Introduction
Non-small cell lung cancer (NSCLC) accounts for ~80%
of all lung cancer cases in worldwide (1). Epidermal growth factor receptor
(EGFR)-targeted therapy is one of the main first-line treatments
for NSCLC (2-4);
however, almost all patients eventually develop acquired resistance
to EGFR-tyrosine kinase inhibitors (TKIs) (5,6). The
acquisition of EGFR-TKI resistance is associated with the
EGFR-T790M gatekeeper mutation in patients with NSCLC treated with
first-generation EGFR-TKIs, such as gefitinib or erlotinib
(7-9).
Notably, multiple mechanisms of acquired resistance to EGFR-TKIs
have been reported, including MET receptor gene amplification
(10), epithelial-mesenchymal
transition (EMT) (11) and the
pathological transformation of NSCLC to SCLC (12,13).
However, other mechanisms underlying the acquired resistance to
EGFR inhibitors remain to be discovered (12,14).
To improve the effectiveness of EGFR-TKI treatment, a greater
understanding of the unknown mechanisms underlying acquired
resistance is urgently needed.
Glycogen synthase kinase 3β (GSK3β) is an isoform of
the GSK3 family of kinases that promotes tumor development and
progression in lung cancer (15,16).
The tumor-promoting role of GSK3β is upregulated in NSCLC cells
(17,18). Previous studies have shown that
silencing GSK3β reduces the proliferation, migration and invasion,
and increases the apoptosis of NSCLC cells (18,19).
GSK3β also serves a pivotal role in the development of
EGFR-resistant lung cancer. A previous study demonstrated that
GSK3β inhibition suppresses the proliferation of cells with
acquired resistance to osimertinib in EGFR-mutant NSCLC (20). However, little is currently known
about the role of GSK3β in gefitinib-resistant NSCLC.
The present study aimed to investigate the role of
GSK3β in gefitinib resistance in the PC-9 NSCLC cell line. Notably,
GSK3β activity is often regulated by the PI3K/AKT or Wnt signaling
pathways in NSCLC (21). A
previous study demonstrated that AKT activity is upregulated in
cancer cells that are resistant to EGFR-TKIs (22). Furthermore, PI3K/AKT signaling is
known to serve an important role in acquired resistance to EGFR-TKI
in NSCLC (23-25).
Therefore, the current study also examined the role of the PI3K/AKT
pathway to explore whether it regulates GSK3β in EGFR-TKI-resistant
cells.
Materials and methods
Cell culture and reagents
The human NSCLC cell lines PC-9 (exon 19 deletion)
and H1975 (L858R/T790M) were kindly provided by The Cell Bank of
Type Culture Collection of the Chinese Academy of Sciences.
Gefitinib-resistant PC-9 cells were induced by gradually increasing
drug concentrations. Briefly, 5x105 cells in logarithmic
phase were placed in RPMI-1640 medium (cat. no. C11875500BT; Gibco;
Thermo Fisher Scientific, Inc.) supplemented with 10% fetal bovine
serum (FBS; cat. no. ST30-3302; PAN-Biotech GmbH) and 1%
penicillin-streptomycin (cat. no. 15140-122; Gibco; Thermo Fisher
Scientific, Inc.) at 37˚C with 5% CO2. After 24 h of
adherence, 100 nmol/l gefitinib (cat. no. HY-50895; MedChemExpress)
was used as the initial concentration to treat cells, and the
medium containing gefitinib was changed daily. After 3-4 weeks, the
cells gradually showed tolerance and stable growth, were cultured
in normal medium for 3-5 days, then passaged, and the concentration
of gefitinib was doubled again until the concentration had been
doubled five times. The final drug concentration was 3.2 µmol/l,
which was maintained for 1 month, after which, the medium was
replaced with drug-free medium for 1 month. The whole process took
7 months to complete, and the established resistant cell line was
named PC-9G. The cells were cultured in RPMI-1640 medium (cat. no.
C11875500BT; Gibco; Thermo Fisher Scientific, Inc.) supplemented
with 10% fetal bovine serum (FBS; cat. no. ST30-3302; PAN-Biotech
GmbH) and 1% penicillin-streptomycin (cat. no. 15140-122; Gibco;
Thermo Fisher Scientific, Inc.). All cells were maintained in a
37˚C incubator with 5% CO2. SC79 (AKT activator, 5
nmol/l; cat. no. HY-18749; MedChemExpress) and MK2206 (AKT
antagonist, 5 nmol/l; cat. no. HY-10358; MedChemExpress) were
dissolved in 0.5% DMSO. The images of cell fluorescence were
obtained using a fluorescence microscope (Olympus IX73; Olympus
Corporation). Cell morphology was assessed by light microscopy
(Olympus BX53; Olympus Corporation).
Cell proliferation assay
The human NSCLC cell lines PC-9, PC-9G and H1975
were seeded into 96-well plates (1.5x103 cells/well) and
were treated with gefitinib (0, 1, 5, 10, 50, 100, 1,000, 5,000,
10,000 or 50,000 nmol/l). Following incubation for 72 h at 37˚C
with 5% CO2, a Cell Counting Kit-8 (CCK8) kit (Vazyme
Biotech Co., Ltd.) was used to determine cell viability and
proliferation. Fresh medium was used to replace the old medium, and
10 µl CCK-8 reagent was added to each well. Cell viability was
measured at a wavelength of 450 nm, and cell proliferation was
calculated as optical density (OD) value of the experimental
well/OD value of control well (day 1). The results were obtained
using five replicates per experiment from three separate
experiments.
Colony formation assay
The human NSCLC cell lines PC-9 and PC-9G were
seeded into six-well plates (1x104 cells/well) and were
treated with gefitinib (300 nmol/l) or 0.3% DMSO. The drugs were
changed every 3 days and cells were cultured at 37˚C with 5%
CO2. After 10 days, cell colonies were fixed with 4%
paraformaldehyde (MilliporeSigma) at room temperature for 30 min,
and then stained with 0.05% violet crystal (MilliporeSigma) at room
temperature for 30 min. After washing three times with PBS, the
number of colonies >1 mm in size was counted manually in each
group.
Migration assays
Following treatment with MK2206 (AKT inhibitor, 5
nmol/l) or SC79 (AKT activator, 5 nmol/l) at 37˚C for 24 h, PC-9
and PC-9G (6x104/well), and H1975
(3x104/well) cells were seeded in 24-well migration
chambers (pore size, 8-µm; Corning, Inc.) containing RPMI-1640
without FBS, and 400 µl RPMI-1640 supplemented with 10% FBS was
added to the lower chamber to stimulate cell migration. After
incubation at 37˚C for 18-24 h, non-migratory cells were removed
from the upper chamber with a cotton swab, and the cells that had
migrated to the lower chamber were fixed with 20% methanol at room
temperature for 30 min, and then stained with 0.1% crystal violet
at room temperature for 30 min. Migratory cells were counted using
a light microscope and analyzed based on four randomly selected
fields.
Invasion assays
Following treatment with MK2206 (5 nmol/l) or SC79
(5 nmol/l) at 37˚C for 24 h, PC-9 and PC-9G
(1.5x105/well), and H1975 (1x105/well) cells
were seeded in 24-well Transwell units (pore size, 8-µm; Corning,
Inc.) containing RPMI-1640 without FBS, and 400 µl RPMI-1640
supplemented with 10% FBS was added to the lower chamber to
stimulate cell invasion. After precoating the chambers with 3 mg/ml
Matrigel (Corning, Inc.) at 37˚C for 30 min, cells were added to
24-well Transwell chambers. After incubation at 37˚C for 18-24 h,
non-invasive cells were removed from the upper chamber with a
cotton swab, and the cells that had invaded the lower chamber were
fixed with 20% methanol at room temperature for 30 min, and then
stained with 0.1% crystal violet at room temperature for 30 min.
Invasive cells were counted using a light microscope and analyzed
based on four randomly selected fields.
Apoptosis assay
PC-9, PC-9G and H1975 cells were seeded into 60-mm
dishes (5x105 cells) and treated with gefitinib (300 nM)
or DMSO (negative control). After incubation at 37˚C for 48 h, the
cells were harvested and stained with annexin V-fluorescein
isothiocyanate (FITC) and propidium iodide (PI) (Vazyme Biotech
Col., Ltd.) at room temperature for 15 min in the dark. BD
FACSCanto (BD Biosciences) was used to measure the fluorescence
intensity in the FITC (FL1, 533 nm) and PI (FL2, 585 nm) channels.
Early apoptotic cells (annexin V-positive only) and late apoptotic
cells (annexin V- and PI-positive) were quantified and analyzed
using FlowJo 10.0.7 software (FlowJo LLC).
