Psoriasis (PS), a chronic T cell-mediated
inflammatory disease, affects ~3% of the general population
worldwide, and has increased in recent years (1). Systemic lupus erythematosus (SLE) is
a recurrent and remitting autoimmune disease that occurs in a
number of organs and systems, for instance, skin, blood system and
kidneys (2). Environmental and
genetic variables have been considered as dominant causes to induce
autoimmune responses, resulting in the overproduction of
inflammatory cytokines, for instance IL-6, and autoantibodies from
B cells, especially in SLE. Indeed, the presence of antibodies
against nuclear and cytoplasmic antigens is a diagnostic indication
of SLE (3). PS and SLE affect the
appearance and quality of life of the patients. Both autoimmune
disorders manifest as chronic inflammatory conditions, which cause
skin lesions and damage to the joints and other organs, such as
those in the cardiovascular system. However, these two disorders
have long been considered as distinct diseases on the basis of
their relatively disparate pathologic mechanisms. While the chronic
inflammatory condition has been associated to T helper (Th)1 and
Th17 cell activation in PS (4,5),
overacted B cells and Th2 cell-associated abnormalities are
associated to SLE development (6,7). It
has also been hypothesized that the pathogenic mechanisms for SLE
and PS are opposite (8); however,
this cannot explain the numerous published cases of comorbid PS and
SLE (9-14).
The key factors that cause autoimmunity in SLE
include the overproduction of autoantibodies, complement activation
and immune-complex deposition (15-19).
A number of recent studies have observed that patients with PS not
only have affected skin but also have other accompanying autoimmune
conditions, such as rheumatoid arthritis, alopecia areata, celiac
disease, systemic sclerosis, Crohn's disease, Sjögren syndrome,
vitiligo, ulcerative colitis, giant cell arthritis and SLE
(20-22).
PS is considered an autoimmune condition that may be induced by the
activation of T cells and B cells in the absence of persistent
infection or other discernible causes (23).
In the present review, a number of developments in
humoral and cellular immunities that occur in patients with
comorbid SLE and PS are presented and discussed. The present review
aimed to find the possible links between PS and SLE by reviewing
the epidemiological data, immunopathogenic mechanisms, genetic
traits and therapeutic efficacies.
Two types of comorbid diseases have been reported in
patients with PS: Those diseases sharing the main pathogenic
mechanisms and those not sharing pathogenesis but with clinically
severe chronic inflammatory conditions (24). SLE belongs to the latter category
(25). According to an
epidemiological study of PS comorbidity, patients with PS were at
greater risk of developing an immune-mediated inflammatory disease
(IMID) compared with general population controls (26). The majority of concurrent IMIDs
appeared before the diagnosis of PS, indicating that there could be
a pathophysiologic mechanism underlying PS and concurrent IMIDs
(27-33).
A population-based case-controlled study of the coexistence of PsA
and SLE in Israel revealed a 2.3-fold increase in the prevalence of
SLE among patients with PsA compared with age and sex-matched
controls from the general population (34). According to research reports, when
analyzed by level of severity, severe PS demonstrated a 3- to
7-fold increased risk for SLE compared with mild PS, especially in
Asian patients (28,35,36).
The explanation was that patients with moderate to severe PS may be
more likely to be receiving additional phototherapy. Both of these
treatments increase the risk for the development of SLE (37,38).
A 10-year retrospective study identified 42 cases of SLE in 9,420
patients with PS (39). In the
same study, the prevalence rate of PS that coexists with SLE was
~1.1%, and was slightly higher in female patients due to the fact
that prevalence of SLE is higher in women compared with men.
Therefore, when treating female patients with PS, caution should be
exercised in medication and vigilance should be exercised against
the occurrence of SLE.
A previous study, investigating the prevalence of PS
in patients with principally diagnosed SLE, analyzed a large
national population database for admission probabilities of
patients with SLE, and demonstrated that 150 of a total of 20,630
hospitalized patients with SLE (0.7%) had a co-existing PS
condition (40). In another study,
it was reported that 0.6% of 520 patients with SLE had PS (41). Patients have a sequential
occurrence of PS and SLE, but the probability of both occurring
together is <1.2%, and the lower incidences in both cases
suggest that comorbidity between two diseases could be an
accidental event. According to reports, patients with SLE
experienced a psoriatic flare that was likely due to the use of
antimalarial drugs such as hydroxychloroquine (10,42).
Therefore, when treating patients with SLE and a family history of
PS, an alternative drug to hydroxychloroquine would be more
appropriate, such as mycophenolate mofetil.
Prior to the discovery of comorbid PS and SLE, it
was hypothesized that the pathological mechanisms of PS and SLE
were different; PS is a systemic inflammatory reaction caused by
Th1 cell activation while abnormalities of SLE are due to highly
reactive Th2 responses (43,44).
To initiate an inflammation, exposure to external infectious or
non-infectious substances, such as bacteria, viruses and
ultraviolet radiation, damage the host cells to form an antigen
complex with released nucleotides and antimicrobial peptides in the
epidermis. Antigen-presenting cells, such as plasmacytoid dendritic
cells (DCs), identify this complex and stimulate antigen-specific T
cell growth in the skin and lymph nodes (45). The plasmacyte DCs secrete type I
interferon that increases the production of IL-23 and TNF-α in
myeloid DCs (46). These cytokines
promote Th17 cell differentiation, which together with IL-1
stimulation, produce IL-17 and IL-22 that further increase the
expression of TNF-α, C-C motif chemokine ligand 20 (CCL20) and
antimicrobial peptides such as LL37(47), leading to an inflammatory response
in the skin and to keratinocyte proliferation (48). Cytokines associated with SLE
include IFN-α, IL-6 and IL-17(49). These inflammatory cytokines, in
particular B-cell activating factor of the TNF family (BAFF), also
serve roles in autoimmunity and autoantibody production in SLE.
A number of B cell subsets may be strongly
associated with SLE. Currently, there are three known B cell
effectors involved in the pathogenesis of SLE: i) Pathogenic
plasmablasts may be produced without the assistance of T cells, as
demonstrated in the BAFF transgenic model (50); ii) autoreactive B cells and
CD4+ T cells interact at the T cell: B cell boundary
after initial autoantigen recognition; and iii) co-stimulatory
signals and cytokine crosstalk activate B cells and autoantibody
production through a T cell-dependent extrafollicular route or
inside spontaneous, autoimmune germinal centers (51,52).
Therefore, the aberrant activation of human B cells is a phenotypic
hallmark of SLE and is associated with the progress of the
disease.
Th17 cells are associated with the pathogenesis of
various autoimmune and inflammatory diseases, such as rheumatoid
arthritis, SLE, multiple sclerosis, PS, inflammatory bowel disease
and allergy and asthma (53), by
producing several effector molecules. They are characterized by the
expression of the orphan nuclear factor receptor retinoic acid
receptor-related orphan receptor-γ-t (RORγt), the cytokines IL-17
and IL-22, the chemokine CCL20, and the inflammatory chemokine
receptor C-C motif chemokine receptor 6 (CCR6) (54-56).
IL-17 is a potent proinflammatory cytokine produced by highly
activated Th17 cells and is known to serve a role in maintaining
chronic inflammation in PS. Indeed, highly expressed IL-17, IL-22
and IL-23 have been demonstrated in skin biopsies from patients
with PS (57,58). IL-22 is also known to be essential
for maintaining the immune barrier within the epidermis and is able
to induce the release of antimicrobial agents and β-defensins from
keratinocytes and promote epidermal hyperplasia (57). In the recruitment of Th17 cells to
local tissues, the CCL20/CCR6 axis has been demonstrated to serve a
crucial role (59). Finally,
several other factors associated to the Th17 response also engage
in the vascular inflammatory pathway by recruiting leukocytes,
activating B cells and producing autoantibodies, and therefore may
contribute to the occurrence and development of SLE and PS
(60,61). The current data indicates the
factors that are common between SLE and PS are an increase in the
number of Th17 lymphocytes and an increase in the serum levels of
IL-17 and IL-23, in which IL-17 is a main proinflammatory cytokine
that serves a crucial role in the pathogenesis of various
inflammatory diseases, including PS and SLE (62-64).
Patients with SLE have higher serum levels of IL-17
and IL-23 compared with healthy controls (65). Furthermore, IL-17 levels in the
plasma are correlated with the severity of SLE (66). Compared with a healthy control
group, the concentration of IL-17 in the serum of patients with SLE
and the expression of IL-17 mRNA in activated peripheral blood
mononuclear cells were increased, which were positively correlated
with the Systemic Lupus Erythematosus Disease Activity Index
(67-69).
The skin biopsy examination of patients with SLE and skin
involvement demonstrated that the expression level of IL-17 was
increased compared with that of normal individuals, confirming that
IL-17 is involved in the immune pathogenesis of SLE (70). IL-17 promotes inflammation and
tissue damage in the context of SLE by recruiting neutrophils and
monocytes, facilitating T-cell tissue infiltration and promoting
antibody production (71). By
contrast, IL-17 also facilitates T-cell activation and infiltration
into the tissues along with increased expression levels of
intercellular adhesion molecule-1 (ICAM-1) and matrix
metalloproteinase (MMPs) (72,73).
The IL-23/Th17 axis has previously been suggested to
be essential in developing lupus nephritis, both in mice models and
in patients with SLE (74,75). In mouse models, IL-17 is associated
not only to T cell-mediated tissue injury, but also to the
production of pathogenic autoantibodies and it was demonstrated
that Th17 cells were increased in a MRL/lpr lupus nephritis mouse
model (76). The IL-23/IL-17 axis
is involved in the pathogenesis of SLE where activated DCs produce
the inflammatory cytokines IL-6 and IL-23, which then stimulate
Th17 cells to produce IL-17(77).
In two in vitro studies, T cells from patients with SLE
increased their IL-17 production and concomitantly limited
production of the regulatory cytokine IL-2 in the presence of
IL-23, leading to exacerbated inflammation (56,57).
Additionally, an attenuated inflammation with a striking decrease
in the accumulation of double-negative T cells were revealed in the
kidneys and secondary lymphoid organs when a IL-23 receptor
deficient MRL/lpr mouse SLE model was used (78,79).
