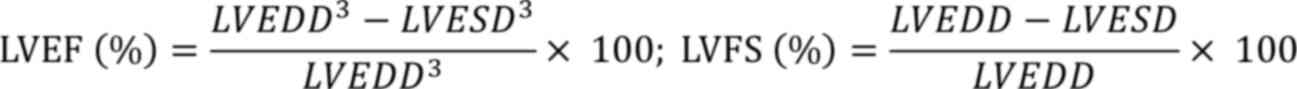Introduction
As a classical antitumor drug, doxorubicin (DOX)
serves a key role in chemotherapy against a number of tumor types
such as leukemia, lymphoma and breast cancer (1,2).
However, DOX treatment may also induce cardiomyopathy, which
adversely affects patient outcome (3,4).
DOX-induced cardiomyopathy causes severe cardiac dysfunction, which
mainly manifests as a decrease in cardiac ejection fraction
(5). DOX-induced cardiomyopathy is
an acute complication of long-term chemotherapy and can lead to an
increased mortality rate in patients (6). The pathogenesis of DOX-induced
cardiomyopathy is complex and remains unclear. Due to the lack of
specific therapeutic drugs, patients with DOX-induced
cardiomyopathy can only been given symptomatic relief and
supportive treatment (7).
Therefore, it is necessary to explore the pathophysiological
processes of DOX-induced cardiomyopathy and investigate potential
efficacious approaches for treatment.
1,25(OH)2D3 is a metabolic
derivative of vitamin D3 that is also a steroid hormone (8). 25-hydroxyvitamin D3
[25(OH)D3] is the major circulating form of vitamin D.
25(OH)D3 metabolizes to
1,25(OH)2D3 by catalysis of the renal
25(OH)D1α hydroxylase (CYP27B1) (9). 1,25(OH)2D3
serves an important role in regulating calcium homeostasis, bone
metabolism and the immune system (10,11).
Previous studies have reported that CYP27B1 is mainly expressed in
the kidney, but an increasing number of studies have reported that
CYP27B1 is widely expressed in multiple organs and various cell
types, such as the lungs, intestines, skin, keratinocytes,
macrophages and osteoblasts, indicating that the targets of
1,25(OH)2D3 are extensive and complex
(12,13). Vitamin D has also been reported to
exert protective effects against cardiovascular disease (14) and diabetic cardiomyopathy (15). Patients with dilated cardiomyopathy
tend to have lower levels of 25-hydroxyvitamin D3 in the serum
compared with those subjects without dilated cardiomyopathy
(16). The mechanism by which
vitamin D protects against cardiomyopathy is mainly attributed to
its antioxidant, anti-inflammatory and inhibitory effects on the
renin-angiotensin system (17,18).
However, whether 1,25(OH)2D3 can protect
against the pathophysiological processes of DOX-induced
cardiomyopathy and its mechanisms remain unknown.
The nod-like receptor family pyrin domain containing
3 (NLRP3) inflammasome is a protein complex that recognizes a
diverse array of extracellular and intracellular signals, including
damage-associated molecular patterns and pathogen-associated
molecular patterns (19).
Activation of the NLRP3 inflammasome pathway induces production of
proinflammatory cytokines, such as IL-1β and IL-18, to generate a
proinflammatory microenvironment and inflamm-ageing (20). However, the relationship between
1,25(OH)2D3 and the NLRP3 inflammasome
pathway in DOX-induced cardiomyopathy remains unclear.
Oxidative stress has recently been widely recognized
to be an important factor for DOX-induced cardiomyopathy (21). An imbalance between reactive oxygen
species (ROS) and the antioxidant pathway may lead to ROS
accumulation in cardiomyocytes. This can then induce damage to
mitochondrial membrane proteins and DNA, thereby inhibiting ATP
production in mitochondria by disrupting mitochondrial oxidative
phosphorylation (22). Increased
levels of ROS can cause lipid peroxidation in mitochondria,
apoptosis and an excessive inflammatory response in cardiomyocytes,
ultimately resulting in cardiac dysfunction (23). A previous study has demonstrated
the antioxidant effect of 1,25(OH)2D3 in a
number of disease settings, such as osteoporosis (24) and chronic kidney disease (25), in addition to skin senescence
(18). Vitamin D3 1α(OH)ase
knockout mice demonstrated a significant increase in ROS and
H2O2 levels, in addition to aggravation of
colon inflammation due to the extensive secretion of
senescence-associated inflammatory cytokines (26). A previous study has reported that
1,25(OH)2D3 can activate nuclear erythroid
2-related factor 2 (Nrf2) pathways to activate enhancer of zeste
homolog 2-mediated H3K27me3 histone modification,
mediated by EZH2, to regulate downstream target genes (such as p16,
p21, p53 and p19) transcriptional expression (27). However, little is known about the
mechanism by which 1,25(OH)2D3 regulates
DOX-induced cardiomyopathy.
To investigate this issue and its underlying
molecular mechanisms, 10-week-old wild-type mice were administered
DOX to construct a DOX-induced cardiomyopathy model in the present
study. These mice were also administered with
1,25(OH)2D3. Echocardiography, histological
and molecular analysis were used to investigate the effect of
1,25(OH)2D3 on DOX-induced cardiomyopathy and
its potential mechanism.
Materials and methods
Drugs
DOX (cat. no. A603456) was purchased from Sangon
Biotech Co., Ltd. DOX was diluted with normal saline to a
concentration of 2 mg/ml for the in vivo study and to 43 µM
for in vitro study. 1,25(OH)2D3 (cat.
no. D1530) was purchased from Sigma-Aldrich; Merck KGaA.
1,25(OH)2D3 was diluted with normal saline to
a concentration of 0.25 µg/ml for the in vivo study and 24
µM for the in vitro study.
Animals and experimental
procedure
All animal studies were approved by Animal Ethical
and Welfare Committee of Nanjing Medical University (approval no.
IACUC-1707002) and were performed in accordance with the
institutional and national regulations.
C57BL/6J mice (male; n=30; 10 weeks old; 23.81±0.34
g) were purchased from the Model Animal Research Center of Nanjing
University and housed at 22-24˚C and in a relative humidity of
50±10% under a 12-h light/dark cycle. All the mice had free access
to standard chow and water.
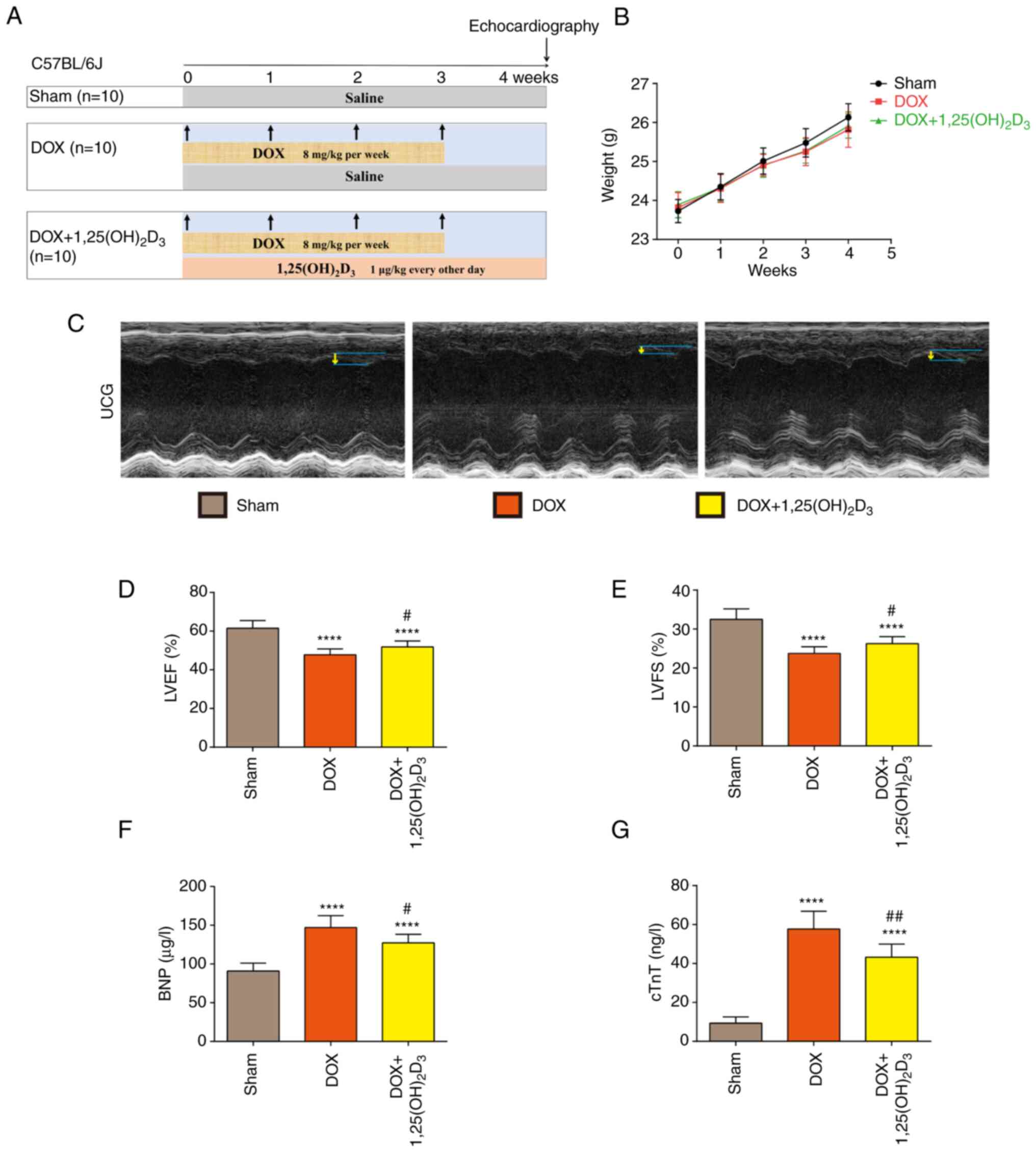
The schematic diagram of experimental procedure in
the present study is presented in Fig.
