Introduction
Bone tissue homeostasis is maintained by
osteoclast-mediated bone resorption and osteoblast-mediated bone
formation (1,2). This continuous regenerative process
is generally recognized as bone remodeling, an imbalance in which
can cause metabolic bone diseases, such as osteoporosis or
poor/inappropriate fracture healing (2). Bone remodeling is tightly controlled
by various factors, including hormones, autacoids and cytokines
(1,2). Because receptors for bone resorptive
factors, such as parathyroid hormone and vitamin D, are not found
on osteoclasts but on osteoblasts, osteoblasts serve a central role
in the regulation of bone resorption (1,2).
Macrophage colony-stimulating factor (M-CSF), which is released
from osteoblasts, is a hematopoietic growth factor that promotes
the proliferation and differentiation of osteoclast progenitor
cells (3,4). M-CSF also stimulates the
differentiation of osteoclast precursors into mature osteoclasts
with bone resorptive activity and activates bone resorption by
cooperating with the receptor activator of nuclear factor-κB (RANK)
ligand (RANKL) expressed on osteoblasts, which binds to RANK on the
surface of osteoclast precursors (5,6). On
the other hand, osteoprotegerin released from osteoblasts is a
glycoprotein belonging to the tumor necrosis factor receptor family
(7), which binds to RANKL as a
decoy receptor and inhibits RANKL-RANK binding, thereby reducing
bone resorption via the suppression of osteoclast differentiation
(7,8). Thus, M-CSF and osteoprotegerin
released from osteoblasts serve pivotal roles in the regulation of
bone remodeling.
Osteal macrophages located on the surface of bone
remodeling sites have been reported to play diverse roles in
skeletal homeostasis (9). The
removal of apoptotic osteoblasts by osteal macrophage phagocytosis
generates specific proteins, such as transforming growth factor
(TGF)-β, to promote the differentiation of progenitor cells into
osteoblasts, leading to osteogenesis (9). Oncostatin M (OSM), which is secreted
by osteal macrophages, is a member of the IL-6 family that shares
gp130 as a common subunit of the IL-6 family receptor (10). OSM is expressed in several cell
types, including osteoblast lineage cells, and has its effects
through multiple receptors, such as OSM receptor (OSMR) and
leukemia inhibitory factor receptor; the roles of both pathways
have previously been explored in osteoblast regulation (11). Regarding bone metabolism, it has
been reported that OSM stimulates the activation of osteoblasts and
inhibits bone resorption (12). It
has also been demonstrated that OSMR-deficient mice exhibit
osteopetrosis with a reduced number of osteoblasts and osteoclasts
(13). Additionally, the lack of
OSM function reportedly leads to delayed bone fracture healing in
mouse models (14). These findings
suggest that OSM may act as an essential modulator of bone
remodeling.
Basic fibroblast growth factor (bFGF), which is
embedded in the bone matrix, is released from bone remodeling sites
by the process of bone resorption and induces osteoblast lineage
cells to promote osteogenesis (15,16).
Thus, bFGF is considered to serve a role in the regulation of bone
remodeling. Regarding the effects on osteoblast lineage cells, bFGF
reportedly induces the expression of M-CSF mRNA, with an increase
of M-CSF secretion from murine bone marrow stromal cells,
precursors of osteoblasts (17).
The binding of bFGF to the corresponding receptor, fibroblast
growth factor receptor (FGFR), triggers the dimerization and
activation of FGFRs, which causes the phosphorylation of FGFR
substrates and subsequent activation of downstream effectors,
including mitogen-activated protein kinases (MAPKs), via
Grb2(18). Notably, our previous
study reported that bFGF elicits FGFR autophosphorylation due to
dimerization in osteoblast-like MC3T3-E1 cells (19). In addition, our previous study
demonstrated that bFGF stimulates the synthesis of osteoprotegerin
via activation of p38 MAPK and stress-activated protein
kinase/c-Jun N-terminal kinase (SAPK/JNK) in osteoblast-like
MC3T3-E1 cells (20). As for the
intracellular signaling mechanism of bFGF, it has been indicated
that bFGF activates p44/p42 MAPK, in addition to p38 MAPK and
SAPK/JNK, resulting in the upregulation of vascular endothelial
growth factor (VEGF) synthesis in these cells (21,22).