RNA-sequencing (RNA-seq)
To obtain the differential gene expression profile
between PC-9 and PC-9G cells, total RNA was extracted from PC-9 and
PC-9G cells (105-106 cells) using
TRIzol® reagent (cat. no. 15596026CN; Invitrogen; Thermo
Fisher Scientific, Inc.) according to the manufacturer's
instructions. The quantity and quality of RNA were evaluated using
an Agilent 2100 Bioanalyzer (Agilent Technologies, Inc.). Samples
(1 µg) with an RNA integrity number score of >7 and RNA
concentration of >1,000 ng were selected for library
preparation. After quantification of nucleic acid extraction, the
NEB Next rRNA Depletion Kit (cat. no. E6310S; New England BioLabs,
Inc.) was used to remove rRNA. The rRNA-depleted product was
subjected to reverse transcription (RT) and double-stranded cDNA
synthesis using M-MLV Reverse Transcriptase, RNase H Minus, Point
Mutant (cat. no. M3683; Promega Corporation) and NEBNext Ultra II
Non-Directional RNA Second Stand Synthesis Module (cat. no. E6111S;
New England BioLabs, Inc). The double-stranded cDNA was then
fragmented using Covaris LE220-plus shearing instrument (Covaris,
LLC). Library construction was performed using the KAPA Hyper Prep
kit (cat. no. KK8504; Kapa Biosystems; Roche Diagnostics), and the
library was quantified using Qubit® dsDNA HS Assay Kit
(Thermo Fisher Scientific, Inc.). The size distribution of library
fragments was analyzed using LabChip GX Touch HT DNA High
Sensitivity Assay Kit (PerkinElmer, Inc.). The whole transcriptome
sequencing library was denatured and diluted to a concentration of
200-250 pM and sequenced on Illumina NovaSeq 6000 sequencing
platform (cat. no. 20012850; Illumina, Inc.). The sequencing
strategy was paired-end 150 bp, and the sequencing chip used was
NovaSeq 6000 S1 Reagent Kit (300 cycles) (cat. no. 20012863;
Illumina, Inc.). The required data output was 10 M reads. The raw
data obtained from sequencing were evaluated with FastQC (0.11.5;
https://github.com/pnnl/fqc). After
removing adapters and low-quality sequences using AdapterRemoval
(2.2.2; https://github.com/MikkelSchubert/adapterremoval),
clean reads were obtained. Subsequently, STAR (2.5.3a; https://github.com/alexdobin/STAR) was used to
align the clean reads to the hg19 whole transcriptome sequence.
RSEM (1.3.0; http://deweylab.biostat.wisc.edu/rsem) was used for
gene expression quantification. Differential expression analysis
was performed using edgeR (3.28.1; http://bioconductor.org/packages/3.2/bioc/html/edgeR.html)
with default parameters, and differentially expressed genes (DEGs)
were identified as|log2(fold change)|>1 and FDR<0.05.
Finally, the pheatmap (V 1.0.12; https://cran.r-project.org/web/packages/pheatmap/index.html)
was used to create a heatmap. KEGG functional enrichment analysis
was performed using org.hs.eg. DB (26). To process the functional enrichment
analysis, when the corrected P-value, that is q-value, was ≤0.05,
the KEGG function was considered to be significantly enriched.
ggplot2 (3.3.5; https://github.com/JLSteenwyk/ggpubfigs.) was used for
plotting the enriched signaling pathway.
Lentiviral packaging and stable cell
line establishment
The gene sequences of GSK3β were synthesized and
cloned into pCDH-GFP-puro (pCDH) lentiviral vectors (Generay
Biotech Co., Ltd.). To produce the lentivirus, 1 µg pMD2.G and 3 µg
psPAX2 were cotransfected with PCDH-GSK3β (4 µg) or the vehicle
plasmid (PCDH, 4 µg) into 293T cells using the Nano293T
Transfection Reagent (cat. no. C500T-1; NCM Biotech) at 37˚C for 12
h. Lentiviral particles (PCDH and PCDH-GSK3β) were harvested at
three timepoints (24, 48 and 72 h) after transfection and all of
the individual harvests were pooled for infection. The multiplicity
of infection for PC-9, PC-9G and H1975 cells was 20, 40 and 10,
respectively. After 24 h infection with the cells, the medium was
changed. PC-9, PC-9G and H1975 cells were screened with 6 µg/ml
puromycin (Beijing Solarbio Science & Technology Co., Ltd.) for
2-3 times to obtain a stably transduced cell line. After puromycin
screening for 3 weeks, stable cell lines PC-9/PCDH, PC-9/GSK3β,
PC-9G/PCDH, PC-9G/GSK3β, H1975/PCDH and H1975/GSK3β were
established and used in subsequent experiments.
RT-quantitative PCR (RT-qPCR)
Total RNA was isolated from PC-9, PC-9G and H1975
cells, as well as cells stably overexpressing GSK3β (PC-9/GSK3β,
PC-9G/GSK3β, H1975/GSK3β) and those infected with an empty vector
(PC-9/PCDH, PC-9G/PCDH, and H1975/PCDH) using the FastPure Cell RNA
Isolation Kit V2 (cat. no. RC112; Vazyme Biotech Co., Ltd.). RNA
was reverse transcribed into cDNA using a RT kit (cat. no. R323-01;
Vazyme Biotech Co., Ltd.) according to the manufacturer's
instructions. Subsequently cDNA was subjected to qPCR using SYBR
Green Mix (Vazyme Biotech Co., Ltd.). The qPCR protocol was as
follows: 95˚C for 5 min; followed by 40 cycles of denaturation at
95˚C for 10 sec, and annealing and extension at 60˚C for 30 sec;
with a final fluorescence measurement at 60˚C. The relative
expression levels of the target genes were determined using the
comparative cycle threshold method (2-ΔΔCq) (27). The primer sequences were as
follows: GSK3β forward, 5'-AGAGACAAGGACGGCAGCAAG-3' and reverse,
5'-GATGGCGACCAGTTCTCCTG-3'; GAPDH forward,
5'-GTCTCCTCTGACTTCAACAGCG-3' and reverse,
5'-ACCACCCTGTTGCTGTAGCCAA-3'.
Nuclear/cytoplasmic protein separation
and extraction
Nuclear and cytosolic proteins were extracted using
a nucleocytoplasmic separation and extraction kit (Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocols. The
supernatants were collected and then analyzed by western
blotting.
Protein extraction and western blot
analysis
Following 24 h of treatment with the AKT activator
or inhibitor at 37˚C, PC-9, PC-9G and H1975 cells were harvested,
and total protein was extracted using radioimmunoprecipitation
assay lysis buffer (Beyotime Institute of Biotechnology)
supplemented with protease inhibitors for 15 min. Proteins were
then centrifuged at 16,000 x g for 15 min at 4˚C. These steps were
all performed on ice. The supernatants were collected and protein
concentration was determined via bicinchoninic acid assay (Thermo
Scientific Fisher Scientific, Inc.). Subsequently, the proteins
were subjected to western blot analysis according to the
manufacturer's instructions (CWBio), after which an ECL Detection
System (Thermo Fisher Scientific, Inc.) was used for signal
detection. Protein bands were semi-quantified using ImageJ analysis
software (version 1.8.0; National Institutes of Health). Equal
amounts of heat-denatured protein (30 µg/lane) were separated by
SDS-PAGE on 10% gels and transferred onto nitrocellulose membranes,
which were blocked with blocking buffer containing 5% non-fat milk
in 1X TBS-0.1% Tween-20 for 2 h at room temperature and incubated
with primary antibodies at 4˚C overnight, followed by incubation
with an appropriate secondary antibody at 37˚C for 2 h. The
following primary antibodies (1:1,000 dilution; all from Cell
Signaling Technology, Inc.) were used: c-Myc (cat. no. 5605),
E-cadherin (cat. no. 3195), vimentin (cat. no. 5741), GAPDH (cat.
no. 5174), β-catenin (cat. no. 8480), Lamin A (cat. no. 86846),
GSK3β (cat. no. 12456), phosphorylated (p)GSK3β (cat. no. 9322),
AKT (cat. no. 4691), pAKT (cat. no. 4060), MET (cat. no. 8198),
pMET (cat. no. 3077), and β-actin (cat. no. 8457). The secondary
antibodies used were horseradish peroxidase-conjugated goat
anti-rabbit IgG secondary antibody (cat. no. L3012; 1:5,000
dilution; SAB Biotherapeutics, Inc.) and anti-mouse IgG secondary
antibody (cat. no. L3032; 1:5,000 dilution; SAB Biotherapeutics,
Inc.).
Immunohistochemistry
Immunohistochemistry was performed to determine
E-cadherin and vimentin expression levels in PC-9 and PC-9G cells
without any treatment. PC-9 and PC-9G cells were fixed with Bouin's
solution containing 5% acetic acid, 9% formaldehyde and 0.9% picric
acid (Millipore Sigma) at room temperature for 24 h, washed with
70% ethanol and embedded in paraffin. The sections (5 µm) were then
deparaffinized in xylene, hydrated in a graded series of ethanol
and placed in a solution of sodium citrate (pH 6.0) under
high-pressure heat-mediated antigen retrieval for 3 min.