It is well known that B cells and autoantibodies
directed against numerous nuclear and cell surface antigens serve
roles in SLE immunopathogenesis. Given the fact that SLE-derived B
cells would increase anti-DNA production in the presence of
IL-17(66), an extensive body of
data obtained in mice models and humans in terms of the T cell-B
cell interactions have revealed that T cells aid the activation of
the autoantibody-producing B cells in SLE (80-82).
However, the exact function of IL-17 that leads to SLE remains
unknown. In the SLE development process, B lymphocyte stimulator
(BLyS) can become upregulated and it may act as a survival factor
to inhibit B cell apoptosis, to stimulate B cell proliferation and
differentiation through an interaction with IL-17, and ultimately
to increase the production of autoantibodies (83,84).
In addition, increased levels of BLyS would promote the
proliferation of Th17 cells leading to increased levels of IL-17,
which in turn could act in conjunction with BAFF to promote the
survival and proliferation of human B cells and their
differentiation into antibody-producing cells (81). In addition to IL-17, the roles of
other cytokines in the T cell-B cell interaction have been noted
with regard to SLE pathogenesis. For example, IL-21, produced by
Th17 cells, stimulates CD8+ T cell proliferation and B
cell differentiation for immunoglobulin production (66,85,86).
These roles of IL-21 have been validated in a lupus-prone mouse
model, in which the IL-21/IL-21 receptor pathway was blocked by the
administration of a fusion protein, resulting in an alleviated
disease progression (87,88). Finally, the production of
autoantibodies by activated B cells leads to the activation of DCs
and the secreted IL-23 can increase production of IL-17(89) (Fig.
1).
Collectively, this data indicates that the
multifarious functions of the Th17 cells and B cells as well as the
inflammatory environment created by the T cell-B cell interaction
all function together to lead to SLE, as demonstrated in other
human IMIDs.
TNF-α is a pleiotropic cytokine that affects the
activities of variant cell types in various physiological and
pathological conditions, such as in the development of T cells, B
cells and DCs. This cytokine is a potent inflammatory mediator and
also an apoptosis inducer. Overexpression of TNF-α has been
observed in patients with PS for two decades and it was revealed to
be distributed throughout the epidermis and specifically localized
to the upper dermal blood vessels (107).
The significance of the involvement of TNF-α in the
pathogenesis of SLE remains controversial. Previous evidence has
suggested that this cytokine serves a dualistic, proinflammatory,
and an immune- or disease suppressive role in SLE progress
(108). TNF-α was reported to be
increased in patients with SLE and was correlated with disease
course, and the immunopathogenesis of SLE (109,110). Anti-TNF-α administration also
demonstrated that this treatment can suppress the inflammatory
responses in an experimental SLE model, which was induced by the
injection of human anti-DNA autoantibodies in mice (111). However, the use of an anti-TNF-α
agent can also lead to increased levels of autoantibodies for
double-stranded DNA (dsDNA) and cardiolipin (112). Higher levels of TNF-α have been
reported in patients with PS than in healthy individuals (113); therefore, the pathogenic roles of
TNF-α in both diseases should be further classified.
Different subpopulations of T cells (Th1, Th2, Th9,
Th17, Th22 and Treg cells) and their corresponding proinflammatory
cytokines are all involved in the pathophysiology of PS and SLE
(114). Activated T cells secrete
proinflammatory cytokines, which in turn stimulate the resident
tissue cells to recruit immune cells and further increase the
secretion of IL-2, IL-4, IL-9, IL-17 A, IL-22, TNF-α, IFN-γ and
GM-CSF in perivascular and renal systems of affected patients with
PS or SLE (84,115-118).
Nevertheless, chronic inflammation due to abnormal immune responses
is the pathogenic bases of SLE and PS. In addition to the
aforementioned cytokines and effector cells, other inflammatory
mediators, such as those from fibroblast-like synoviocytes (FLSs),
could also crosstalk with these factors to lead to
pathophysiological conditions and to accelerate inflammation in PS
and SLE. Previously, local stromal cells, such as FLSs, have been
revealed as inflammatory effectors that affect the phenotype and
function of different organs, such as kidney, gastrointestinal
tract, and joints, in autoimmune disease (119). For example, the involvement of
FLS in inflammation and cartilage destruction has been observed in
PsA; however, how activated T cells modulate the release of the
inflammatory mediators is not fully understood in both
diseases.
Genetic studies on patients with autoimmune
conditions have identified the susceptibility loci for a number of
diseases, including for PS and SLE (120-123).
A number of loci identified by genome-wide association studies have
been associated with both PS and SLE, such as protein tyrosine
phosphatase non-receptor type 22 (PTPN22), TNF receptor associated
factor 3 interacting protein 2 (TRAF3IP2), signal transducer and
activator of transcription 4 (STAT4) and TNF-α-induced protein 3
interacting protein 1 (TNIP1) (121,123-128).
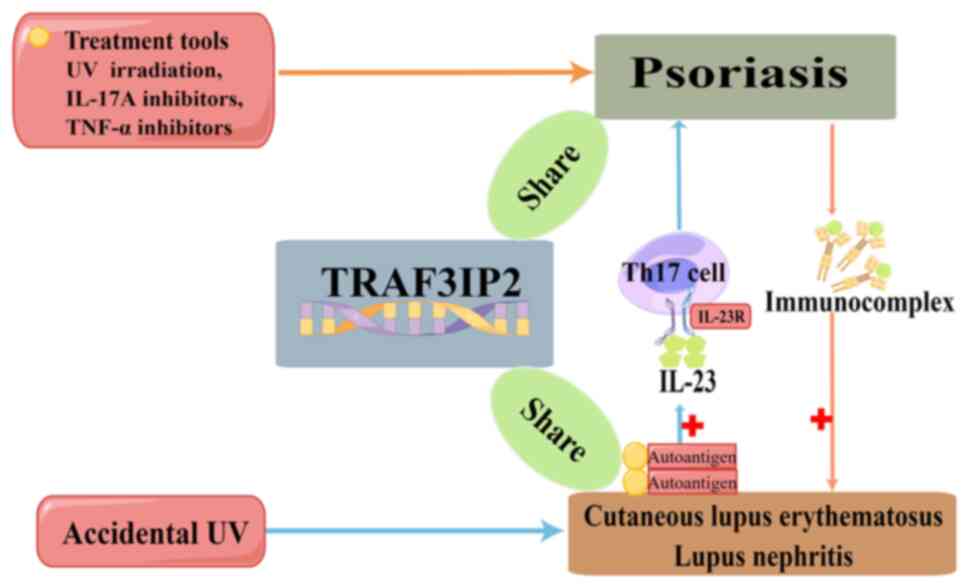
The TRAF3IP2 locus, located on chromosome 6q21, encodes NF-κB
activator 1, which is both a negative regulator of the humoral
immunity and a positive signaling adaptor of the IL-17-dependent
NF-κB activation. As a downstream target of the IL-17 receptor, it
may have a pivotal role in the IL-23/IL-17 axis in the pathogenesis
of PS (129). The PTPN22 gene
encodes lymphoid tyrosine phosphatase, a lymphoid-specific tyrosine
phosphatase that acts as a negative regulator of T cell signaling.
A gain of function for PTPN22 could allow it to participate in the
release of autoantibodies and increase the formation and deposition
of immune complexes, which would trigger an inflammatory response
resulting in the possible development of SLE and its clinical
manifestations (130,131). A number of studies have revealed
an association between PS and rs1217414 located in intron 1 of
PTPN22 (122,132). The level of PTPN22 transcription
could negatively regulate T-cell function and thereby changes
susceptibility to PS (122). The
STAT4 protein is located in T and B lymphocytes, monocytes,
macrophages, natural killer cells, and DCs. Its expression may be
associated to immune cell differentiation into inflammatory
subsets, the production of inflammatory cytokines and
autoantibodies, the suppression of apoptosis, and the presentation
of autoantigens, all of which may promote the development of SLE
and PS (133). TNIP1 also serves
a critical role in immunological homeostasis and autoimmunity
prevention, since mice with TNIP1 knocked out have been
demonstrated to acquire almost all autoimmune characteristics,
including spontaneous germinal center development, isotype
switching and autoantibody production (134,135). Additionally, the protein level of
TNIP1 was negatively associated with the disease activity of SLE
and was decreased in the peripheral blood mononuclear cells of
patients with SLE compared with in that of healthy controls
(136). In an imiquimod-induced
mouse model of dermatitis, downregulation of the TNIP1 expression
levels resulted in an increased proliferation of human
keratinocytes and a more severe PS-like condition (137).
According to a study in China, the NF-κB-inhibitor α
(NFKBIA) and IL-28 receptor α (IL-28RA) loci occur at increased
frequencies in Chinese Han populations with PS compared with
Chinese Han populations without PS. In this study, the susceptible
loci, NFKBIA and IL28RA, for SLE in the Chinese Han population were
also identified (138). NFKBIA is
an inhibitor of NF-κB signaling, acting to inhibit Th17 cell
activity and IL-17 expression in a healthy individuals (139). In patients with PS and/or SLE,
the insufficient levels of NFKBIA will in turn increase the levels
of IL-17, which has been revealed in the skin lesions of patients
with PS and/or SLE (140).
Although IL-28RA mRNA expression levels are increased in the
peripheral blood mononuclear cells of patients with SLE, they are
decreased in the lesional tissues from individuals with PS plaques.
After the cause of PS has been confirmed as due to the reduced
expression levels of IL-28RA, IL-28RA could be a useful
pharmacological target, at least for the therapy of PS (138,141).
These genetic predispositions may provide hints for
the early inflammatory mechanism of the coexistence of PS and SLE.
The present review considers that identification of new common
susceptible genes for both diseases may provide an understanding of
the immunopathogenesis of the coexistence of SLE and PS and may
inspire further treatment options for these two conditions.
The different drugs that are used to treat SLE and
PS, with regard to treatable and inducible disease are summarized
in Table I.