These findings led to the hypothesis that osteoblast functions are
finely tuned by MAPKs stimulated by bFGF. Furthermore, our recent
study reported that OSM suppresses TGF-β-stimulated syntheses of
M-CSF and VEGF in these cells (23). However, the details of the
molecular mechanisms underlying how OSM affects osteoblast
functions remain to be elucidated.
In the present study, the effects and the underlying
mechanisms of OSM on the bFGF-induced synthesis of osteoprotegerin
and M-CSF were investigated in osteoblast-like MC3T3-E1 cells.
Materials and methods
Cell culture
Clonal osteoblast-like MC3T3-E1 cells derived from
newborn mouse calvariae (24) were
donated by Dr Masayoshi Kumegawa (Graduate School of Dentistry,
Department of Dentistry, Meikai University, Sakado, Japan) and
maintained as previously described (20). Mouse MC3T3-E1 cells were cultured
in α-minimum essential medium (α-MEM) (MilliporeSigma) supplemented
with 10% fetal bovine serum (FBS; Gibco; Thermo Fisher Scientific,
Inc.) at 37˚C in a humidified atmosphere with 5% CO2/95%
air. The cells were seeded onto 35-mm diameter dishes
(5x104 cells/dish) for enzyme-linked immunosorbent assay
(ELISA) and reverse transcription-quantitative polymerase chain
reaction (RT-qPCR) analysis, or 90-mm diameter dishes
(2x105 cells/dish) for western blot analysis in α-MEM
supplemented with 10% FBS. After 5 days, the medium was replaced
with α-MEM supplemented with 0.3% FBS. After 48 h, the cells were
used for experiments. It is well known that the addition of
ascorbate and β-glycerophosphate (BGP) practically induces the
differentiation of MC3T3-E1 cells into osteoblasts (25). In the present study, α-MEM
containing 50 mg/l ascorbate, but no BGP, was used, thus
experiments were performed on a model of pre-osteoblasts.
ELISA
To assess osteoprotegerin, the cultured MC3T3-E1
cells were pretreated with 0, 3, 10, 30 or 50 ng/ml OSM for 60 min
at 37˚C, and then stimulated with 30 ng/ml bFGF or vehicle (mast
cell medium; 150 mM NaCl, 5 mM KCl, 5.5 mM glucose, 0.8 mM
MgSO4, 1 mM CaCl2, 5 mM HEPES, pH 7.4) in 1
ml α-MEM supplemented with 0.3% FBS for 48 h at 37˚C. Pretreatment
with 30 µM SB203580 or vehicle was performed for 60 min prior to
pretreatment with OSM. To assess M-CSF, the cultured MC3T3-E1 cells
were pretreated with 0, 3, 10, 30 or 50 ng/ml OSM, 50 µM PD98059,
10 µM SB203580, 3 µM SP600125 or vehicle (50 µl mast cell medium)
for 60 min at 37˚C, and then stimulated with 30 ng/ml bFGF or
vehicle (50 µl of mast cell medium) in 1 ml α-MEM supplemented with
0.3% FBS for 48 h at 37˚C. Recombinant mouse OSM, and ELISA kits
for mouse osteoprotegerin (cat. no. MOP00) and M-CSF (cat. no.
MMC00) were obtained from R&D Systems, Inc. Recombinant human
bFGF was purchased from Gibco; Thermo Fisher Scientific, Inc.
PD98059, SB203580 and SP600125 were purchased from Calbiochem;
Merck KGaA. OSM and bFGF were dissolved in mast cell medium.
PD98059, SB203580 and SP600125 were dissolved in dimethyl
sulfoxide. The conditioned medium was then collected, and the
concentrations of osteoprotegerin and M-CSF were measured using the
mouse ELISA kits for osteoprotegerin or M-CSF, in accordance with
the manufacturer's protocols.
