Introduction
Maffucci syndrome, also known as chondrodysplasia
with hemangioma or enchondroma with multiple cavernous hemangiomas,
was first reported by Maffucci in 1881 as a rare disorder (1). There are <300 cases of Maffucci
syndrome reported globally to date, however, the number of case
reports is likely an underestimate due to the economic and
scientific research expertise of the regions where the majority of
case studies have been reported (1,2). In
addition, medical professionals in certain regions may lack
awareness of the disease. Maffucci syndrome is characterized by
vascular and bone lesions, with patients reporting multiple
enchondromas and vascular malformations, mostly affecting the skin,
particularly on the upper and lower extremities (3). Previous studies have shown that
non-hereditary mutations in isocitrate dehydrogenase 1 (IDH1) and
isocitrate dehydrogenase 2 (IDH2) genes are closely related to the
pathogenesis of Maffucci syndrome (4,5).
These mutations serve an important role in the occurrence of
numerous types of malignant tumors, including intrahepatic
cholangiocarcinoma, acute myeloid leukemia, glioma and oral
squamous cell carcinoma (5).
Maffucci syndrome is extremely rare and there is
limited knowledge about this disease. There are a few hundred cases
in the world and there is no definitive agreement on the
pathogenesis and main treatment methods for this disease (1,2). The
present case study provides information for clinicians about
Maffucci syndrome by reporting imaging results, physical
examinations, clinical manifestations and the strategy used to
treat the patient. The present study aimed to establish a worldwide