Subsequently, the slides were probed with E-cadherin (cat. no.
3195, 1:400 dilution; Cell Signaling Technology, Inc.) and vimentin
(cat. no. 5741, 1:100 dilution; Cell Signaling Technology, Inc.)
antibodies overnight at room temperature. After incubation with
HRP-conjugated secondary antibody (cat. no. #K8000; 1:1,000;
Agilent Technologies, Inc.) at room temperature for 30 min, the
signal was developed using the DAB Histochemistry Kit (Invitrogen;
Thermo Fisher Scientific, Inc.). The images were visualized using a
light microscope.
Statistical analysis
Data are presented as the mean ± SD of at least
three independent experiments. Statistical differences were
analyzed by an independent samples t-test. All data were analyzed
using GraphPad Prism version 8 software (Dotmatics). P<0.05 was
considered to indicate a statistically significant difference.
Results
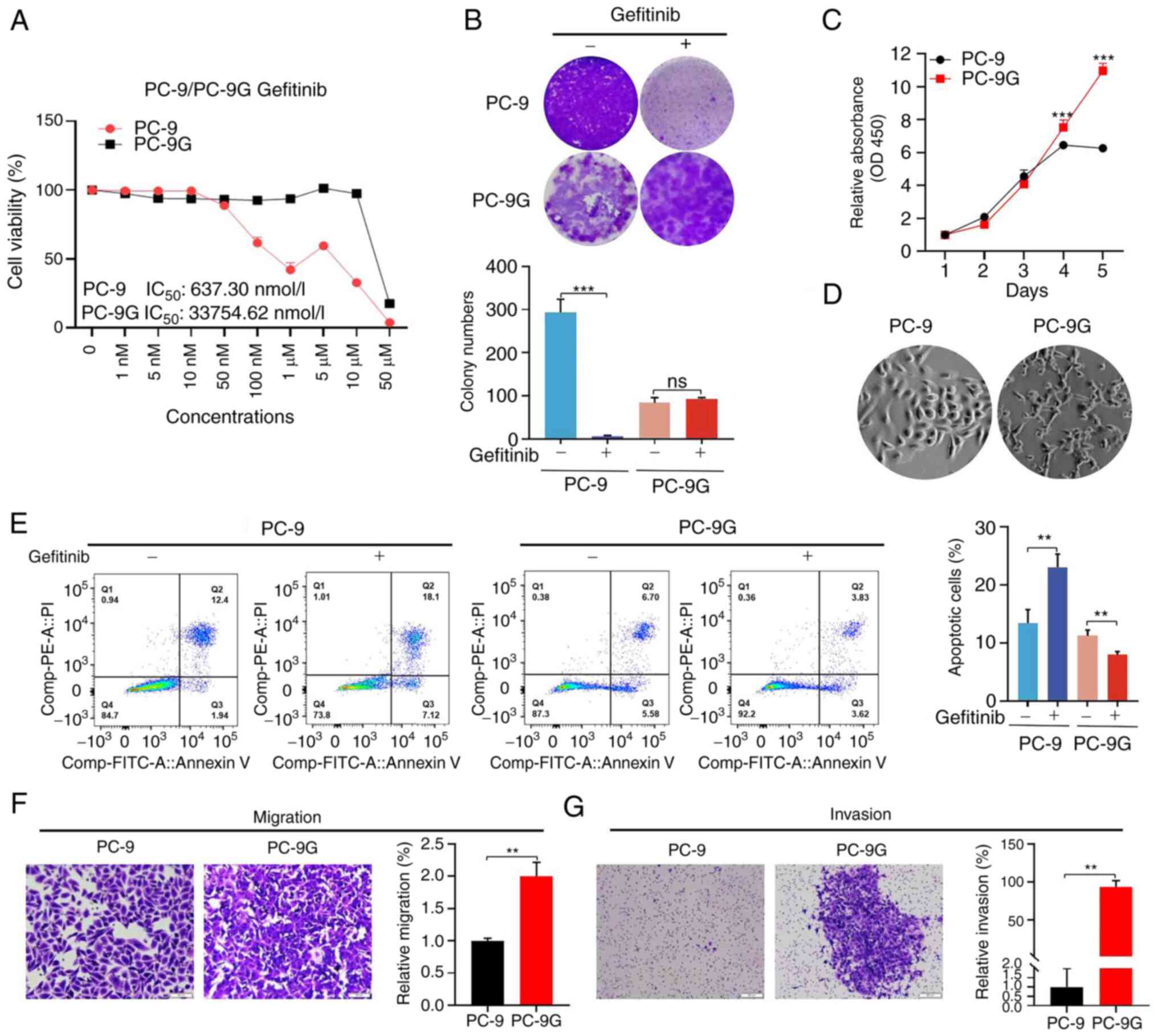
Successful generation of PC-9G cells
in vitro
PC-9G cells were successfully constructed in
vitro by increasing the drug concentration for 10 months. The
results of the CCK8 assay showed that the half maximal inhibitory
concentration (IC50) values of PC-9 and PC-9G cells were
637.3±0.21 nmol/l and 3.37±0.13 µmol/l, respectively, and the
resistant index was 52.91 (Fig.
1A). Notably, neither EGFR mutations or MET exon 14 skipping
mutation were detected in PC-9G cells (Fig. S1). To further elucidate the effect
of gefitinib on the colony formation of PC-9 and PC-9G cells, 300
nmol/l gefitinib was used to treat the cells. The results showed
that the colony formation of PC-9 cells was significantly decreased
following gefitinib treatment; however, the clonogenic ability of
PC-9G cells was not significantly affected by gefitinib (Fig. 1B). The proliferation of the cells
was also assessed using the CCK8 kit and was significantly higher
in PC-9G cells than in PC-9 cells without gefitinib treatment
(Fig. 1C). PC-9G cells were well
defined and showed a mesenchymal phenotype, including a spindle
appearance and loss of intercellular connections, and some cells
had prominent pseudopodia compared with PC-9 cells (Fig. 1D). Furthermore, the apoptotic rate
of PC-9G cells was lower than that of PC-9 cells. It was further
verified that 300 nmol/l gefitinib significantly increased the
apoptosis of PC-9 cells, but did not significantly affect the
apoptosis of PC-9G cells (Fig.
1E). Migration and invasion assays showed that PC-9G cells had
significantly enhanced migration (Fig.
1F) and invasion (Fig. 1G)
compared with PC-9 cells without gefitinib treatment. These results
indicated that PC-9G cells were successfully established in
vitro.
RNA-seq of PC-9 and PC-9G cells
To further investigate the molecular mechanism
underlying PC-9 resistance to gefitinib, RNA-seq was performed to
compare PC-9 and PC-9G cells. KEGG pathway enrichment analysis
showed that DEGs between PC-9 and PC-9G cells were enriched in
‘PI3K-AKT signaling pathway’, ‘herpes simplex virus 1 infection’
and ‘MAPK signaling pathway’ (Fig.
2A). In addition, heatmap clustering analysis of PI3K-AKT
signaling pathway-related genes was conducted and the gene that was
the most downregulated in PC-9G cells compared with in PC-9 cells,
GSK3β, was selected for further analysis (Fig. 2B). The phosphorylation and
expression of GSK3β and AKT were assessed in PC-9 and PC-9G cells
following treatment with 300 nmol/l gefitinib (Figs. 2C and S2). The protein expression levels of
pGSK3β and total GSK3β were lower in PC-9G cells than those in PC-9
cells. Following gefitinib treatment, the expression levels of
pGSK3β in PC-9 cells were increased, whereas the phosphorylation of
GSK3β in PC-9G cells was decreased. Furthermore, AKT
phosphorylation in PC-9G cells was markedly decreased compared with
that in PC-9 cells. The expression of c-Myc was higher in PC-9G
cells than that in PC-9 cells, but was decreased by treatment with
gefitinib in both PC-9 and PC-9G cells; notably, c-Myc is known to
positively regulate cell proliferation (28). Furthermore, the expression levels
of E-cadherin were lower in PC-9G cells than those in PC-9 cells,
whereas the expression levels of vimentin were higher in PC-9G
cells than those in PC-9 cells; these proteins are closely related
to metastasis (29). These results
indicated that the EMT of PC-9G cells was increased, which was
consistent with the results of the migration and invasion assays
(Fig. 2C). Immunohistochemistry
results also confirmed that E-cadherin expression was strongly
membrane-positive in PC-9 cells and negative in PC-9G cells. By
contrast, vimentin expression was negative in PC-9 cells and
strongly positive in PC-9G cells (Fig.
2D). These results indicated that GSK3β was downregulated in
gefitinib-resistant cells, which is related to cell proliferation
and metastasis.
