Introduction
Dystrophic epidermolysis bullosa pruriginosa
(DEB-Pr) is a rare clinical subtype of inherited DEB and there is
currently no specific treatment (1). All DEB-Pr variants reported so far
are caused by mutations in the collagen type VII (COL7) gene, whose
encoded protein is a major component of the anchoring fibrils in
the dermo-epidermal junction. In addition, DEB-Pr shows genetic
heterogeneity, as autosomal dominant [dominant DEB (DDEB)] and
recessive [recessive DEB (RDEB)] inheritance patterns have been
previously reported and current studies summarized that dominant
DEB tends to have heterozygous glycine substitutions and recessive
DEB have nonsense, frameshift or splice site mutations on both
COL7, α1 (COL7A1) alleles (2). To
date, >60 variants have been reported to be associated with
DEB-Pr and glycine substitutions are the most common variants
(3). Although all forms of DEB
result from COL7A1 mutations, there is phenotypic variability and
both intra- and inter-family variability have been reported
(4-8).
Compared to skin microscopy and classical Sanger
sequencing, next-generation sequencing-based whole-exome sequencing
(WES) is a more accurate and sensitive technique and has shown
promise in improving the diagnosis of DEB, particularly in the
differential diagnosis from other types of EB and other genetic
diseases (9-13).
The existence of cell-free DNA (cfDNA) in human blood was first
reported in 1948(14). CfDNA and
cell-free fetal DNA (cffDNA) are always present in plasma and there
has been growing interest in prenatal diagnostics, as well as
cancer diagnosis and surveillance (15,16).
However, there are limited studies using cfDNA for DEB-Pr
diagnosis.
Molecular investigation is extremely important for
the comprehensive diagnosis of EB subtypes, disease prognosis,
genetic counseling, and patient management. In the present study,
WES technology was applied in a Chinese family with a
four-generation pedigree of DDEB, and a heterozygous variation was
identified in the COL7A1 gene. Using cfDNA as a template, it was
possible to confirm the disease-causing variant by Sanger
sequencing. To further understand the prevalence of the variants
identified to date and provide an overview of DEB-Pr-causing
variants, we summarized all variants associated with DEB-Pr
reported.
Patients and methods
Ethical approval
This research project was conducted after receiving
approval from the Institutional Review Board (IRB) at the
Dermatology Institute of Jiangxi Province, The Affiliated
Dermatology Hospital of Nanchang University (Nanchang, China), and
is in accordance with the Helsinki Declaration. All participants,
including their parents or legal guardians, provided written
informed consent. Use of the clinical picture of the proband was
not permitted.
Patients
A total of 46 individuals from a DEB-Pr family were
recruited for a comprehensive questionnaire and physical
examination in this study at the Dermatology Institute of Jiangxi
Province, The Affiliated Dermatology Hospital of Nanchang
University between July 2020 and March 2021. Among them, 18
individuals donated their blood, 12 individuals agreed to the use
of their samples for WES and all donors agreed to the use of their
genomic material for COL7A1 PCR amplification. In addition, in
order to have non-DEB controls, blood was also collected from 50
healthy controls who visited hospitals for annual physical
examination and had a confirmed lack of DEB-associated genetic
disease in the family history (median age 45.5 years; age range,
26-76 years; 32% women), and blood from one 32-year-old pregnant
woman at week 37 who also presented as healthy and with a lack of
DEB-related disease was collected in order to test whether cffDNA
could be used for COL7A1 PCR amplification.
WES
The DNA was extracted by DNA isolation kit (cat. no.
DC111; Vazyme Biotech Co., Ltd.) and DNA was measured by
Nanodrop2000 (Thermo Fisher Scientific, Inc.). Genomic DNA (1 µg)
was sheared to 150-250 bp using a Covaris LE220 instrument with the
following parameters: Duty cycle, 20%; intensity, 5; cycles per
burst, 200; time, 90 sec; and shearing tubes, Crimp-Cap microtubes
with AFA fibers (Covaris Inc.). The DNA fragments were then
selected by VAHTSTM DNA Clean Beads (cat. no. N411; Vazyme Biotech
Co., Ltd.). The library was constructed using the MGIEasy Exome FS
Library Prep Set kit v2.0 (cat. no. 1000009658; MGI Tech Co., Ltd.)
following the manufacturer's instructions. Briefly, the end-repair
for DNA fragments was performed by adding an ‘A’ nucleotide to the
3' end of each strand. Adapters were then ligated to both ends of
the end-repaired/dA-tailed DNA fragments for amplification and
sequencing. Size-selected DNA fragments were amplified by
ligation-mediated PCR, purified and hybridized to the exome array
for enrichment. Non-hybridized fragments were then washed out.
Finally, the captured products were then circularized by MGIEasy
circularization commercial kit (cat. no. 1000005259; MGI Tech Co.,
Ltd.). Rolling circle amplification was performed to produce DNA
Nanoballs. The library was sequenced on the BGISEQ-500 sequencing
platform (MGI Tech Co., Ltd.) and the overall quality control data
is presented in Table SI. The raw
sequence data reported in this paper were deposited in the National
Institute of Biotechnology Information (NCBI) Sequence Read Archive
database (accession no. SAMN36949954-SAMN36949963) and are
accessible through https://www.ncbi.nlm.nih.gov/sra/PRJNA1000907.
Variant calling
The reads were mapped to the hg19/GRCh37 human
reference genome by the BWA.v0.7.12 tool and Picard was used to
mark the duplicates. A Genome Analysis Toolkit v 3.7 and Samtools
(http://www.htslib.org/) were used to detect
single nucleotide variants and short insertions/deletions (Indels).
Short Indels in the repeat regions and within the 10 bp range from
the start and end of the read were also excluded. The remaining
variants were filtered from public databases comprising 1000
Genomes (ftp://ftp-trace.ncbi.nih.gov/1000genomes/ftp/release) and
gnomAD (The Genome Aggregation Database; https://gnomad.broadinstitute.org/).
Variant annotation and prediction
The variants were annotated with the ANNOVAR program
(https://annovar.openbioinformatics.org/en/latest/).