In the lupus-induced mouse model, IL-17 production
was positively associated with disease progression since the
manifestation could be eliminated by reducing IL-17 production and
IL 17-/- mice did not develop anti-dsDNA, anti-single
stranded DNA, anti-nuclear ribonucleoprotein and anti-chromatin
autoantibodies (78). In mouse
models, overexpression of IL-17 using an adenovirus increased the
severity of lupus nephritis, while inhibition of IL-17 using
neutralizing antibodies resulted in a reduced severity of lupus
nephritis (142). These results
suggest that IL-17 is involved in the pathogenesis of SLE (71). Currently, a number of IL-17
inhibitors, including the anti-IL-17 monoclonal antibodies
secukinumab, ixekizumab and bimekizumab, and the anti-IL-17
receptor A monoclonal antibody brodalumab, have been demonstrated
to be effective as PS treatments (143-145).
Furthermore, a case report described the efficacy of an IL-17
inhibitor, secukinumab, in a patient with SLE (146). The use of an IL-17 monoclonal
antibody to treat lupus was approved by the US Food and Drug
Administration. Additionally, a double-blind phase II study has
demonstrated the efficacy and safety of ustekinumab (an IL-12 and
IL-23 antagonist) in patients with active SLE (147). Ustekinumab has been demonstrated
to improve a number of mucocutaneous and musculoskeletal diseases,
such as atopic dermatitis, Crohn's disease and ankylosing
spondylitis, perhaps by decreasing the levels of anti-dsDNA titers
and complement 3. It was also revealed to improve the renal
function of the patients with SLE (146,148). After receiving secukinumab
treatment, a patient with PsA combined with SLE had improved
clinical symptoms along with decreased levels of IL-17 in the serum
and renal tissue (149). The
success in treating this patient with both SLE and PsA suggests
that the IL-17/IL-23 axis is the common immunogenic mechanism for
developing both diseases.
Cytokines that activate B cells and promote the
interaction between B and T cells contribute to the production of
autoantibodies (150,151). Inhibition of B cell-associated
molecules should downregulate the overactive immune responses in
SLE. B-lymphocyte antigen (126,127) (called CD20) is highly expressed
on the surface of all B-cells and also serves critical roles in
cell cycle progression during human B cell proliferation and
activation. Anti-CD20 antibodies have been employed in treatment of
a number of diseases (152). In
patients with rheumatic arthritis, anti-CD20 therapy achieved a
rapid and almost complete B-cell depletion in the peripheral blood
and suppressed the generation of plasma blasts, sustainable for at
least 6 months (153,154). The disappearance of the
anti-neutrophilscytoplasmic antibody (ANCA) in ANCA-associated
vasculitis or anti-desmoglein antibodies in pemphigus confirmed the
efficacy of the anti-CD20 monoclonal antibody (155,156). In addition, a study revealed that
an anti-CD20 antibody, Rituximab, can reduce the expression levels
of RORγt and IL-22 and decrease the numbers of Th17-positive cells
(157). In a case report of
palmoplantar pustulosis, a less severe and localized variant of
pustular PS, after treatment failure with an TNF-α blocker, an
improvement was demonstrated with rituximab treatment (158). The present review considered that
rituximab might reverse the effects of TNF-α via the
antigen-antibody complex. However, contradictory effects of
anti-CD20 are also observed in the literaturs. For example, the use
of anti-CD20 therapy (Rituximab) for autoimmune diseases such as
SLE may be the cause of the development of PS (159,160). It is likely that the depletion of
B-lymphocytes in these patients caused an imbalance between the T
and B cells and this interaction may promote a hyperactive T cells
response.
Unlike Th17 antagonists, the efficacy of IFN-α
antagonists on PS is uncertain. Collamer et al (161) found that the number of patients
with new onset or exacerbation of preexisting PS is increasing due
to TNF therapy. This does not seem to be consistence with the fact
that IFN-α is a key element in the early phase of psoriatic skin
lesion induction (46). It was
suspected that an IFN-α antagonist may also be involved in other
mechanisms for PS induction (161), and inhibition of TNF-α has been
demonstrated in turn to induce the overexpression of IFN-α
(162). Increased expression
levels of IFN-α in the psoriatic lesions of patients that have been
administered with anti-TNF-α therapeutics were also reported
(163). The increased production
of IFN-α will stimulate myeloid DCs, promote the polarization of
Th1 cells and lead to an excessive proliferation of keratinocytes
via IL-15(164).
Likely acting via the same mechanism, treatment with
anti-TNF-α agents is controversial in SLE as well, since it may
further induce antinuclear antibodies, anti-dsDNA and
anticardiolipin antibodies. Indeed, cases of drug-induced lupus
have been observed in patients with rheumatoid arthritis (165).
Treatment of patients with both PS and SLE or
prevention of the occurrence of comorbidity is challenging. The
onset of PS and SLE could appear in a different order of
precedence, which may not only affect the profiles of the
inflammatory cytokines (such as IL-17, IL-10 and IL-23) but also
may change the efficacy of the treatment in each individual
patient. For example, phototherapy for PS can exacerbate SLE and
hydroxychloroquine or systemic corticosteroids for SLE treatment
would exacerbate PS (42).
When ultraviolet (UV) light is used to control PS
through triggering keratinocyte apoptosis, it also promotes an
immunopathogenesis in lupus (166). As a result, nucleoprotein
autoantigens will be transported to the surface of keratinocytes to
stimulate the release of further inflammatory cytokines, including
IFN-γ, TNF-α, IL-1, IL-6, IL-8, IL-10 and IL-17 (167-170).
The accumulation of these apoptotic keratinocytes would increase
the aforementioned changes to cause a secondary necrotic process
and to amplify the release of proinflammatory cytokines and
potential autoantigens (171).
These factors would recruit inflammatory cells into the skin and
cause tissue inflammation.
By contrast, after the patients with a genetic
predisposition to PS and lupus receive UV treatment, the skin cells
of the patients will be more likely to have thymine dimmers in
their DNA (172). The
photo-induced thymine-dimmers in the DNA are then the target
antigens of the autoimmune response, thus causing lupus. However,
the pathophysiology of induced lupus is still poorly understood. In
addition, the mechanism of drug-induced PS is not completely
understood. A study concluded that hydroxychloroquine disrupts the
barrier of the epidermis by inhibiting transglutaminase activity
(173,174). Following this initial break in
the skin barrier, the epidermis undergoes a physiological
proliferation in an attempt to restore the integrity of the
barrier. In a genetically predisposed individual, this damage to
the skin barrier may be sufficient to initiate a non-specific
stimulus-induced epidermal proliferation (173).
At present, TNF-α inhibitors have been widely
investigated for the treatment of PS (175), and anti-TNF-α drugs are
frequently reported to induce systemic drug-induced lupus
erythematosus (176,177). A number of hypotheses have been
proposed for the mechanism of autoantibody induction by anti-TNF-α
agents as follows: i) An imbalance between IFN-α and TNF-α induces
an apoptosis in inflammatory cells; ii) decreased expression levels
of CD44 causes nucleosome accumulation in apoptotic cells and leads
to the production of DNA and other nuclear antigens; iii)
infections in patients receiving anti-TNF-α can lead to lymphocyte
activation and subsequently to polyclonal B lymphocytes production;
and iv) suppression of the Th1 response caused by the anti-TNF-α
and Th2 response, IL-10, and INF-α, promotes humoral autoimmunity
and autoantibody production and suppresses cytotoxic T-lymphocytes
(178-181)
(Fig. 2).
In a case report, the symptoms of lupus nephritis in
patients with both PS and SLE diseases worsened after secukinumab
treatment (182). Certainly,
caution is necessary when administering drugs to patients with PS
and SLE, since it may effective for one of these diseases but it
may not be effective for the other disease, particularly when TNF-α
antagonists are used. As aforementioned, TNF-α inhibitors have well
been documented to cause lupus-like syndromes with the onset of
antinuclear antibodies and anti-dsDNA, as well as an exacerbation
of PS. Therefore, when treating patients with biological agents,
their immunological profiles and family history should be evaluated
in detail in order to avoid a deterioration in the inflammation of
both diseases.
PS is a disease that occurs worldwide and its
prevalence varies from 2-11% according to the region. The global
prevalence of SLE ranges from 13-7,713.5 per 100,000 individuals
(183). A 40-year follow-up study
that was carried out in the US, revealed that the incidence and
prevalence of SLE had increased each year (overall prevalence
increased from 30.6 in 1985 to 97.4 in 2015), and PS and SLE
occurred in all age groups and in both men and women (184). At present, the comorbidity of PS
and SLE is still a rare skin condition. In the present review, the
animal and clinical evidence to support the possibility of SLE
coexisting with PS has been summarized. Firstly, both diseases
share the same susceptibile gene loci (138,185-188),
which are associated with IL-17 signal transduction (186,189), and the interactions with Treg
cells and B lymphocytes that form the foundation of pathogenesis of
PS and SLE. Secondly, biological agents demonstrated efficacy in
patients with PS and SLE. For example, IL-17 inhibitors that have
been widely used for PS, are now being tested as treatments for SLE
in clinical trials (190).
Nevertheless, current evidence cannot completely exclude that this
is simply a coincidence; however, caution may be needed when
dealing with immunotherapy treatments for patients with single PS,
SLE or both conditions.
Not applicable.
Funding: This work was supported by grants from the National
Nature Science Foundation of China (grant no. NM 82272358), the Key
Research and Development Plan of Jining (grant no. NM 2021YXNS121)
and the Traditional Chinese Medicine Science and Technology Program
of Shandong Province (grant no. NM 2021M080).
Not applicable.
DS contributed to the conception of the present
work. YQ and DS examined the literature and drafted the manuscript.
YQ and DS produced the figures. DS, WL, DL and YQ made critical
revisions to the manuscript. All authors read and approved the
final version of the manuscript. Data authentication is not
applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
AlQassimi S, AlBrashdi S, Galadari H and
Hashim MJ: Global burden of psoriasis-comparison of regional and
global epidemiology, 1990 to 2017. Int J Dermatol. 59:566–571.