1A. After a 1-week acclimation period, mice were randomly
divided into three groups (n=10/group). Mice in the DOX-treated
group (DOX) were injected intraperitoneally with DOX at a dose of 8
mg/kg per week for 4 weeks and with saline every other day for 4
weeks (28). Mice in the DOX +
1,25(OH)2D3-treated group [DOX +
1,25(OH)2D3] were injected intraperitoneally
with DOX at the aforementioned dose and with
1,25(OH)2D3 at a dose of 1 µg/kg every other
day for 4 weeks (29). Mice in the
Sham group were injected intraperitoneally with saline every other
day for 4 weeks. The body weight of each mouse in the Sham, DOX or
DOX + 1,25(OH)2D3 groups was measured every
week. All mice were sacrificed by CO2 (50% volume
replacement rate/min) 4 weeks after the first injection of DOX when
echocardiography and serum collection was complete.
The humane endpoints used to determine when animals
should be immediately euthanized prior to the scheduled
experimental endpoint were as follows: Weight loss of 20%; appetite
loss for 24 h or poor appetite (<50% of normal appetite) for 3
days; weakness (inability to eat, drink or stand or extreme
difficulty standing); impending death (depressive state with
hypothermia); severe diseases of the cutaneous system (uncurable
confrontation wound or repetitive self-injury); and severe diseases
of the circulatory system (severe hemorrhage or rapid heart rate
with marked dyspnea). No mice reached these humane endpoints or
died prior to the scheduled experimental endpoint during this
4-week period.
Echocardiography
Echocardiography was performed in the Animal Core
Facility of Nanjing Medical University (Nanjing, China) using the
high-frequency color ultrasound system (40 µm resolution, 500
frames per second) Vevo 2100 (VisualSonics, Inc.). Cardiac function
was evaluated in week 4 after the first injection of DOX was
administered. Mice were anesthetized by inhaling isoflurane at a
1:1 concentration with oxygen (4% induction concentration, 1.5%
maintenance concentration). Anesthetized mice were fixed in the
supine position on a constant-temperature workbench at 25-26˚C.
Hair on the chest area was removed using a leather knife and a
simulated electrocardiogram was linked. Left ventricular
end-diastolic diameter (LVEDD) and left ventricular end-systolic
diameter (LVESD) were measured in the long axis and short axis,
respectively. The following formulas were used to calculate left
ventricular ejection fraction (LVEF) and left ventricular
fractional shortening (LVFS), respectively.
Enzyme-linked immunosorbent assay
(ELISA)
Serum samples of mice were collected in the Animal
Core Facility of Nanjing Medical University in week 4 after the
first injection of DOX. Briefly, mice were anesthetized by the
intraperitoneal injection of 1% pentobarbital sodium (50 mg/kg),
the hair around the eyeball was clipped and blood was then
collected by removing the eyeball. A 0.75-ml volume of blood was
collected from each mouse. Subsequently, mice were euthanized using
CO2. Serum was obtained by centrifugation (1,500 x g for
10 min at 4˚C) after the blood was left for 1 h at 4˚C for
component separation. Brain natriuretic peptide (BNP; cat. no.
YFXEM00760) and cardiac troponin T (cTnT; cat. no. YFXEM00789)
levels in serum were quantified using ELISA kits (Nanjing Yifeixue
Biotechnology Co., Ltd.) according to the manufacturer's
protocols.
Measurement of antioxidant enzymes and
lipid peroxidation
Myocardial tissues from the left ventricle were
prepared as a 10% homogenate with normal saline, followed by
centrifugation at 4˚C at 2,000 x g for 15 min. Malondialdehyde
(MDA; cat. no. A003-1-1), glutathione peroxidase (GSH-Px; cat. no.
A005-1-1) and total superoxide dismutase (T-SOD; cat. no. A001-1-1)
in the tissue supernatants were measured using assay kits according
to the manufacturer's protocols (Nanjing Jiancheng Bioengineering
Institute).
Masson's trichrome staining
Hearts were harvested and fixed in 4%
paraformaldehyde at room temperature for 24 h. After standard
paraffin embedding (75% alcohol for 4 h, 85% alcohol for 2 h, 90%
alcohol for 2 h, 95% alcohol for 1 h, ethyl alcohol I for 30 min,
ethyl alcohol II for 30 min, ethyl alcohol and xylene for 10 min,
xylene I for 10 min, xylene II for 10 min, 65˚C melted paraffin I
for 1 h, 65˚C melted paraffin II for 1 h and 65˚C melted paraffin
III for 1 h), heart tissues were cut into 4-µm sequential sections.
Masson's trichrome staining (iron hematoxylin for 3 min, lichun red
acid fuchsin for 8 min and aniline blue for 5 min, at room
temperature) was used to evaluate the degree of myocardial
fibrosis. The representative histopathological samples were imaged
using a light microscope (Nikon Corporation) and analyzed using
Image Pro-Plus software (version 6.0; Media Cybernetics, Inc.). A
total of three images from randomly selected non-overlapping fields
were captured per section at x400 magnification and the collagen
tissue area was expressed as the percentage of the full ventricle
area.
Immunohistochemistry
The 4-µm heart paraffin sections were
deparaffinized, rehydrated in alcohol (ethyl alcohol for 1 min, 95%
alcohol I for 1 min, 95% alcohol II for 1 min and 70% alcohol for 1
min) and washed in tap water. The tissue sections were then boiled
for antigen retrieval in a citrate-EDTA antigen retrieval solution
(cat. no. P0083; Beyotime Institute of Biotechnology) in a steamer
for 30 min at 95-100˚C. Endogenous peroxidase activity was then
blocked with methanol and hydrogen peroxide (1:10) for 10 min at
room temperature. After washing with TBS (pH 7.6), the sections
were blocked with 10% normal goat serum (cat. no. C0265; Beyotime
Institute of Biotechnology) for 1 h at room temperature, then
incubated with the corresponding primary antibodies overnight at
4˚C. The dilutions of primary antibodies were as follows: IL-1β
(cat. no. ab9722; 1 µg/ml; Abcam), IL-6 (cat. no. A0286; 1:200;
ABclonal Biotech Co., Ltd.) and TNF-α (cat. no. GTX110520; 1:200;
GeneTex, Inc.). After washing with PBS, the sections were incubated
with a secondary antibody (MaxVision™ HRP-polymer anti-rabbit IHC
kit; ready to use; cat. no. KIT-5004; Fuzhou Maixin Biotech Co.,
Ltd.) for 30 min at room temperature. Subsequently,
3,3'-diaminobenzidine was used as a chromogen. Finally, tissue
sections were counterstained with hematoxylin at room temperature
for 3 min and sealed with neutral balsam. The representative
histopathological samples (magnification, x400, three random
slides/sample) were imaged using a light microscope (Nikon
Corporation) and analyzed using Fiji (version 2.3.0), an image
processing package based on ImageJ (National Institutes of Health)
(30).
Western blotting
Total protein was obtained from left ventricular
myocardial tissues by sonication, centrifugation and heat
denaturation. Briefly, heart tissues were placed into 4˚C RIPA
lysis buffer (cat. No. P0013B; Beyotime Institute of Biotechnology)
containing proteinase and phosphatase inhibitors (cat. No.
4906845001; Roche Diagnostics) and phenylmethanesulfonyl fluoride
(cat. No. ST506; Beyotime Institute of Biotechnology) for protein
extraction. Subsequently, the tissues were homogenized using a
FastPrep-24 5G homogenizer (MP Biomedicals, LLC) at 4˚C for 2 min
with 4.0-10 m/sec oscillation speed, and was centrifuged at 12,000
x g at 4˚C for 10 min. After quantification using a Pierce BCA
Protein Assay Kit (cat. no. PI23225; Thermo Fisher Scientific,
Inc.), the supernatant was mixed with loading buffer and heated to
95-100˚C for 5 min. An equal amount (30 µg/lane) of protein was
used for western blotting. Proteins were resolved by SDS-PAGE using
8-15 % gradient gels and then transferred onto PVDF membranes.
Membranes were blocked with 5 % non-fat milk for 60 min at room
temperature and then incubated with the corresponding primary
antibodies overnight at 4˚C. The dilutions of primary antibodies
used were as follows: Collagen I (cat. No. GTX26308; 1:1,000;
GeneTex, Inc.), α-SMA (α-smooth muscle actin; cat. No. ab5694;
1:1,000; Abcam), IL-1β (cat. no. ab9722; 0.25 µg/ml; Abcam), IL-6
(cat. No. A0286; 1:1,000; Abclonal Biotech Co., Ltd.), TNF-α (cat.