case database and present a summarize of existing research
regarding the characteristics, etiology, provisional diagnosis,
differential diagnosis, treatment and prevention of Maffucci
syndrome, to improve understanding of this disease.
Case report
A 15-year-old male presented to Changzhi Yunfeng
hospital in August 2018 (Changzhi, China) with a 9 year history of
multiple nodules on both upper limbs and left hand nodule bleeding
for 1 day. There was no obvious cause for the initial occurrence of
multiple nodules in the bilateral upper limbs, particularly in the
left limb, which presented without itching and pain. Having visited
a number of hospitals, the patient had not received a clear
diagnosis. The patient was treated in Changzhi Traditional Chinese
medicine hospital (Changzhi, China) with oral Traditional Chinese
Medicine (dosage unknown). The patient was admitted to Changzhi
Yunfeng hospital due to bleeding of the left palm and finger
nodules. Upon further diagnostic tests, the patient was diagnosed
with Maffucci syndrome.
The patient was born to healthy, non-consanguineous
parents and was a first-time full-term delivery, with no
deformities at birth. The patient works in customer service, denied
smoking and drinking and had no family medical history of Maffucci
syndrome. Physical examination of the patient on admission
indicated short stature, valgus deformity of the left knee joint,
varus deformity of the right knee joint and 15 cm shortening of the
left lower limb compared with the right lower limb. The patient had
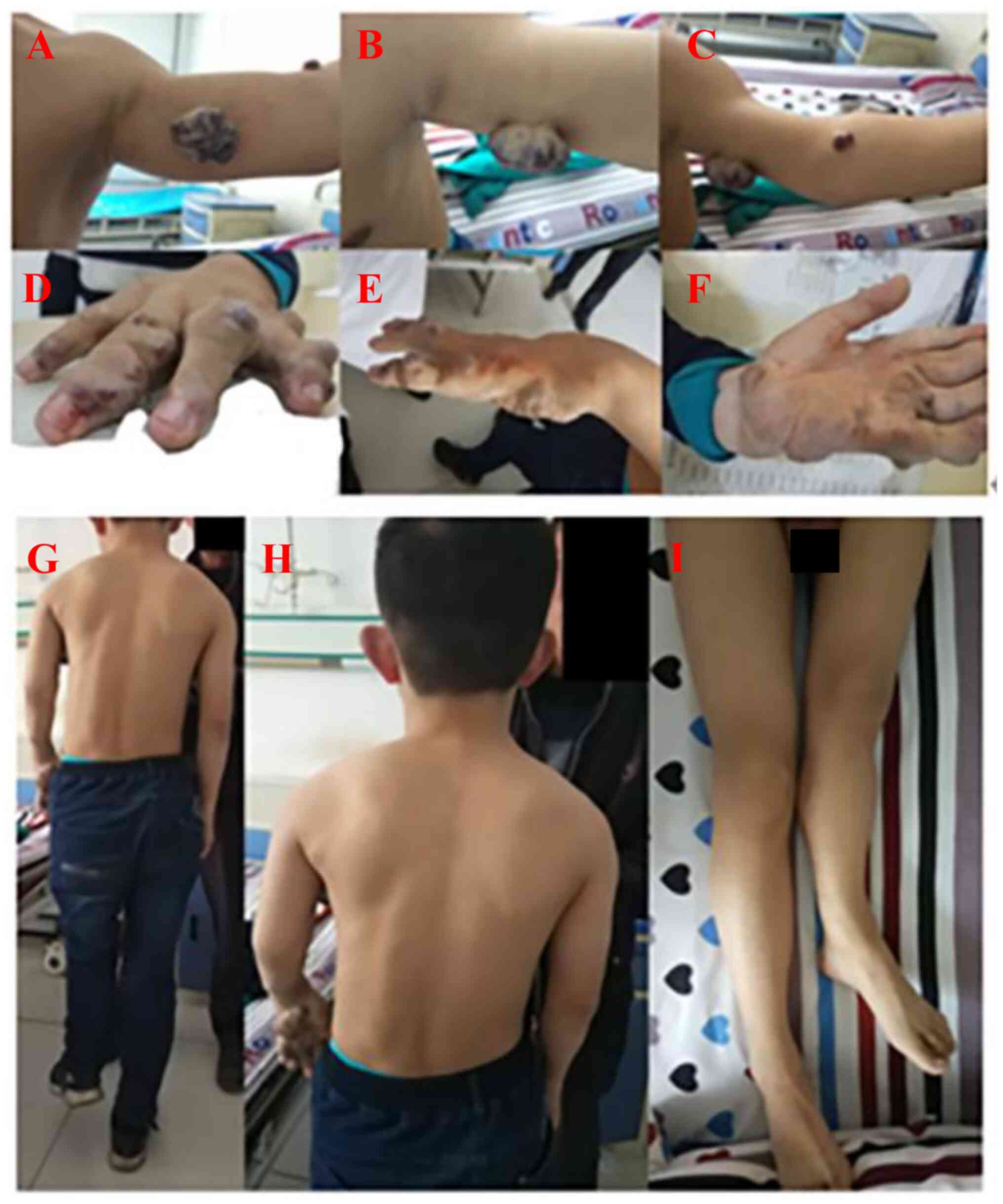