RT-qPCR
The cultured MC3T3-E1 cells were pretreated with 50
ng/ml OSM or vehicle (50 µl of mast cell medium) for 60 min at 37˚C
and were then stimulated with 30 ng/ml bFGF or vehicle (50 µl of
mast cell medium) in α-MEM containing 0.3% FBS for 4 or 6 h at
37˚C. TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.) and Omniscript Reverse Transcriptase kit (Qiagen,
Inc.) were used to isolate total RNA and transcribe it into cDNA,
respectively. qPCR was performed using a LightCycler 2 Real-Time
PCR system and software (version 3.5; Roche Diagnostics) with the
LightCycler FastStart DNA Master SYBR Green I (Roche Diagnostics)
according to the manufacturer's protocol. Samples were subjected to
thermocycling conditions as follows: Initial denaturation at 95˚C
for 10 min, followed by 40 cycles of denaturation at 95˚C for 1
sec, annealing at 60˚C for 5 sec and elongation at 72˚C for 7 sec.
Forward and reverse primers for mouse osteoprotegerin (primer set
ID: MA026526) and M-CSF mRNA (primer set ID: MA171365) were
obtained from Takara Bio Inc. The primer sequences were as follows:
Osteoprotegerin, forward 5'-CAATGGCTGGCTTGGTTTCATAG-3', reverse
5'-CTGAACCAGACATGACAGCTGGA-3'; M-CSF, forward
5'-CATGTGGAGCAGCATGAGG-3' and reverse 5'-CAATGTCTGAGGGTCTCGATGG-3'.
Forward and reverse primers for mouse GAPDH mRNA were synthesized
based on the report of Simpson et al (26). The primer sequences for GAPDH were
as follows: Forward 5'-AACGACCCCTTCATTGAC-3' and reverse
5'-TCCACGACATACTCAGCAC-3'. The amplified products were determined
by melting curve analysis in accordance with the system protocol.
The mRNA expression levels of osteoprotegerin and M-CSF were
normalized to those of GAPDH using SPSS Statistics (version 22;
IBM, Corp.), and the relative mRNA expression levels were analyzed
using the 2-ΔΔCq method (27), which was created automatically with
the LightCycler software in each run (20,23).
Western blot analysis
The cultured MC3T3-E1 cells were pretreated with 0,
30, 50 or 70 ng/ml OSM for 60 min at 37˚C, and then stimulated with
30 ng/ml bFGF or vehicle (200 µl of mast cell medium) in 4 ml α-MEM
containing 0.3% FBS for 10 or 20 min. The cells were then lysed,
homogenized and sonicated (output 20W, 1 sec x20 cycles at 4˚C) in
a lysis buffer containing 62.5 mM Tris/HCl (pH 6.8), 2% sodium
dodecyl sulfate (SDS), 50 mM dithiothreitol and 10% glycerol. The
concentration of protein in the samples was assessed using a Pierce
BCA protein kit (cat. no. 23225; Thermo Fisher Scientific, Inc.).
Proteins (20 µg per lane) were separated by SDS-polyacrylamide
electrophoresis, which was performed in accordance with the method
of Laemmli using 10% polyacrylamide gels (28). The proteins were then transferred
onto Immuno-Blot PVDF membranes (Bio-Rad Laboratories, Inc.), which
were blocked with 5% fat-free dry milk in Tris-buffered
saline-Tween (TBS-T: 20 mM Tris-HCl, pH 7.6; 137 mM NaCl; 0.1%
Tween 20) for 1 h at room temperature before incubation with
primary antibodies. The membrane was subsequently incubated at 4˚C
overnight with primary antibodies (1:1,000) followed by incubation
with the appropriate secondary antibodies (1:1,000) at room
temperature for 1 h. Western blot analysis was performed as
described previously (20) using
primary antibodies against phosphorylated (p)-p38 MAPK (cat. no.
4511), p38 MAPK (cat. no. 9212), p-SAPK/JNK (cat. no. 4668),
SAPK/JNK (cat. no. 9252), p-p44/p42 MAPK (cat. no. 9101), p44/p42
MAPK (cat. no. 9102) (all from Cell Signaling Technology Inc.) or
GAPDH (cat. no. 60004-1-lg; Proteintech Group, Inc.). KPL
horseradish peroxidase (HRP)-labeled goat anti-rabbit IgG (cat. no.