No. GTX110520; 1:500; GeneTex, Inc.), SOD1 (cat. No. ab16831;
1:2,000; Abcam), SOD2 (cat. No. NB100-1992; 1:2,000; Novus
Biologicals, LLC), Nrf2 (cat. No. GTX135165; 1:1,000; GeneTex,
Inc.), KEAP1 (Kelch-like ECH-associated protein 1; cat. No.
ab119403; 1:1,000; Abcam), Caspase-1 (cat. No. 83383; 1:500; Cell
Signaling Technology, Inc.), NLRP3 (cat. No. ab214185; 1:1,000;
Abcam), ASC (caspase recruitment domain; cat. No. GTX55818;
1:1,000; GeneTex, Inc.) and GAPDH (cat. No. 2118; 1:1,000; Cell
Signaling Technology, Inc.). After washing with TBST (containing
0.25% Tween20), the membrane was incubated with HRP-conjugated goat
anti-mouse or anti-rabbit secondary antibodies (cat. No. SA00001-1
or cat. No. SA00001-2; 1:2,000; ProteinTech Group Inc.) for 60 min
at room temperature. The membrane was washed three times in TBS
with 0.1 % Tween20 (cat. No. ST671; Beyotime Institute of
Biotechnology), before being visualized with an enhanced
chemiluminescent solution (cat. No. WBKLS0050; MilliporeSigma) and
imaged using an Uvitec Alliance mini-chemiluminescence device
(Uvitec). GAPDH was used as a loading control. The intensity of the
protein bands was quantified using ImageJ software (version 1.44;
National Institutes of Health).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA was extracted from left ventricular myocardial
tissues using TRIzol™ Reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and reverse transcription was performed using the
5X All-In-One RT MasterMix (Applied Biological Materials Inc.)
according to the manufacturer's protocols. qPCR was performed using
the Hieff™ qPCR SYBR® Green Master Mix (Shanghai Yeasen
Biotechnology Co., Ltd.) on StepOnePlus Real-Time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol (a two-step amplification: Initial
denaturation at 95˚C for 5 min, followed by 40 cycles of 95˚C for
10 sec and 60˚C for 30 sec). Quantification of mRNA was carried out
using the 2-ΔΔCq method and normalized to GAPDH
(31). Primer sequences used for
the RT-qPCR reactions are presented in Table I.
 | Table IPrimer sequences used for reverse
transcription-quantitative PCR. |
Table I
Primer sequences used for reverse
transcription-quantitative PCR.
| Gene target | Forward sequence
(5'-3') | Reverse sequence
(5'-3') |
|---|
| GAPDH |
CCCTTAAGAGGGATGCTGCC |
TACGGCCAAATCCGTTCACA |
| α-SMA |
ACATCAAGGAGAAGCTGTGCT |
TTTCGTGGATGCCCGCTG |
| POSTN |
TCCCTGATGTCATTACTGATCC |
AACCATCTTCAGCCCTGAGC |
| Fibronectin |
ATGAGAAGCCTGGATCCCCT |
GGAAGGGTAACCAGTTGGGG |
| Vimentin |
TTCTCTGGCACGTCTTGACC |
CTTTCATACTGCTGGCGCAC |
Cell culture and treatment
Human cardiomyocyte-like AC16 cells were purchased
from MilliporeSigma (cat. no. SCC109) and cultured in DMEM (Gibco;
Thermo Fisher Scientific, Inc.) containing 10% FBS (Gibco; Thermo
Fisher Scientific, Inc.) and 1% penicillin/streptomycin and
incubated in 5% CO2 at 37˚C. To model cardiotoxicity
in vitro, AC16 cells were treated with 1 µM DOX (32) in the absence or presence of
1,25(OH)2D3 for 24 h at 37˚C after serum
starvation [DOX treatment group or DOX +
1,25(OH)2D3 treatment group, respectively].
Untreated AC16 cells served as the Control group.
Cell viability assays
Cell viability was assayed with the Enhanced Cell
Counting Kit (CCK)-8 (Beyotime Institute of Biotechnology)
according to the manufacturer's protocols. AC16 cells were seeded
into a 96-well plate (2x104 cells/well) and incubated
until adherence was achieved. Cell culture medium was then replaced
using DMEM. To evaluate the potential toxicity of
1,25(OH)2D3 towards AC16 cells, AC16 cells
were treated with 1,25(OH)2D3 in triplicate
at various concentrations (0, 25, 50, 100, 200 and 400 nM) for 24 h
at 37˚C. To evaluate the protective effects of
1,25(OH)2D3 against DOX-induced cytotoxicity,
AC16 cells were treated with 1 µM DOX and various concentrations of
1,25(OH)2D3 (0, 25, 50, 100 and 200 nM) in
triplicate for 24 h at 37˚C. Subsequently, 10 µl enhanced CCK-8
solution was added into each well, cells were incubated for 2 h at
37˚C and the absorbance at 450 nm was measured.
ROS analysis
ROS production in AC16 cells was assessed using
2',7'-dichlorofluorescin diacetate (DCFH-DA; cat. no. 35845;
MilliporeSigma). Following starvation treatment and induction of
cardiotoxicity, AC16 cells were incubated with 10 µM DCFH-DA in the
dark at 37˚C for 30 min and then washed twice with PBS. Oxidized
2',7'-dichlorofluorescin fluorescence (representing ROS level) was
detected immediately by fluorescent microscopy (Guangzhou
Micro-shot Technology Co., Ltd.). A total of three images from
randomly selected non-overlapping fields were captured and the
intensity of fluorescence was quantified using ImageJ software
(version 1.44; National Institutes of Health).
Chromatin immunoprecipitation (ChIP)
assay
AC16 cells were collected for ChIP assay using the
High-Sensitivity ChIP kit (cat. no. 9003; Cell Signaling
Technology, Inc.) following the manufacturer's protocol. H2AK119Ub
(cat. no. 8240), H3K4me3 (cat. no. 9751) and
H3K27me3 (cat. no. 9733) were purchased from Cell
Signaling Technology Inc., which were used for the ChIP assay.
Briefly, two 10-cm dishes containing 1x107 cells in 10
ml culture media were used for each ChIP experiment. Cells were
crosslinked with 1% formaldehyde for 10 min at room temperature on
an orbital shaker (60 rpm). The formaldehyde was then quenched by
adding glycine (75 mM), the cells were incubated at room
temperature for 10 min and rinsed twice with PBS. Cells were
scraped and transferred into 15-ml Falcon tubes, before being
centrifuged at 350 x g for 5 min at 4˚C. Cells were resuspended in
15 ml Buffer A [20 mM HEPES (pH=7.4), 10 mM EDTA, 0.5 mM EGTA and
0.25% Triton X-100] and incubated at 4˚C for 5 min. The cells were
then centrifuged at 350 x g for 5 min at 4˚C and resuspended in 15
ml Buffer B [20 mM HEPES (pH=7.4), 150 mM NaCl, 10 mM EDTA and 0.5
mM EGTA] and incubated at 4˚C for 5 min on a rotating wheel. Cells
were centrifuged again at 350 x g for 5 min at 4˚C and resuspended
in 1 ml Buffer C [20 mM HEPES (pH=7.4), 10 mM EDTA, 0.5 mM EGTA,
0.1% SDS and 1X Protease Inhibitor Cocktail (cat. no. #5871; Cell
Signaling Technology). After incubating for 10 min on ice, the
cells were transferred into a 15-ml sonication tube. Sonication was
performed using a Bioruptor (Bioruptor PLUS; Diagenode S.A.) using
the following protocol: 10 Cycles of 30-sec sonication (150 Hz, at
4˚C), followed by 20 sec off, at high power for a total of 20
cycles with a 10-min pause and incubation on ice every 10 cycles.
The sonicated chromatin was centrifuged at 16,000 x g for 10 min at
4˚C. For each immunoprecipitation reaction, chromatin (10 µg DNA),
measured via Nanodrop2000 (Thermo Fisher Scientific, Inc.), was
diluted to a total of 500 µl with 1X ChIP Buffer + PIC. Next, 5 µg
antibodies were added into chromatin (10 µg DNA) in 500 µl 1X ChIP
Buffer + PIC. Immunoprecipitated samples were incubated on an
oscillating platform at 4˚C overnight. The following day, 30 µl
Protein G Magnetic Beads were added immediately into each
immunoprecipitation reaction and incubated on an oscillating
platform at 4˚C for 2 h. Each sample tube was placed on a magnetic
separator for 2 min to precipitate the Protein G Magnetic Beads
until the solution was cleared and the supernatant was removed. The
protein G magnetic beads were then washed using low- and high-salt
wash. They were then re-suspended in 150 µl 1X ChIP eluting buffer
before the chromatin was eluted from the samples using a vortex
(200 x g at 65˚C for 30 min). Samples were centrifuged at 10,000 x
g at 4˚C for 10 sec to remove trace amounts of evaporated sample
from the centrifuge tube cap. The eluted chromatin supernatant was
then transferred to a new tube. Un-crosslinking was performed by
adding 6 µl 5M NaCl and 2 µl Proteinase K to each sample and
incubating at 65˚C for 2 h. DNA Binding Buffer (750 µl) was added
to each DNA sample and swirled to mix, before 450 µl sample was
transferred to a DNA centrifuge column in the collection tube and
centrifuged at 16,000 x g at 4˚C for 30 sec. DNA Wash Buffer (750
µl) was added to the centrifuge column in the collection tube and
the samples were centrifuged again at 16,000 x g at 4˚C for 30 sec.