2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Gómez-Bañuelos E, Fava A and Andrade F: An
update on autoantibodies in systemic lupus erythematosus. Curr Opin
Rheumatol. 35:61–67. 2023.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Karrar S and Cunninghame Graham DS:
Abnormal B cell development in systemic lupus erythematosus: What
the genetics tell us. Arthritis Rheumatol. 70:496–507.
2018.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zaba LC, Fuentes-Duculan J, Eungdamrong
NJ, Abello MV, Novitskaya I, Pierson KC, Gonzalez J, Krueger JG and
Lowes MA: Psoriasis is characterized by accumulation of
immunostimulatory and Th1/Th17 cell-polarizing myeloid dendritic
cells. J Invest Dermatol. 129:79–88. 2009.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Kagami S, Rizzo HL, Lee JJ, Koguchi Y and
Blauvelt A: Circulating Th17, Th22, and Th1 cells are increased in
psoriasis. J Invest Dermatol. 130:1373–1383. 2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Funauchi M, Ikoma S, Enomoto H and
Horiuchi A: Decreased Th1-like and increased Th2-like cells in
systemic lupus erythematosus. Scand J Rheumatol. 27:219–224.
1998.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Richaud-Patin Y, Alcocer-Varela J and
Llorente L: High levels of TH2 cytokine gene expression in systemic
lupus erythematosus. Rev Invest Clin. 47:267–272. 1995.PubMed/NCBI
|
|
8
|
Redisch W, Messina EJ, Hughes G and McEwen
C: Capillaroscopic observations in rheumatic diseases. Ann Rheum
Dis. 29:244–253. 1970.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Tselios K, Yap KS, Pakchotanon R, Polachek
A, Su J, Urowitz MB and Gladman DD: Psoriasis in systemic lupus
erythematosus: A single-center experience. Clin Rheumatol.
36:879–884. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Shindo E, Shikano K, Kawazoe M, Yamamoto
T, Kusunoki N, Hashimoto Y and Nanki T: A case of generalized
pustular psoriasis caused by hydroxychloroquine in a patient with
systemic lupus erythematosus. Lupus. 28:1017–1020. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Akaji K, Nakagawa Y, Kakuda K, Takafuji M,
Kiyohara E, Murase C, Takeichi T, Akiyama M and Fujimoto M:
Generalized pustular psoriasis associated with systemic lupus
erythematosus successfully treated with secukinumab. J Dermatol.
48:e43–e44. 2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Varada S, Gottlieb AB, Merola JF, Saraiya
AR and Tintle SJ: Treatment of coexistent psoriasis and lupus
erythematosus. J Am Acad Dermatol. 72:253–260. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Millns JL and Muller SA: The coexistence
of psoriasis and lupus erythematosus. An analysis of 27 cases. Arch
Dermatol. 116:658–663. 1980.PubMed/NCBI
|
|
14
|
Wang Y, Da G, Yu Y, Han J and Li H:
Coincident systemic lupus erythematosus and psoriasis vulgaris: A
case report. G Ital Dermatol Venereol. 150:749–751. 2015.PubMed/NCBI
|
|
15
|
Gaber W, Sayed S, Rady HM and Mohey AM:
Interleukin-27 and its relation to disease parameters in SLE
patient. Egypt Rheumatol. 34:99–105. 2012.
|
|
16
|
Fu SM, Dai C, Zhao Z and Gaskin F:
Anti-dsDNA Antibodies are one of the many autoantibodies in
systemic lupus erythematosus. F1000Res. 4(939)2015.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Pisetsky DS: Anti-DNA
antibodies-quintessential biomarkers of SLE. Nat Rev Rheumatol.
12:102–110. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Trouw LA, Pickering MC and Blom AM: The
complement system as a potential therapeutic target in rheumatic
disease. Nat Rev Rheumatol. 13:538–547. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Qi S, Chen Q, Xu D, Xie N and Dai Y:
Clinical application of protein biomarkers in lupus erythematosus
and lupus nephritis. Lupus. 27:1582–1590. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Weber B, Merola JF, Husni ME, Di Carli M,
Berger JS and Garshick MS: Psoriasis and cardiovascular disease:
Novel mechanisms and evolving therapeutics. Curr Atheroscler Rep.
23(67)2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Bu J, Ding R, Zhou L, Chen X and Shen E:
Epidemiology of Psoriasis and comorbid diseases: A narrative
review. Front Immunol. 13(880201)2022.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Yamazaki F: Psoriasis: Comorbidities. J
Dermatol. 48:732–740. 2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Davidson A and Diamond B: Autoimmune
diseases. N Engl J Med. 345:340–350. 2001.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Kommoss KS, Enk A, Heikenwälder M, Waisman
A, Karbach S and Wild J: Cardiovascular comorbidity in
psoriasis-psoriatic inflammation is more than just skin deep. J
Dtsch Dermatol Ges. 21:718–725. 2023.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Christophers E: Comorbidities in
psoriasis. Clin Dermatol. 25:529–534. 2007.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Davidovici BB, Sattar N, Prinz J, Puig L,
Emery P, Barker JN, van de Kerkhof P, Ståhle M, Nestle FO,
Girolomoni G and Krueger JG: Psoriasis and systemic inflammatory
diseases: Potential mechanistic links between skin disease and
co-morbid conditions. J Invest Dermatol. 130:1785–1796.
2010.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wu JJ, Nguyen TU, Poon KY and Herrinton
LJ: The association of psoriasis with autoimmune diseases. J Am
Acad Dermatol. 67:924–930. 2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Edson-Heredia E, Zhu B, Lefevre C, Wang M,
Barrett A, Bushe CJ, Cox A, Wu JJ and Maeda-Chubachi T: Prevalence
and incidence rates of cardiovascular, autoimmune, and other
diseases in patients with psoriatic or psoriatic arthritis: A
retrospective study using clinical practice research datalink. J
Eur Acad Dermatol Venereol. 29:955–963. 2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Sticherling M: Psoriasis and autoimmunity.
Autoimmun Rev. 15:1167–1170. 2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Prinz JC: Melanocytes: Target cells of an
HLA-C*06:02-Restricted autoimmune response in psoriasis. J Invest
Dermatol. 137:2053–2058. 2017.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Furue K, Ito T, Tsuji G, Kadono T,
Nakahara T and Furue M: Autoimmunity and autoimmune co-morbidities
in psoriasis. Immunology. 154:21–27. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Prinz JC: Human leukocyte antigen-class I
alleles and the autoreactive T cell response in psoriasis
pathogenesis. Front Immunol. 9(954)2018.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Andersen YMF, Wu JJ, Thyssen JP and
Egeberg A: Chronologic order of appearance of immune-mediated
inflammatory diseases relative to diagnosis of psoriasis. J Am Acad
Dermatol. 81:1283–1291. 2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Korkus D, Gazitt T, Cohen AD, Feldhamer I,
Lavi I, Haddad A, Greenberg-Dotan S, Batat E and Zisman D:
Increased prevalence of systemic lupus erythematosus comorbidity in
patients with psoriatic arthritis: A population-based case-control
study. J Rheumatol. 48:207–213. 2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Kaslow RA: High rate of death caused by
systemic lupus erythematosus among U.S. residents of Asian descent.
Arthritis Rheum. 25:414–418. 1982.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Tsai TF, Wang TS, Hung ST, Tsai PI,
Schenkel B, Zhang M and Tang CH: Epidemiology and comorbidities of
psoriasis patients in a national database in Taiwan. J Dermatol
Sci. 63:40–46. 2011.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Mohan AK, Edwards ET, Coté TR, Siegel JN
and Braun MM: Drug-induced systemic lupus erythematosus and
TNF-alpha blockers. Lancet. 360(646)2002.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Debandt M, Vittecoq O, Descamps V, Le Loët
X and Meyer O: Anti-TNF-alpha-induced systemic lupus syndrome. Clin
Rheumatol. 22:56–61. 2003.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Zalla MJ and Muller SA: The coexistence of
psoriasis with lupus erythematosus and other photosensitive
disorders. Acta Derm Venereol Suppl (Stockh). 195:1–15.
1996.PubMed/NCBI
|
|
40
|
Ojemolon PE, Unadike CE and Uwumiro F:
Psoriasis is associated with an increased risk of hospitalization
for systemic lupus erythematosus: Analysis of the national
inpatient sample database. Cureus. 12(e11771)2020.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Astudillo L, Sailler L, Carreiro M, Dahan
S, Ollier S and Arlet P: Psoriasis and systemic lupus
erythematosus: A rare association with specific therapeutic
problems. Ann Med Interne (Paris). 154:3–6. 2003.PubMed/NCBI(In French).
|
|
42
|
Wang WM, Wang KY, Wang T, Jin HZ and Fang
K: Hydroxychloroquine-induced psoriasis-form erythroderma in a
patient with systemic lupus erythematosus. Chin Med J (Engl).
131:1887–1888. 2018.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Sharabi A and Tsokos GC: T cell
metabolism: New insights in systemic lupus erythematosus
pathogenesis and therapy. Nat Rev Rheumatol. 16:100–112.
2020.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Brembilla NC and Boehncke WH: Revisiting
the interleukin 17 family of cytokines in psoriasis: Pathogenesis
and potential targets for innovative therapies. Front Immunol.
14(1186455)2023.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Lande R, Gregorio J, Facchinetti V,
Chatterjee B, Wang YH, Homey B, Cao W, Wang YH, Su B, Nestle FO, et
al: Plasmacytoid dendritic cells sense self-DNA coupled with
antimicrobial peptide. Nature. 449:564–569. 2007.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Nestle FO, Conrad C, Tun-Kyi A, Homey B,
Gombert M, Boyman O, Burg G, Liu YJ and Gilliet M: Plasmacytoid
predendritic cells initiate psoriasis through interferon-alpha
production. J Exp Med. 202:135–143. 2005.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Lande R, Ganguly D, Facchinetti V, Frasca
L, Conrad C, Gregorio J, Meller S, Chamilos G, Sebasigari R,
Riccieri V, et al: Neutrophils activate plasmacytoid dendritic
cells by releasing self-DNA-peptide complexes in systemic lupus
erythematosus. Sci Transl Med. 3(73ra19)2011.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Yoshiki R, Kabashima K, Honda T, Nakamizo
S, Sawada Y, Sugita K, Yoshioka H, Ohmori S, Malissen B, Tokura Y
and Nakamura M: IL-23 from Langerhans cells is required for the
development of imiquimod-induced psoriasis-like dermatitis by
induction of IL-17A-producing γδ T cells. J Invest Dermatol.