numerous light blue subcutaneous nodules of different sizes with a
soft, clear boundary and no pain in his left hand. On the left
upper arm, a dark red nodular lesions (~4x3 cm) and an elbow
nodular lesions (~1x1 cm) could be observed. Numerous subcutaneous
nodules could also be observed in the thenar of the right palm,
scrotum and perineum (Fig. 1).
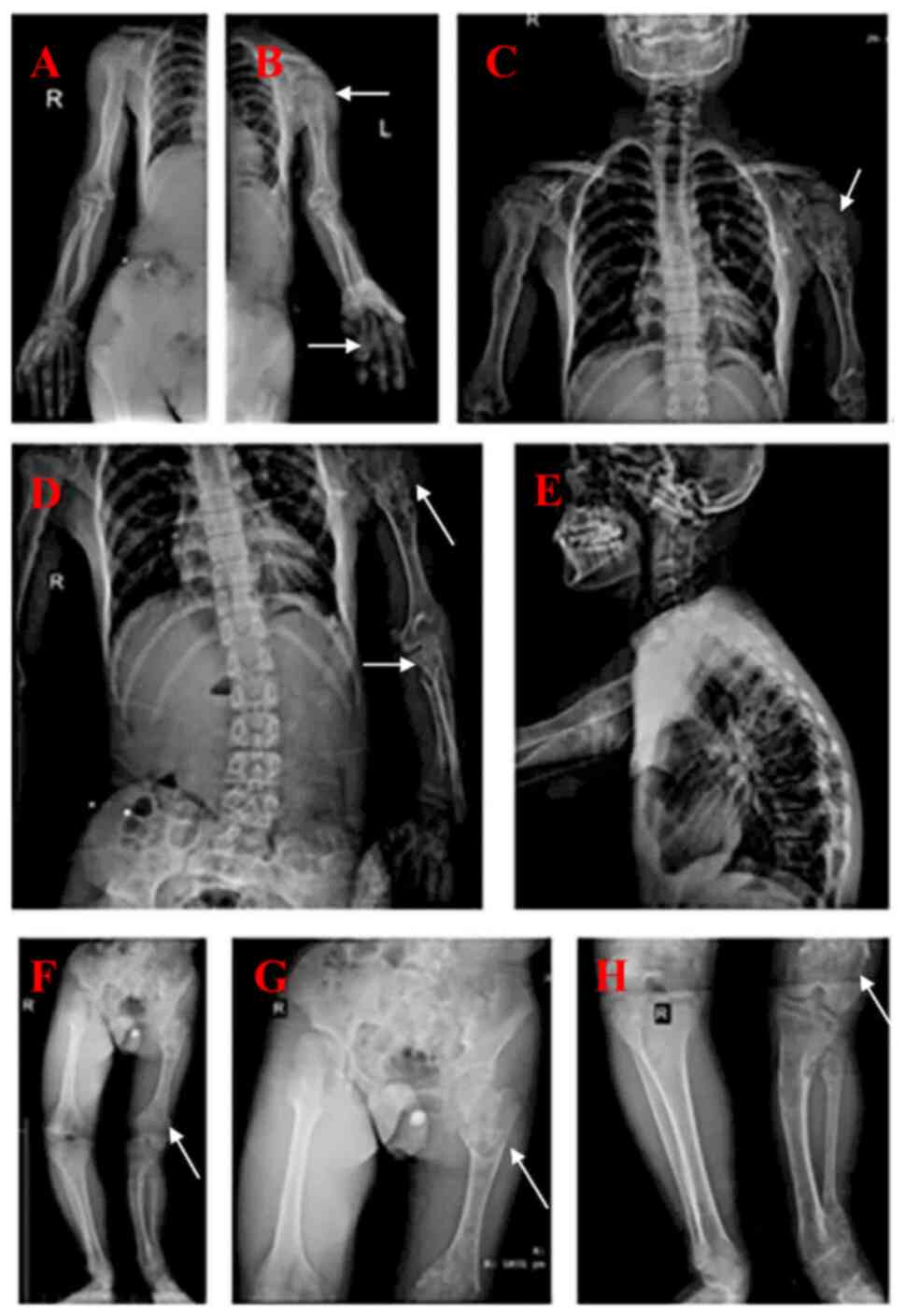
Radiographs demonstrated that the lower limbs, upper
limbs and shoulder blades had multiple enchondromas, in addition to
developmental malformations of the left humerus, radius, femur,
tibia and fibula. The patient had significant bone deformities and
solitary hemangioma (Fig. 2). CT
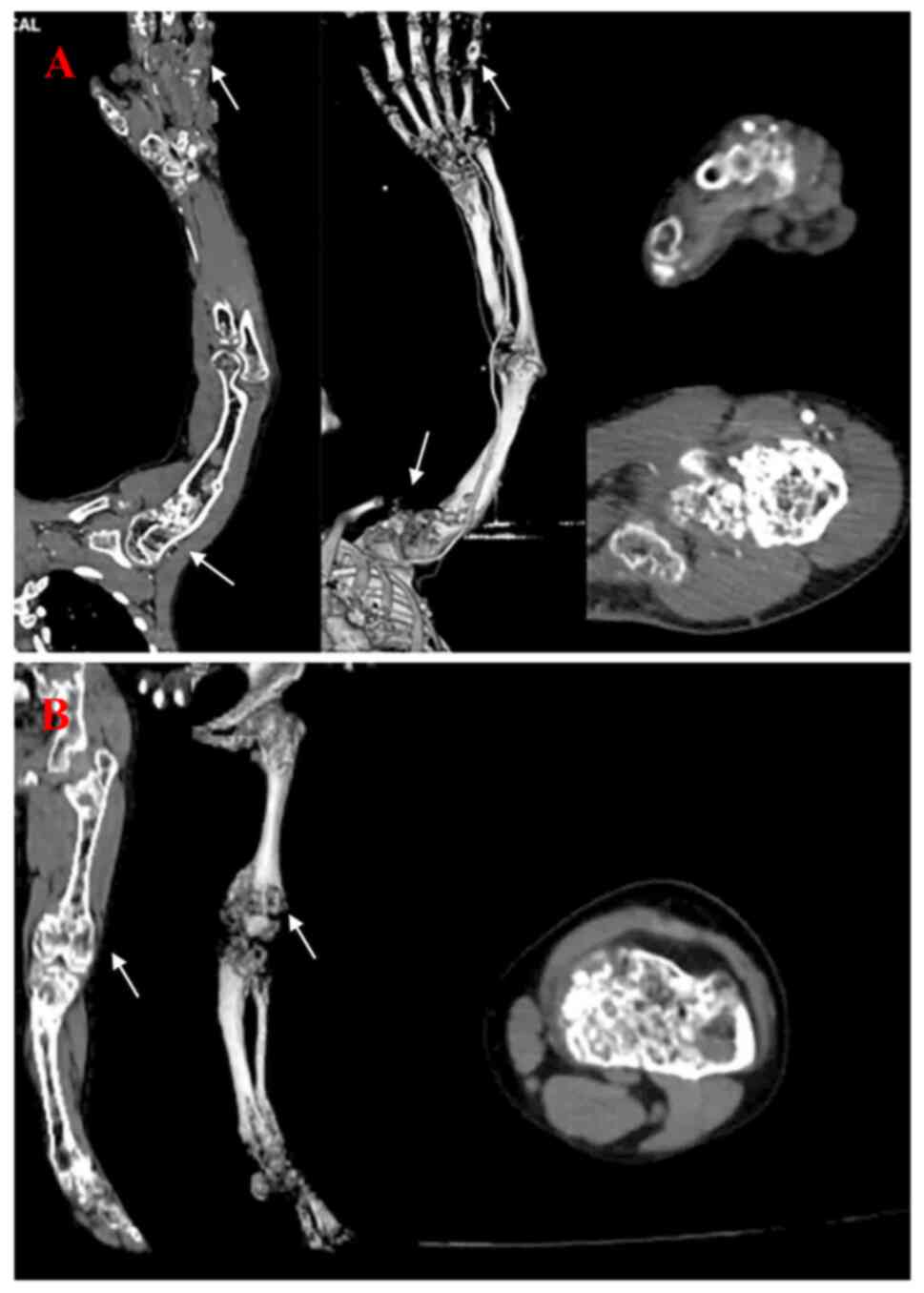
scans demonstrated that the left upper and lower limbs had multiple
endophytic chondromas, developmental malformations and a venous
stone that could be observed in the hemangioma of the hand
(Fig. 3). Surgical resection of
the diseased tissue and pathological examination were recommended.
The patient refused orthopaedic surgery to remove the focus and
requested conservative treatment. The patient's hemangioma involved
a large amount of skin and soft tissue, but there was no pain or
limb dysfunction reported. Since conservative treatment options for
Maffucci syndrome are currently lacking, it was recommended for the
patient to be admitted to a hospital with a higher level of
expertise for futher diagnosis and treatment. The patient was
instructed to undergo regular medical review, closely monitor the
signs of malignant transformation of endophytic chondroma and
multiple hemangioma, seek early treatment and prevent pathological
fractures.