DNA Elution Buffer (50 µl) was added to each centrifuge column,
placed in a clean 1.5-ml microcentrifuge tube and centrifuged at
16,000 x g at 4˚C for 30 sec to elute the DNA. The eluate was
purified for subsequent qPCR assay. DNA purification was performed
as described in the Chip kit instructions (cat. no. 9003; Cell
Signaling Technology, Inc.). qPCR was performed using the Hieff™
qPCR SYBR® Green Master Mix (Shanghai Yeasen
Biotechnology Co., Ltd.) on StepOnePlus Real-Time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol (a two-step amplification: Initial
denaturation at 95˚C for 5 min, followed by 40 cycles of 95˚C for
10 sec and 60˚C for 30 sec). Quantification of mRNA was carried out
using the 2-ΔΔCq method and normalized to input. In this
experiment, the qPCR method was used to quantify the Chip
experiment. The enrichment level was calculated by the formula 2% x
2 [C(T) 2% input-C(T) IP sample] with reference to operating
instructions of the Chip kit (cat. no. 9003; Cell Signaling
Technology, Inc.). For DNA gel assay, 2 X Taq Master Mix (Dye Plus)
(Vazyme, P112-01) was used for PCR assay. The following DNA
amplification procedure was applied: i) Initial denaturation 95˚C
for 5 min; ii) denaturation 95˚C for 30 sec; ii) refolding at 62˚C
for 30 sec; iv) extension at 72˚C for 30 sec; v) repeat of step
ii)-iv) for 34 cycles; and vi) terminal extension at 72˚C for 5
min. DNA products were detected using 2% agarose gel
electrophoresis. GelDoc XR System (Bio-Rad) was used for gel
imaging. Primer sequences used for ChIP-qPCR reactions are
presented in Table II.
| Table IIPrimer sequences used for chromatin
immunoprecipitation. |
Table II
Primer sequences used for chromatin
immunoprecipitation.
| Gene target | Forward sequence
(5'-3') | Reverse sequence
(5'-3') |
|---|
| NLRP3 |
GGGACCAAATTGAGGGCTTC |
TCAACGTCACCAGTCCTCAGA |
| Nrf2 |
AGAATGAATGGCCTCCAGCC |
CATTACCATGCCCCCAGAGG |
Statistical analysis
Data from ≥ three independent experiments were
expressed as the mean ± standard deviation. All statistical
analyses were performed using the GraphPad Prism software (version
7.0; GraphPad Software, Inc.; Dotmatics). Statistical differences
among treatment groups were analyzed using a one-way ANOVA followed
by Tukey's test. P<0.05 was considered to indicate a
statistically significant difference.
Results
1,25(OH)2D3
alleviates DOX-induced cardiac dysfunction and injury
Mice were administered with DOX to construct a
DOX-induced cardiomyopathy model whilst
1,25(OH)2D3 was administered to mice as
previously described (Fig. 1A).
The body weight of each mouse in the Sham, DOX and DOX +
1,25(OH)2D3 groups were measured each week,
which yielded no significant differences among the three treatment
groups (Fig. 1B). Echocardiography
images of mice on week 4 indicated that ventricular wall motion
amplitude markedly decreased after DOX administration, which was
alleviated by 1,25(OH)2D3 (Fig. 1C). LVEF and LVFS were observed to
be significantly decreased after DOX administration compared with
those in the sham group (Fig. 1D
and E). Compared with those in the
DOX-treated group, 1,25(OH)2D3 treatment
significantly increased LVEF and LVFS (Fig. 1D and E). Treatment with
1,25(OH)2D3 also significantly reduced the
serum levels of BNP in mice with DOX-induced cardiomyopathy
(Fig. 1F). Analysis of cTnT levels
in serum was performed to detect signs of myocardial injury, which
demonstrated that cTnT levels were significantly increased after
DOX administration compared with those in the Sham group, which was
in turn significantly reversed by 1,25(OH)2D3
treatment (Fig. 1G). These results
suggest that 1,25(OH)2D3 treatment can
alleviate DOX-induced cardiac dysfunction and injury.
1,25(OH)2D3
inhibits the activation of a pro-fibrotic and inflammatory
microenvironment in DOX-induced cardiomyopathy
As 1,25(OH)2D3 was found to
alleviate DOX-induced cardiac dysfunction and injury, it was
therefore hypothesized that 1,25(OH)2D3 may
reverse the fibrotic and inflammatory microenvironment in a model
of DOX-induced cardiomyopathy. Masson's trichrome staining was used
to evaluate the extent of myocardial fibrosis.
1,25(OH)2D3 was demonstrated to significantly
inhibit collagen deposition in the heart tissues of mice with
DOX-induced cardiac dysfunction (Fig.
2A and B). The mRNA expression
levels of α-SMA, periostin (POSTN), Fibronectin and Vimentin in
mice with DOX-induced cardiomyopathy were also found to be
significantly reduced after 1,25(OH)2D3
treatment (Fig. 2C). In addition,
it was demonstrated through western blotting that
1,25(OH)2D3 significantly downregulated α-SMA
and collagen I protein expression levels compared with those in the
DOX group (Fig. 2D and E). These results suggest the
anti-fibrotic effect of 1,25(OH)2D3 against
DOX-induced cardiomyopathy.
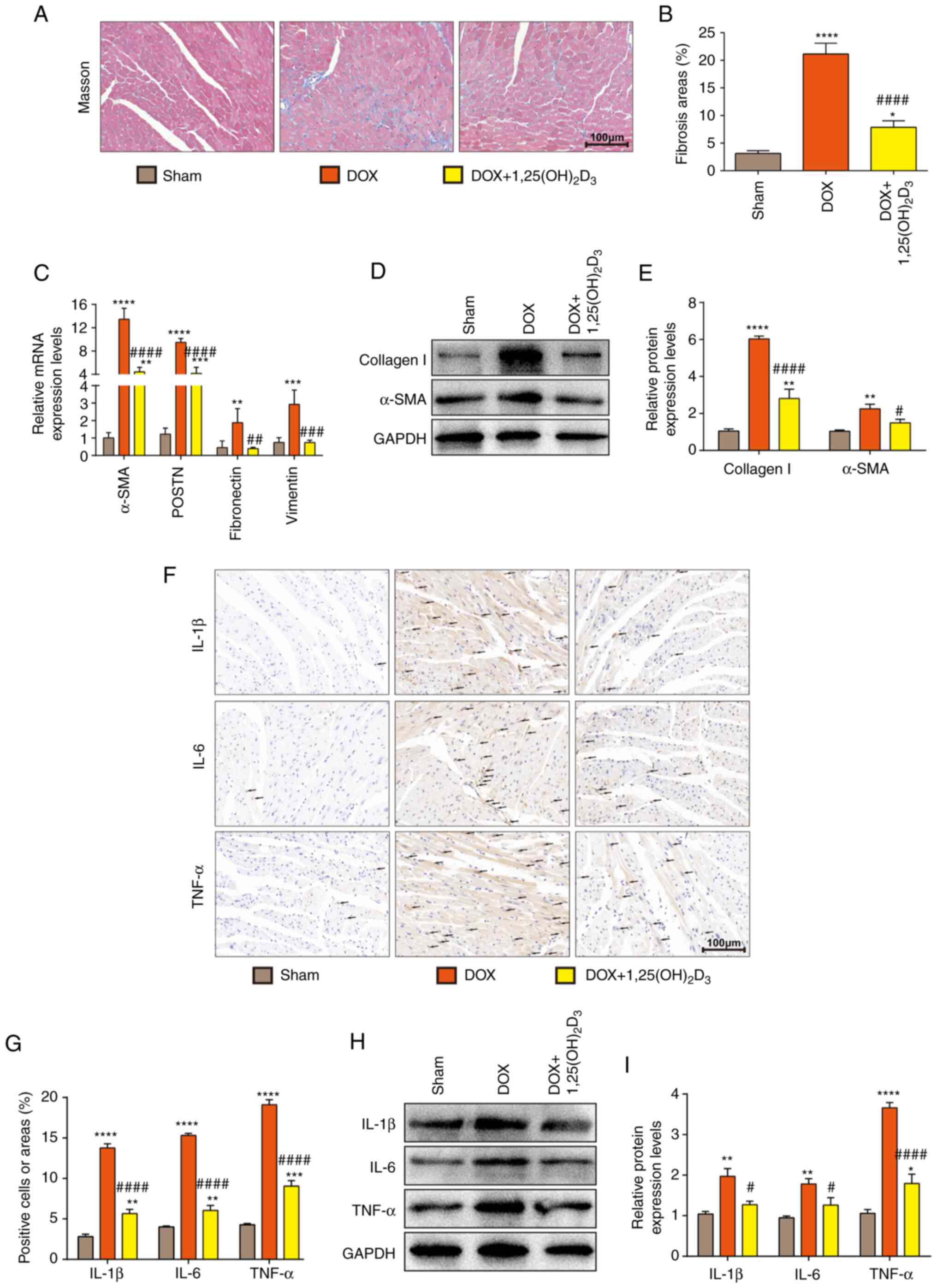 | Figure 21,25(OH)2D3
inhibits the activation of a pro-fibrotic and inflammatory
microenvironments in DOX-induced cardiomyopathy in mice. (A)
Representative images of Masson's trichrome staining of cardiac
tissue and (B) quantification of fibrosis (n=3). (C) Relative mRNA
expression levels of α-SMA, POSTN, fibronectin and vimentin in
cardiac tissues (n=5). (D) Western blotting images of cardiac
tissue extracts for protein expression of collagen I and α-SMA.
GAPDH was used as the loading control. (E) Semi-quantification of
protein expression levels of collagen I and α-SMA. Protein
expression levels relative to Sham group were assessed by
densitometric analysis (n=3). (F) Representative images of
immunohistochemical staining for IL-1β, IL-6 and TNF-α. Black
arrows indicate areas of positive expression. (G) Percentage of
cells or areas positive for IL-1β, IL-6 and TNF-α relative to
number of total cells or areas (n=3). (H) Western blotting images
of cardiac tissue extract for the protein expression of IL-1β, IL-6
and TNF-α. GAPDH was used as the loading control. (I)
Semi-quantification of protein expression levels of IL-1β, IL-6 and
TNF-α. Protein expression levels relative to Sham group were
assessed by densitometric analysis (n=3). *P<0.05,
**P<0.01, ***P<0.001 and
****P<0.0001 vs. Sham; #P<0.05,
##P<0.01, ###P<0.001 and
####P<0.0001 vs. DOX. DOX, doxorubicin hydrochloride;
α-SMA, α-smooth muscle actin; POSTN, periostin. |
Immunohistochemistry staining was subsequently used
to examine whether 1,25(OH)2D3 can reduce the
production of inflammatory mediators in DOX-induced cardiomyopathy.