134:1912–1921. 2014.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Vincent FB, Morand EF, Schneider P and
Mackay F: The BAFF/APRIL system in SLE pathogenesis. Nat Rev
Rheumatol. 10:365–373. 2014.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Jenks SA, Cashman KS, Zumaquero E,
Marigorta UM, Patel AV, Wang X, Tomar D, Woodruff MC, Simon Z,
Bugrovsky R, et al: Distinct effector B cells induced by
unregulated toll-like receptor 7 contribute to pathogenic responses
in systemic lupus erythematosus. Immunity. 49:725–739.e6.
2018.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Jenks SA, Cashman KS, Woodruff MC, Lee FE
and Sanz I: Extrafollicular responses in humans and SLE. Immunol
Rev. 288:136–148. 2019.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Hale M, Rawlings DJ and Jackson SW: The
long and the short of it: Insights into the cellular source of
autoantibodies as revealed by B cell depletion therapy. Curr Opin
Immunol. 55:81–88. 2018.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Maddur MS, Miossec P, Kaveri SV and Bayry
J: Th17 cells: Biology, pathogenesis of autoimmune and inflammatory
diseases, and therapeutic strategies. Am J Pathol. 181:8–18.
2012.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Korn T, Bettelli E, Oukka M and Kuchroo
VK: IL-17 and Th17 cells. Annu Rev Immunol. 27:485–517.
2009.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Miossec P, Korn T and Kuchroo VK:
Interleukin-17 and type 17 helper T cells. N Engl J Med.
361:888–898. 2009.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Meitei HT, Jadhav N and Lal G: CCR6-CCL20
axis as a therapeutic target for autoimmune diseases. Autoimmun
Rev. 20(102846)2021.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Zheng Y, Danilenko DM, Valdez P, Kasman I,
Eastham-Anderson J, Wu J and Ouyang W: Interleukin-22, a T(H)17
cytokine, mediates IL-23-induced dermal inflammation and
acanthosis. Nature. 445:648–651. 2007.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Zaba LC, Cardinale I, Gilleaudeau P,
Sullivan-Whalen M, Suárez-Fariñas M, Fuentes-Duculan J, Novitskaya
I, Khatcherian A, Bluth MJ, Lowes MA and Krueger JG: Amelioration
of epidermal hyperplasia by TNF inhibition is associated with
reduced Th17 responses. J Exp Med. 204:3183–3194. 2007.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Lee AYS and Körner H: The CCR6-CCL20 axis
in humoral immunity and T-B cell immunobiology. Immunobiology.
224:449–454. 2019.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Li D, Guo B, Wu H, Tan L, Chang C and Lu
Q: Interleukin-17 in systemic lupus erythematosus: A comprehensive
review. Autoimmunity. 48:353–361. 2015.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Yang J, Yang X, Zou H and Li M: Oxidative
stress and Treg and Th17 dysfunction in systemic lupus
erythematosus. Oxid Med Cell Longev. 2016(2526174)2016.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Wong CK, Ho CY, Li EK and Lam CW:
Elevation of proinflammatory cytokine (IL-18, IL-17, IL-12) and Th2
cytokine (IL-4) concentrations in patients with systemic lupus
erythematosus. Lupus. 9:589–593. 2000.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Reihani H, Rastin M, Mahmoudi M, Ghoryani
M, Abdollahi N, Tabasi NS, Zamani Taghizadeh Rabe S and Sahebari M:
Influence of 1 alpha, 25-dihydroxyvitamin D3 on T helper 17 cells
and related cytokines in systemic lupus erythematosus. Iran J
Immunol. 12:82–93. 2015.PubMed/NCBI
|
|
64
|
Liu T, Li S, Ying S, Tang S, Ding Y, Li Y,
Qiao J and Fang H: The IL-23/IL-17 pathway in inflammatory skin
diseases: From bench to bedside. Front Immunol.
11(594735)2020.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Mok MY, Wu HJ, Lo Y and Lau CS: The
relation of interleukin 17 (IL-17) and IL-23 to Th1/Th2 cytokines
and disease activity in systemic lupus erythematosus. J Rheumatol.
37:2046–2052. 2010.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Dong G, Ye R, Shi W, Liu S, Wang T, Yang
X, Yang N and Yu X: IL-17 induces autoantibody overproduction and
peripheral blood mononuclear cell overexpression of IL-6 in lupus
nephritis patients. Chin Med J (Engl). 116:543–548. 2003.PubMed/NCBI
|
|
67
|
Bălănescu P, Bălănescu E, Tănăsescu C,
Nicolau A, Tănăsescu R, Grancea C, Vagu C, Ruţă S and Bleoţu C: T
helper 17 cell population in lupus erythematosus. Rom J Intern Med.
48:255–259. 2010.PubMed/NCBI
|
|
68
|
Dolff S, Quandt D, Wilde B, Feldkamp T,
Hua F, Cai X, Specker C, Kribben A, Kallenberg CG and Witzke O:
Increased expression of costimulatory markers CD134 and CD80 on
interleukin-17 producing T cells in patients with systemic lupus
erythematosus. Arthritis Res Ther. 12(R150)2010.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Ballantine LE, Ong J, Midgley A, Watson L,
Flanagan BF and Beresford MW: The pro-inflammatory potential of T
cells in juvenile-onset systemic lupus erythematosus. Pediatr
Rheumatol Online J. 12(4)2014.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Tanasescu C, Balanescu E, Balanescu P,
Olteanu R, Badea C, Grancea C, Vagu C, Bleotu C, Ardeleanu C and
Georgescu A: IL-17 in cutaneous lupus erythematosus. Eur J Intern
Med. 21:202–207. 2010.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Apostolidis SA, Crispín JC and Tsokos GC:
IL-17-producing T cells in lupus nephritis. Lupus. 20:120–124.
2011.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Albanesi C, Cavani A and Girolomoni G:
IL-17 is produced by nickel-specific T lymphocytes and regulates
ICAM-1 expression and chemokine production in human keratinocytes:
Synergistic or antagonist effects with IFN-gamma and TNF-alpha. J
Immunol. 162:494–502. 1999.PubMed/NCBI
|
|
73
|
Schwarzenberger P, Huang W, Ye P, Oliver
P, Manuel M, Zhang Z, Bagby G, Nelson S and Kolls JK: Requirement
of endogenous stem cell factor and granulocyte-colony-stimulating
factor for IL-17-mediated granulopoiesis. J Immunol. 164:4783–4789.
2000.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Puwipirom H, Hirankarn N, Sodsai P,
Avihingsanon Y, Wongpiyabovorn J and Palaga T: Increased
interleukin-23 receptor(+) T cells in peripheral blood mononuclear
cells of patients with systemic lupus erythematosus. Arthritis Res
Ther. 12(R215)2010.PubMed/NCBI View
Article : Google Scholar
|
|
75
|
Izati AF, Mohd Shukri ND, Wan Ghazali WS,
Che Hussin CM and Wong KK: Increased IL-23R+ Th cells
population exhibits higher SLEDAI-2K scores in systemic lupus
erythematosus patients. Front Immunol. 12(690908)2021.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Koga T, Ichinose K and Tsokos GC: T cells
and IL-17 in lupus nephritis. Clin Immunol. 185:95–99.
2017.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Martin JC, Baeten DL and Josien R:
Emerging role of IL-17 and Th17 cells in systemic lupus
erythematosus. Clin Immunol. 154:1–12. 2014.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Amarilyo G, Lourenço EV, Shi FD and La
Cava A: IL-17 promotes murine lupus. J Immunol. 193:540–543.
2014.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Dai H, He F, Tsokos GC and Kyttaris VC:
IL-23 limits the production of IL-2 and promotes autoimmunity in
lupus. J Immunol. 199:903–910. 2017.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Lanzavecchia A: Antigen-specific
interaction between T and B cells. Nature. 314:537–539.
1985.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Doreau A, Belot A, Bastid J, Riche B,
Trescol-Biemont MC, Ranchin B, Fabien N, Cochat P, Pouteil-Noble C,
Trolliet P, et al: Interleukin 17 acts in synergy with B
cell-activating factor to influence B cell biology and the
pathophysiology of systemic lupus erythematosus. Nat Immunol.
10:778–785. 2009.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Yap DYH and Lai KN: Cytokines and their
roles in the pathogenesis of systemic lupus erythematosus: From
basics to recent advances. J Biomed Biotechnol.
2010(365083)2010.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Liu Y and La Cava A: Targeting BLyS in
systemic lupus erythematosus. Recent Pat Inflamm Allergy Drug
Discov. 6:91–96. 2012.PubMed/NCBI View Article : Google Scholar
|
|
84
|
López P, Rodríguez-Carrio J,
Caminal-Montero L, Mozo L and Suárez A: A pathogenic IFNα, BLyS and
IL-17 axis in systemic lupus erythematosus patients. Sci Rep.
6(20651)2016.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Ozaki K, Spolski R, Feng CG, Qi CF, Cheng
J, Sher A, Morse HC III, Liu C, Schwartzberg PL and Leonard WJ: A
critical role for IL-21 in regulating immunoglobulin production.
Science. 298:1630–1634. 2002.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Kuchen S, Robbins R, Sims GP, Sheng C,
Phillips TM, Lipsky PE and Ettinger R: Essential role of IL-21 in B
cell activation, expansion, and plasma cell generation during CD4+
T cell-B cell collaboration. J Immunol. 179:5886–5896.
2007.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Fina D, Sarra M, Fantini MC, Rizzo A,
Caruso R, Caprioli F, Stolfi C, Cardolini I, Dottori M, Boirivant
M, et al: Regulation of gut inflammation and Th17 cell response by
interleukin-21. Gastroenterology. 134:1038–1048. 2008.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Herber D, Brown TP, Liang S, Young DA,
Collins M and Dunussi-Joannopoulos K: IL-21 has a pathogenic role
in a lupus-prone mouse model and its blockade with IL-21R.Fc
reduces disease progression. J Immunol. 178:3822–3830.
