2004)2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Yang Z, Jiang B, Wang Y, Ni H, Zhang J,
Xia J, Shi M, Hung LM, Ruan J, Mak TW, et al: 2-HG inhibits
necroptosis by Stim-ulating dnmt1-dependent hypermethylation of the
RIP3 promoter. Cell Rep. 19:1846–1857. 2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Xu Y, Li Y, Jin J, Han G, Sun C, Pizzi MP,
Huo L, Scott A, Wang Y, Ma L, et al: LncRNA PVT1 up-regulation is a
poor prognosticator and serves as a therapeutic target in
esophageal adenocarcinoma. Mol Cancer. 18(141)2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Nugues A, ElBouazzati H, Hétuin D, Berthon
C, Loyens A, Bertrand E, Jouy N, Idziorek T and Quesnel B: RIP3 is
downregulated in human myeloid leukemia cells and modulates
apoptosis and caspase-mediated p65/RelA cleavage. Cell Death Dis.
5(e1384)2014.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Höckendorf U, Yabal M, Herold T,
Munkhbaatar E, Rott S, Jilg S, Kauschinger J, Magnani G, Reisinger
F, Heuser M, et al: RIPK3 restricts myeloid leukemogenesis by
promoting cell death and differentiation of leukemia initiating
Cells. Cancer Cell. 30:75–91. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Koo G, Morgan MJ, Lee D, Kim WJ, Yoon JH,
Koo JS, Kim SI, Kim SJ, Son MK, Hong SS, et al:
Methylation-dependent loss of RIP3 expression in cancer represses
programmed necrosis in response to chemotherapeutics. Cell Re.
25:707–725. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Bozec D, luga AC, Roda G, Dahan S and
Yeretssian G: Critical function of thenecroptosis adaptor RIPK3 in
protecting from intestinal tumorigenesis. Oncotarget.
7:46384–46400. 2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Liu X, Zhou M, Mei L, Ruan J, Hu Q, Peng
J, Su H, Liao H, Liu S, Liu W, et al: Key roles of necroptotic
factors in promoting tumor growth. Oncotarget. 7:22219–22233.
2016.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Schmidt SV, Seibert S, Walch-Ruckheim B,
Vicinus B, Kamionka EM, Pahne-Zeppenfeld J, Solomayer EF, Kim YJ,
Bohle RM, Smola S, et al: RIPK3 expression in cervical cancer cells
is required for PolyIC-induced necroptosis, IL-1α release, and
efficient paracrine dendritic cell activation. Oncotarget.
6:8635–8647. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Takemura R, Takaki H, Okada S, Shime H,
Akazawa T, Oshiumi H, Matsumoto M, Teshima T and Seya T: Poly1:
C-induced, TLR3 RIP3-dependent necroptosis backs up immune
effector-mediated tumor elimination in vivo. Cancer Immunol Res.
3:902–914. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Yatim N, Jusforgues-Saklani H, Orozco S,
Schulz O, Barreira da Silva R, Reis e Sousa C, Green DR, Oberst A
and Albert ML: RIPKI and NF-κB signaling in dying cells determines
cross-priming of CD8+Tcells. Science. 350:328–334.
2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kang YJ, Bang BR, Han KH, Hong L, Shim EJ,
Ma J, Lerner RA and Otsuka M: Regulation of NKT cell mediated
immune responses to tumours and liver inflammation by mitochondrial
PGAM5-Drpl signalling. Nat Commun. 6(8371)2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Strilic B, Yang L, Albarrán-Juárez J,
Wachsmuth L, Han K, Müller UC, Pasparakis M and Offermanns S:
Tumour-cell-induced endothelial cell necroptosis via death receptor
6 promotes metastasis. Nature. 536:215–218. 2016.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Seifert L, Werba G, Tiwari S, Giao Ly NN,
Alothman S, Alqunaibit D, Avanzi A, Barilla R, Daley D, Greco SH,
et al: The necrosome promotes pancreatic oncogenesis via CXCL1 and
Mincle-induced immune suppression. Nature. 532:245–249.