The results demonstrated that the expression levels of IL-1β, IL-6
and TNF-α were significantly decreased in the DOX +
1,25(OH)2D3-treated group compared with those
in the DOX-treated group (Fig. 2F
and G). These findings were
confirmed by western blotting, which demonstrated significantly
reduced IL-1β, IL-6 and TNF-α protein expression levels in mice
with DOX-induced cardiomyopathy treated with
1,25(OH)2D3 (Fig. 2H and I). These results suggest that
1,25(OH)2D3 inhibited the activation of a
pro-fibrotic and inflammatory microenvironment in a mouse model of
DOX-induced cardiomyopathy.
Increases in oxidative stress and the
NLRP3 inflammasome pathways are ameliorated by
1,25(OH)2D3 treatment in DOX-induced
cardiomyopathy
Although 1,25(OH)2D3 inhibited
the activation of a pro-fibrotic and inflammatory microenvironment,
the detailed mechanism underlying the regulation of this process
remain poorly understood. Previous studies have reported that
mitochondrial dysfunction and excessive activation of oxidative
stress can aggravate DOX-induced cardiomyopathy (33,34).
Furthermore, another previous study demonstrated that
1,25(OH)2D3 can significantly inhibit the
production of ROS in skin and liver senescent cells (18). It was therefore hypothesized that
1,25(OH)2D3 administration could inhibit
oxidative stress. Consequently, the associated pathway in
DOX-induced cardiomyopathy was next assessed. To clarify this
hypothesis, cell viability was analyzed using the CCK-8 kit to
determine the minimal treatment concentration of
1,25(OH)2D3 that can cause toxicity in AC16
cells. CCK-8 results demonstrated that cell viability was not
affected upon treatment with different concentrations of
1,25(OH)2D3 (0, 25, 50, 100, 200 and 400 nM),
suggesting that no cytotoxicity to
1,25(OH)2D3 was demonstrated in AC16 cells up
to a dose of 400 nM (Fig. 3A).
Cell viability was found to be significantly decreased when the
AC16 cells were treated with DOX (Fig.
3B). Doses of 50, 100 and 200 nM
1,25(OH)2D3 exhibited a significant
protective effect against DOX-induced cytotoxicity in AC16 cells
(Fig. 3B). In particular, the 100
and 200 nM doses of 1,25(OH)2D3 exhibited
significantly increased protective effects on cell viability
compared with that after treatment with the 50 nM dose of
1,25(OH)2D3 (Fig. 3B). Doubling the
1,25(OH)2D3 concentration to 200 nM had no
significant effects on cell viability compared with that after 100
nM 1,25(OH)2D3 treatment (Fig. 3B). Therefore, 100 nM
1,25(OH)2D3 was selected as the safe and
effective treatment dose for subsequent experiments.
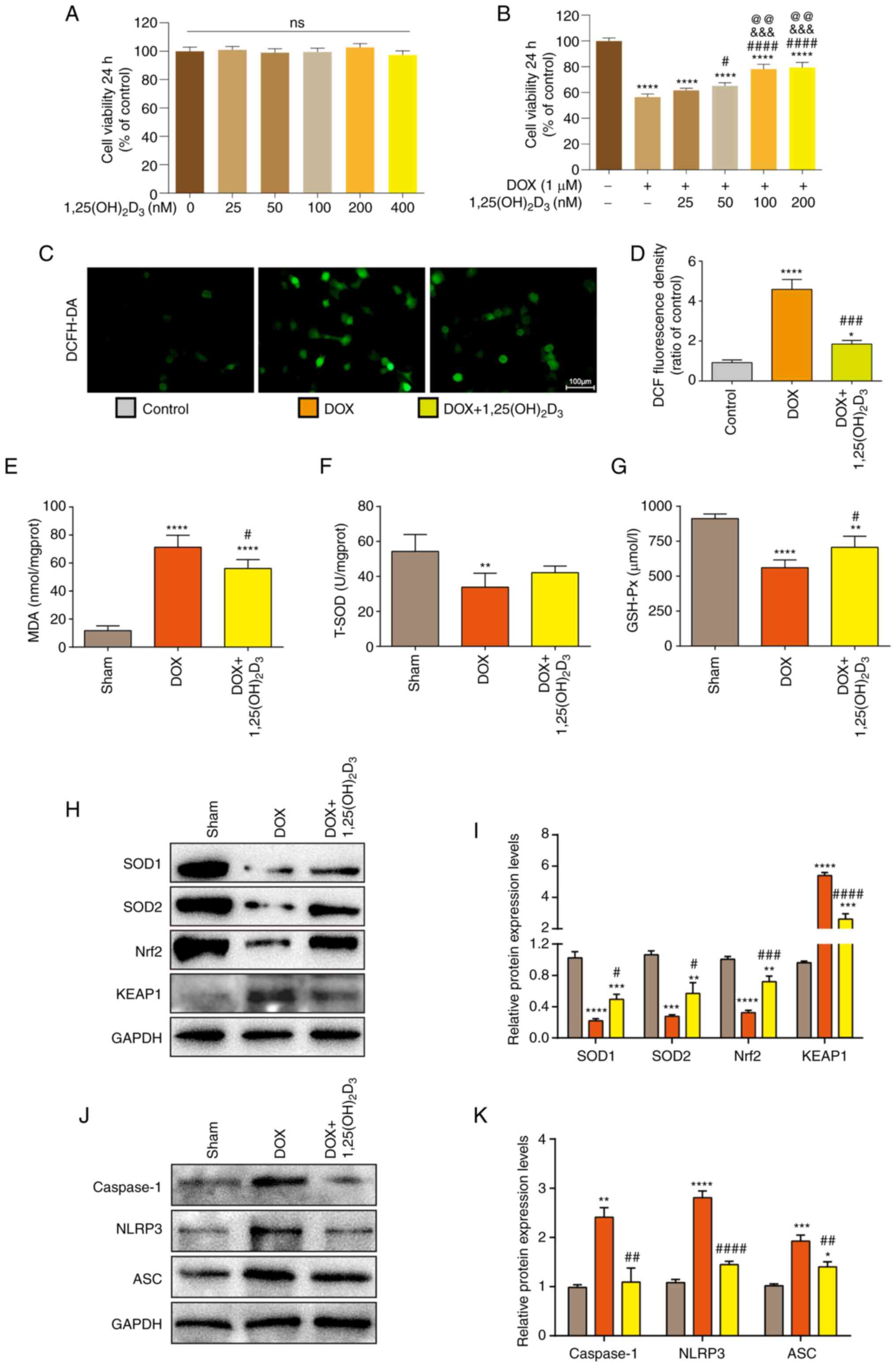 | Figure 3Increased oxidative stress and NLRP3
inflammasome pathways are ameliorated by
1,25(OH)2D3 treatment in DOX-induced
cardiomyopathy in mice. (A) AC16 cells treated with different
concentrations of 1,25(OH)2D3 (0, 25, 50,
100, 200 and 400 nM) for 24 h. Cell viability was measured using
CCK-8 assay (n=3). (B) AC16 cells were treated with 1 µM DOX and
different concentrations of 1,25(OH)2D3 (0,
25, 50, 100 and 200 nM) for 24 h. Cell viability was measured using
CCK-8 assay (n=3). ****P<0.0001 vs. Control group;
#P<0.05 and ####P<0.0001 vs. DOX-only
group; &&&P<0.001 vs. DOX + 25 nM
1,25(OH)2D3-treated group;
@@P<0.01 vs. DOX + 50 nM
1,25(OH)2D3-treated group. (C) Levels of
reactive oxygen species and (D) densitometric analysis in AC16
cells after treatment with DOX or DOX +
1,25(OH)2D3 via DCFH-DA staining (n=5).
*P<0.05 and ****P<0.0001 vs. Control;
###P<0.001 vs. DOX group. Levels of (E) MDA, (F)
T-SOD and (G) GSH-Px in cardiac tissue 4 weeks after the first
injection of DOX (n=5). (H) Western blotting images of cardiac
tissue extracts analyzing protein expression of SOD1, SOD2, Nrf2
and KEAP1. GAPDH was used as the loading control. (I)
Semi-quantification of protein expression levels of SOD1, SOD2,
Nrf2 and KEAP1. Protein expression levels relative to the Sham
group were assessed by densitometric analysis (n=3). (J) Western
blotting images of cardiac tissue extracts analyzing protein
expression of NLRP3, ASC and Caspase-1. GAPDH was used as the
loading control. (K) Semi-quantification of protein expression
levels NLRP3, ASC and Caspase-1. Protein expression levels relative
to the Sham group were assessed by densitometric analysis (n=3).
*P<0.05, **P<0.01,
***P<0.001 and ****P<0.0001 vs. Sham;
#P<0.05, ##P<0.01,
###P<0.001 and ####P<0.001 vs. DOX.