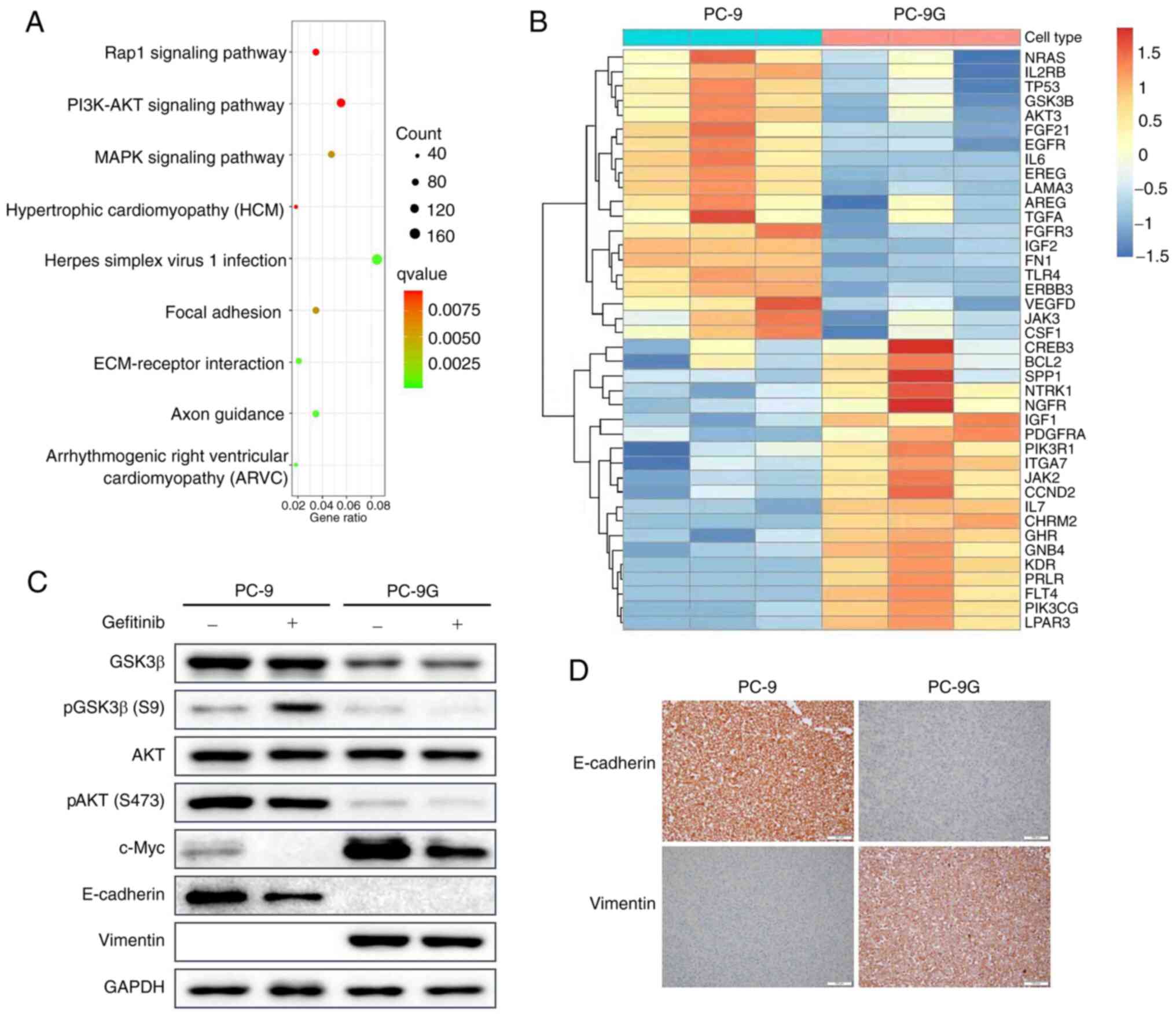 | Figure 2GSK3β is involved in gefitinib
resistance in PC-9G cells. (A) Genes related to the PI3K-AKT
signaling pathway were differentially expressed between PC-9 and
PC-9G cells. (B) Heatmap clustering analysis of differentially
expressed genes between PC-9 and PC-9G cells. (C) Protein
expression levels of GSK3β, pGSK3β (Ser9), AKT, pAKT (Ser473),
c-Myc, E-cadherin and vimentin were detected via western blot
analysis in PC-9 and PC-9G cells treated with DMSO or 300 nmol/l
gefitinib. (D) Immunohistochemical analysis showed that the
expression of E-cadherin in PC-9 cells was higher than that in
PC-9G cells, whereas the expression of vimentin in PC-9 cells was
lower than that in PC-9G cells (magnification, x20). GSK3β,
glycogen synthase kinase 3β; p, phosphorylated; PC-9G,
gefitinib-resistant PC-9. |
Effects of GSK3β overexpression on the
proliferation, apoptosis, migration and invasion of PC-9, PC-9G and
H1975 cells
To determine the function of GSK3β in PC-9G cells,
GSK3β was overexpressed in PC-9, PC-9G and H1975 cells using
lentiviral vectors. Cell fluorescence analysis showed that the
overexpression was successful in PC-9, PC-9G and H1975 cells
(Fig. 3A). The protein and mRNA
expression levels of GSK3β were markedly increased in PC-9, PC-9G
and H1975 cells post-infection (Fig.
3B and C), indicating that
infection with lentiviral vectors stably expressing GSK3β was
successful compared with the empty vector control. In addition, the
expression levels of c-Myc were reduced by GSK3β overexpression in
PC-9, PC-9G and H1975 cells compared with those in the empty vector
control (Figs. 3C and S3). However, the expression of vimentin
was not induced by GSK3β overexpression in PC-9, PC-9G and H1975
cells compared with that in the empty vector control. In addition,
GSK3β overexpression did not induce the expression of E-cadherin in
PC-9 and PC-9G cells, but its overexpression inhibited E-cadherin
expression in H1975 cells compared with that in the empty vector
control. Moreover, cytoplasmic and nuclear β-catenin expression was
reduced by overexpression of GSK3β in PC-9 and H1975 cells, but not
in PC-9G cells, compared with that in the control group (Fig. 3C). Furthermore, overexpression of
GSK3β increased the cell proliferation, invasion and migration of
both PC-9 and H1975 cells compared with that in the empty vector
control group, whereas it suppressed the proliferation, invasion
and migration of PC-9G cells (Fig.
3D-F). Taken together, these results suggested that GSK3β
overexpression exhibited opposite effects between resistant and
parental cell lines.
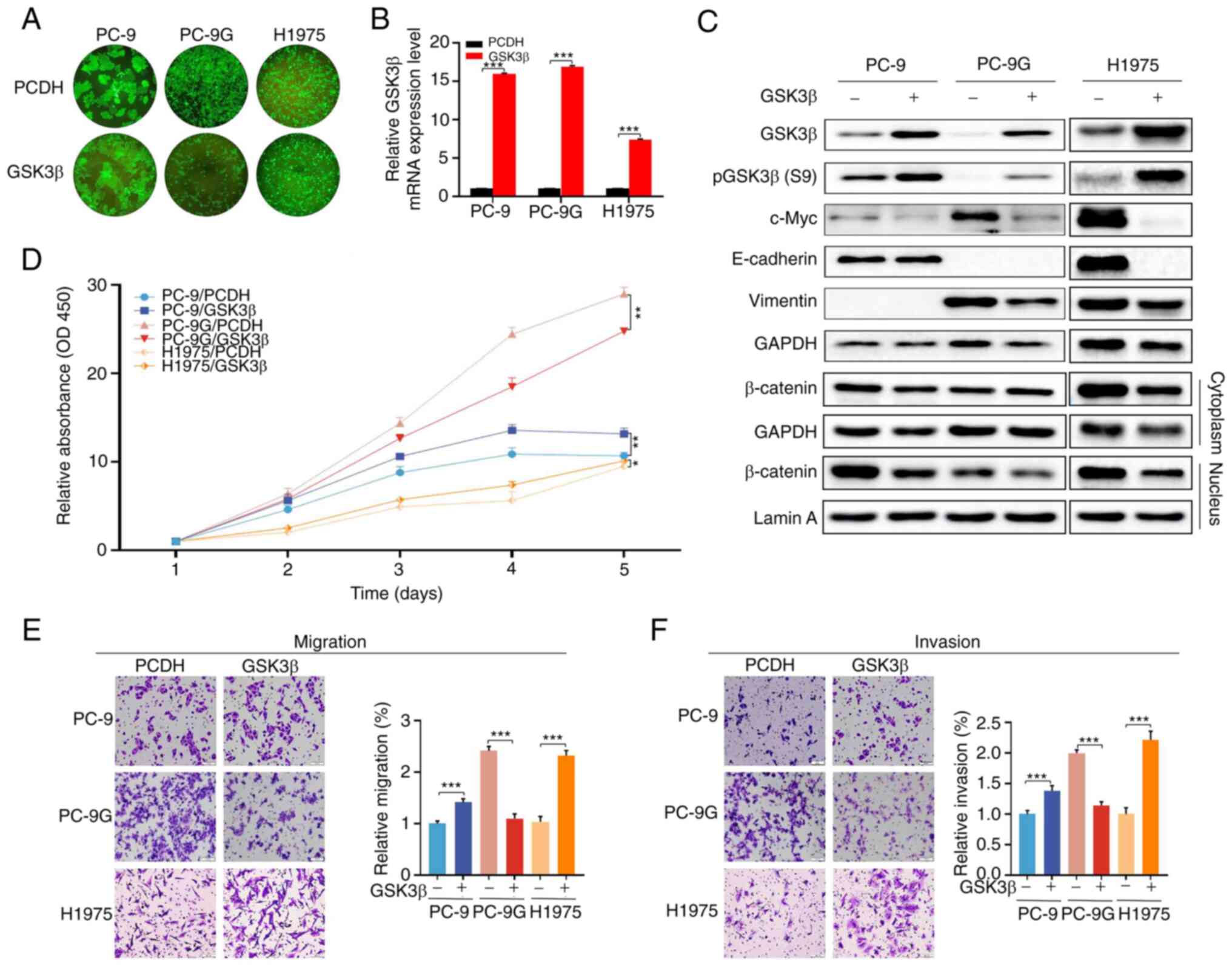 | Figure 3Effects of GSK3β overexpression on
the proliferation, migration and invasion of PC-9, PC-9G and H1975
cells. (A) Fluorescence microscopy showed that stable GSK3β
overexpression was successfully achieved in PC-9, PC-9G and H1975
cells (magnification, x10). (B) mRNA expression levels of GSK3β
were upregulated in PC-9, PC-9G and H1975 cell lines with stable
GSK3β overexpression. (C) Protein expression levels of GSK3β,
pGSK3β (Ser9), c-Myc, E-cadherin, vimentin and β-catenin was
determined via western blot analysis in PC-9, PC-9G and H1975 cells
following overexpression of GSK3β. (D) GSK3β overexpression
enhanced the proliferation of PC-9 and H1975 cells, but not that of
PC-9G cells. GSK3β overexpression reduced the (E) migration and (F)
invasion of PC-9G cells, but enhanced the migration and invasion of
PC-9 and H1975 cells (magnification, x20). Data are presented as
the mean ± SD of three independent experiments.