The in-silico analysis was performed by Sorting Intolerant
From Tolerant (SIFT) (https://sift.bii.a-star.edu.sg/), Polymorphism
Phenotyping v2 (PolyPhen2) (http://genetics.bwh.harvard.edu/pph2/), mutation
assessor (http://mutationassessor.org/r3/), mutation taster
(https://www.mutationtaster.org/), FATHMM
(http://fathmm.biocompute.org.uk/) and
CADD (https://cadd.gs.washington.edu/score) to anticipate
the functional effect of missense and nonsense variants. Mega
version 11 (11.0.13), Jalview Version 2 (2.11.2.7) and BioEdit
software (v7.2) were used to align the sequences against the COL7A1
reference sequence (accession no. NC_000003).
ELISA
ELISAs for desmogelin (Dsg)1 (cat. no. 7880E), Dsg3
(cat. no. 7885E), BP180 (cat. no. 7695E), BP230 (cat. no. 7613E)
and Type VII collagen (cat. no. 7845R2) were performed using
commercial kits purchased from Medical and Biological laboratories,
Co., Ltd., and the procedures were performed according to the
manufacturer's instructions.
Immunoblotting
For immunoblotting, recombinant proteins including
LM332 (cat. no. LN332-0502, Biolamina AB), LM111 (cat. no.
LN111-0501, Biolamina AB) and integrin α6β4 (cat. no. 5497-A6-050;
BD Biosciences) were used for the antigen source (100 ng per
reaction), and final blots were incubated with patients' sera at
1:5-1:800 dilution. The detailed method was performed as previously
described (17-19).
Indirect immunofluorescence (IF)
Indirect IF studies of normal human skin and 1M
NaCl-split normal human skin were performed as previously described
(19).
CfDNA and cffDNA extraction
Both cfDNA and cffDNA were extracted from plasma
using a QIAamp DNA Blood Mini extraction kit (Qiagen GmbH)
according to the manufacturer's instructions. The concentration of
the DNA was measured by a Nanodrop 2000 (Thermo Fisher Scientific,
Inc.) and the quality of the isolated DNA was checked by 1% agarose
gel electrophoresis.
PCR and Sanger sequencing
The candidate variants were validated by direct
Sanger sequencing. Primers for PCR and sequencing were provided by
Tsingke Biological Technology. For PCR amplification, 10 ng of
total genomic DNA was used as a template in 20 µl of the reaction
mixture using Vazyme Taq DNA polymerase as previously described
(20). The primers used in this
study and listed in Table SII.
The thermocycling conditions were as follows: 95˚C for 3 min,
followed by 35 cycles of 95˚C for 15 sec, 61˚C for 15 sec and 72˚C
for 30 sec, and a final extension at 72˚C for 3 min. The PCR
products were checked by DNA electrophoresis and sent to Tsignke
Biological Technology for sequencing. The COL7A1 consensus sequence
generated from sanger sequencing results from 50 healthy controls,
the reference sequence from NC_000003 and the sequences from
affected individuals were also aligned, as shown in Fig. S1.
Software predication
Three-dimensional structure prediction of selected
proteins was performed using I-TASSER (https://zhanggroup.org/I-TASSER/) (21). In details, the complete gene
sequence of COL7A1 was retrieved from the NCBI GenBank (https://www.ncbi.nlm.nih.gov/gene/1294)
under accession no. NC_000003. The wild-type protein of COL7A1 with
an exonic region of 70 to 118 was used for 3D structure prediction.
The C-score=-1.33, TM-score=0.55±0.15 and root-mean-square
deviation (RMSD)=12.2±4.4 Å were selected, where C-score is a
confidence score for estimating the quality of predicted models by
I-TASSER and TM-score is a metric for assessing the topological
similarity of protein structures; it is designed to solve two major
problems in traditional metrics such as RMSD.
Literature review
A review of the literature was performed using
PubMed and Google search with the search terms ‘epidermolysis
bullosa pruriginosa’, ‘DEB pruriginosa’, ‘EB pruriginosa’ and
‘COL7A1’ covering the period of 1994 to November 2022, and the
systematic reviews were limited to studies published in
English.
Results
Case presentation
A 50-year-old female proband presented to the
Dermatology Hospital of Jiangxi Province (Nanchang, China) with
complaints of itching, skin nodules and blackish discoloration of
the skin on both lower limbs for >32 years. Upon physical
examination, depigmentation at the center with scarring was seen
over the larger nodules and multiple lichenified papules to nodules
were present over the lower limbs extending from the knee to the
ankle joint. Routine investigations, including chest X-ray,
computed tomography, electrocardiogram, upper and lower
gastrointestinal endoscopy, complete blood count, liver and renal
function tests, as well as serum IgE, ferritin and thyroid function
tests, all showed normal results. It was further confirmed that
direct immunofluorescence using a biopsy of lesioned skin of the
left leg showed no IgG, IgA, IgM and complement C3 deposition at
the basement membrane zone or on keratinocyte cell surfaces. This
patient's serum showed negative IgG, IgA and IgM results on
indirect immunofluorescence using normal human skin and indirect
immunofluorescence using salt-split skin (22,23).
According to ELISAs using commercially available kits (Medical and
Biological Laboratories, Co., Ltd.), IgG autoantibodies against
Dsg1, Dsg3, BP180, BP230 and COL7 were all negative. Other
diagnostic tests, including immunoblotting of normal human
epidermal extract, dermal extract and recombinant proteins of
laminin 332, laminin 111 and integrin α6β4 (24-26),
which are routinely used in our laboratory for the detection of
known autoimmune bullous disease (AIBD) autoantibodies (18,27),
showed negative results for this patient's serum. Based on all of
these clinical and laboratory data, the diagnosis of AIBD was ruled
out for this patient.
Identification of disease-causing
variant by WES
The proband and her family under investigation
comprised 46 individuals and a questionnaire revealed the absence
of consanguineous marriage. Among the first three generations, 14
individuals had similar mild clinical disease phenotypes, as
indicated in the pedigree chart (Fig.
1A). Based on the clinical examinations and questionnaires
completed for the proband's family, the diagnosis of DEB-Pr was
suspected.