5220-0336; SeraCare Life Sciences, Inc.) or HRP-labeled anti-mouse
IgG antibodies (cat. no. 7076; Cell Signaling Technology, Inc.)
were used as a secondary antibody. The ECL western blot detection
system was purchased from Cytiva. The primary and secondary
antibodies were diluted in TBS-T with 5% fat-free dry milk to
optimal concentrations. An X-ray film with the ECL western blot
detection system was used to visualize peroxidase activity on the
membrane, and different membranes were used for every single
protein. A densitometric analysis was performed using a scanner and
image analysis program (ImageJ version 1.48; National Institutes of
Health). The background-subtracted signal intensity of each
phosphorylation signal was normalized to the respective intensity
of the total protein, and then plotted as the fold increase
compared to that of control cells treated without stimulation
(20).
Statistical analysis
All data were analyzed using Mini StatMate (version
2.01; ATMS Co., Ltd.). Data are presented as the mean ± SEM of at
least triplicate determinations from independent cell preparations.
The statistical significance of the data was analyzed using one-way
or two-way analysis of variance, as appropriate, followed by the
Bonferroni method for multiple comparisons between pairs. P<0.05
was considered to indicate a statistically significant
difference.
Results
Effects of OSM on the bFGF-induced
release of osteoprotegerin and M-CSF from MC3T3-E1 cells
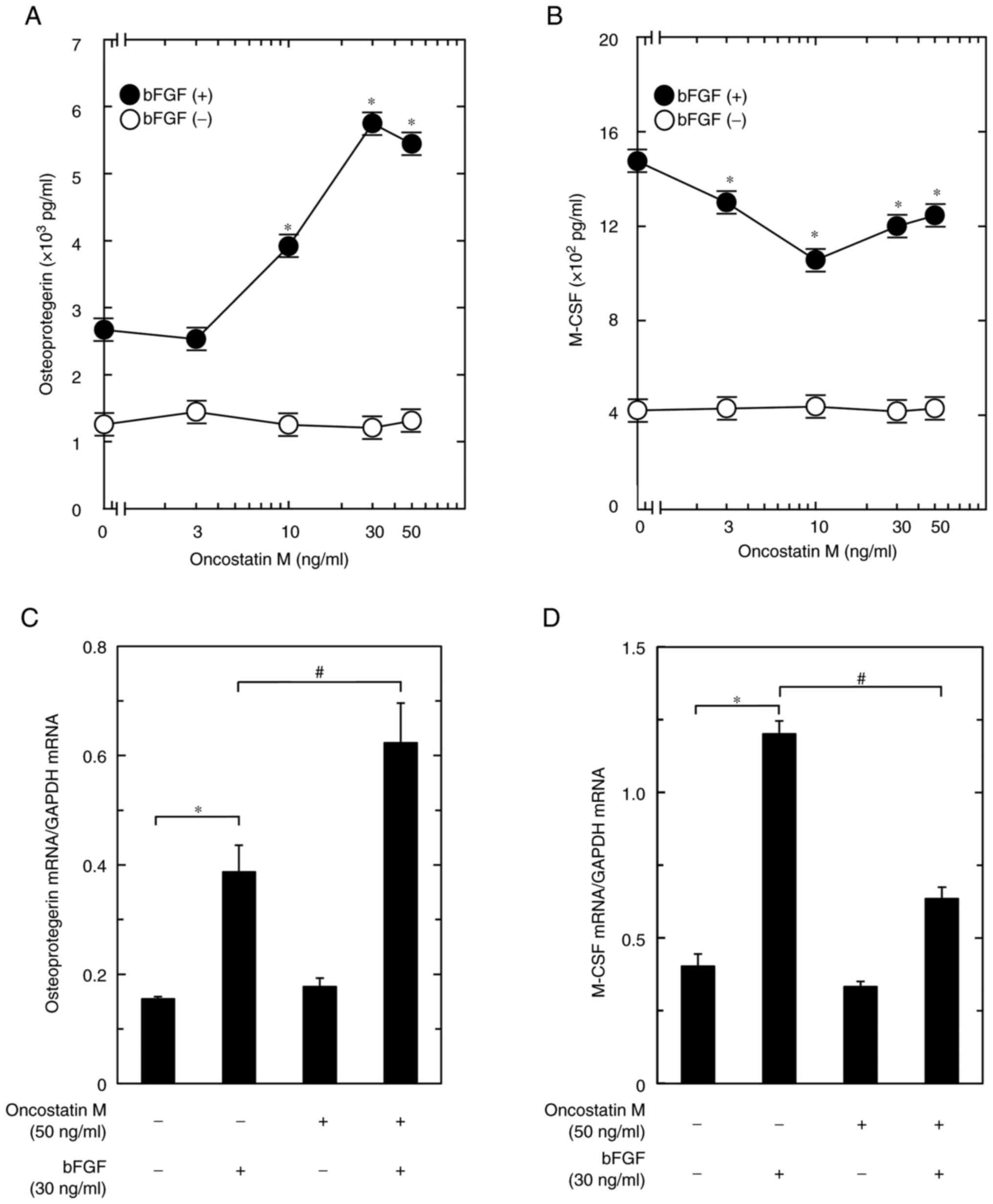
To investigate the effect of OSM on osteoblasts, the
present study examined the effect of OSM on bFGF-induced
osteoprotegerin release from these cells. OSM, which by itself did
not affect osteoprotegerin release, significantly enhanced the
bFGF-stimulated osteoprotegerin release observed within a range of
3-50 ng/ml (Fig. 1A). The maximum
effect of OSM observed at 30 ng/ml was ~320% amplification of the
bFGF effect.