*P<0.05, **P<0.01,
***P<0.001. GSK3β, glycogen synthase kinase 3β; p,
phosphorylated; PC-9G, gefitinib-resistant PC-9. |
Effects of an AKT inhibitor on the
proliferation, apoptosis, migration and invasion of PC-9, PC-9G and
H1975 cells
To further determine whether GSK3β overexpression
suppressed the proliferation, migration and invasion of PC-9G cells
via the PI3K/AKT signaling pathway, PC-9, PC-9G and H1975 cells
were treated with 5 nM MK2206 (AKT antagonist). Treatment with
MK2206 significantly increased the phosphorylation of GSK3β in
PC-9, PC-9G and H1975 cells compared with that in the control group
(Figs. 4A and S4). In addition, the expression of c-Myc
was decreased by MK2206 treatment in PC-9, PC-9G and H1975 cells.
However, MK2206 treatment did not alter the expression levels of
E-cadherin or vimentin in PC-9, PC-9G and H1975 cells compared with
those in the control group. Moreover, MK2206 treatment increased
the apoptotic rate of PC-9, PC-9G and H1975 cells (Fig. 4B and D). Additionally, MK2206 treatment
inhibited the proliferation of H1975 cells, and it reduced the
proliferation of PC-9 cells on day 5 of treatment (Fig. 4C). Furthermore, treatment with 2206
reduced the migration and invasion of PC-9, PC-9G and H1975 cells
compared with the control group (Fig.
4E and F). These findings
indicated that AKT inhibition could increase the apoptosis, and
reduce the migration and invasion of PC-9, PC-9G and H1975
cells.
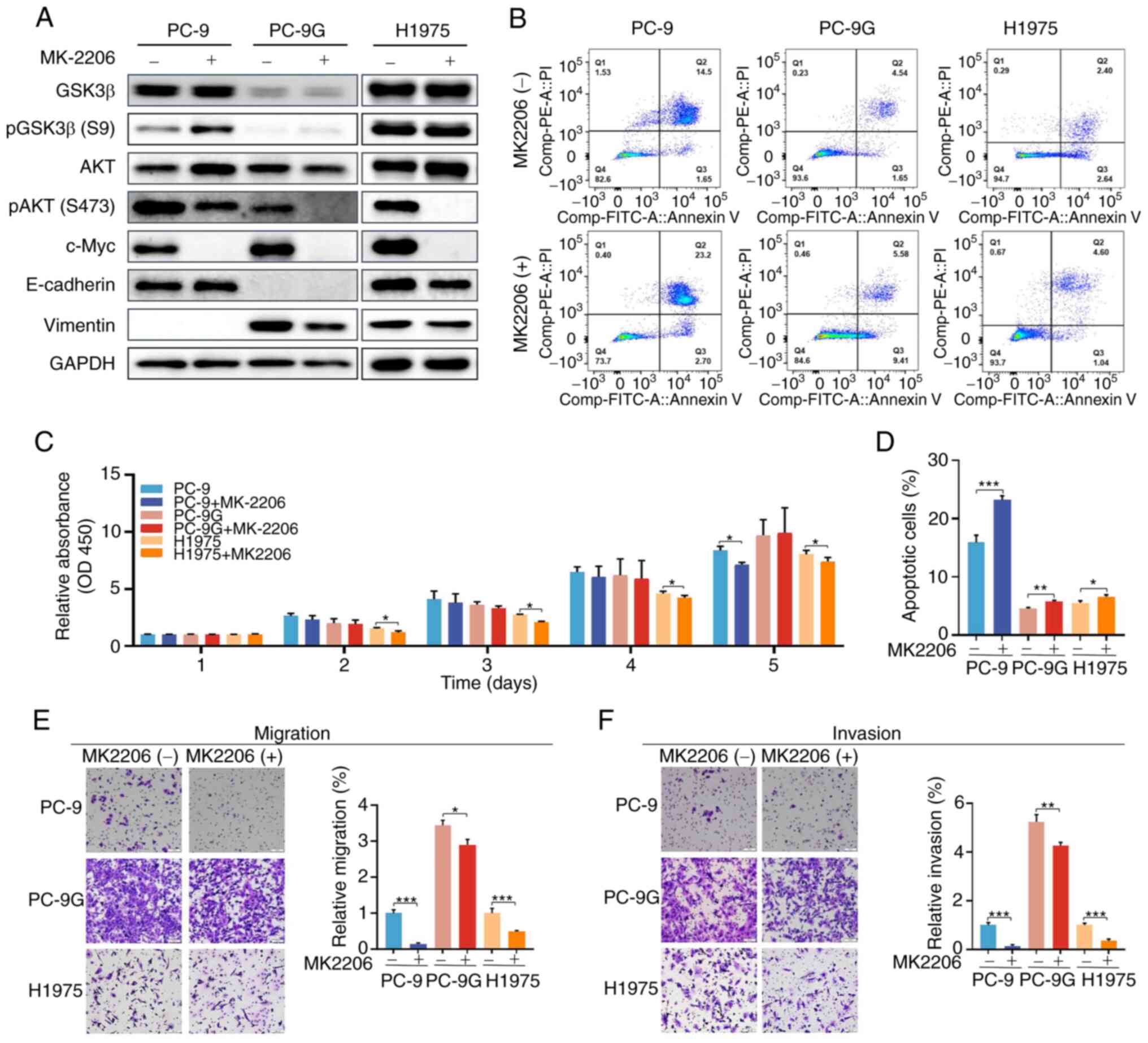 | Figure 4Effects of an AKT inhibitor on the
proliferation, apoptosis, migration and invasion of PC-9, PC-9G and
H1975 cells. (A) Protein expression levels of GSK3β, pGSK3β (Ser9),
AKT, pAKT (Ser473), c-Myc, E-cadherin and vimentin were determined
via western blot analysis in PC-9, PC-9G and H1975 cells treated
with DMSO or 5 nmol/l MK2206. (B) Apoptotic rate of PC-9, PC-9G and
H1975 cells was increased after MK2206 treatment. (C) MK2206
treatment inhibited the proliferation of H1975 cells from day 2 to
5 and reduced the proliferation of PC9 cells at day 5. (D)
Quantification of apoptosis results. MK2206 treatment inhibited the
(E) migration and (F) invasion of PC-9, PC-9G and H1975 cells
(magnification, x20). Data are presented as the mean ± SD of three
independent experiments. *P<0.05,
**P<0.01, ***P<0.001. FITC, fluorescein
isothiocyanate; GSK3β, glycogen synthase kinase 3β; p,
phosphorylated; PC-9G, gefitinib-resistant PC-9; PI, propidium
iodide. |
Effects of an AKT activator on the
proliferation, apoptosis, migration and invasion of PC-9, PC-9G and
H1975 cells
To further examine whether AKT activation could
affect the proliferation, migration and invasion of
gefitinib-resistant cells, PC-9, PC-9G and H1975 cells were treated
with 5 nM SC79 (AKT activator). Phosphorylation of AKT was
increased by treatment with SC79 in PC-9, PC-9G and H1975 cells
(Figs. 5A and S5). However, GSK3β phosphorylation was
not altered by treatment with SC79 in PC-9, PC-9G and H1975 cells
compared with that in the control group. The expression of c-Myc in
PC-9 and H1975 cells was significantly increased following SC79
treatment, whereas that in PC-9G cells was significantly decreased.