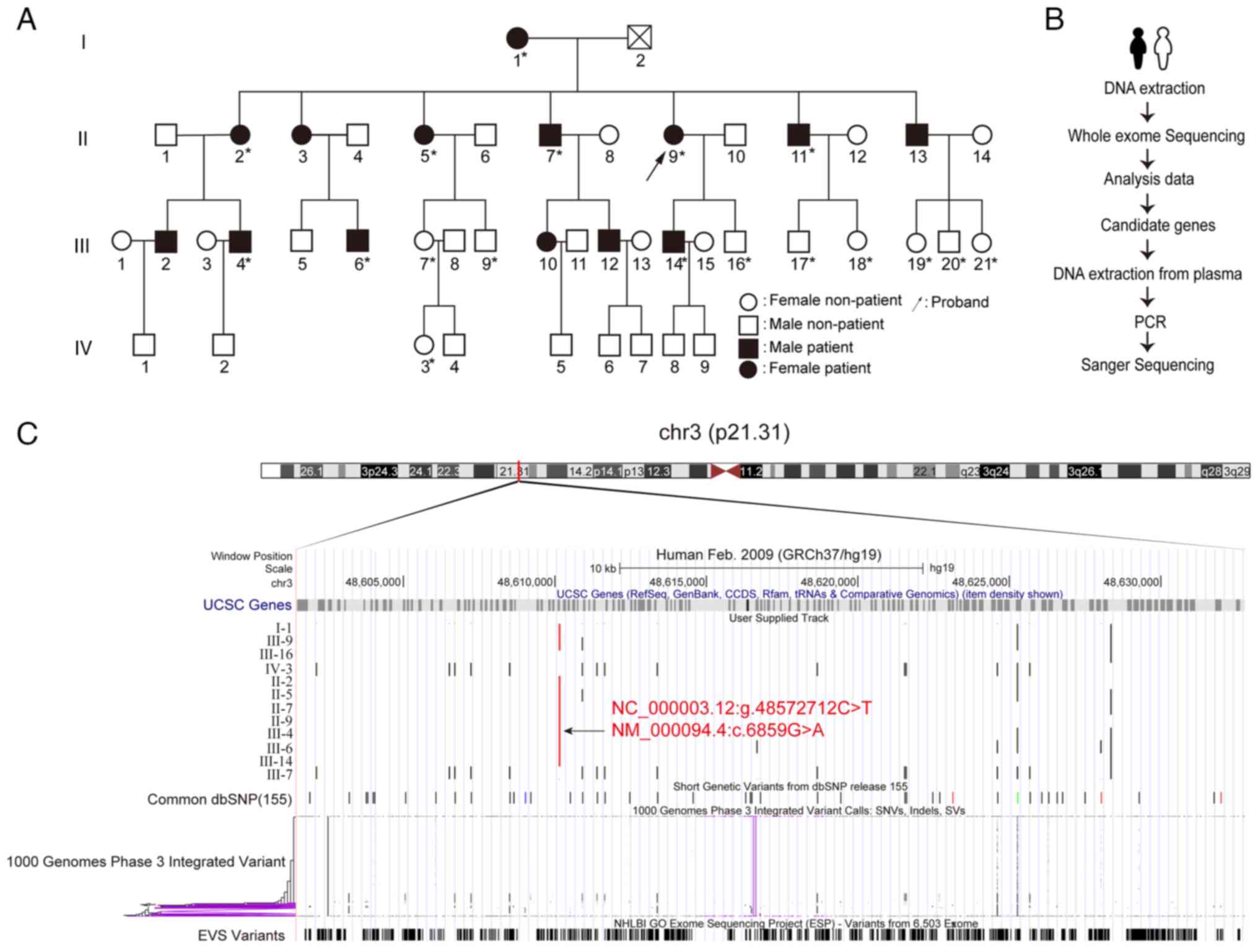 | Figure 1Molecular diagnosis of DEB-Pr. (A)
Pedigree of the DEB-Pr family spanning 4 generations. The squares
indicate male and circles female individuals; black filling denotes
affected individuals; the slash indicates a deceased individual;
the arrow points out the proband; asterisks indicate that the
sample was used for sanger sequencing. (B) Overview of the
experimental design. (C) Graphic view of COL7A1 generated by the
UCSC genome browser. A total of 12 individual whole-exome
sequencing results were aligned and compared with common dbSNP,
1000 genomes variant and the EVS variants datasets. The candidate
Chr3:48610145 C>T position in the genome was marked in red.
COL7A1, collagen type VII, α1; DEB-Pr, dystrophic epidermolysis
bullosa pruriginosa; EVS, Exome Variant Server; UCSC, University of
California Santa Cruz; SNV, single nucleotide variant; dbSNP,
single nucleotide polymorphism database; Chr, chromosome; Indel,
insertion and deletion; SV, structural variation. |
To thoroughly investigate the potential genetic
basis of DEB-Pr, WES of 12 individuals from the proband's family
was performed, including the proband, affected individuals and
unaffected individuals (Fig. 1B
and C). Analysis of the WES data
led to the identification of a G-to-A transition at the first base
of codon 6,859 in exon 87 of the COL7A1 gene, resulting in the
substitution of Gly (GGA) with Arg (AGA) (Fig. 1C). This variant (c.6859 G>A) in
the COL7A1 gene has been reported in the DEB-Pr by Japanese studies
(28-30).
To predict the potential impact of the variant on the protein
functions, different bioinformatic analysis tools were used and
returned with predicted values, including the SIFT (value: 0.0),
PolyPhen2 (value: 0.998), mutation assessor (value: 4.32), mutation
taster (value: 0.999), FATHMM (value: -6.29) and CADD (value:
6.70). Overall, the variant was predicted to be ‘deleterious’ with
high confidence, suggesting that COL7A1 mutation is the genetic
basis of DEB-Pr.
Pathogenic variant validation using
cfDNA
To confirm the results of the WES analysis and
explore the potential use of cfDNA for DEB-Pr diagnosis, target DNA
amplification and Sanger sequencing were next conducted. The
dominant peaks observed in the cfDNA samples were ~166 and 300 bp,
which aligns with the size range reported in previous studies
(31,32) (Fig.
2A). The targeted region in exon 87 of COL7A1 was amplified and
Sanger sequencing was performed in patients and 50 ethnically
matched healthy controls (Fig.