The present study next examined the effect of OSM on
bFGF-induced M-CSF release from osteoblast-like MC3T3-E1 cells.
OSM, which by itself did not affect M-CSF release, significantly
suppressed the bFGF-stimulated M-CSF release within a range of 3-50
ng/ml (Fig. 1B). The maximum
effect of OSM observed at 10 ng/ml was ~35% attenuation of the bFGF
effect.
Effects of OSM on the mRNA expression
levels of osteoprotegerin and M-CSF in MC3T3-E1 cells
To elucidate whether the OSM-induced increase in the
release of osteoprotegerin stimulated by bFGF was mediated via
transcriptional events, the present study examined the effect of
OSM on the bFGF-induced mRNA expression of osteoprotegerin in
osteoblast-like MC3T3-E1 cells. Although OSM by itself did not have
any significant effect on the mRNA expression levels of
osteoprotegerin, it significantly enhanced the bFGF-upregulated
mRNA expression levels of osteoprotegerin when used at a dose of 50
ng/ml (Fig. 1C).
To evaluate whether OSM suppressed the
bFGF-stimulated M-CSF release through transcriptional events, the
present study next examined the effect of OSM on the bFGF-induced
mRNA expression of M-CSF in these cells. OSM alone did not have any
significant effect on the mRNA expression levels of M-CSF; however,
the bFGF-upregulated mRNA expression levels of M-CSF were
significantly suppressed by OSM at a concentration of 50 ng/ml
(Fig. 1D).
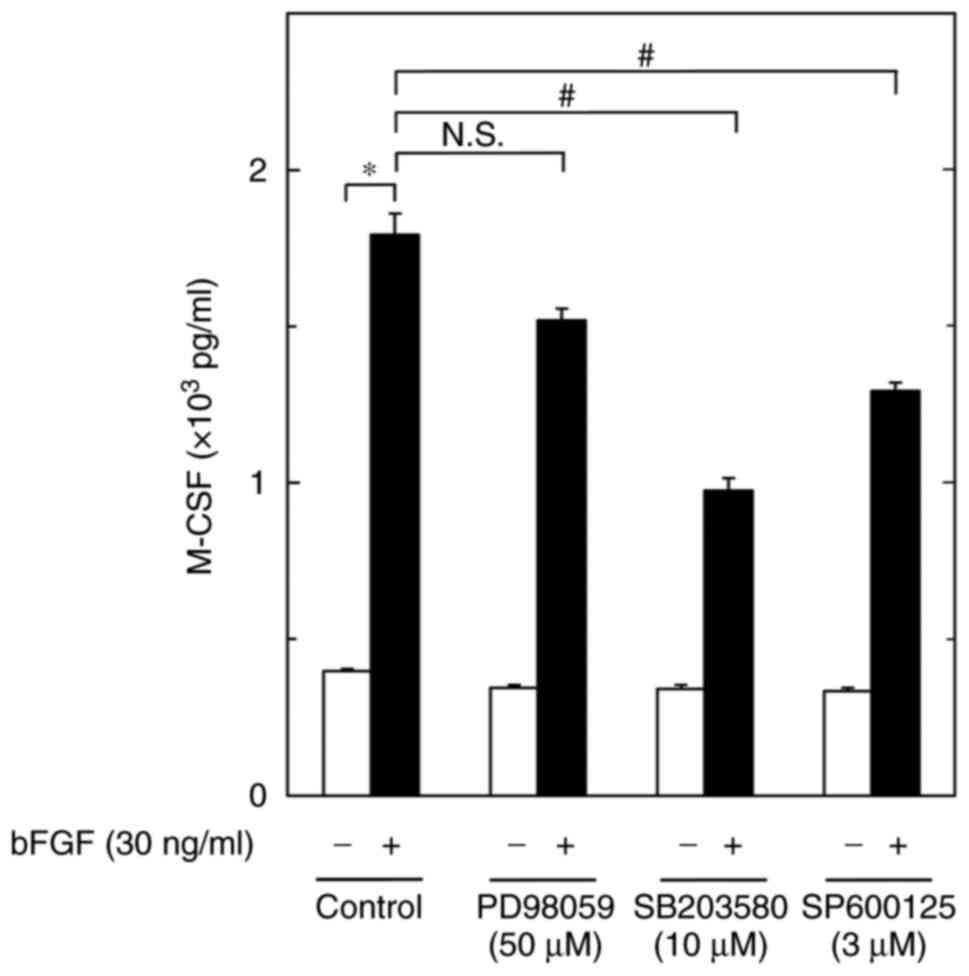
Effects of PD98059, SB203580 or
SP600125 on the bFGF-induced M-CSF release from MC3T3-E1 cells
To investigate the intracellular signaling mechanism
underlying the bFGF-induced synthesis of M-CSF in MC3T3-E1 cells,
the present study examined the effects of PD98059, an inhibitor of
MEK1/2 which is the upstream kinase of p44/p42 MAPK (29), SB203580, a specific inhibitor of
p38 MAPK (30), or SP600125, a