DOX, doxorubicin hydrochloride; CCK-8, Cell Counting Kit-8; ns, no
significant difference; DCFH-DA, 2',7'-dichlorofluorescin
diacetate; MDA, malondialdehyde; T, total; GSH-Px, glutathione
peroxidase; SOD, superoxide dismutase; Nrf2, nuclear erythroid
2-related factor 2; KEAP1, Kelch-like ECH-associated protein 1;
ASC, caspase recruitment domain; NLRP3, nod-like receptor family
pyrin domain-containing 3 inflammasome. |
Intracellular ROS levels in AC16 cells were analyzed
using DCFH-DA staining, which demonstrated that DOX treatment
significantly increased the intracellular ROS levels compared with
those in control cells (Fig. 3C
and D). Treatment of AC16 cells
with 100 nM 1,25(OH)2D3 significantly
decreased intracellular ROS levels compared with cells treated with
DOX only (Fig. 3C and D). Oxidative stress was also measured in
cardiac tissue homogenates of mice on the 4th week after the first
injection of DOX was administered. Compared with those in the Sham
group, cardiac tissues from DOX-treatment mice demonstrated a
significantly increased MDA level but decreased T-SOD and GSH-Px
levels (Fig. 3E-G). Mice in the
DOX + 1,25(OH)2D3-treated group demonstrated
significantly lower MDA levels, but markedly higher T-SOD and
significantly higher GSH-Px levels, compared with those in the
DOX-treated group (Fig. 3E-G). To
further determine the potential mechanism of action underlying this
process, protein expression levels of KEAP1, Nrf2, SOD1 and SOD2
were analyzed using western blotting. Compared with those in the
DOX-treated group, 1,25(OH)2D3 treatment
significantly increased SOD1, SOD2 and Nrf2 protein expression,
whilst significantly decreasing KEAP1 expression (Fig. 3H and I). Taken together, these results suggest
that 1,25(OH)2D3 reduced oxidative stress in
DOX-induced cardiomyopathy by inhibiting the KEAP1/Nrf2
pathway.
The NLRP3 inflammasome has received considerable
attention in the field of inflammation research (35,36).
Levels of NLRP3 inflammasome-associated proteins in DOX-induced
cardiomyopathy mice were therefore next analyzed, which
demonstrated that the protein expression levels of NLRP3, ASC and
Caspase-1 were significantly decreased with
1,25(OH)2D3 treatment compared with those in
the DOX-only treatment group (Fig.
3J and K).
Taken together, these results suggested that
1,25(OH)2D3 ameliorated oxidative stress and
inhibited the NLRP3 inflammasome pathway to alleviate the
pathophysiological processes associated with DOX-induced
cardiomyopathy.
1,25(OH)2D3
treatment affects histone modification levels in NLRP3 and Nrf2
promoters in DOX-induced cardiomyopathy
Previous studies have reported that
1,25(OH)2D3 can affect histone modifications
to regulate downstream target gene expression and impact
transcriptional regulation (37).
To clarify the effect of 1,25(OH)2D3 on the
level of histone modification in the NLRP3 promoter, ChIP assay was
used to assess the levels of H3K4 (H3K4me3) and H3K27
(H3K27me3) trimethylation, in addition to H2AK119
monoubiquitination (H2AK119Ub), in the NLRP3 promoter region. The
results demonstrated that 1,25(OH)2D3
treatment significantly decreased the level of H3K4me3
whilst significantly increasing the levels of H3K27me3
and H2AK119Ub in the NLRP3 promoter region, compared with that in
the DOX-treated group (Figs. 4A-C
and S1A). H3K4me3
modification may loosen chromatin and activate transcriptional
expression (38). By contrast,
H3K27me3 and H2AK119Ub modification may cause chromatin
compression and inhibit the transcription of downstream genes
(39). These results suggested
that the inhibition of NLRP3 transcription by
1,25(OH)2D3 may have occurred due to a
decrease in the levels of histone modification that is conducive to
transcription activation and an increase in the levels of histone
modifications that favor transcriptional inhibition in the NLRP3
promoter region.
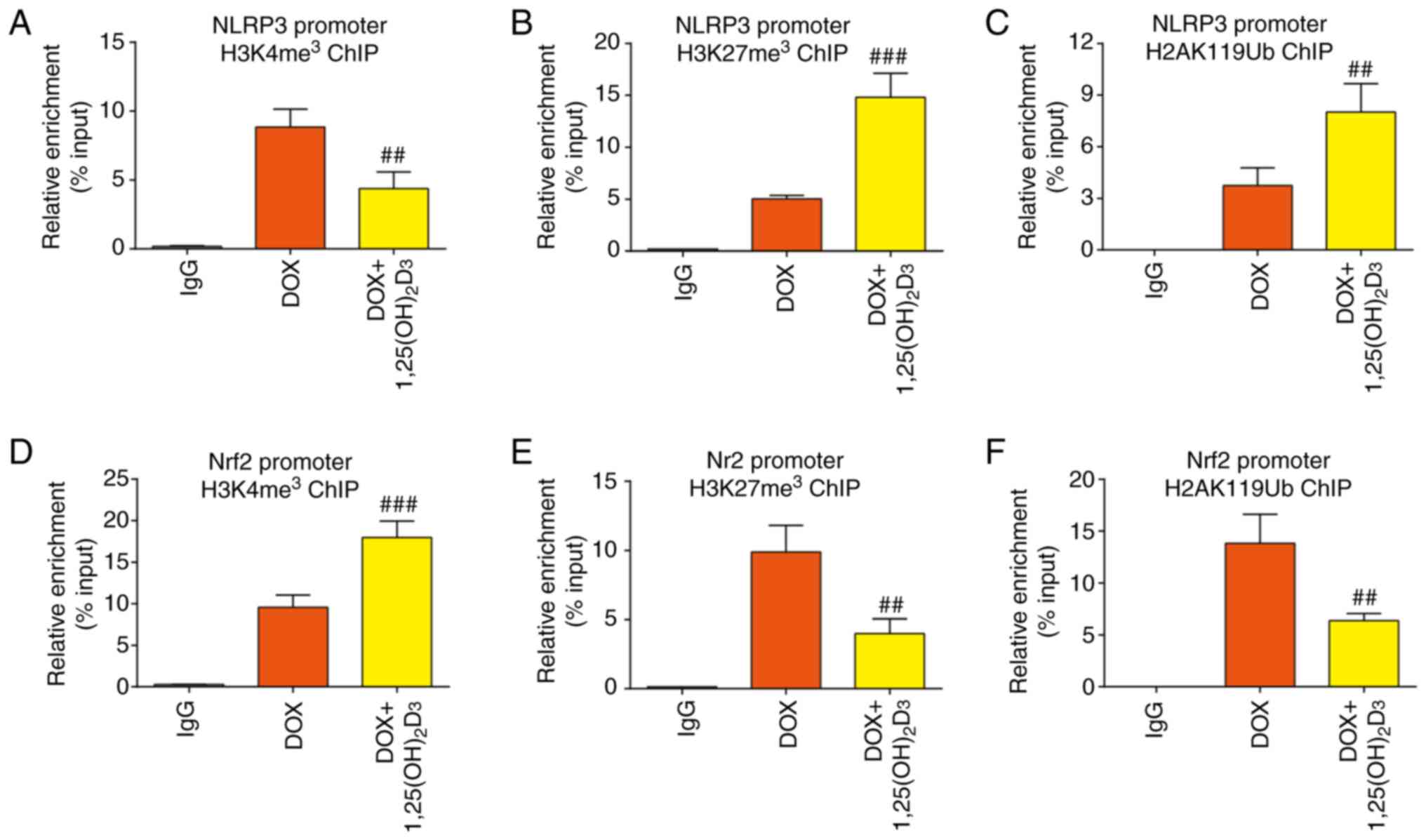 | Figure 41,25(OH)2D3
treatment affects histone modification levels in NLRP3 and Nrf2
promoters in DOX-induced cardiomyopathy. Binding levels of (A)
H3K4me3, (B) H3K27me3 and (C) H2AK119Ub in
the NLRP3 promoter region in AC16 cells after treatment with DOX or
DOX + 1,25(OH)2D3, detected using ChIP assay
(n=5). Binding levels of (D) H3K4me3, (E)
H3K27me3 and (F) H2AK119Ub in the Nrf2 promoter region
in AC16 cells after treatment with DOX or DOX +
1,25(OH)2D3, detected using ChIP assay (n=5).
##P<0.01 and ###P<0.001 vs. DOX. DOX,
doxorubicin hydrochloride; NLRP3, nod-like receptor family pyrin
domain-containing 3; Nrf2, nuclear erythroid 2-related factor 2;
ChIP, chromatin immunoprecipitation. |
The levels of different histone modifications in
the Nrf2 promoter region were also evaluated. Treatment with
1,25(OH)2D3 significantly increased the level
of H3K4me3 whilst significantly decreasing the levels of
H3K27me3 and H2AK119Ub in the Nrf2 promoter region in a
model of DOX-induced cardiomyopathy, suggesting that
1,25(OH)2D3 stimulation increased the
transcriptional level of Nrf2 (Figs.
4D-F and S1B). These results
contrast with those observed in the NLRP3 promoter. Therefore,
these data suggest that 1,25(OH)2D3 treatment
could significantly affect chromatin accessibility in
cardiomyocytes to participate in the progression of cardiomyopathy,
but the regulation of this process is likely to be complex.
Discussion
DOX, an anthracycline antibiotic derived from
Streptomyces, is widely used for the treatment of multiple
types of cancer, such as leukemia, lung and breast cancer (40). However, the toxic effects of DOX on
cardiomyocytes limits its clinical application (3). The pathogenesis of DOX-induced
cardiomyopathy remains poorly understood and there is a lack of
effective therapeutic drugs (7).
Therefore, it is particularly important to explore its mechanism
and search for potential and effective treatment.
1,25(OH)2D3 is a metabolic
derivative of vitamin D3 and its production needs catalysis by the
CYP27B1 enzyme (9).
1,25(OH)2D3 binds to the vitamin D receptor
(VDR) to induce downstream effects, such as anti-oxidative stress
and anti-cell senescence (17,18).
VDR and CYP27B1 are expressed in multiple organ and cell types,
including the heart, suggesting that the targets of
1,25(OH)2D3 are wide-ranging (41). Previous studies have reported that
1 µg/kg 1,25(OH)2D3 exhibits antioxidant and
anti-inflammatory effects on certain organs, such as the skin,
liver and kidneys, in addition to impacting a number of diseases,
such as arthrosis (by promoting apoptosis of fibroblast-like
synoviocytes) and breast cancer (by inhibiting oxidative stress and
inducing tumor cellular senescence) in mice (18,29,42).