2B-D). The results showed that the affected individuals carried
AGA, whereas the unaffected individuals and healthy controls were
GGA at this position, further supporting the association between
genotype and phenotype. Of note, one affected individual (III-9)
carried heterozygous variants according to both WES (Fig. 1C) and Sanger sequencing (Fig. 2D). However, clinical examination
indicated that this individual did not show any symptoms of DEB-Pr,
which is consistent with other reports, suggesting other factors
may also have an important role in disease progression, such as
metabolic, genetic, epigenetic or environmental factors (33,34),
and follow-up of this individual may be recommended. To further
determine whether the cfDNA may be used as a template to amplify
other regions, pairs of primers for 12 out of 118 exons were
randomly selected. The results indicated that all amplicons may be
amplified with target sizes, implying that cfDNA may be used for
genetic testing (Fig. 2E). To
further extend its potential for prenatal diagnosis, cffDNA was
subsequently isolated from a pregnant woman (Fig. 2F) and amplification of the COL7A1
gene were performed. Successful amplification of the COL7A1 gene
from cffDNA suggested that cffDNA may be used as a non-invasive
means of detecting the COL7A1 variants in suspected hereditary
epidermolysis bullosa (Fig.
2G).
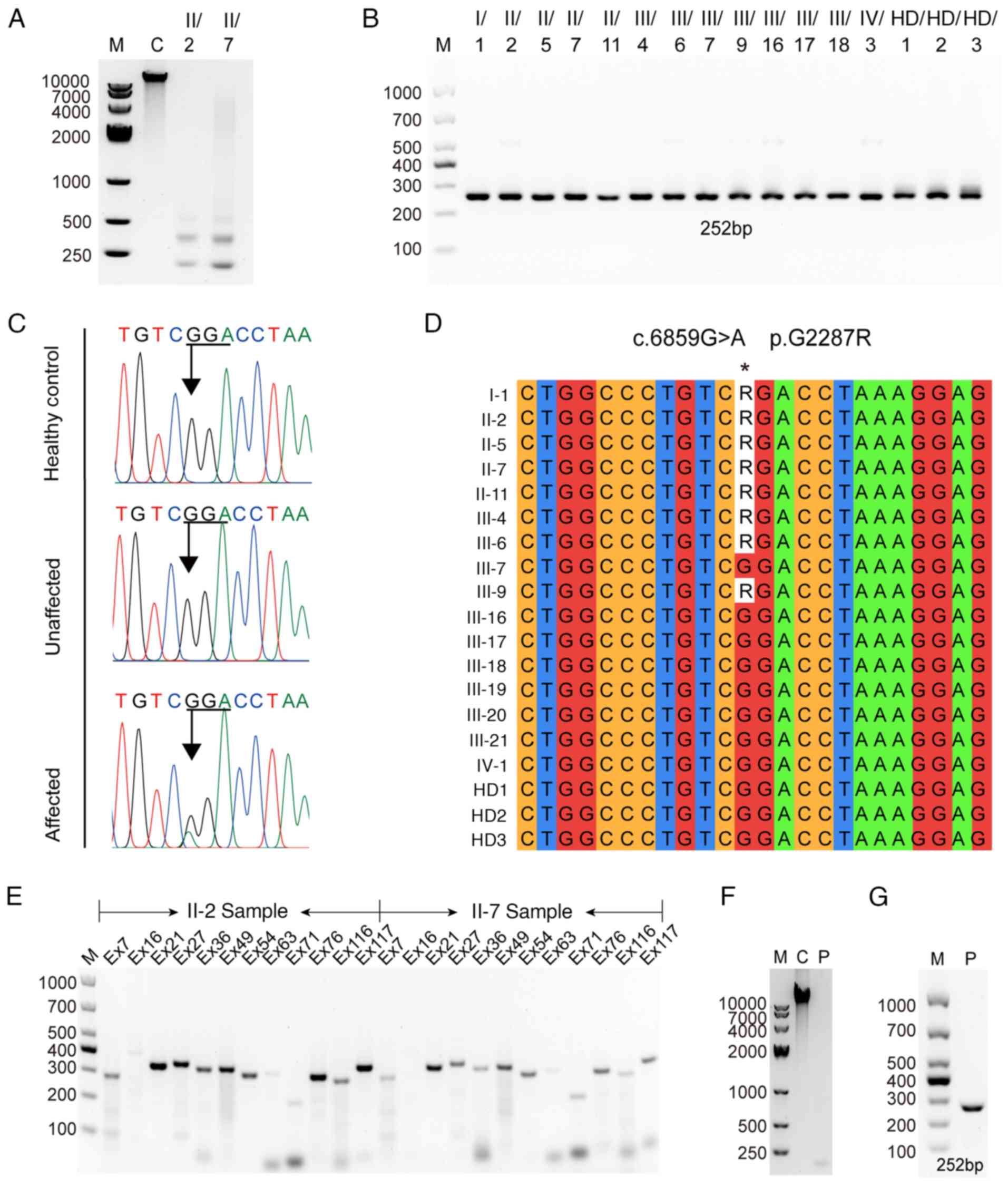 | Figure 2Molecular basis of dystrophic
epidermolysis bullosa pruriginosa confirmed by Sanger sequencing.
(A) CfDNA was extracted and the size distribution was checked by 1%
DNA gel electrophoresis. (B) The target region-carrying variant was
amplified from 13 members mentioned in Figs. 1A and 3 representative healthy controls. The PCR
product was visualized by 1% DNA gel electrophoresis. (C)
Representative data of direct nucleotide sequencing indicating a
heterozygous, single nucleotide substitution in exon 87,
c.6859G>A. (D) Sequence alignment of tested family members and
healthy controls. R represents a heterozygous G/A of the variant.