In addition, the expression levels of E-cadherin and vimentin were
not altered by SC79 treatment in PC-9, PC-9G and H1975 cells when
compared with those in the control group. Moreover, SC79 treatment
reduced the apoptotic rate in PC-9 cells, but not in PC-9G and
H1975 cells, compared with that in the control group (Fig. 5B and D). The proliferation of H1975 cells
increased from day 3 to 5 after SC79 treatment, whereas the
proliferation of PC-9 cells, but not PC-9G cells, increased on day
5 after SC79 treatment (Fig. 5D).
Furthermore, SC79 treatment increased the migration and invasion of
PC-9, PC-9G and H1975 cells compared with those in the control
group (Fig. 5E and F). Collectively, these results suggested
that AKT activation attenuated the apoptosis of PC-9 cells, and
enhanced the migration and invasion of PC-9G cells, and GSK3β was
not associated with the AKT pathway in PC-9G cells.
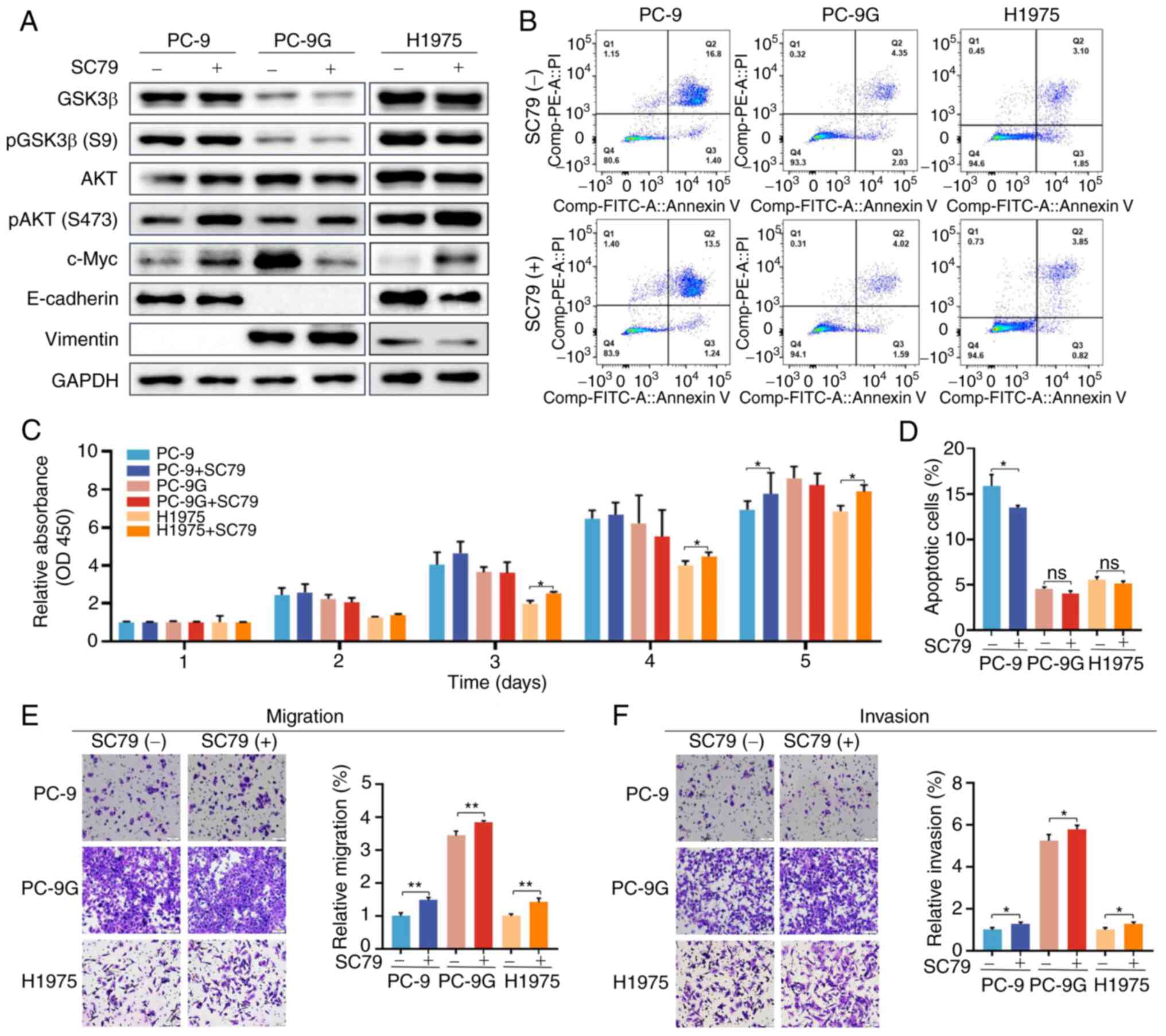 | Figure 5Effects of an AKT activator on the
proliferation, apoptosis, migration and invasion of PC-9, PC-9G and
H1975 cells. (A) Protein expression levels of GSK3β, pGSK3β (Ser9),
AKT, pAKT (Ser473), c-Myc, E-cadherin and vimentin was determined
via western blot analysis in PC-9, PC-9G and H1975 cells treated
with DMSO or 5 nmol/l SC79. (B) Apoptotic rate of PC-9 cells was
decreased after SC79 treatment. (C) SC79 treatment promoted the
proliferation of PC-9 and H1975 cells. (D) Quantification of
apoptosis results. SC79 treatment promoted the (E) migration and
(F) invasion of PC-9, PC-9G and H1975 cells (magnification, x20).
Data are presented as the mean ± SD of three independent
experiments. *P<0.05, **P<0.01. FITC,
fluorescein isothiocyanate; GSK3β, glycogen synthase kinase 3β; p,
phosphorylated; PC-9G, gefitinib-resistant PC-9; PI, propidium
iodide. |
Discussion
In previous studies, PC-9 cells with acquired
resistance to gefitinib exhibited an EGFR mutation (deletion of
exon 19) or generated a second EGFR-T790M mutation (30,31).
However, in the present study, the PC-9G gefitinib-resistant cell
line did not exhibit EGFR mutations, thus indicating that the
mechanisms of acquired resistance to EGFR-TKIs are not always
associated with EGFR mutations and suggesting that different
molecular mechanisms may be involved in the resistance to
gefitinib. Analysis of differentially expressed genes between PC-9
and PC-9G cell lines revealed that GSK3β was downregulated in PC-9G
cells compared with that in PC-9 cells. In addition, overexpression
of GSK3β decreased c-Myc expression in PC-9G cells, which was
associated with cell proliferation, and the expression of the
EMT-associated proteins vimentin was reduced by overexpression of
GSK3β. Furthermore, overexpression of GSK3β suppressed the
proliferation, migration and invasion of PC-9G cell lines, whereas
it promoted the proliferation, migration and invasion of the PC-9
and H1975 cell lines. In general, GSK3β is upregulated as a tumor
promoter in NSCLC cells (17,18).
A previous study demonstrated that silencing GSK3β can
significantly decrease the proliferation of resistant cells
harboring the EGFR T790M and L858R mutations (20). In the present study, GSK3β was
identified as a tumor suppressor in PC-9G cells without EGFR
mutations, suggesting that the role of GSK3β may vary according to
whether EGFR is mutated or not in gefitinib-resistant NSCLC.
The PI3K/AKT pathway serves a key role in cancer
cells that are resistant to EGFR-TKIs (22,32),
and AKT phosphorylates GSK3β at serine 9 (33,34).
In the present study, AKT inhibition induced the apoptosis, and
decreased the migration and invasion of PC-9, PC-9G and H1975
cells. By contrast, AKT activation increased the migration and
invasion of PC-9, PC-9G and H1975 cells, and reduced the apoptosis
of PC-9 cells. However, this pattern of AKT activation was
inconsistent with that of GSK3β in PC-9G cells; thus, the
activation of GSK3β may not be related to the AKT signaling
pathway.