Oxidative stress and inflammation can also serve vital roles in
DOX-induced cardiomyopathy (34).
However, a potential risk of vitamin D overproduction is that it
can cause high calcium and phosphorus levels, which are harmful to
health, resulting in hyperparathyroidism, vascular calcification
and arrhythmia (43). Previous
studies have reported that 1 µg/kg
1,25(OH)2D3 has no effect on serum levels of
calcium and phosphorus in mice, suggesting that 1 µg/kg can be used
as a safe dose in mouse studies (18,29).
Considering the impact of 1,25(OH)2D3 on
multiple organ systems and the safety data reported in previous
studies (18,29,42),
the aforementioned dose (1 µg/kg) was used to study the potential
effects of 1,25(OH)2D3 in a mouse model of
DOX-induced cardiomyopathy.
The present study demonstrated that
1,25(OH)2D3 treatment could increase the
levels of H3K27me3 and H2AK119Ub and decrease the level
of H3K4me3 in the NLRP3 promoter region in DOX-induced
cardiomyopathy. By contrast, treatment with
1,25(OH)2D3 decreased the levels of
H3K27me3 and H2AK119Ub and increased the level of
H3K4me3 in the Nrf2 promoter region. Therefore,
1,25(OH)2D3 may inhibit the activation of the
NLRP3 inflammasome and KEAP1/Nrf2-induced oxidative stress, in turn
alleviating the pathophysiological processes of DOX-induced
cardiomyopathy.
The NLRP3 inflammasome is an important part of the
inflammasome family and serves a critical role in inflammatory
responses to multiple exogenous and endogenous factors (35). The NLRP3 inflammasome contains an
NLRP3 protein that interacts with its adapter apoptosis-associated
speck-like protein containing an ASC to recruit and activate
caspase-1, which processes pro-IL-1β to mature IL-1β to trigger an
inflammatory response (44). Cao
et al (45) previously
reported that 1,25(OH)2D3 can alleviate
colitis disease progression by inhibiting the NLRP3 pathway, which
is consistent with findings of the present study. The present study
also demonstrated the inhibitory effect of
1,25(OH)2D3 on the NLRP3 inflammasome pathway
in DOX-induced cardiomyopathy. However, Tulk et al (46) reported that
1,25(OH)2D3 can enhance the secretion of
IL-1β in THP-1 cells by activating the NLRP3 inflammasome pathway
after phorbol 12-myristate 13-acetate stimulation. Conflicting
results obtained using different cell lines suggest that
1,25(OH)2D3 may serve different roles
depending on the cell type or under different pathological
conditions, further indicating the complex role of
1,25(OH)2D3 in different cell types.
Epigenetic modification serves as another
regulatory process of gene transcription (47,48).
Epigenetic regulation mechanisms, including CpG island DNA
methylation, histone modification and non-coding RNA regulation,
are considered key to regulating the transcription of the NLRP3
inflammasome (49). Lecoeur et
al (50) reported that NLRP3
promoters exhibit a high level of histone H3K9/14 deacetylation and
a high level of histone H3K4 demethylation during Leishmania
amazonensis infection. As an important type of histone
modification, histone ubiquitination also serves an important role
in transcriptional regulation (39). Histone ubiquitination has been
reported to serve an important role in DNA damage repair (51), stem cell regulation (52) and DNA replication (53). Previous studies have reported that
1,25(OH)2D3 can increase the level of
H3K9me2 in the bone morphogenic protein 2 (BMP2)
promoter region and regulates the transcriptional expression of the
BMP2 gene through binding to VDR (54,39).
However, to the best of our knowledge, no studies have assessed
whether 1,25(OH)2D3 can affect histone
modifications in the NLRP3 promoter region. In the present study,
it was demonstrated that 1,25(OH)2D3
decreased H3K4me3 levels whilst increasing
H3K27me3 and H2AK119Ub levels in the NLRP3 promoter
region. A decrease in the levels of histone modifications favoring
transcriptional activation and an increase in the levels of histone
modifications favoring transcriptional inhibition can lead to
significant chromatin compression in the NLRP3 promoter region to
inhibit transcription.
In conclusion, the present study demonstrated that
1,25(OH)2D3 can regulate histone modification
in NLRP3 and Nrf2 promoters, inhibit activation of the NLRP3
inflammasome pathway and oxidative stress in cardiomyocytes, in
addition to alleviating the pathophysiological processes of
DOX-induced cardiomyopathy. Therefore,
1,25(OH)2D3 may serve to be a future
potential therapeutic drug for the treatment of DOX-induced
cardiomyopathy.
Supplementary Material
1,25(OH)2D3
treatment affects histone modification levels in NLRP3 and Nrf2
promoters in DOX-induced cardiomyopathy. The DNA gels of different
histone modifications in the (A) NLRP3 and (B) Nrf2 promoter
regions in AC16 cells after treating with DOX or DOX +
1,25(OH)2D3. NLRP3, nod-like receptor family
pyrin domain containing 3; Nrf2, nuclear erythroid 2-related factor
2; DOX, doxorubicin hydrochloride.
Acknowledgements
The authors would like to thank Professor Zhijian
Yang (Nanjing Medical University, Nanjing, China) for his
assistance in ethics writing.
Funding
Funding: The present study was supported by the Natural Science
Foundation of Jiangsu Province (grant no. BK20210101), the
Scientific Research Project of Gusu Health Talent Plan (grant no.
GSWS2022067), the General Project of Suzhou Science and Technology
Administration (grant no. SKYD2022131), the Research Project of
Gusu School of Nanjing Medical University (grant no. GSKY20210214),
the Key Project of Jiangsu Provincial Health Commission (grant no.
ZDA2020023), the General Project of Wuxi Traditional Chinese
Medicine Administration (grant no. ZYKJ202013) and the General
Project of Wuxi Science and Technology Administration (grant no.
N20202019).
Availability of data and materials
The datasets used and/or analyzed during the
current study are available from the corresponding author on
reasonable request.
Authors' contributions
XG, XW, YD and JZ contributed to study conception
and design. XG, LZ, JY, LC, CS and BL performed experiments. XG,
LZ, JY and XW generated figures and performed data analysis. XG and
YD confirm the authenticity of all the raw data. All authors read
and approved the final version of the manuscript.
Ethics approval and consent to
participate
All animal studies were approved by Animal Ethical
and Welfare Committee of Nanjing Medical University (approval no.
IACUC-1707002).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang AJ, Tang Y, Zhang J, Wang BJ, Xiao M,
Lu G, Li J, Liu Q, Guo Y and Gu J: Cardiac SIRT1 ameliorates
doxorubicin-induced cardiotoxicity by targeting sestrin 2. Redox
Biol. 52(102310)2022.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kabir S, Lingappa N and Mayrovitz H:
Potential therapeutic treatments for doxorubicin-induced
cardiomyopathy. Cureus. 14(e21154)2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Schirone L, D'Ambrosio L, Forte M,
Genovese R, Schiavon S, Spinosa G, Iacovone G, Valenti V, Frati G
and Sciarretta S: Mitochondria and doxorubicin-induced
cardiomyopathy: A complex interplay. Cells. 11(2000)2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Baker LH, Boonstra PS, Reinke DK, Antalis
E, Zebrack BJ and Weinberg RL: Burden of chronic diseases among
sarcoma survivors treated with anthracycline chemotherapy: Results
from an observational study. J Cancer Metastasis Treat.
6(24)2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Suzuki K, Murtuza B, Suzuki N, Smolenski
RT and Yacoub MH: Intracoronary infusion of skeletal myoblasts
improves cardiac function in doxorubicin-induced heart failure.
Circulation. 104:I213–I217. 2001.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Deng S, Kulle B, Hosseini M, Schluter G,
Hasenfuss G, Wojnowski L and Schmidt A: Dystrophin-deficiency
increases the susceptibility to doxorubicin-induced cardiotoxicity.
Eur J Heart Fail. 9:986–994. 2007.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Chen Y, Shi S and Dai Y: Research progress
of therapeutic drugs for doxorubicin-induced cardiomyopathy. Biomed
Pharmacother. 156(113903)2022.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Christakos S, Dhawan P, Verstuyf A,
Verlinden L and Carmeliet G: Vitamin D: Metabolism, molecular
mechanism of action, and pleiotropic effects. Physiol Rev.
96:365–408. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Carlberg C and Munoz A: An update on
vitamin D signaling and cancer. Semin Cancer Biol. 79:217–230.
2022.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Malluche HH, Henry H, Meyer-Sabellak W,
Sherman D, Massry SG and Norman AW: Effects and interactions of
24R,25(OH)2D3 and 1,25(OH)2D3 on bone. Am J Physiol. 238:E494–E498.
1980.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Carlberg C: Vitamin D signaling in the
context of innate immunity: Focus on human monocytes. Front
Immunol. 10(2211)2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Chanakul A, Zhang MY, Louw A, Armbrecht
HJ, Miller WL, Portale AA and Perwad F: FGF-23 regulates CYP27B1
transcription in the kidney and in extra-renal tissues. PLoS One.
8(e72816)2013.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ismailova A and White JH: Vitamin D,
infections and immunity. Rev Endocr Metab Disord. 23:265–277.
2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
de la Guia-Galipienso F, Martinez-Ferran
M, Vallecillo N, Lavie CJ, Sanchis-Gomar F and Pareja-Galeano H:
Vitamin D and cardiovascular health. Clin Nutr. 40:2946–2957.