According to patient follow-up, the patient had
undergone resection of the focus at Shanghai traditional Chinese
Medicine Hospital (Shanghai, China) and postoperative nodule
bleeding was well controlled. As the patient underwent the removal
of skin nodules, these images were not re-photographed.
Pathological results reported cavernous hemangioma with thrombosis
in the patient. However, no IDH1 or IDH2 gene mutations were
detected in peripheral blood or tumor tissue DNA. The patient had
two major clinical symptoms of multiple endophytic chondroma and
hemangioma, therefore Maffucci syndrome was diagnosed. The patient
presented with significant skeletal deformity, but their current
condition was relatively stable and the patient had no intention of
orthopedic surgery. It was reccommended to the patient to engage in
follow-up studies to monitor the changes in their condition.
Discussion
Literature search
Certain types of malignant tumor have a risk of
being associated with Maffucci syndrome, according to previously
published case reports (3,4). The present study analyzed Maffucci
syndrome according to case reports published in the Web of Science
Core Collection database (webofscience.com/wos/woscc/basic-search; accessed July
20, 2022). The keywords used in the literature search were
‘Maffucci’ and ‘case’.
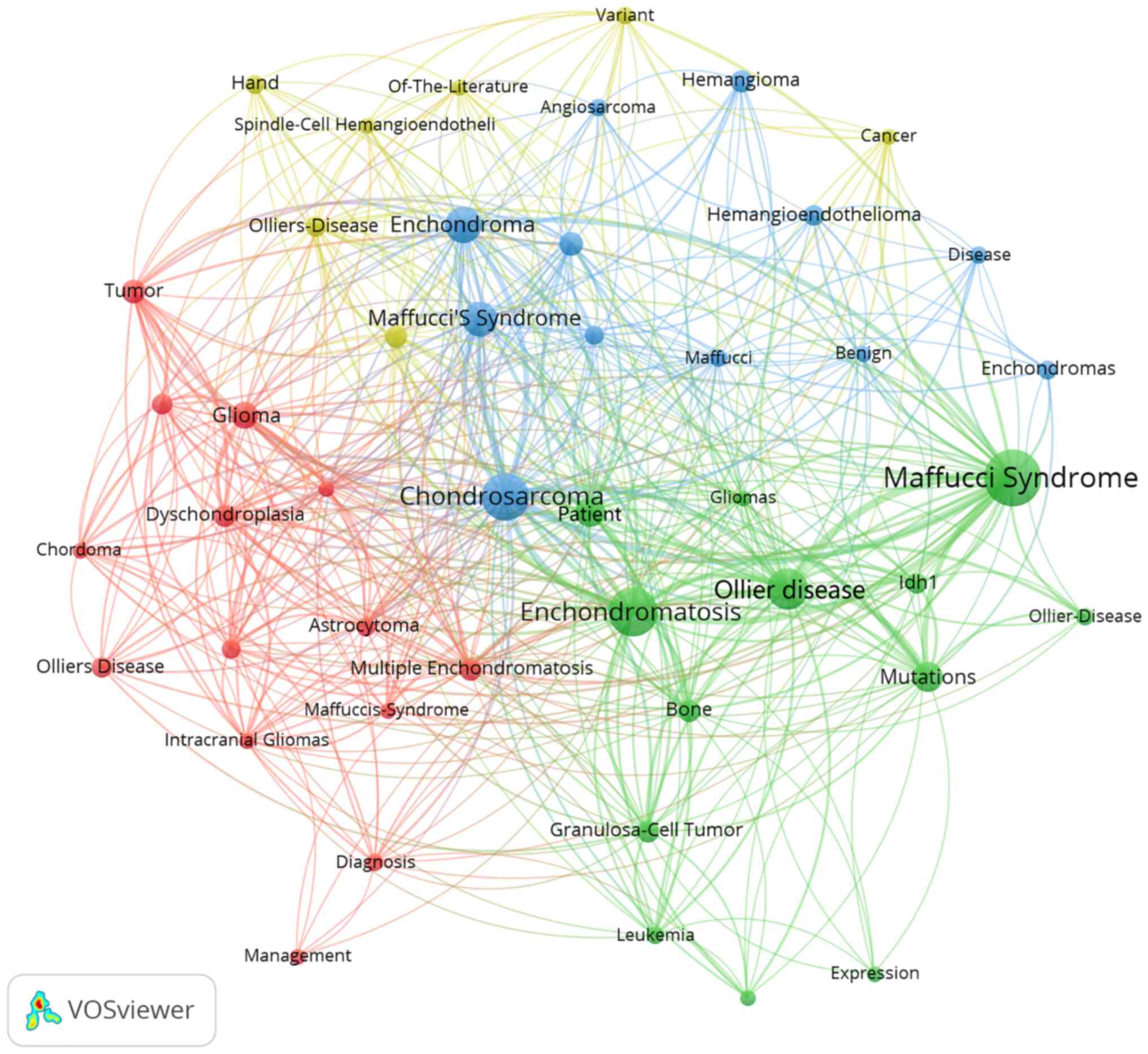