The Wnt/β-catenin pathway has a promotive effect on
NSCLC cell proliferation and metastasis, leading to an increase in
EGFR-TKI resistance in erlotinib-resistant NSCLC HCC827 cells
(35,36). In the present study, cytoplasmic
β-catenin and E-cadherin expression levels were reduced by
overexpression of GSK3β in both PC-9 and H1975 cells. These
findings are consistent with those of a previous study that
reported that activation of the Wnt/β-catenin pathway leads to a
decrease in cytoplasmic β-catenin expression (37), thereby promoting EMT and metastasis
of NSCLC cells (30,38). A recent study demonstrated that
GSK3β promotes gefitinib resistance by activating Wnt/β-catenin in
PC-9 cells (39). However, in the
present study, β-catenin expression was decreased in PC-9G cells
compared with that in PC-9 cells, and GSK3β overexpression was
insufficient to attenuate the already decreased cytoplasmic or
nuclear β-catenin expression in PC-9G cells. It is difficult to
determine whether GSK3β acts through the Wnt/β-catenin pathway in
EGFR-resistant lung cancer without EGFR mutations, thus further
studies are required.
In the present study, overexpression of GSK3β
increased the proliferation, migration and invasion of PC-9 and
H1975 cells. It has previously been reported that a GSK3 antagonist
(CHIR99021), which inhibits both GSK3α and GSK3β, can inhibit the
proliferation of human H1975 and H1299 NSCLC cell lines (40). However, knockdown of GSK3β, but not
GSK3α, has been shown to induce a decrease in the proliferation of
osimertinib-resistant H1975 cells (20), thus indicating that the functions
of GSK3 isoforms are different. These findings indicated that GSK-3
isoforms exhibit important cell type-specific functions, which
require further clarification.
A limitation of the present study is that it did not
access the function of GSK3β in a gefitinib-resistant PC-9G
xenograft model for in vivo functional validation. In
vivo experiments are necessary steps for investigating the
potential of a therapeutic strategy prior to clinical trials. In
future, in vivo experiments should be performed to explore
the suppressive role of GSK3β in gefitinib-resistant NSCLC.
In conclusion, the present study demonstrated that
overexpression of GSK3β increased the proliferation, migration and
invasion of gefitinib-resistant H1975 cells harboring the T790M and
L858R mutations. Conversely, GSK3β overexpression reduced the
proliferation, migration and invasion of PC-9G cells without EGFR
mutations, suggesting that GSK3β serves a dynamic tumor-promoting
or tumor-suppressive role in gefitinib-resistant lung
adenocarcinoma cells depending on the type of EGFR mutation.
Additionally, AKT inhibition induced the apoptosis, and reduced the
migration and invasion of PC-9, PC-9G and H1975 cells, and these
effects were reversed following AKT activation; the function of
GSK3β was not dependent on the tumor promotor role of the AKT
pathway in PC-9G cells without EGFR mutations. Notably, the
molecular mechanisms underlying the diverse effects of GSK3β among
different EGFR mutation types in NSCLC remain to be elucidated, and
the properties of GSK3β need to be carefully evaluated when
developing a therapeutic strategy for EGFR-mutant NSCLC that
targets GSK3β.
Supplementary Material
Analysis of mutations in PC-9, PC-9G
and H1975 cells. (A) The exon 19 deletion mutation was detected in
the parental PC-9 cell line, but no EGFR mutations were detected in
PC-9G cells. In addition, the EGFR T790M and L858R mutations were
detected in H1975 cells. (B) MET amplification or MET exon 14
mutations were not detected in PC-9G cells. EGFR, epidermal growth
factor receptor. p, phosphorylated; PC-9G, gefitinib-resistant
PC-9; Ct, cycle threshold; FAM, 5/6-carboxyfluorescein; HEX,
6-chloro-6 methylfluorescein.
Protein expression and phosphorylation
of GSK3β and its upstream protein AKT in PC-9 and PC-9G cells
following DMSO or 300 nM gefitinib treatment. (A) Raw western blot
analysis data for Fig. 2.
Semi-quantified immunoblots of (B) GSK3β, (C) pGSK3β, (D) pAKT, (E)
c-Myc, (F) E-cadherin and (G) vimentin, normalized against their
corresponding protein level or GAPDH signals.
*P<0.05, **P<0.01 and
***P<0.001. GSK3β, glycogen synthase kinase 3β; p,
phosphorylated; PC-9G, gefitinib-resistant PC-9.
Effect of GSK3β overexpression on the
protein expression and phosphorylation of GSK3β and its downstream
proteins in PC-9, PC-9G and H1975 cells. (A) Raw western blot
analysis data for Fig. 3.
Semi-quantified immunoblots of (B) GSK3β, (C) pGSK3β, (D) c-Myc,
(E) E-cadherin, (F) vimentin, and (G) cytoplasmic and (H) nuclear
β-catenin, normalized against their corresponding protein level,
GAPDH or Lamin A signals. *P<0.05,
**P<0.01, ***P<0.001. GSK3β, glycogen
synthase kinase 3β; p, phosphorylated; PC-9G, gefitinib-resistant
PC-9.
Protein expression and phosphorylation
of GSK3β and its upstream protein AKT in PC-9, PC-9G and H1975
cells following the treatment with an AKT inhibitor. (A) Raw
western blot analysis data for Fig.
4. Semi-quantified immunoblots of (B) GSK3β, (C) pGSK3β, (D)
pAKT, (E) c-Myc, (F) E-cadherin and (G) vimentin, normalized
against their corresponding protein level or GAPDH signals.
*P<0.05, **P<0.01,
***P<0.001. GSK3β, glycogen synthase kinase 3β; p,
phosphorylated; PC-9G, gefitinib-resistant PC-9.
Protein expression and phosphorylation
of GSK3β and its upstream protein AKT in PC-9, PC-9G and H1975
cells following the treatment with an AKT activator. (A) Raw
western blot analysis data for Fig.
5. Semi-quantified immunoblots of (B) GSK3β, (C) pGSK3β, (D)
pAKT, (E) c-Myc, (F) E-cadherin and (G) vimentin, normalized
against their corresponding protein level or GAPDH signals.
*P<0.05, **P<0.01,
***P<0.001. GSK3β, glycogen synthase kinase 3β; p,
phosphorylated; PC-9G, gefitinib-resistant PC-9.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 8200103816
and 81860414), and support was also provided by Hainan Cancer
Hospital (grant no. 2022BS05).
Availability of data and materials
The RNA-seq data are available in the Figshare
repository (https://doi.org/10.6084/m9.figshare.23575992). The
other datasets used and/or analyzed during the current study are
available from the corresponding author on reasonable request.
Author's contributions
JL, SX and XX conceptualized and designed the study.
XW and XJ acquired the data and drafted the manuscript. XJ and CH
performed data analysis. JL, XW, XJ, SX and XX wrote the
manuscript. SX and XX revised the manuscript. JL, SX and XX confirm
the authenticity of all the raw data. All authors read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Osmani L, Askin F, Gabrielson E and Li QK:
Current WHO guidelines and the critical role of immunohistochemical
markers in the subclassification of non-small cell lung carcinoma
(NSCLC): Moving from targeted therapy to immunotherapy. Semin
Cancer Biol. 52:103–109. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Lynch TJ, Bell DW, Sordella R,
Gurubhagavatula S, Okimoto RA, Brannigan BW, Harris PL, Haserlat
SM, Supko JG, Haluska FG, et al: Activating mutations in the
epidermal growth factor receptor underlying responsiveness of
non-small-cell lung cancer to gefitinib. N Engl J Med.
350:2129–2139. 2004.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: Correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500.
2004.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zhou C, Wu YL, Chen G, Feng J, Liu XQ,
Wang C, Zhang S, Wang J, Zhou S, Ren S, et al: Erlotinib versus
chemotherapy as first-line treatment for patients with advanced
EGFR mutation-positive non-small-cell lung cancer (OPTIMAL,
CTONG-0802): A multicentre, open-label, randomised, phase 3 study.
Lancet Oncol. 12:735–742. 2011.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Mitsudomi T, Morita S, Yatabe Y, Negoro S,
Okamoto I, Tsurutani J, Seto T, Satouchi M, Tada H, Hirashima T, et
al: Gefitinib versus cisplatin plus docetaxel in patients with
non-small-cell lung cancer harbouring mutations of the epidermal
growth factor receptor (WJTOG3405): An open label, randomised phase
3 trial. Lancet Oncol. 11:121–128. 2010.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Riely GJ, Politi KA, Miller VA and Pao W:
Update on epidermal growth factor receptor mutations in non-small
cell lung cancer. Clin Cancer Res. 12:7232–7241. 2006.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kobayashi S, Boggon TJ, Dayaram T, Jänne
PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG and Halmos
B: EGFR mutation and resistance of non-small-cell lung cancer to
gefitinib. N Engl J Med. 352:786–792. 2005.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Pao W, Miller VA, Politi KA, Riely GJ,
Somwar R, Zakowski MF, Kris MG and Varmus H: Acquired resistance of
lung adenocarcinomas to gefitinib or erlotinib is associated with a
second mutation in the EGFR kinase domain. PLoS Med.