(E) PCR amplification of randomly selected exons in the COL7A1 gene
using two individual-derived cfDNA. (F) Visualization of cffDNA
extracted from a plasma sample. (G) COL7A1 exon 87 amplification
using cffDNA. M, DNA marker; C, control DNA extracted from Jurkat T
cells; P, DNA extracted from plasma derived from a pregnant
subject; COL7A1, collagen type VII, α1; Ex, exon; HD, healthy
donor; cffDNA, cell-free fetal DNA; cfDNA, cell-free DNA. |
Structural prediction of the variant
in COL7A1 gene causing DEB-Pr
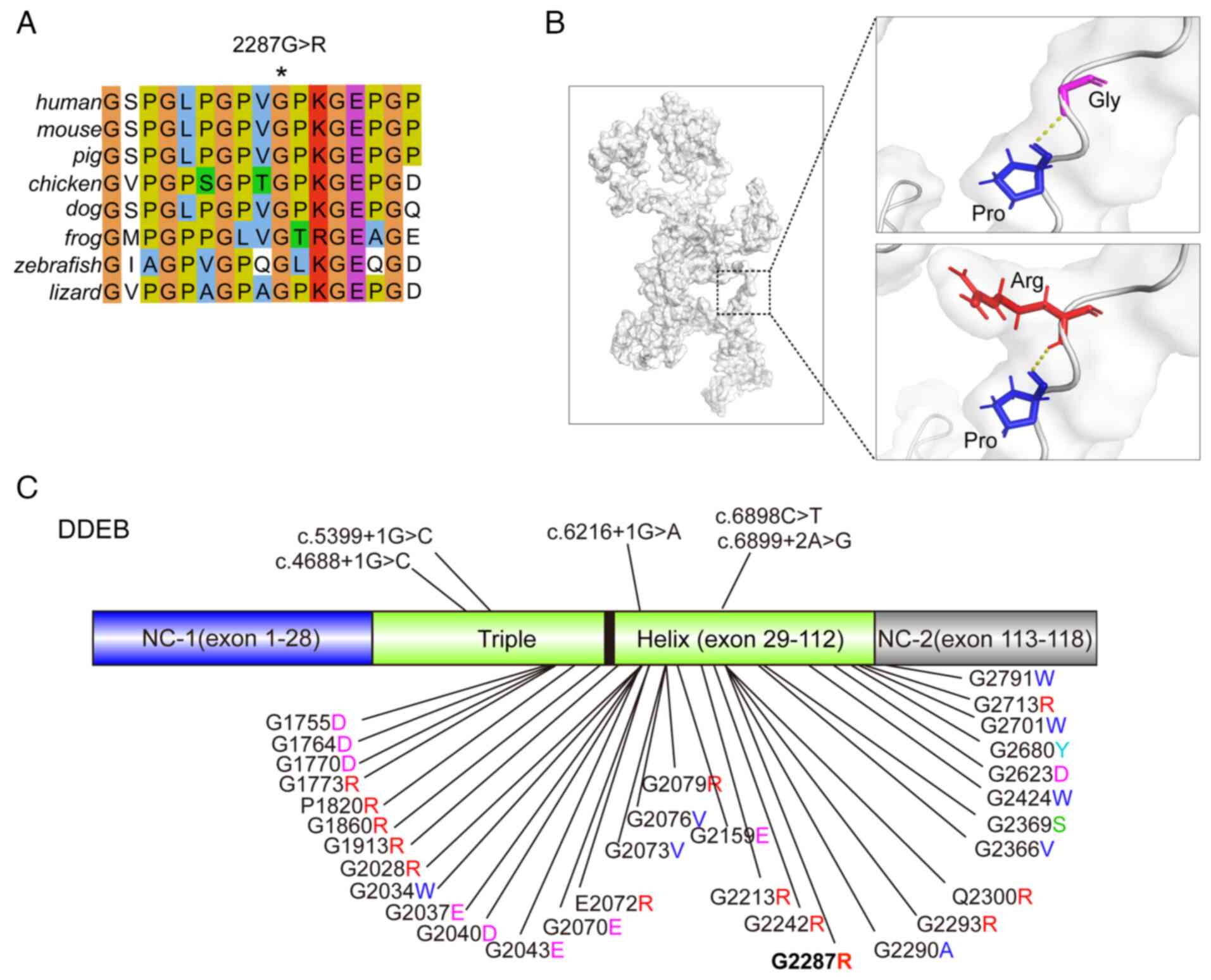
Multi-alignments analysis indicated high
evolutionary conservation of p2287 glycine and the Gly-X-Y motif
across several vertebrates, including human, mouse, pig, chicken,
dog, frog, zebrafish and lizard, suggesting critical roles of the
amino acid in maintaining normal protein function (Fig. 3A). Next, the possible molecular
mechanism of the variant on the COL7A1 gene causing DEB-Pr was
investigated using the structure prediction tool I-TASSER, which is
considered the finest tool for predicting the tertiary structure of
a protein (https://seq2fun.dcmb.med.umich.edu/I-TASSER/)
(21). Analysis of the predicted
3D protein structures did not reveal any difference in the hydrogen
bond interactions between wild-type and mutant residues. However,
the residue size and charge differences may impact the protein
folding and thus overall protein stability (Fig. 3B). To gain an overview of the
COL7A1 mutations causing DEB-Pr, a total of 65 unique mutations in
COL7A1gene were summarized, of which 37 were specifically reported
in DEB-Pr cases (Fig. 3C). This
summary demonstrated that the major cause of DEB-Pr involves
glycine substitutions (86.5%), although deletions or splice-site
mutations may appear in certain cases, consistent with previous
findings (2,6). Notably, all variants are located
within the triple helix of COL7A1 in DDEB. By contrast, the
mutations include nonsense, splice site, exon skipping and
non-glycine missense mutations within the triple helix or
non-collagenous NC-2 domain in RDEB.
Discussion
DEB-Pr represents a rare variant within the broad
spectrum of DEB and was first reported by McGrath et al
(1) in 1994. It has been
established that mutations in the COL7A1 gene, which encodes the
COL7 protein, underlie all subtypes of DEB. WES provides a
promising tool for disease diagnosis. In the present study, on the
basis of clinical and genetic examinations, a large family was
diagnosed with DEB-Pr using WES, and this was further confirmed by
Sanger sequencing. In this family, DEB-Pr was caused by the COL7A1
variant at position 6859 with G to A substitution, and an amino
acid level change of Gly to Arg. In addition, it was also
demonstrated that plasma cfDNA can be used as a source for
diagnosis.
In the present study, it was observed that G2287R
resulted in mild disease manifestations in the proband and related
family members, which is consistent with previous studies in both
Japanese and Chinese patients (28,29,35).
These findings support the notion that specific glycine
substitutions in the collagenous domain of COL7A1 relate to mild
diseases. Previous studies have indicated that the onset of DEB-Pr
varies from infancy to late adulthood. Typically, clinical
manifestations begin to appear during the first two decades of
life. However, in certain patients, the onset may be delayed to
adulthood, with the eldest case reported being that of a
71-year-old male (29,36). Of note, in the present study, an
individual (III-9) in their teens who carried the disease-causing
variants but did not exhibit any disease manifestations was
identified. Conversely, it was confirmed that III-19, 20 and 21
carried the wild-type allele, despite their father being affected
by DEB-Pr. The underlying causes of the phenotypic disparity
observed in DEB-Pr remain elusive. It has been proposed that
additional factors, such as genetic, epigenetic, metabolic,
immunological or environmental factors, may also contribute to the
DEB-Pr phenotype (33,37,38).