2007.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Tsokos GC, Lo MS, Costa Reis P and
Sullivan KE: New insights into the immunopathogenesis of systemic
lupus erythematosus. Nat Rev Rheumatol. 12:716–730. 2016.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Stiehm ER: Joseph A: Bellanti (ed)
immunology IV: Clinical applications in health and disease. J Clin
Immunol. 32(647)2012.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Vieira PL, Christensen JR, Minaee S,
O'Neill EJ, Barrat FJ, Boonstra A, Barthlott T, Stockinger B,
Wraith DC and O'Garra A: IL-10-secreting regulatory T cells do not
express Foxp3 but have comparable regulatory function to naturally
occurring CD4+CD25+ regulatory T cells. J Immunol. 172:5986–5993.
2004.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Bellanti JA and Li D: Treg cells and
epigenetic regulation. Adv Exp Med Biol. 1278:95–114.
2021.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Sakaguchi S, Yamaguchi T, Nomura T and Ono
M: Regulatory T cells and immune tolerance. Cell. 133:775–787.
2008.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Lim HW, Hillsamer P, Banham AH and Kim CH:
Cutting edge: Direct suppression of B cells by CD4+ CD25+
regulatory T cells. J Immunol. 175:4180–4183. 2005.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Quaglino P, Ortoncelli M, Comessatti A,
Ponti R, Novelli M, Bergallo M, Costa C, Cicchelli S, Savoia P and
Bernengo MG: Circulating CD4+CD25 bright FOXP3+ T cells are
up-regulated by biological therapies and correlate with the
clinical response in psoriasis patients. Dermatology. 219:250–258.
2009.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Yang HX, Zhang W, Zhao LD, Li Y, Zhang FC,
Tang FL, He W and Zhang X: Are CD4+CD25-Foxp3+ cells in untreated
new-onset lupus patients regulatory T cells? Arthritis Res Ther.
11(R153)2009.PubMed/NCBI View
Article : Google Scholar
|
|
97
|
Wehrens EJ, Prakken BJ and van Wijk F: T
cells out of control-impaired immune regulation in the inflamed
joint. Nat Rev Rheumatol. 9:34–42. 2013.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Shevach EM: Regulatory T cells in
autoimmmunity*. Annu Rev Immunol. 18:423–449. 2000.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Crispín JC, Kyttaris VC, Terhorst C and
Tsokos GC: T cells as therapeutic targets in SLE. Nat Rev
Rheumatol. 6:317–325. 2010.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Scheinecker C, Bonelli M and Smolen JS:
Pathogenetic aspects of systemic lupus erythematosus with an
emphasis on regulatory T cells. J Autoimmun. 35:269–275.
2010.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Hagiwara E, Gourley MF, Lee S and Klinman
DK: Disease severity in patients with systemic lupus erythematosus
correlates with an increased ratio of interleukin-10:
Interferon-gamma-secreting cells in the peripheral blood. Arthritis
Rheum. 39:379–385. 1996.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Rousset F, Garcia E, Defrance T, Péronne
C, Vezzio N, Hsu DH, Kastelein R, Moore KW and Banchereau J:
Interleukin 10 is a potent growth and differentiation factor for
activated human B lymphocytes. Proc Natl Acad Sci USA.
89:1890–1893. 1992.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Llorente L, Zou W, Levy Y, Richaud-Patin
Y, Wijdenes J, Alcocer-Varela J, Morel-Fourrier B, Brouet JC,
Alarcon-Segovia D, Galanaud P and Emilie D: Role of interleukin 10
in the B lymphocyte hyperactivity and autoantibody production of
human systemic lupus erythematosus. J Exp Med. 181:839–844.
1995.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Itoh K and Hirohata S: The role of IL-10
in human B cell activation, proliferation, and differentiation. J
Immunol. 154:4341–4350. 1995.PubMed/NCBI
|
|
105
|
Ohl K and Tenbrock K: Regulatory T cells
in systemic lupus erythematosus. Eur J Immunol. 45:344–355.
2015.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Zhao DM, Thornton AM, DiPaolo RJ and
Shevach EM: Activated CD4+CD25+ T cells selectively kill B
lymphocytes. Blood. 107:3925–3932. 2006.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Kristensen M, Chu CQ, Eedy DJ, Feldmann M,
Brennan FM and Breathnach SM: Localization of tumour necrosis
factor-alpha (TNF-alpha) and its receptors in normal and psoriatic
skin: Epidermal cells express the 55-kD but not the 75-kD TNF
receptor. Clin Exp Immunol. 94:354–362. 1993.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Kollias G and Kontoyiannis D: Role of
TNF/TNFR in autoimmunity: Specific TNF receptor blockade may be
advantageous to anti-TNF treatments. Cytokine Growth Factor Rev.
13:315–321. 2002.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Aringer M, Feierl E, Steiner G, Stummvoll
GH, Höfler E, Steiner CW, Radda I, Smole JS and Graninger WB:
Increased bioactive TNF in human systemic lupus erythematosus:
Associations with cell death. Lupus. 11:102–108. 2002.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Kollias G, Kontoyiannis D, Douni E and
Kassiotis G: The role of TNF/TNFR in organ-specific and systemic
autoimmunity: Implications for the design of optimized ‘anti-TNF’
therapies. Curr Dir Autoimmun. 5:30–50. 2002.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Segal R, Dayan M, Zinger H and Mozes E:
Suppression of experimental systemic lupus erythematosus (SLE) in
mice via TNF inhibition by an anti-TNFalpha monoclonal antibody and
by pentoxiphylline. Lupus. 10:23–31. 2001.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Aringer M, Graninger WB, Steiner G and
Smolen JS: Safety and efficacy of tumor necrosis factor alpha
blockade in systemic lupus erythematosus: An open-label study.
Arthritis Rheum. 50:3161–3169. 2004.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Takahashi H, Tsuji H, Hashimoto Y,
Ishida-Yamamoto A and Iizuka H: Serum cytokines and growth factor
levels in Japanese patients with psoriasis. Clin Exp Dermatol.
35:645–649. 2010.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Moulton VR, Suarez-Fueyo A, Meidan E, Li
H, Mizui M and Tsokos GC: Pathogenesis of human systemic lupus
erythematosus: A cellular perspective. Trends Mol Med. 23:615–635.
2017.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Crispín JC, Oukka M, Bayliss G, Cohen RA,
Van Beek CA, Stillman IE, Kyttaris VC, Juang YT and Tsokos GC:
Expanded double negative T cells in patients with systemic lupus
erythematosus produce IL-17 and infiltrate the kidneys. J Immunol.
181:8761–8766. 2008.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Yu JJ and Gaffen SL: Interleukin-17: A
novel inflammatory cytokine that bridges innate and adaptive
immunity. Front Biosci. 13:170–177. 2008.PubMed/NCBI View
Article : Google Scholar
|
|
117
|
Henriques A, Inês L, Couto M, Pedreiro S,
Santos C, Magalhães M, Santos P, Velada I, Almeida A, Carvalheiro
T, et al: Frequency and functional activity of Th17, Tc17 and other
T-cell subsets in systemic lupus erythematosus. Cellular
Immunology. 264:97–103. 2010.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Suárez-Fueyo A, Bradley SJ, Klatzmann D
and Tsokos GC: T cells and autoimmune kidney disease. Nat Rev
Nephrol. 13:329–343. 2017.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Ospelt C: Synovial fibroblasts in 2017.
RMD Open. 3(e000471)2017.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Kyogoku C, Langefeld CD, Ortmann WA, Lee
A, Selby S, Carlton VE, Chang M, Ramos P, Baechler EC, Batliwalla
FM, et al: Genetic association of the R620W polymorphism of protein
tyrosine phosphatase PTPN22 with human SLE. Am J Hum Genet.
75:504–507. 2004.PubMed/NCBI View
Article : Google Scholar
|
|
121
|
International Consortium for Systemic
Lupus Erythematosus Genetics (SLEGEN). Harley JB, Alarcón-Riquelme
ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ,
Langefeld CD, et al: Genome-wide association scan in women with
systemic lupus erythematosus identifies susceptibility variants in
ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 40:204–210.
2008.PubMed/NCBI View
Article : Google Scholar
|
|
122
|
Smith RL, Warren RB, Eyre S, Ke X, Young
HS, Allen M, Strachan D, McArdle W, Gittins MP, Barker JN, et al:
Polymorphisms in the PTPN22 region are associated with psoriasis of
early onset. Br J Dermatol. 158:962–968. 2008.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Li Y, Liao W, Chang M, Schrodi SJ, Bui N,
Catanese JJ, Poon A, Matsunami N, Callis-Duffin KP, Leppert MF, et
al: Further genetic evidence for three psoriasis-risk genes:
ADAM33, CDKAL1, and PTPN22. J Invest Dermatol. 129:629–634.
2009.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Remmers EF, Plenge RM, Lee AT, Graham RR,
Hom G, Behrens TW, de Bakker PI, Le JM, Lee HS, Batliwalla F, et
al: STAT4 and the risk of rheumatoid arthritis and systemic lupus
erythematosus. N Engl J Med. 357:977–986. 2007.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Nair R, Duffin KC, Helms C, Ding J, Stuart
PE, Goldgar D, Gudjonsson JE, Li Y, Tejasvi T, Feng BJ, et al:
Genome-wide scan reveals association of psoriasis with IL-23 and
NF-kappaB pathways. Nat Genet. 41:199–204. 2009.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Zervou MI, Goulielmos GN, Castro-Giner F,
Tosca AD and Krueger-Krasagakis S: STAT4 gene polymorphism is
associated with psoriasis in the genetically homogeneous population
of Crete, Greece. Hum Immunol. 70:738–741. 2009.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Genetic Analysis of Psoriasis Consortium
& the Wellcome Trust Case Control Consortium 2. Strange A,
Capon F, Spencer CC, Knight J, Weale ME, Allen MH, Barton A, Band
G, Bellenguez C, et al: A genome-wide association study identifies
new psoriasis susceptibility loci and an interaction between HLA-C
and ERAP1. Nat Genet. 42:985–990. 2010.PubMed/NCBI View
Article : Google Scholar
|
|
128
|
Ellinghaus E, Ellinghaus D, Stuart PE,
Nair RP, Debrus S, Raelson JV, Belouchi M, Fournier H, Reinhard C,
Ding J, et al: Genome-wide association study identifies a psoriasis
susceptibility locus at TRAF3IP2. Nat Genet. 42:991–995.