The VOSviewer (version 1.6.17) software was used for
constructing and visualizing bibliometric networks (vosviewer.com). Based on literature search results,
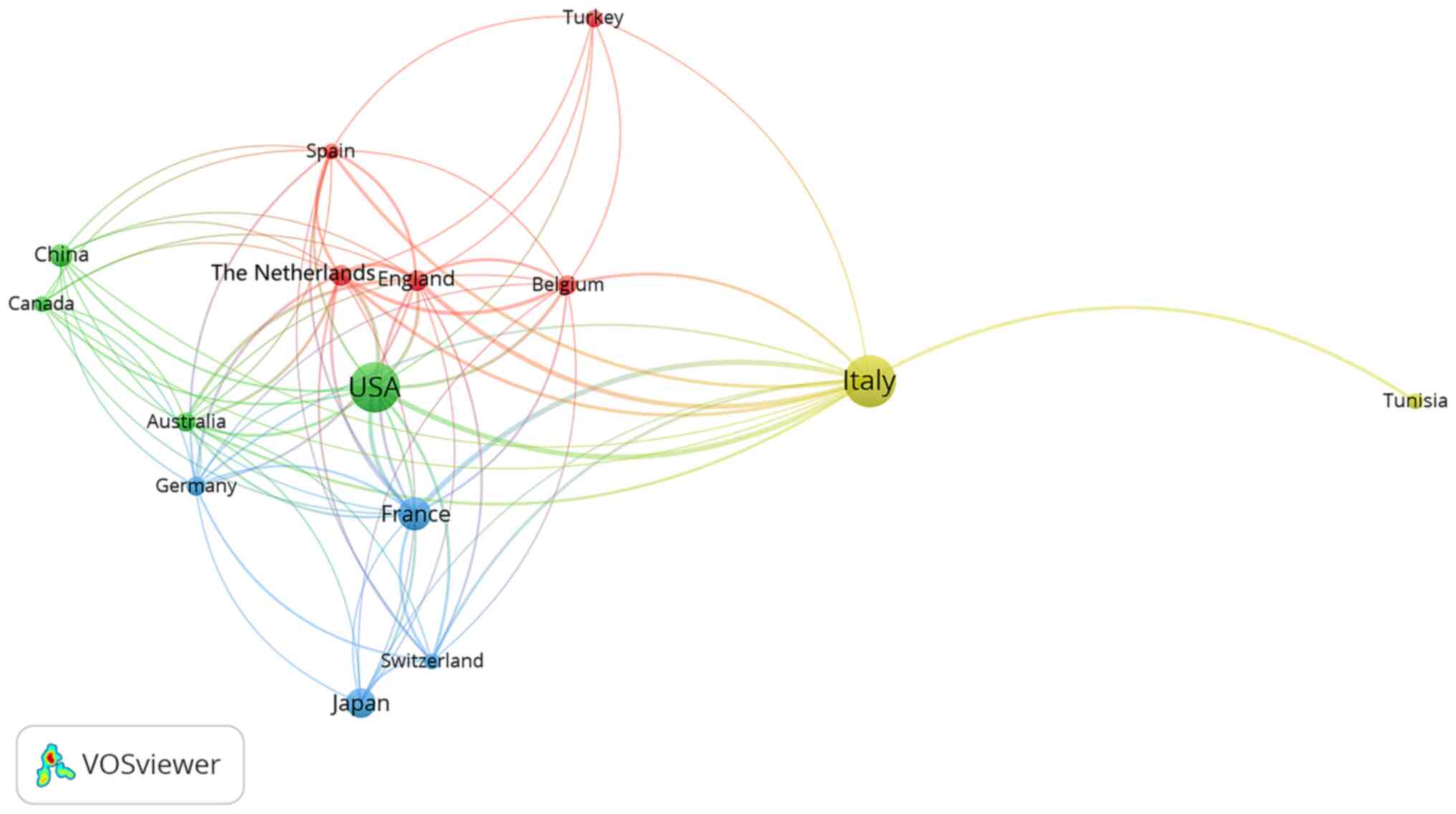
Maffucci syndrome was reported in 248 case reports in 45 countries,
with the highest number of reports conducted in Italy (n=66) and
the USA (n=63). The majority of the top 10 countries that reported
cases of Maffucci syndrome in the literature review were located in
Europe and the USA, which may indicate that Maffucci syndrome is
more likely to occur in Caucasian populations in these countries
(Table I, Fig. 4). However, the publication of case
reports can be related to the economic status and scientific
research expertise of the region. Therefore, the absence of reports
of Maffucci syndrome in certain regions does not mean there is no
incidence of disease in these regions, as case studies require a
large amount of data to support publication. The present study used
VOSviewer to analyze the frequency of keywords used by previous
case studies reporting Maffucci syndrome The high-frequency subject
terms were extracted using the Bibliographic Items Co-occurrence
Matrix Builder (BICOMB), and a core subject term co-occurrence
matrix was established. Through VOSviewer statistics of keywords in
the field of Maffucci syndrome to form a co-word network diagram
composed of core subject terms (Fig.