2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Guo X, Lin H, Liu J, Wang D, Li D, Jiang
C, Tang Y, Wang J, Zhang T, Li Y, et al: 1,25-Dihydroxyvitamin D
attenuates diabetic cardiac autophagy and damage by vitamin D
receptor-mediated suppression of FoxO1 translocation. J Nutr
Biochem. 80(108380)2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Samefors M, Scragg R, Lanne T, Nyström FH
and Östgren CJ: Association between serum 25(OH)D3 and
cardiovascular morbidity and mortality in people with type 2
diabetes: A community-based cohort study. Diabet Med. 34:372–379.
2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Cui C, Xu P, Li G, Qiao Y, Han W, Geng C,
Liao D, Yang M, Chen D and Jiang P: Vitamin D receptor activation
regulates microglia polarization and oxidative stress in
spontaneously hypertensive rats and angiotensin II-exposed
microglial cells: Role of renin-angiotensin system. Redox Biol.
26(101295)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Chen L, Yang R, Qiao W, Zhang W, Chen J,
Mao L, Goltzman D and Miao D: 1,25-Dihydroxyvitamin D exerts an
antiaging role by activation of Nrf2-antioxidant signaling and
inactivation of p16/p53-senescence signaling. Aging Cell.
18(e12951)2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Qin Q, Liu H, Shou J, Jiang Y, Yu H and
Wang X: The inhibitor effect of RKIP on inflammasome activation and
inflammasome-dependent diseases. Cell Mol Immunol. 18:992–1004.
2021.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Sharma BR and Kanneganti TD: NLRP3
inflammasome in cancer and metabolic diseases. Nat Immunol.
22:550–559. 2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhang X, Hu C, Kong CY, Song P, Wu HM, Xu
SC, Yuan YP, Deng W, Ma ZG and Tang QZ: FNDC5 alleviates oxidative
stress and cardiomyocyte apoptosis in doxorubicin-induced
cardiotoxicity via activating AKT. Cell Death Differ. 27:540–555.
2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Nolfi-Donegan D, Braganza A and Shiva S:
Mitochondrial electron transport chain: Oxidative phosphorylation,
oxidant production, and methods of measurement. Redox Biol.
37(101674)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Byrne NJ, Rajasekaran NS, Abel ED and
Bugger H: Therapeutic potential of targeting oxidative stress in
diabetic cardiomyopathy. Free Radic Biol Med. 169:317–342.
2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yang R, Zhang J, Li J, Qin R, Chen J, Wang
R, Goltzman D and Miao D: Inhibition of Nrf2 degradation alleviates
age-related osteoporosis induced by 1,25-Dihydroxyvitamin D
deficiency. Free Radic Biol Med. 178:246–261. 2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Bosworth CR, Levin G, Robinson-Cohen C,
Hoofnagle AN, Ruzinski J, Young B, Schwartz SM, Himmelfarb J,
Kestenbaum B and de Boer IH: The serum 24,25-dihydroxyvitamin D
concentration, a marker of vitamin D catabolism, is reduced in
chronic kidney disease. Kidney Int. 82:693–700. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhang W, Chen L, Zhang L, Xiao M, Ding J,
Goltzman D and Miao D: Administration of exogenous 1,25(OH)2D3
normalizes overactivation of the central renin-angiotensin system
in 1alpha (OH)ase knockout mice. Neurosci Lett. 588:184–189.
2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Yang R, Chen J, Zhang J, Qin R, Wang R,
Qiu Y, Mao Z, Goltzman D and Miao D: 1,25-Dihydroxyvitamin D
protects against age-related osteoporosis by a novel VDR-Ezh2-p16
signal axis. Aging Cell. 19(e13095)2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Tomczyk MM, Cheung KG, Xiang B, Tamanna N,
Fonseca TA, Agarwal P, Kereliuk SM, Spicer V, Lin L, Treberg J, et
al: Mitochondrial sirtuin-3 (SIRT3) prevents doxorubicin-induced
dilated cardiomyopathy by modulating protein acetylation and
oxidative stress. Circ Heart Fail. 15(e8547)2022.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Gu X, Gu B, Lv X, Yu Z, Wang R, Zhou X,
Qiao W, Mao Z, Zuo G, Li Q, et al: 1, 25-dihydroxy-vitamin D3 with
tumor necrosis factor-alpha protects against rheumatoid arthritis
by promoting p53 acetylation-mediated apoptosis via Sirt1 in
synoviocytes. Cell Death Dis. 7(e2423)2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Schindelin J, Arganda-Carreras I, Frise E,
Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S,
Schmid B, et al: Fiji: An open-source platform for biological-image
analysis. Nat Methods. 9:676–682. 2012.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative RCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Chen B and Zhang JP: Bcl-xL is required
for the protective effects of low-dose berberine against
doxorubicin-induced cardiotoxicity through blocking apoptosis and
activating mitophagy-mediated ROS elimination. Phytomedicine.
101(154130)2022.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Bi Y, Xu H, Wang X, Zhu H, Ge J, Ren J and
Zhang Y: FUNDC1 protects against doxorubicin-induced cardiomyocyte
PANoptosis through stabilizing mtDNA via interaction with TUFM.
Cell Death Dis. 13(1020)2022.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Shi S, Chen Y, Luo Z, Nie G and Dai Y:
Role of oxidative stress and inflammation-related signaling
pathways in doxorubicin-induced cardiomyopathy. Cell Commun Signal.
21(61)2023.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Coll RC, Schroder K and Pelegrin P: NLRP3
and pyroptosis blockers for treating inflammatory diseases. Trends
Pharmacol Sci. 43:653–668. 2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Harris J and Borg NA: The multifaceted
roles of NLRP3-modulating proteins in virus infection. Front
Immunol. 13(987453)2022.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Moena D, Nardocci G, Acevedo E, Lian J,
Stein G, Stein J and Montecino M: Ezh2-dependent H3K27me3
modification dynamically regulates vitamin D3-dependent epigenetic
control of CYP24A1 gene expression in osteoblastic cells. J Cell
Physiol. 235:5404–5412. 2020.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Park S, Kim GW, Kwon SH and Lee JS: Broad
domains of histone H3 lysine 4 trimethylation in transcriptional
regulation and disease. FEBS J. 287:2891–2902. 2020.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Kang SJ and Chun T: Structural
heterogeneity of the mammalian polycomb repressor complex in immune
regulation. Exp Mol Med. 52:1004–1015. 2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Rawat PS, Jaiswal A, Khurana A, Bhatti JS
and Navik U: Doxorubicin-induced cardiotoxicity: An update on the
molecular mechanism and novel therapeutic strategies for effective
management. Biomed Pharmacother. 139(111708)2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Wang Y, Zhu J and DeLuca HF: Where is the
vitamin D receptor? Arch Biochem Biophys. 523:123–133.
2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Chen L, Yang R, Qiao W, Yuan X, Wang S,
Goltzman D and Miao D: 1,25-Dihydroxy vitamin D prevents
tumorigenesis by inhibiting oxidative stress and inducing tumor
cellular senescence in mice. Int J Cancer. 143:368–382.
2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Spasovski G: Advances in pharmacotherapy
for hyperphosphatemia in renal disease. Expert Opin Pharmacother.
16:2589–2599. 2015.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Sutterwala FS, Haasken S and Cassel SL:
Mechanism of NLRP3 inflammasome activation. Ann N Y Acad Sci.
1319:82–95. 2014.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Cao R, Ma Y, Li S, Shen D, Yang S, Wang X,
Cao Y, Wang Z, Wei Y, Li S, et al: 1,25(OH)(2) D(3) alleviates
DSS-induced ulcerative colitis via inhibiting NLRP3 inflammasome
activation. J Leukoc Biol. 108:283–295. 2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Tulk SE, Liao KC, Muruve DA, Li Y, Beck PL
and MacDonald JA: Vitamin D(3) metabolites enhance the
NLRP3-dependent secretion of IL-1β from human THP-1 monocytic
cells. J Cell Biochem. 116:711–720. 2015.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Poli G, Fabi C, Bellet MM, Costantini C,
Nunziangeli L, Romani L and Brancorsini S: Epigenetic mechanisms of
inflammasome regulation. Int J Mol Sci. 21(5758)2020.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Dai X, Liao R, Liu C, Liu S, Huang H, Liu
J, Jin T, Guo H, Zheng Z, Xia M, et al: Epigenetic regulation of
TXNIP-mediated oxidative stress and NLRP3 inflammasome activation
contributes to SAHH inhibition-aggravated diabetic nephropathy.
Redox Biol. 45(102033)2021.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Lecoeur H, Prina E, Rosazza T, Kokou K,
N'Diaye P, Aulner N, Varet H, Bussotti G, Xing Y, Milon G, et al:
Targeting macrophage histone H3 modification as a leishmania
strategy to dampen the NF-kappaB/NLRP3-mediated inflammatory
response. Cell Rep. 30:1870–1882. 2020.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Lecoeur H, Rosazza T, Kokou K, Varet H,
Coppee JY, Lari A, Commere PH, Weil R, Meng G, Milon G, et al:
Leishmania amazonensis subverts the transcription factor landscape
in dendritic cells to avoid inflammasome activation and stall
maturation. Front Immunol. 11(1098)2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Uckelmann M and Sixma TK: Histone
ubiquitination in the DNA damage response. DNA Repair (Amst).
56:92–101. 2017.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Chan HL and Morey L: Emerging roles for
polycomb-group proteins in stem cells and cancer. Trends Biochem
Sci. 44:688–700. 2019.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Barbour H, Daou S, Hendzel M and Affar EB:
Polycomb group-mediated histone H2A monoubiquitination in epigenome
regulation and nuclear processes. Nat Commun.
11(5947)2020.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Han X, Zhu N, Wang Y and Cheng G:
1,25(OH)2D3 inhibits osteogenic differentiation through activating
beta-catenin signaling via downregulating bone morphogenetic
protein 2. Mol Med Rep. 22:5023–5032. 2020.PubMed/NCBI View Article : Google Scholar
|