2010.PubMed/NCBI View
Article : Google Scholar
|
|
129
|
Capon F, Burden AD, Trembath RC and Barker
JN: Psoriasis and other complex trait dermatoses: From Loci to
functional pathways. J Invest Dermatol. 132:915–922.
2012.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Gregersen PK: Gaining insight into PTPN22
and autoimmunity. Nat Genet. 37:1300–1302. 2005.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Siggs OM, Miosge LA, Yates AL, Kucharska
EM, Sheahan D, Brdicka T, Weiss A, Liston A and Goodnow CC:
Opposing functions of the T cell receptor kinase ZAP-70 in immunity
and tolerance differentially titrate in response to nucleotide
substitutions. Immunity. 27:912–926. 2007.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Wang H, Wang Z, Rani PL, Fu X, Yu W, Bao
F, Yu G, Li J, Li L, Sun L, et al: Identification of PTPN22,
ST6GAL1 and JAZF1 as psoriasis risk genes demonstrates shared
pathogenesis between psoriasis and diabetes. Exp Dermatol.
26:1112–1117. 2017.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Piotrowski P, Lianeri M, Wudarski M,
Olesińska M and Jagodziński PP: Contribution of STAT4 gene
single-nucleotide polymorphism to systemic lupus erythematosus in
the Polish population. Mol Biol Rep. 39:8861–8866. 2012.PubMed/NCBI View Article : Google Scholar
|
|
134
|
Shamilov R and Aneskievich BJ: TNIP1 in
autoimmune diseases: Regulation of toll-like receptor signaling. J
Immunol Res. 2018(3491269)2018.PubMed/NCBI View Article : Google Scholar
|
|
135
|
He CF, Liu YS, Cheng YL, Gao JP, Pan TM,
Han JW, Quan C, Sun LD, Zheng HF, Zuo XB, et al: TNIP1, SLC15A4,
ETS1, RasGRP3 and IKZF1 are associated with clinical features of
systemic lupus erythematosus in a Chinese Han population. Lupus.
19:1181–1186. 2010.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Nanda SK, Venigalla RK, Ordureau A,
Patterson-Kane JC, Powell DW, Toth R, Arthur JS and Cohen P:
Polyubiquitin binding to ABIN1 is required to prevent autoimmunity.
J Exp Med. 208:1215–1228. 2011.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Chen Y, Yan H, Song Z, Chen F, Wang H, Niu
J, Shi X, Zhang D, Zhang N, Zhai Z, et al: Downregulation of TNIP1
expression leads to increased proliferation of human keratinocytes
and severer psoriasis-like conditions in an imiquimod-induced mouse
model of dermatitis. PLoS One. 10(e0127957)2015.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Li Y, Cheng H, Zuo XB, Sheng YJ, Zhou FS,
Tang XF, Tang HY, Gao JP, Zhang Z, He SM, et al: Association
analyses identifying two common susceptibility loci shared by
psoriasis and systemic lupus erythematosus in the Chinese Han
population. J Med Genet. 50:812–818. 2013.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Coto-Segura P, Coto E, González-Lara L,
Alonso B, Gómez J, Cuesta-Llavona E and Queiro R: Gene variant in
the NF-κB pathway inhibitor NFKBIA distinguishes patients with
psoriatic arthritis within the spectrum of psoriatic disease.
Biomed Res Int. 2019(1030256)2019.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Martin DA, Towne JE, Kricorian G, Klekotka
P, Gudjonsson JE, Krueger JG and Russell CB: The emerging role of
IL-17 in the pathogenesis of psoriasis: Preclinical and clinical
findings. J Invest Dermatol. 133:17–26. 2013.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Yin X, Zhang S, Li B, Zhang Y and Zhang X:
IL28RA inhibits human epidermal keratinocyte proliferation by
inhibiting cell cycle progression. Mol Biol Rep. 46:1189–1197.
2019.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Wen Z, Xu L, Xu W, Yin Z, Gao X and Xiong
S: Interleukin-17 expression positively correlates with disease
severity of lupus nephritis by increasing anti-double-stranded DNA
antibody production in a lupus model induced by activated
lymphocyte derived DNA. PLoS One. 8(e58161)2013.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Langley RG, Elewski BE, Lebwohl M, Reich
K, Griffiths CE, Papp K, Puig L, Nakagawa H, Spelman L,
Sigurgeirsson B, et al: Secukinumab in plaque psoriasis-results of
two phase 3 trials. N Engl J Med. 371:326–338. 2014.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Griffiths CE, Reich K, Lebwohl M, van de
Kerkhof P, Paul C, Menter A, Cameron GS, Erickson J, Zhang L,
Secrest RJ, et al: Comparison of ixekizumab with etanercept or
placebo in moderate-to-severe psoriasis (UNCOVER-2 and UNCOVER-3):
Results from two phase 3 randomised trials. Lancet. 386:541–551.
2015.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Lebwohl M, Strober B, Menter A, Gordon K,
Weglowska J, Puig L, Papp K, Spelman L, Toth D, Kerdel F, et al:
Phase 3 studies comparing brodalumab with ustekinumab in psoriasis.
N Engl J Med. 373:1318–1328. 2015.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Satoh Y, Nakano K, Yoshinari H, Nakayamada
S, Iwata S, Kubo S, Miyagawa I, Yoshikawa M, Miyazaki Y, Saito K
and Tanaka Y: A case of refractory lupus nephritis complicated by
psoriasis vulgaris that was controlled with secukinumab. Lupus.
27:1202–1206. 2018.PubMed/NCBI View Article : Google Scholar
|
|
147
|
van Vollenhoven RF, Hahn BH, Tsokos GC,
Wagner CL, Lipsky P, Touma Z, Werth VP, Gordon RM, Zhou B, Hsu B,
et al: Efficacy and safety of ustekinumab, an IL-12 and IL-23
inhibitor, in patients with active systemic lupus erythematosus:
Results of a multicentre, double-blind, phase 2, randomised,
controlled study. Lancet. 392:1330–1339. 2018.PubMed/NCBI View Article : Google Scholar
|
|
148
|
Raychaudhuri SK, Saxena A and Raychaudhuri
SP: Role of IL-17 in the pathogenesis of psoriatic arthritis and
axial spondyloarthritis. Clin Rheumatol. 34:1019–1023.
2015.PubMed/NCBI View Article : Google Scholar
|
|
149
|
Sato K, Aizaki Y, Yoshida Y and Mimura T:
Treatment of psoriatic arthritis complicated by systemic lupus
erythematosus with the IL-17 blocker secukinumab and an analysis of
the serum cytokine profile. Mod Rheumatol Case Rep. 4:181–185.
2020.PubMed/NCBI View Article : Google Scholar
|
|
150
|
Tanaka Y, Kubo S, Iwata S, Yoshikawa M and
Nakayamada S: B cell phenotypes, signaling and their roles in
secretion of antibodies in systemic lupus erythematosus. Clin
Immunol. 186:21–25. 2018.PubMed/NCBI View Article : Google Scholar
|
|
151
|
Yap DYH and Chan TM: B cell abnormalities
in systemic lupus erythematosus and lupus nephritis-role in
pathogenesis and effect of immunosuppressive treatments. Int J Mol
Sci. 20(6231)2019.PubMed/NCBI View Article : Google Scholar
|
|
152
|
Guidelli GM, Fioravanti A, Rubegni P and
Feci L: Induced psoriasis after rituximab therapy for rheumatoid
arthritis: A case report and review of the literature. Rheumatol
Int. 33:2927–2930. 2013.PubMed/NCBI View Article : Google Scholar
|
|
153
|
Edwards JCW, Szczepanski L, Szechinski J,
Filipowicz-Sosnowska A, Emery P, Close DR, Stevens RM and Shaw T:
Efficacy of B-cell-targeted therapy with rituximab in patients with
rheumatoid arthritis. N Engl J Med. 350:2572–2581. 2004.PubMed/NCBI View Article : Google Scholar
|
|
154
|
Leandro MJ, Cambridge G, Ehrenstein MR and
Edwards JCW: Reconstitution of peripheral blood B cells after
depletion with rituximab in patients with rheumatoid arthritis.
Arthritis Rheum. 54:613–620. 2006.PubMed/NCBI View Article : Google Scholar
|
|
155
|
Dumoitier N, Terrier B, London J, Lofek S
and Mouthon L: Implication of B lymphocytes in the pathogenesis of
ANCA-associated vasculitides. Autoimmun Rev. 14:996–1004.
2015.PubMed/NCBI View Article : Google Scholar
|
|
156
|
Joly P, Maho-Vaillant M, Prost-Squarcioni
C, Hebert V, Houivet E, Calbo S, Caillot F, Golinski ML, Labeille
B, Picard-Dahan C, et al: First-line rituximab combined with
short-term prednisone versus prednisone alone for the treatment of
pemphigus (Ritux 3): A prospective, multicentre, parallel-group,
open-label randomised trial. Lancet. 389:2031–2040. 2017.PubMed/NCBI View Article : Google Scholar
|
|
157
|
van de Veerdonk FL, Lauwerys B,
Marijnissen RJ, Timmermans K, Di Padova F, Koenders MI,
Gutierrez-Roelens I, Durez P, Netea MG, van der Meer JW, et al: The
anti-CD20 antibody rituximab reduces the Th17 cell response.