5). Meanwhile the article lists the first 10 high-frequency
keywords (Table II).
 | Table IDistribution of research on Maffucci
Syndrome by country of origin |
Table I
Distribution of research on Maffucci
Syndrome by country of origin
| Country of
origin | Case reports, n |
|---|
| Italy | 66 |
| USA | 63 |
| France | 27 |
| Japan | 21 |
| China | 14 |
| The Netherlands | 12 |
| England | 12 |
| Belgium | 10 |
| Germany | 9 |
| Australia | 9 |
| Table IIHigh-frequency keywords used in
previously published case studies of Maffucci Syndrome. |
Table II
High-frequency keywords used in
previously published case studies of Maffucci Syndrome.
| Keyword | Frequency, n |
|---|
| Maffucci
syndrome | 61 |
| Enchondromatosis | 44 |
| Chondrosarcoma | 42 |
| Ollier disease | 33 |
| Enchondroma | 26 |
| Maffucci's
syndrome | 24 |
| Patient | 18 |
| Mutations | 18 |
| Glioma | 14 |
| Tumor | 11 |
Clinical characteristics and
etiology
Maffucci syndrome is a rare mesodermal dysplastic
disease characterized by multiple enchondromas and vascular
malformations, particularly fusiform cell hemangiomas. Growth and
developmental problems can result from vascular and bone
malformations as early as childhood (1). Enchondroma is a common benign bone
tumor in the distal extremities, characterized by chondrocyte
involvement in the development of the long bones, limb shortening,
pelvic tilt and scoliosis. Typically, enchondroma appears as
solitary lesions, but Maffucci syndrome can cause the occurrence of
multiple lesions. Enchondroma tends to be recurrent, which can lead
to local bone destruction, pain and fractures, amongst other
complications. Vascular lesions in patients with Maffucci syndrome
are located in the subcutaneous tissue, usually distal to the
extremities and demonstrate a lateral distribution, but can also
involve mucosal tissues, such as the oral cavity (5). Additionally, these patients may
develop secondary central chondrosarcomas and are more likely to
develop non-skeletal malignancies in addition to bony masses, bone
deformities and pathological fractures. In addition to causing the
malignant transformation of visceral chondromas and hemangiomas,
Maffucci syndrome has been reported to be linked to brain and
ovarian tumors, leukemia and oral squamous cell carcinoma (1,5-7).
The pathogenesis of Maffucci syndrome is currently
unclear, but it is generally believed to be caused by
non-hereditary mutations in IDH1 and IDH2 (1,7,8).
Amary et al (1) and
Pansuriya et al (8)
reported that Maffucci syndrome is caused by somatic mosaic IDH1/2
mutations, which can also be related to cases of isolated
enchondroma and chondrosarcoma. >70% of reported Maffucci
syndrome cases have a IDH1/2 mutation, which strongly suggests that
these mutations are responsible for its pathogenesis (9). In addition, IDH1/2 mutations can
cause a range of cancers, such as glioma, cholangiocarcinoma,
chondrosarcoma, and acute myeloid leukemia (4). Mutations in IDH1/2 causes production
of the tumor metabolite 2-hydroxyglutaric acid, which restricts
cell differentiation by inhibiting the activity of
chromatin-modified histones and DNA demethylation (5). Moreover, previous studies have
reported mutations in the parathyroid hormone receptor 1 (PTH1R)
gene in patients with multiple endophytic chondromatosis (10,11).
It has been reported that a small number of patients with multiple
endochondromatosis have mutations in PTH1R and these mutations lead
to a decline in receptor function, which may also be a cause of
Maffucci syndrome (12,13).
Diagnosis
Currently, there are no unified criteria for the
diagnosis of Maffucci syndrome; however, there are a number of
clinical features that can be useful for diagnosis. The development
of specific diagnostic criteria need to be verified using data from
a large number of case studies. The first step towards diagnosis
should analyze clinical features of disease combined with patient
imaging results. The clinical features were described previously in
this study. When combined with imaging results, indicated that
there were spots, longitudinal strips and honeycomb in the center,
or side, of the bone in addition to cystic transparent areas,
either presenting alone or mixed. In multiple enchondromas, it is
possible to see spots of calcification in the transparent area,
sometimes radial dense stripes, and often long tubular bone
shortening. Small hemangiomas are difficult to observe through
imaging studies, as they may only exhibit increased local soft
tissue density on X-ray films, while larger multiple hemangiomas
exhibit irregular thickening or nodular protrusions of soft tissue,
and there are often venous stones of different sizes present, which
may suggest Maffucci syndrome (14,15).
In addition, it is possible to locally remove an isolated mass for
pathological analysis. Pathological examinations found mainly
endophytic chondroma and cavernous angioma, with or without
thrombosis (3). However,
performing a biopsy of diseased tissue may be unsafe and cause
serious complications, such as severe bleeding, so the decision to
remove diseased tissue for pathological examination should be based
on the clinical situation (3,16).
Molecular detection methods are used to analyze peripheral blood
and tumor tissue to detect mutations in IDH1, IDH2,
ELKS/RAB6-interacting/CAST family member 2 (ERC2) gene or PTH1R.