Further research is needed to elucidate the mechanism underlying
this phenotypic disparity.
cfDNA analysis has emerged as a game-changing tool
in genetic disease diagnosis, offering significant advantages over
traditional diagnostic methods. Its non-invasive nature, early
detection capabilities and high accuracy have propelled the field
of precision medicine forward, improving patient outcomes and
quality of life (32,39). However, it is important to note
that the level of cfDNA in peripheral blood is relatively low.
Typically, the content of cffDNA reaches 4% after 10-12 weeks of
pregnancy and various factors, such as body weight and gestational
age, may influence the content of cffDNA, which may limit its
application and accuracy (39-44).
Using I-TASSER, the finest structure prediction
tool, the molecular basis of the influence of variants on
functional changes was analyzed. The collagen molecule requires a
glycine residue to maintain its specific triple helical
conformation (45,46). In the present cases, arginine
residues were substituted for a glycine residue. In general, the
characteristics of the amino acids have an important impact on the
conformation of a protein. Although there is no difference in
hydrogen bond interaction in the predicted model, arginine is a
basic amino acid and has a more bulky side chain compared to
glycine and it may alter protein stability as well as structural
conformation. Therefore, different glycine substitutions may result
in different characteristics of COL7 and may thus explain the
differences in the clinical features. Further structure analysis
will need to confirm this hypothesis and guide drug
development.
The identification of variations in the COL7A1 gene
has a critical role in diagnosing DEB and its specific subtypes.
Genetic testing is utilized to detect mutations in the gene,
allowing clinicians to confirm the presence of the disease and
provide appropriate genetic counseling to affected families.
Certain studies suggest that specific mutations in the COL7A1 gene
may be associated with varying disease severities in individuals
with DEB. Understanding this genotype-phenotype correlation may
assist in predicting disease progression and guiding treatment
strategies (1,3). For families with a history of DEB or
known COL7A1 gene mutations, prenatal testing may be conducted to
determine if a fetus carries the genetic variation. This
information enables parents and healthcare providers to make
informed decisions about the pregnancy and plan appropriate care
after birth (47,48). Research focused on the COL7A1 gene
and its variants has opened up new possibilities for gene therapy
and other targeted treatments for DEB. Scientists are actively
exploring strategies to correct or replace the defective gene as
potential therapeutic options (8).
Studying the COL7A1 gene and its protein product, type VII
collagen, provides valuable insight into the molecular mechanisms
underlying DEB. This knowledge is crucial for the development of
drugs aimed at alleviating symptoms, promoting wound healing and
improving the quality of life for affected individuals.
The present study's main strength lies in the
extensive collection of individuals from the same family, allowing
us to confirm the association between the genotype COL7A1 G2287R
and the clinical phenotype. In addition, the potential application
of cfDNA in genetic diagnosis was successfully demonstrated.
Furthermore, identifying the specific variants responsible for the
disease will markedly benefit disease management.
However, it is essential to acknowledge the
limitations of the present study. First, the sample size of cffDNA
was relatively small, prompting the need for further research with
a larger sample to fully explore the application of cffDNA for
prenatal diagnosis. Secondly, the COL7A1 protein structure is not
available yet. We predicted and interpreted the impact of residue
changes on protein structure and the consequences of the protein
structure changes, which may affect downstream molecular mechanisms
or the function of collagen as a cellular component. While the
exact molecular mechanisms remain elusive. Further investigation is
warranted to gain a more comprehensive understanding. Finally, we
encountered an exceptional case where an individual carried the
variant but did not exhibit any clinical manifestations. The reason
behind this observation remains elusive and requires further
investigation.
In conclusion, in the present study, the rare
heterozygous variant c.6859G>A (p.Gly2287Arg) in the COL7A1 gene
was identified through WES together with Sanger sequencing using
individual plasma cfDNA in patients with DEB-Pr. Overall, the
present study provides compelling evidence supporting the
involvement of the COL7A1 G2287R gene variant in the development of
DEB-Pr. Furthermore, the present findings underscore the potential
of utilizing cfDNA in genetic disease diagnosis, offering new
avenues for improved diagnostics and personalized disease
management in the future.
Supplementary Material
Consensus sequence of collagen type
VII, α1 in the present study. The first rows are for the members of
the pedigree as indicated in the pedigree chart in Fig. 1A. The COL7A1 exon 87 was amplified,
sequenced, and aligned by Bioedit v7.2 software. The color is also
defined by Bioedit. * indicates the position of the
variant. HD, healthy donor.
Summary statistics of quality control
for whole-exome sequencing.
Primer pairs used in the present
study.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by Hangzhou Normal
University (grant no. 4255C50222204027).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author upon reasonable
request.
Authors' contributions
XS and FH conceived and designed the study. YY, YG,
WW, HQ, KZ, YM, MZ and DZ performed data analysis and conducted
experiments. XS and XL wrote and edited the manuscript. FH and XL
organized and provided clinical samples. All authors have read and
approved the final manuscript. YY and XS confirm the authenticity
of all the raw data.
Ethics approval and consent to
participate
The present study was approved by the IRB of the
Dermatology Hospital of Jiangxi Province (Nanchang, China).
Patient consent for publication
All patients or their legal guardians provided
written informed consent for genetic testing and publication of
their results.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
McGrath JA, Schofield OM and Eady RA:
Epidermolysis bullosa pruriginosa: Dystrophic epidermolysis bullosa
with distinctive clinicopathological features. Br J Dermatol.
130:617–625. 1994.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bardhan A, Bruckner-Tuderman L, Chapple
ILC, Fine JD, Harper N, Has C, Magin TM, Marinkovich MP, Marshall
JF, McGrath JA, et al: Epidermolysis bullosa. Nat Rev Dis Primers.