Arthritis Rheum. 63:1507–1516. 2011.PubMed/NCBI View Article : Google Scholar
|
|
158
|
Chang YS, Lee HT, Chen WS, Hsiao KH, Chen
MH, Tsai CY and Chou CT: Treatment of psoriasis with rituximab. J
Am Acad Dermatol. 66:e184–e185. 2012.PubMed/NCBI View Article : Google Scholar
|
|
159
|
Dass S, Vital EM and Emery P: Development
of psoriasis after B cell depletion with rituximab. Arthritis
Rheum. 56:2715–2718. 2007.PubMed/NCBI View Article : Google Scholar
|
|
160
|
Markatseli TE, Kaltsonoudis ES, Voulgari
PV, Zioga A and Drosos AA: Induction of psoriatic skin lesions in a
patient with rheumatoid arthritis treated with rituximab. Clin Exp
Rheumatol. 27:996–998. 2009.PubMed/NCBI
|
|
161
|
Collamer AN, Guerrero KT, Henning JS and
Battafarano DF: Psoriatic skin lesions induced by tumor necrosis
factor antagonist therapy: A literature review and potential
mechanisms of action. Arthritis Rheum. 59:996–1001. 2008.PubMed/NCBI View Article : Google Scholar
|
|
162
|
Palucka AK, Blanck JP, Bennett L, Pascual
V and Banchereau J: Cross-regulation of TNF and IFN-alpha in
autoimmune diseases. Proc Natl Acad Sci USA. 102:3372–3377.
2005.PubMed/NCBI View Article : Google Scholar
|
|
163
|
Schmitt J, Zhang Z, Wozel G, Meurer M and
Kirch W: Efficacy and tolerability of biologic and nonbiologic
systemic treatments for moderate-to-severe psoriasis: Meta-analysis
of randomized controlled trials. Br J Dermatol. 159:513–526.
2008.PubMed/NCBI View Article : Google Scholar
|
|
164
|
Tracey D, Klareskog L, Sasso EH, Salfeld
JG and Tak PP: Tumor necrosis factor antagonist mechanisms of
action: A comprehensive review. Pharmacol Ther. 117:244–279.
2008.PubMed/NCBI View Article : Google Scholar
|
|
165
|
Joseph A, Brasington R, Kahl L,
Ranganathan P, Cheng TP and Atkinson J: Immunologic rheumatic
disorders. J Allergy Clin Immunol. 125 (2 Suppl 2):S204–S215.
2010.PubMed/NCBI View Article : Google Scholar
|
|
166
|
Norris DA, Whang K, David-Bajar K and
Bennion SD: The influence of ultraviolet light on immunological
cytotoxicity in the skin. Photochem Photobiol. 65:636–646.
1997.PubMed/NCBI View Article : Google Scholar
|
|
167
|
Köck A, Schwarz T, Kirnbauer R, Urbanski
A, Perry P, Ansel JC and Luger TA: Human keratinocytes are a source
for tumor necrosis factor alpha: Evidence for synthesis and release
upon stimulation with endotoxin or ultraviolet light. J Exp Med.
172:1609–1614. 1990.PubMed/NCBI View Article : Google Scholar
|
|
168
|
Takashima A and Bergstresser PR: Impact of
UVB radiation on the epidermal cytokine network. Photochem
Photobiol. 63:397–400. 1996.PubMed/NCBI View Article : Google Scholar
|
|
169
|
Meller S, Winterberg F, Gilliet M, Müller
A, Lauceviciute I, Rieker J, Neumann NJ, Kubitza R, Gombert M,
Bünemann E, et al: Ultraviolet radiation-induced injury,
chemokines, and leukocyte recruitment: An amplification cycle
triggering cutaneous lupus erythematosus. Arthritis Rheum.
52:1504–1516. 2005.PubMed/NCBI View Article : Google Scholar
|
|
170
|
Kuhn A and Sontheimer RD: Cutaneous lupus
erythematosus: Molecular and cellular basis of clinical findings.
Curr Dir Autoimmun. 10:119–140. 2008.PubMed/NCBI View Article : Google Scholar
|
|
171
|
Muñoz LE, Lauber K, Schiller M, Manfredi
AA and Herrmann M: The role of defective clearance of apoptotic
cells in systemic autoimmunity. Nat Rev Rheumatol. 6:280–289.
2010.PubMed/NCBI View Article : Google Scholar
|
|
172
|
Kochevar IE: Action spectrum and
mechanisms of UV radiation-induced injury in lupus erythematosus. J
Invest Dermatol. 85 (1 Suppl):140s–143s. 1985.PubMed/NCBI View Article : Google Scholar
|
|
173
|
Wolf R, Schiavo AL, Lombardi ML, de
Angelis F and Ruocco V: The in vitro effect of hydroxychloroquine
on skin morphology in psoriasis. Int J Dermatol. 38:154–157.
1999.PubMed/NCBI View Article : Google Scholar
|
|
174
|
Harrison CA, Layton CM, Hau Z, Bullock AJ,
Johnson TS and MacNeil S: Transglutaminase inhibitors induce
hyperproliferation and parakeratosis in tissue-engineered skin. Br
J Dermatol. 156:247–257. 2007.PubMed/NCBI View Article : Google Scholar
|
|
175
|
Tzu J and Kerdel F: From conventional to
cutting edge: The new era of biologics in treatment of psoriasis.
Dermatol Ther. 21:131–141. 2008.PubMed/NCBI View Article : Google Scholar
|
|
176
|
Costa MF, Said NR and Zimmermann B:
Drug-induced lupus due to anti-tumor necrosis factor alpha agents.
Semin Arthritis Rheum. 37:381–387. 2008.PubMed/NCBI View Article : Google Scholar
|
|
177
|
Grönhagen CM, Fored CM, Linder M, Granath
F and Nyberg F: Subacute cutaneous lupus erythematosus and its
association with drugs: A population-based matched case-control
study of 234 patients in Sweden. Br J Dermatol. 167:296–305.
2012.PubMed/NCBI View Article : Google Scholar
|
|
178
|
Ramos-Casals M, Brito-Zerón P, Muñoz S,
Soria N, Galiana D, Bertolaccini L, Cuadrado MJ and Khamashta MA:
Autoimmune diseases induced by TNF-targeted therapies: Analysis of
233 cases. Medicine (Baltimore). 86:242–251. 2007.PubMed/NCBI View Article : Google Scholar
|
|
179
|
Williams VL and Cohen PR: TNF alpha
antagonist-induced lupus-like syndrome: Report and review of the
literature with implications for treatment with alternative TNF
alpha antagonists. Int J Dermatol. 50:619–625. 2011.PubMed/NCBI View Article : Google Scholar
|
|
180
|
Mudduluru BM, Shah S, Shamah S and
Swaminath A: TNF-alpha antagonist induced lupus on three different
agents. Postgrad Med. 129:304–306. 2017.PubMed/NCBI View Article : Google Scholar
|
|
181
|
Wetter DA and Davis MDP: Lupus-like
syndrome attributable to anti-tumor necrosis factor alpha therapy
in 14 patients during an 8-year period at Mayo clinic. Mayo Clin
Proc. 84:979–984. 2009.PubMed/NCBI View Article : Google Scholar
|
|
182
|
Hsieh CY and Tsai TF: Aggravation of
discoid lupus erythematosus in a patient with psoriasis and
psoriatic arthritis during treatment of secukinumab: A case report
and review of literature. Lupus. 31:891–894. 2022.PubMed/NCBI View Article : Google Scholar
|
|
183
|
Anstey NM, Bastian I, Dunckley H and
Currie BJ: Systemic lupus erythematosus (SLE): Different
prevalences in different populations of Australian aborigines. Aust
N Z J Med. 25(736)1995.PubMed/NCBI View Article : Google Scholar
|
|
184
|
Duarte-García A, Hocaoglu M,
Valenzuela-Almada M, Osei-Onomah SA, Dabit JY, Sanchez-Rodriguez A,
Duong SQ, Giblon RE, Langenfeld HE, Alarcón GS, et al: Rising
incidence and prevalence of systemic lupus erythematosus: A
population-based study over four decades. Ann Rheum Dis.
(annrheumdis-2022-222276)2022.PubMed/NCBI View Article : Google Scholar : (Epub ahead of
print).
|
|
185
|
Helms C, Cao L, Krueger JG, Wijsman EM,
Chamian F, Gordon D, Heffernan M, Daw JA, Robarge J, Ott J, et al:
A putative RUNX1 binding site variant between SLC9A3R1 and NAT9 is
associated with susceptibility to psoriasis. Nat Genet. 35:349–356.
2003.PubMed/NCBI View
Article : Google Scholar
|
|
186
|
Hüffmeier U, Uebe S, Ekici AB, Bowes J,
Giardina E, Korendowych E, Juneblad K, Apel M, McManus R, Ho P, et
al: Common variants at TRAF3IP2 are associated with susceptibility
to psoriatic arthritis and psoriasis. Nat Genet. 42:996–999.
2010.PubMed/NCBI View
Article : Google Scholar
|
|
187
|
Uddin M, Sturge M, Rahman P and Woods MO:
Autosome-wide copy number variation association analysis for
rheumatoid arthritis using the WTCCC high-density SNP genotype
data. J Rheumatol. 38:797–801. 2011.PubMed/NCBI View Article : Google Scholar
|
|
188
|
Perricone C, Ciccacci C, Ceccarelli F, Di
Fusco D, Spinelli FR, Cipriano E, Novelli G, Valesini G, Conti F
and Borgiani P: TRAF3IP2 gene and systemic lupus erythematosus:
Association with disease susceptibility and pericarditis
development. Immunogenetics. 65:703–709. 2013.PubMed/NCBI View Article : Google Scholar
|
|
189
|
Wu L, Wang C, Boisson B, Misra S, Rayman
P, Finke JH, Puel A, Casanova JL and Li X: The differential
regulation of human ACT1 isoforms by Hsp90 in IL-17 signaling. J
Immunol. 193:1590–1599. 2014.PubMed/NCBI View Article : Google Scholar
|
|
190
|
Petrić M and Radić M: Is Th17-targeted
therapy effective in systemic lupus erythematosus? Curr Issues Mol
Biol. 45:4331–4343. 2023.PubMed/NCBI View Article : Google Scholar
|