For example, as an alternative diagnostic method to tissue biopsy,
detection of cell-free DNA (cfDNA) can provide a highly accurate
diagnostic method for Maffucci syndrome and can avoid the
complications associated with tissue biopsy procedures (13). It has previously been reported that
low-frequency somatic IDH1p.Arg132Cys mutations are consistently
detected in hemangioma tissues and cystic blood-derived (13). Therefore, it has been suggested
that genetic diagnosis of Maffucci syndrome may be improved with
cfDNA sequencing, a reliable and sensitive diagnostic approach
(1,8,16,17).
Differential diagnosis
A Maffucci syndrome diagnosis typically occurs for a
patient during adolescence (2), as
a result of the clinical manifestations of disease coupled with
imaging results, as demonstrated by the patient in the present
study. Combining the results from imaging studies, pathological
analysis and molecular analysis, indicated that the provisional
diagnosis of Maffucci syndrome was likely, however, a differential
diagnosis was still required (18,19).
It is important to distinguish endophytic chondroma from bone
cysts, giant cell tumors of the bone, bone fibrous dysplasia and
other conditions with a comparable imaging presentation.
Enchondromatosis can be a symptom of either Maffucci syndrome or
Ollier disease, which is also caused by IDH gene mutation (17,18).
Compared with Maffucci syndrome, Ollier disease lacks the clinical
manifestation of hemangioma and typically presents unilaterally.
Imaging studies of patients with Ollier disease have demonstrated
that there is generally no venous stone in the hemangioma and
previously published studies reported that the probability of
chondroma malignant transformation in Ollier disease was low
(20,21). Furthermore, Maffucci syndrome needs
to be distinguished from other diseases characterized by similar
vascular malformations, such as blue rubber bleb nevus syndrome,
Klippel-Trenaunay syndrome and glomuvenous malformations.
Treatment and prognosis
Currently, there is no standard treatment plan for
Maffucci syndrome; however, specific inhibitors of IDH1/2 have
demonstrated to be beneficial for the treatment of specific
malignant tumor (16). Acute
myeloblastic leukemia can be effectively treated using ivosidenib
or enasidenib, which inhibit IDH1 and IDH2 protein expression
(22). Current research into
treatments for Maffucci syndrome has been initiated by the
discovery of IDH1/2 inhibitors, an important step forward for
treatment of this disease, particularly for patients suffering from
severe symptoms such as rapid tumor progression, dysfunction of
blood system and motor system and malignant tumors (16).
At present, there is no effective treatment
available for vascular lesions of Maffucci syndrome. Sirolimus, a
mTOR inhibitor, can effectively treat various vascular anomalies,
but further research must be performed to determine its full
effectiveness (23). In general,
hemangiomas should be treated to prevent patient pain, swelling and
dysfunction of blood system and motor system. Currently, sclerosing
agents, surgical resection, radiotherapy, embolization and chemical
therapy are the most common treatments (24). Enchondromatosis could cause
skeletal deformity and dysfunction, which may require orthopedic
surgery, therefore malignant tumors should be resected as soon as
possible (25).
To conclude, Maffucci syndrome is relatively rare,
presents with diverse clinical features and has an unknown
pathogenesis, therefore individual institutions are unlikely to
have sufficient resources to study this disease. Therefore, there
is an urgent need to summarize the existing cases around the world
and produce a set of effective guidelines for the diagnosis,
treatment and prevention of Maffucci syndrome, in order to gain a
better understanding of this disease.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by Chang Zhi Medical
College's 2020 Doctoral Starting Fund (grant no. BS202004).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YPW, WJD, SY and ZW wrote the manuscript and
analyzed patient data. YPW, SY and ZW confirm the authenticity of
all the raw data. YFX, PFH and SLQ designed the study. All authors
have read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The patient's guardian provided written informed
consent to participate in this study.
Patient consent for publication
The patient's guardian provided written informed
consent for the publication of this case report and all
accompanying images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Amary MF, Damato S, Halai D, Eskandarpour
M, Berisha F, Bonar F, McCarthy S, Fantin VR, Straley KS, Lobo S,
et al: Ollier disease and Maffucci syndrome are caused by somatic
mosaic mutations of IDH1 and IDH2. Nat Genet. 43:1262–1265.
2011.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
El Abiad JM, Robbins SM, Cohen B, Levin
AS, Valle DL, Morris CD and de Macena Sobreira NL: Natural history
of Ollier disease and Maffucci syndrome: Patient survey and review
of clinical literature. Am J Med Genet A. 182:1093–1103.
2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ichimura N, Yamamoto N, Toyama N and Hibi
H: A case of Maffucci syndrome with a buccal hemangioma harboring a
mutation in IDH1. Oral Oncol. 122(105553)2021.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Yilmaz-Gulec E, Marzin P, Huber-Lequesne C
and Cormier-Daire V: An intermediate phenotype in IDH related
enchondromatosis spectrum. Eur J Med Genet.