6(78)2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Dang N and Murrell DF: Mutation analysis
and characterization of COL7A1 mutations in dystrophic
epidermolysis bullosa. Exp Dermatol. 17:553–568. 2008.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Bremer J, van der Heijden EH, Eichhorn DS,
Meijer R, Lemmink HH, Scheffer H, Sinke RJ, Jonkman MF, Pasmooij
AMG and Van*den*Akker PC: Natural exon skipping sets the stage for
exon skipping as therapy for dystrophic epidermolysis Bullosa. Mol
Ther Nucleic Acids. 18:465–475. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Pulkkinen L and Uitto J: Mutation analysis
and molecular genetics of epidermolysis bullosa. Matrix Biol.
18:29–42. 1999.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Kim WB, Alavi A, Walsh S, Kim S and Pope
E: Epidermolysis bullosa pruriginosa: A systematic review exploring
genotype-phenotype correlation. Am J Clin Dermatol. 16:81–87.
2015.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Fine JD: Inherited epidermolysis bullosa.
Orphanet J Rare Dis. 5(12)2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Uitto J, McGrath JA, Rodeck U,
Bruckner-Tuderman L and Clare*Robinson E: Progress in epidermolysis
bullosa research: Toward treatment and cure. J Invest Dermatol.
130:1778–1784. 2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Takeichi T, Liu L, Fong K, Ozoemena L,
McMillan JR, Salam A, Campbell P, Akiyama M, Mellerio JE, McLean
WH, et al: Whole-exome sequencing improves mutation detection in a
diagnostic epidermolysis bullosa laboratory. Br J Dermatol.
172:94–100. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Fozia F, Nazli R, Alrashed MM, Ghneim HK,
Haq ZU, Jabeen M, Alam Khan S, Ahmad I, Bourhia M and Aboul-Soud
MAM: Detection of novel biallelic causative variants in COL7A1 gene
by whole-exome sequencing, resulting in congenital recessive
dystrophic epidermolysis bullosa in three unrelated families.
Diagnostics (Basel). 12(1525)2022.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Fozia F, Nazli R, Bibi N, Khan SA,
Muhammad N, Shakeeb N, Khan S, Jelani M and Wasif N: Whole exome
sequencing confirms molecular diagnostics of three pakhtun families
with autosomal recessive epidermolysis Bullosa. Front Pediatr.
9(727288)2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Albertsen HM, Matalliotaki C,
Matalliotakis M, Zervou MI, Matalliotakis I, Spandidos DA, Chettier
R, Ward K and Goulielmos GN: Whole exome sequencing identifies
hemizygous deletions in the UGT2B28 and USP17L2 genes in a
three-generation family with endometriosis. Mol Med Rep.
19:1716–1720. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hashmi JA, Safar RA, Afzal S, Albalawi AM,
Abdu-Samad F, Iqbal Z and Basit S: Whole exome sequencing
identification of a novel insertion mutation in the phospholipase C
ε-1 gene in a family with steroid resistant inherited nephrotic
syndrome. Mol Med Rep. 18:5095–5100. 2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Mandel P and Metais P: Nuclear acids in
human blood plasma. C R Seances Soc Biol Fil. 142:241–243.
1948.PubMed/NCBI(In French).
|
|
15
|
de Miranda FS, Barauna VG, dos Santos L,
Costa G, Vassallo PF and Campos LCG: Properties and application of
cell-free DNA as a clinical biomarker. Int J Mol Sci.
22(9110)2021.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Han DSC and Lo YMD: The nexus of cfDNA and
nuclease biology. Trends Genet. 37:758–770. 2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Li X, Qian H, Sogame R, Hirako Y, Tsuruta
D, Ishii N, Koga H, Tsuchisaka A, Jin Z, Tsubota K, et al: Integrin
β4 is a major target antigen in pure ocular mucous membrane
pemphigoid. Eur J Dermatol. 26:247–253. 2016.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Liu W, Sun X, Gao Y, Li H, Shi L, Cheng L,
Zhou Z, Li X and Qian H: A Chinese case of concurrent anti-laminin
γ1 pemphigoid and anti-laminin 332-type mucous membrane pemphigoid.
J Dermatol. 50:e69–e71. 2023.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Dainichi T, Kurono S, Ohyama B, Ishii N,
Sanzen N, Hayashi M, Shimono C, Taniguchi Y, Koga H, Karashima T,
et al: Anti-laminin gamma-1 pemphigoid. Proc Natl Acad Sci USA.
106:2800–2805. 2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Christiano AM, Hoffman GG, Zhang X, Xu Y,
Tamai Y, Greenspan DS and Uitto J: Strategy for identification of
sequence variants in COL7A1 and a novel 2-bp deletion mutation in
recessive dystrophic epidermolysis bullosa. Hum Mutat. 10:408–414.
1997.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Yang J, Yan R, Roy A, Xu D, Poisson J and
Zhang Y: The I-TASSER Suite: Protein structure and function
prediction. Nat Methods. 12:7–8. 2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Chai ZT, Lim YL, Busmanis I and Pang SM:
Anti-laminin-γ1 pemphigoid in an epitope spreading phenomenon,
successfully treated with rituximab. Int J Dermatol. 61:508–510.
2022.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Li S, Xiang R, Jing K, Li Z, Wang Y, Zhang
H, Li X and Feng S: Diagnostic values of indirect
immunofluorescence using salt-split skin, direct immunofluorescence
and BP180 NC16A ELISA on bullous pemphigoid. Chin Med J (Engl).
135:1379–1380. 2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Li X, Tsuchisaka A, Qian H, Teye K, Ishii
N, Sogame R, Harada K, Nakagomi D, Shimada S, Tateishi C, et al:
Linear IgA/IgG bullous dermatosis reacts with multiple laminins and
integrins. Eur J Dermatol. 25:418–423. 2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhou Y, Zhou X, Feng X, Xia D, Qian H, Liu
H, Li X and Li W: Case Report: Prurigo nodularis-like linear
IgA/IgG bullous dermatosis: A case report and literature review.
Front Immunol. 14(1201163)2023.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Shi L, Li X and Qian H: Anti-Laminin
332-type mucous membrane pemphigoid. Biomolecules.
12(1461)2022.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Liu W, Li H, Jin Y, Cheng L, Shi L, Gao Y,
Zhou Z, Feng S, Qian H, Hashimoto T and Li X: Case report: Mucous
membrane pemphigoid with complicated autoantibody profile
indicating the necessity of comprehensive diagnostic methods and
the contribution of IgA autoantibodies. Front Immunol.