2(e73)2005.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Adams E, Sepich-Poore GD,
Miller-Montgomery S and Knight R: Using all our genomes:
Blood-based liquid biopsies for the early detection of cancer. View
(Beijing). 3(20200118)2022.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Engelman JA, Zejnullahu K, Mitsudomi T,
Song Y, Hyland C, Park JO, Lindeman N, Gale CM, Zhao X, Christensen
J, et al: MET amplification leads to gefitinib resistance in lung
cancer by activating ERBB3 signaling. Science. 316:1039–1043.
2007.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Thomson S, Petti F, Sujka-Kwok I, Epstein
D and Haley JD: Kinase switching in mesenchymal-like non-small cell
lung cancer lines contributes to EGFR inhibitor resistance through
pathway redundancy. Clin Exp Metastasis. 25:843–854.
2008.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Sequist LV, Waltman BA, Dias-Santagata D,
Digumarthy S, Turke AB, Fidias P, Bergethon K, Shaw AT, Gettinger
S, Cosper AK, et al: Genotypic and histological evolution of lung
cancers acquiring resistance to EGFR inhibitors. Sci Transl Med.
3(75ra26)2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yano S, Wang W, Li Q, Matsumoto K,
Sakurama H, Nakamura T, Ogino H, Kakiuchi S, Hanibuchi M, Nishioka
Y, et al: Hepatocyte growth factor induces gefitinib resistance of
lung adenocarcinoma with epidermal growth factor
receptor-activating mutations. Cancer Res. 68:9479–9487.
2008.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Oxnard GR, Arcila ME, Chmielecki J,
Ladanyi M, Miller VA and Pao W: New strategies in overcoming
acquired resistance to epidermal growth factor receptor tyrosine
kinase inhibitors in lung cancer. Clin Cancer Res. 17:5530–5537.
2011.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Zheng X, Huang D, Liu X, Liu QY, Gao X and
Liu L: GSK3β/ITCH/c-FLIP axis counteracts TRAIL-induced apoptosis
in human lung adenocarcinoma cells. Protein Pept Lett. 30:242–249.
2023.PubMed/NCBI View Article : Google Scholar
|
|
16
|
He L, Endress J, Cho S, Li Z, Zheng Y,
Asara JM and Blenis J: Suppression of nuclear GSK3 signaling
promotes serine/one-carbon metabolism and confers metabolic
vulnerability in lung cancer cells. Sci Adv.
8(eabm8786)2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Alves M, Borges DP, Kimberly A, Neto FM,
Oliveira AC, de Sousa JC, Nogueira CD, Carneiro BA and Tavora F:
Glycogen synthase kinase-3 beta expression correlates with worse
overall survival in non-small cell lung cancer-A
clinicopathological series. Front Oncol. 11(621050)2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Zeng J, Liu D, Qiu Z, Huang Y, Chen B,
Wang L, Xu H, Huang N, Liu L and Li W: GSK3β overexpression
indicates poor prognosis and its inhibition reduces cell
proliferation and survival of non-small cell lung cancer cells.
PLoS One. 9(e91231)2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Vincent EE, Elder DJ, O'Flaherty L, Pardo
OE, Dzien P, Phillips L, Morgan C, Pawade J, May MT, Sohail M, et
al: Glycogen synthase kinase 3 protein kinase activity is
frequently elevated in human non-small cell lung carcinoma and
supports tumour cell proliferation. PLoS One.
9(e114725)2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Fukuda K, Takeuchi S, Arai S, Kita K,
Tanimoto A, Nishiyama A and Yano S: Glycogen synthase kinase-3
inhibition overcomes epithelial-mesenchymal transition-associated
resistance to osimertinib in EGFR-mutant lung cancer. Cancer Sci.
111:2374–2384. 2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Nagini S, Sophia J and Mishra R: Glycogen
synthase kinases: Moonlighting proteins with theranostic potential
in cancer. Semin Cancer Biol. 56:25–36. 2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Nakata A and Gotoh N: Recent understanding
of the molecular mechanisms for the efficacy and resistance of EGF
receptor-specific tyrosine kinase inhibitors in non-small cell lung
cancer. Expert Opin Ther Targets. 16:771–781. 2012.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Deng QF, Su BO, Zhao YM, Tang L, Zhang J
and Zhou CC: Integrin β1-mediated acquired gefitinib resistance in
non-small cell lung cancer cells occurs via the phosphoinositide
3-kinase-dependent pathway. Oncol Lett. 11:535–542. 2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Wang W, Xia X, Chen K, Chen M, Meng Y, Lv
D and Yang H: Reduced PHLPP expression leads to EGFR-TKI resistance
in lung cancer by activating PI3K-AKT and MAPK-ERK dual signaling.
Front Oncol. 11(665045)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wu SG and Shih JY: Management of acquired
resistance to EGFR TKI-targeted therapy in advanced non-small cell
lung cancer. Mol Cancer. 17(38)2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Carlson M: org.Hs.eg.db: Genome wide
annotation for human. 2019. R package version 3.10.0. https://bioconductor.org/packages/release/data/annotation/html/org.Hs.eg.db.html.
2020.
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Schuhmacher M, Staege MS, Pajic A, Polack
A, Weidle UH, Bornkamm GW, Eick D and Kohlhuber F: Control of cell
growth by c-Myc in the absence of cell division. Curr Biol.
9:1255–1258. 1999.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Tsoukalas N, Aravantinou-Fatorou E, Tolia
M, Giaginis C, Galanopoulos M, Kiakou M, Kostakis ID, Dana E,
Vamvakaris I, Korogiannos A, et al: Epithelial-mesenchymal
transition in non small-cell lung cancer. Anticancer Res.
37:1773–1778. 2017.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Song YA, Ma T, Zhang XY, Cheng XS,
Olajuyin AM, Sun ZF and Zhang XJ: Apatinib preferentially inhibits
PC9 gefitinib-resistant cancer cells by inducing cell cycle arrest
and inhibiting VEGFR signaling pathway. Cancer Cell Int.
19(117)2019.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zhu Y, He W, Gao X, Li B, Mei C, Xu R and
Chen H: Resveratrol overcomes gefitinib resistance by increasing
the intracellular gefitinib concentration and triggering apoptosis,
autophagy and senescence in PC9/G NSCLC cells. Sci Rep.
5(17730)2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Sordella R, Bell DW, Haber DA and
Settleman J: Gefitinib-sensitizing EGFR mutations in lung cancer
activate anti-apoptotic pathways. Science. 305:1163–1167.
2004.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Dajani R, Fraser E, Roe SM, Young N, Good
V, Dale TC and Pearl LH: Crystal structure of glycogen synthase
kinase 3 beta: Structural basis for phosphate-primed substrate
specificity and autoinhibition. Cell. 105:721–732. 2001.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Jope RS and Johnson GV: The glamour and
gloom of glycogen synthase kinase-3. Trends Biochem Sci. 29:95–102.
2004.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Feng S, Liu H, Du P, Dong X, Pang Q and
Guo H: Long non-coding RNA AC122108.1 promotes lung adenocarcinoma
brain metastasis and progression through the Wnt/β-catenin pathway
by directly binding to aldolase A. Ann Transl Med.
9(1729)2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Wang Q, Liao J, He Z, Su Y, Lin D, Xu L,
Xu H and Lin J: LHX6 affects erlotinib resistance and migration of
EGFR-mutant non-small-cell lung cancer HCC827 cells through
suppressing Wnt/β-catenin signaling. Onco Targets Ther.
13:10983–10994. 2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Shang S, Hua F and Hu ZW: The regulation
of β-catenin activity and function in cancer: Therapeutic
opportunities. Oncotarget. 8:33972–33989. 2017.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Roy LD, Sahraei M, Subramani DB, Besmer D,
Nath S, Tinder TL, Bajaj E, Shanmugam K, Lee YY, Hwang SI, et al:
MUC1 enhances invasiveness of pancreatic cancer cells by inducing
epithelial to mesenchymal transition. Oncogene. 30:1449–1459.
2011.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Huang JQ, Duan LX, Liu QY, Li HF, Hu AP,
Song JW, Lin C, Huang B, Yao D, Peng B, et al: Serine-arginine
protein kinase 1 (SRPK1) promotes EGFR-TKI resistance by enhancing
GSK3β Ser9 autophosphorylation independent of its kinase activity
in non-small-cell lung cancer. Oncogene. 42:1233–1246.
2023.PubMed/NCBI View Article : Google Scholar
|
|
40
|
O'Flaherty L, Shnyder SD, Cooper PA, Cross
SJ, Wakefield JG, Pardo OE, Seckl MJ and Tavaré JM: Tumor growth
suppression using a combination of taxol-based therapy and GSK3
inhibition in non-small cell lung cancer. PLoS One.
14(e0214610)2019.PubMed/NCBI View Article : Google Scholar
|