2016.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Dragomir MP, Kopetz S, Ajani JA and Calin
GA: Non-coding RNAs in GI cancers: From cancer hallmarks to
clinical utility. Gut. 69:748–763. 2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Zhao Z, Liu H, Zhou X, Fang D, Ou X, Ye J,
Peng J and Xu J: Necroptosis-Related lncRNAs: Predicting prognosis
and the distinction between the cold and hot tumors in gastric
cancer. J Oncol. 2021(6718443)2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Xue ST, Zheng B, Cao SQ, Ding JC, Hu GS,
Liu W and Chen C: Long non-coding RNA LINC00680 functions as a
ceRNA to promote esophageal squamous cell carcinoma progression
through the miR-423-5p/PAK6 axis. Mol Cancer. 21(69)2022.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Xu JH, Chen RZ, Liu LY, Li XM, Wu CP, Zhou
YT, Yan JD and Zhang ZY: LncRNA ZEB2-AS1 promotes the
proliferation, migration and invasion of esophageal squamous cell
carcinoma cell through miR-574-3p/HMGA2 axis. Eur Rev Med Pharmacol
Sci. 25(3397)2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Wang K, Liu F, Liu CY, An T, Zhang J, Zhou
LY, Wang M, Dong YH, Li N, Gao JN, et al: The long noncoding RNA
NRF regulates programmed necrosis and myocardial injury during
ischemia and reperfusion by targeting miR-873. Cell Death Differ.
23:1394–1405. 2016.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Huang J, Xu Z, Teh BM, Zhou C, Yuan Z, Shi
Y and Shen Y: Construction of a necroptosis-related lncRNA
signature to predict the prognosis and immune microenvironment of
head and neck squamous cell carcinoma. J Clin Lab Anal.
36(e24480)2022.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Luo L, Li L, Liu L, Feng Z, Zeng Q, Shu X,
Cao Y and Li Z: A Necroptosis-Related lncRNA-Based signature to
predict prognosis and probe molecular characteristics of stomach
adenocarcinoma. Front Genet. 13(833928)2022.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Liu L, Huang L, Chen W, Zhang G, Li Y, Wu
Y, Xiong J and Jie Z: Comprehensive Analysis of Necroptosis-Related
Long Noncoding RNA immune infiltration and prediction of prognosis
in patients with colon cancer. Front Mol Biosci.
9(811269)2022.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Chen F, Yang J, Fang M, Wu Y, Su D and
Sheng Y: Necroptosis-related lncRNA to establish novel prognostic
signature and predict the immunotherapy response in breast cancer.
J Clin Lab Anal. 36(e24302)2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Yatim N, Jusforgues-Saklani H, Orozco S,
Schulz O, Barreira da Silva R, Reis e Sousa C, Green DR, Oberst A
and Albert ML: RIPK1 and NF-κB signaling in dying cells determines
cross-priming of CD8 + T cells. Science. 350:328–334.
2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Zhao Z, Liu H, Zhou X, Fang D, Ou X, Ye J,
Peng J and Xu J: Necroptosis-related lncRNAs: Predicting prognosis
and the distinction between the cold and hot tumors in gastric
cancer. J Oncol. 2021(6718443)2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Hong W, Liang L, Gu Y, Qi Z, Qiu H, Yang
X, Zeng W, Ma L and Xie J: Immune-related lncRNA to construct novel
signature and predict the immune landscape of human hepatocellular
carcinoma. Mol Ther Nucleic Acids. 22:937–947. 2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Geeleher P, Cox NJ and Huang RS: Clinical
drug response can be predicted using baseline gene expression
levels and in vitro drug sensitivity in cell lines. Genome Biology.
15(R47)2014.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Yang C and Chen K: Long Non-Coding RNA in
esophageal cancer: A review of research progress. Pathol Oncol Res.
28(1610140)2022.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Jin-Ming T and Wen-Xiang W: The role of
LNCRNA ZFAS1 in esophageal Carcinoma and its mechanism. Chin J
Biol. 762–768. 2020.
|
|
42
|
Tang D, Wang B, Khodahemmati S, Li J, Zhou
Z, Gao J, Sheng W and Zeng Y: A transcriptomic analysis of
malignant transformation of human embryonic esophageal epithelial
cells by HPV18 E6E7. Transl Cancer Res. 9:1818–1832.