66(104697)2023.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Molenaar RJ, Maciejewski JP, Wilmink JW
and van Noorden CJF: Wild-type and mutated IDH1/2 enzymes and
therapy responses. Oncogene. 37:1949–1960. 2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Casal D, Mavioso C, Mendes MM and Mouzinho
MM: Hand involvement in Ollier disease and Maffucci syndrome: A
case series. Acta Reumatol Port. 35:375–378. 2010.PubMed/NCBI
|
|
7
|
Chen C, Li J, Jiang T, Tang J, Zhang Z,
Luo Y, Wang X, Sun K, Jiang Z, Zhou J and Liu Z: IDH mutations are
potentially the intrinsic genetic link among the multiple
neoplastic lesions in Ollier disease and Maffucci syndrome: A
clinicopathologic analysis from a single institute in Shanghai,
China. Diagnostics (Basel). 12(2764)2022.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Pansuriya TC, van Eijk R, d'Adamo P, van
Ruler MA, Kuijjer ML, Oosting J, Cleton-Jansen AM, van Oosterwijk
JG, Verbeke SL, Meijer D, et al: Somatic mosaic IDH1 and IDH2
mutations are associated with enchondroma and spindle cell
hemangioma in Ollier disease and Maffucci syndrome. Nat Genet.
43:1256–1261. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Nejo T, Tanaka S, Ikemura M, Nomura M,
Takayanagi S, Shin M, Ushiku T, Shibahara J, Saito N and Mukasa A:
Maffucci syndrome complicated by three different central nervous
system tumors sharing an IDH1 R132C mutation: Case report. J
Neurosurg. 131:1829–1834. 2018.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Couvineau A, Wouters V, Bertrand G, Rouyer
C, Gérard B, Boon LM, Grandchamp B, Vikkula M and Silve C: PTHR1
mutations associated with Ollier disease result in receptor loss of
function. Hum Mol Genet. 17:2766–2775. 2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Pansuriya TC, Kroon HM and Bovée JVMG:
Enchondromatosis: Insights on the different subtypes. Int J Clin
Exp Pathol. 3:557–569. 2010.PubMed/NCBI
|
|
12
|
Hopyan S, Gokgoz N, Poon R, Gensure RC, Yu
C, Cole WG, Bell RS, Jüppner H, Andrulis IL, Wunder JS and Alman
BA: A mutant PTH/PTHrP type I receptor in enchondromatosis. Nat
Genet. 30:306–310. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Verdegaal SH, Bovée JV, Pansuriya TC,
Grimer RJ, Ozger H, Jutte PC, San Julian M, Biau DJ, van der Geest
IC, Leithner A, et al: Incidence, predictive factors, and prognosis
of chondrosarcoma in patients with Ollier disease and Maffucci
syndrome: An international multicenter study of 161 patients.
Oncologist. 16:1771–1779. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Verma GG, Jain VK and Iyengar KP:
Monomelic Maffucci syndrome. BMJ Case Rep.
14(e239619)2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Lv H, Jiang H, Zhang M, Luo H, Hong Z,
Yang H, Xu W, Shen B, Zhang W, Qiu H and Zhu R: Maffucci syndrome
complicated by giant chondrosarcoma in the left ankle with an IDH1
R132C mutation: A case report. World J Surg Oncol.
20(218)2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Sun Y, Fan X, Rao Y, Wang Z, Wang D, Yang
X, Zheng L, Wen M, Cai R and Su L: Cell-free DNA from plasma as a
promising alternative for detection of gene mutations in patients
with Maffucci syndrome. Hereditas. 159(4)2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Cheng P, Chen K, Zhang S, Mu KT, Liang S
and Zhang Y: IDH1 R132C and ERC2 L309I mutations contribute to the
development of Maffucci's syndrome. Front Endocrinol (Lausanne).
12(763349)2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Noël G, Feuvret L, Calugaru V, Hadadi K,
Baillet F, Mazeron JJ and Habrand JL: Chondrosarcomas of the base
of the skull in Ollier's disease or Maffucci's syndrome-three case
reports and review of the literature. Acta Oncol. 43:705–710.
2004.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Saiji E, Pause FG, Lascombes P, Cerato
Biderbost C, Marq NL, Berczy M, Merlini L and Rougemont AL: IDH1
immunohistochemistry reactivity and mosaic IDH1 or IDH2 somatic
mutations in pediatric sporadic enchondroma and enchondromatosis.
Virchows Arch. 475:625–636. 2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Sharif B, Lindsay D and Saifuddin A:
Update on the imaging features of the enchondromatosis syndromes.
Skeletal Radiol. 51:747–762. 2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Jacobi CMC, Hiranya ES, Gay A, Holzmann D,
Kollias S and Soyka MB: Enchondroma of the nasal septum due to
Ollier disease: A case report and review of the literature. Head
Neck. 37:E30–E33. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Yao K, Liu H, Yu S, Zhu H and Pan J:
Resistance to mutant IDH inhibitors in acute myeloid leukemia:
Molecular mechanisms and therapeutic strategies. Cancer Lett.
533(215603)2022.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Gupta V, Mridha AR and Khaitan BK:
Unsatisfactory response to sirolimus in Maffucci
syndrome-associated spindle cell hemangiomas. Dermatol Ther.
32(e12851)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Mazingi D, Mbanje C, Jakanani G, Muguti
GI, Mandizvidza V and Bopoto S: Maffucci's syndrome in association
with giant tubular adenoma of the breast: Case report and
literature review. Int J Surg Case Rep. 63:147–152. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Koca K, Akpancar S and Yıldız C:
Correction of length discrepancy of radius and ulna with
distraction osteogenesis: Three cases. Case Rep Orthop.
2015(656542)2015.PubMed/NCBI View Article : Google Scholar
|