14(1149119)2023.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Yajima M, Kitoh A, Nakano H, Sawamura D,
Miyachi Y and Kabashima K: Dominant dystrophic epidermolysis
bullosa pruriginosa with the G2287R mutation. Eur J Dermatol.
22:685–687. 2012.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Hayashi M, Kawaguchi M, Hozumi Y, Nakano
H, Sawamura D and Suzuki T: Dystrophic epidermolysis bullosa
pruriginosa of elderly onset. J Dermatol. 38:173–178.
2011.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Chao YC, Hong JB, Liu C, Lee MS and Lin
RY: COL7A1 G2287R mutation with two clinical phenotypes in the same
family: Bullous dermolysis of the newborn and dystrophic
epidermolysis bullosa pruriginosa. J Dermatol. 49:e313–e314.
2022.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Alcaide M, Cheung M, Hillman J, Rassekh
SR, Deyell RJ, Batist G, Karsan A, Wyatt AW, Johnson N, Scott DW
and Morin RD: Evaluating the quantity, quality and size
distribution of cell-free DNA by multiplex droplet digital PCR. Sci
Rep. 10(12564)2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Stawski R, Stec-Martyna E, Chmielecki A,
Nowak D and Perdas E: Current Trends in Cell-Free DNA applications.
scoping review of clinical trials. Biology (Basel).
10(906)2021.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Has C, Bauer JW, Bodemer C, Bolling MC,
Bruckner-Tuderman L, Diem A, Fine JD, Heagerty A, Hovnanian A,
Marinkovich MP, et al: Consensus reclassification of inherited
epidermolysis bullosa and other disorders with skin fragility. Br J
Dermatol. 183:614–627. 2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Almaani N, Liu L, Harrison N, Tanaka A,
Lai-Cheong J, Mellerio JE and McGrath JA: New glycine substitution
mutations in type VII collagen underlying epidermolysis bullosa
pruriginosa but the phenotype is not explained by a common
polymorphism in the matrix metalloproteinase-1 gene promoter. Acta
Derm Venereol. 89:6–11. 2009.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Yu Y, Wang Z, Mi Z, Sun L, Fu X, Yu G,
Pang Z, Liu H and Zhang F: Epidermolysis Bullosa in Chinese
Patients: Genetic analysis and mutation landscape in 57 pedigrees
and sporadic cases. Acta Derm Venereol.
101(adv00503)2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Brick K, Hand JL, Frankel AS, Siegel DH,
Thomas KB, El-Azhary R and Krol A: Epidermolysis bullosa
pruriginosa: Further clarification of the phenotype. Pediatr
Dermatol. 29:732–737. 2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Wagner RN, Piñón Hofbauer J, Wally V,
Kofler B, Schmuth M, De Rosa L, De Luca M and Bauer JW: Epigenetic
and metabolic regulation of epidermal homeostasis. Exp Dermatol.
30:1009–1022. 2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Rami A, Łaczmański Ł, Jacków-Nowicka J and
Jacków J: Reprogramming and differentiation of cutaneous squamous
cell carcinoma cells in recessive dystrophic epidermolysis Bullosa.
Int J Mol Sci. 22(245)2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Taylor-Phillips S, Freeman K, Geppert J,
Agbebiyi A, Uthman OA, Madan J and Clarke A, Quenby S and Clarke A:
Accuracy of non-invasive prenatal testing using cell-free DNA for
detection of Down, Edwards and Patau syndromes: A systematic review
and meta-analysis. BMJ Open. 6(e010002)2016.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Hu P, Liang D, Chen Y, Lin Y, Qiao F, Li
H, Wang T, Peng C, Luo D, Liu H and Xu Z: An enrichment method to
increase cell-free fetal DNA fraction and significantly reduce
false negatives and test failures for non-invasive prenatal
screening: A feasibility study. J Transl Med.
17(124)2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Gerson KD, Truong S, Haviland MJ, O'Brien
BM, Hacker MR and Spiel MH: Low fetal fraction of cell-free DNA
predicts placental dysfunction and hypertensive disease in
pregnancy. Pregnancy Hypertens. 16:148–153. 2019.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Wang E, Batey A, Struble C, Musci T, Song
K and Oliphant A: Gestational age and maternal weight effects on
fetal cell-free DNA in maternal plasma. Prenat Diagn. 33:662–666.
2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Hou Y, Yang J, Qi Y, Guo F, Peng H, Wang
D, Wang Y, Luo X, Li Y and Yin A: Factors affecting cell-free DNA
fetal fraction: Statistical analysis of 13,661 maternal plasmas for
non-invasive prenatal screening. Hum Genomics.
13(62)2019.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Zhou Y, Wang Y, Addai FP, Li X, Zhang X,
Liu H, Yang G, Zeng F, Jiang T and Liu J: Analysis of cell-free
fetal DNA in 16,843 pregnant women from a single center in China
using targeted sequencing approach. Placenta. 122:18–22.
2022.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Khan FF, Khan N, Rehman S, Ejaz A, Ali U,
Erfan M, Ahmed ZM and Naeem M: Identification and computational
analysis of novel pathogenic variants in Pakistani families with
diverse epidermolysis bullosa phenotypes. Biomolecules.
11(620)2021.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Fatima Y, Shabbir MA, Makhdoom SI,
Saleem-ur-Rehman H, Waseem M, Jabeen K, Laila SU, Shahid A and
Naveed M: Pharmacophore based drug designing of COL7A1; The
causative gene of dystrophic epidermolysis Bullosa. BioScientific
Review. 3:11–26. 2021.
|
|
47
|
Klingberg S, Mortimore R, Parkes J, Chick
JE, Clague AE, Murrell D, Weedon D and Glass IA: Prenatal diagnosis
of dominant dystrophic epidermolysis bullosa, by COL7A1 molecular
analysis. Prenat Diagn. 20:618–622. 2000.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Wang Y, Song Z, Zhang L, Li N, Zhao J,
Yang R, Ji S and Sun P: Genetic analysis and prenatal diagnosis of
recessive dystrophic epidermolysis bullosa caused by compound
heterozygous variants of the COL7A1 gene in a Chinese family. Front
Pediatr. 10(941201)2022.PubMed/NCBI View Article : Google Scholar
|