2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Li Z, Zhou Y, Tu B, Bu Y, Liu A and Kong
J: Long noncoding RNA MALAT1 affects the efficacy of radiotherapy
for esophageal squamous cell carcinoma by regulating Cks1
expression. J Oral Pathol Med. 46:583–590. 2017.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Farooqi A, Legaki E, Gazouli M, Rinaldi S
and Berardi R: MALAT1 as a versatile regulator of cancer: Overview
of the updates from predatory role as competitive endogenous RNA to
mechanistic insights. Curr Cancer Drug Targets: Jul 30, 2020. doi:
10.2174/1568009620999200730183110 (Epub ahead of print).
|
|
45
|
Seifert L, Werba G, Tiwari S, Giao Ly NN,
Alothman S, Alqunaibit D, Avanzi A, Barilla R, Daley D, Greco SH,
et al: The necrosome promotes pancreatic oncogenesis via CXCL1 and
Mincle-Induced immune suppression. Nature. 532:245–249.
2016.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Snyder AG, Hubbard NW, Messmer MN, Kofman
SB, Hagan CE, Orozco SL, Chiang K, Daniels BP, Baker D and Oberst
A: Intratumoral activation of the necroptotic pathway components
RIPK1 and RIPK3 potentiates antitumor immunity. Sci Immunol.
4(eaaw2004)2019.PubMed/NCBI View Article : Google Scholar
|
|
47
|
McComb S, Cheung HH, Korneluk RG, Wang S,
Krishnan L and Sad S: cIAP1 and cIAP2 limit macrophage necroptosis
by inhibiting Rip1and Rip3 activation. Cell Death Differ.
19:1791–1801. 2012.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Liu Y, Chen Y, Tan L, Zhao H and Xiao N:
Linc00299/miR-490-3p/AURKA axis regulates cell growth and migration
in atherosclerosis. Heart Vessels. 34:1370–1380. 2019.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Chang M, Liu G, Wang Y, Lv H and Jin Y:
Long non-coding RNA LINC00299 knockdown inhibits ox-LDL-induced T/G
HA-VSMC injury by regulating miR-135a-5p/XBP1 axis in
atherosclerosis. Panminerva Med. 64:38–47. 2022.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Manoochehri M, Jones M, Tomczyk K,
Fletcher O, Schoemaker MJ, Swerdlow AJ, Borhani N and Hamann U: DNA
methylation of the long intergenic noncoding RNA 299 gene in
triple-negative breast cancer: Results from a prospective study.
Sci Rep. 10(11762)2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Talkowski ME, Maussion G, Crapper L,
Rosenfeld JA, Blumenthal I, Hanscom C, Chiang C, Lindgren A,
Pereira S, Ruderfer D, et al: Disruption of a large intergenic
noncoding RNA in subjects with neurodevelopmental disabilities. Am
J Hum Genet. 91:1128–1134. 2012.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Li M, Wang J, Liu D and Huang H: High
throughput sequencing reveals differentially expressed lncRNAs and
circRNAs, and their associated functional network, in human
hypertrophic scars. Mol Med Rep. 18:5669–5682. 2018.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Kakeji Y, Oshikiri T, Takiguchi G, Kanaji
S, Matsuda T, Nakamura T and Suzuki S: Multimodality approaches to
control esophageal cancer: Development of chemoradiotherapy,
chemotherapy, and immunotherapy. Esophagus. 18:25–32.
2021.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Frankel T, Lanfranca MP and Zou W: The
role of tumor microenvironment in cancer immunotherapy. Adv Exp Med
Biol. 1036:51–64. 2017.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Pitt JM, Marabelle A, Eggermont A, Soria
JC, Kroemer G and Zitvogel L: Targeting the tumor microenvironment:
Removing obstruction to anticancer immune responses and
immunotherapy. Ann Oncol. 27:1482–1492. 2016.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Nishikawa H and Koyama S: Mechanisms of
regulatory T cell infiltration in tumors: Implications for
innovative immune precision therapies. J Immunother Cancer.
9(e002591)2021.PubMed/NCBI View Article : Google Scholar
|