specific inhibitor of SAPK/JNK (31), on the bFGF-induced M-CSF release
from osteoblast-like MC3T3-E1 cells. PD98059 failed to suppress
M-CSF release with or without bFGF. By contrast, SB203580 and
SP600125, which by themselves had little effect on M-CSF release,
significantly reduced the bFGF-stimulated M-CSF release (Fig. 2). Regarding the effects of PD98059,
SB203580 and SP600125 on the activities of p44/p42 MAPK, p38 MAPK
and SAPK/JNK, respectively, in these cells, we previously reported
that the bFGF-induced phosphorylation of p44/p42 MAPK, p38 MAPK and
SAPK/JNK was markedly suppressed by PD98059, SB203580 and SP600125,
respectively (21,22). Thus, the present results suggested
that M-CSF synthesis involves the bFGF-elicited activation of not
p44/p42 MAPK, but of p38 MAPK and SAPK/JNK, in MC3T3-E1 cells.
Effects of OSM on the bFGF-induced
phosphorylation of p38 MAPK, SAPK/JNK and p44/p42 MAPK in MC3T3-E1
cells
In order to investigate whether OSM modulates the
activation of p38 MAPK, SAPK/JNK and p44/p42 MAPK, the present
study next examined the effects of OSM on the phosphorylation
levels induced by bFGF in osteoblast-like MC3T3-E1 cells. As
previously reported (21,22), it was confirmed that bFGF
significantly induced the phosphorylation of p38 MAPK, SAPK/JNK and
p44/p42 MAPK (Fig. 3A-C). OSM,
which alone hardly affected the levels of p-p38 MAPK, significantly
enhanced the levels of bFGF-induced phosphorylation at 30, 50 and
70 ng/ml (Fig. 3A). By contrast,
OSM, which by itself hardly affected the phosphorylation levels of
SAPK/JNK, markedly dose-dependently suppressed the levels of
phosphorylation stimulated by bFGF at 30, 50 and 70 ng/ml (Fig. 3B). Furthermore, OSM hardly affected
the levels of p44/p42 MAPK phosphorylation when used at doses up to
70 ng/ml with or without bFGF stimulation (Fig. 3C).
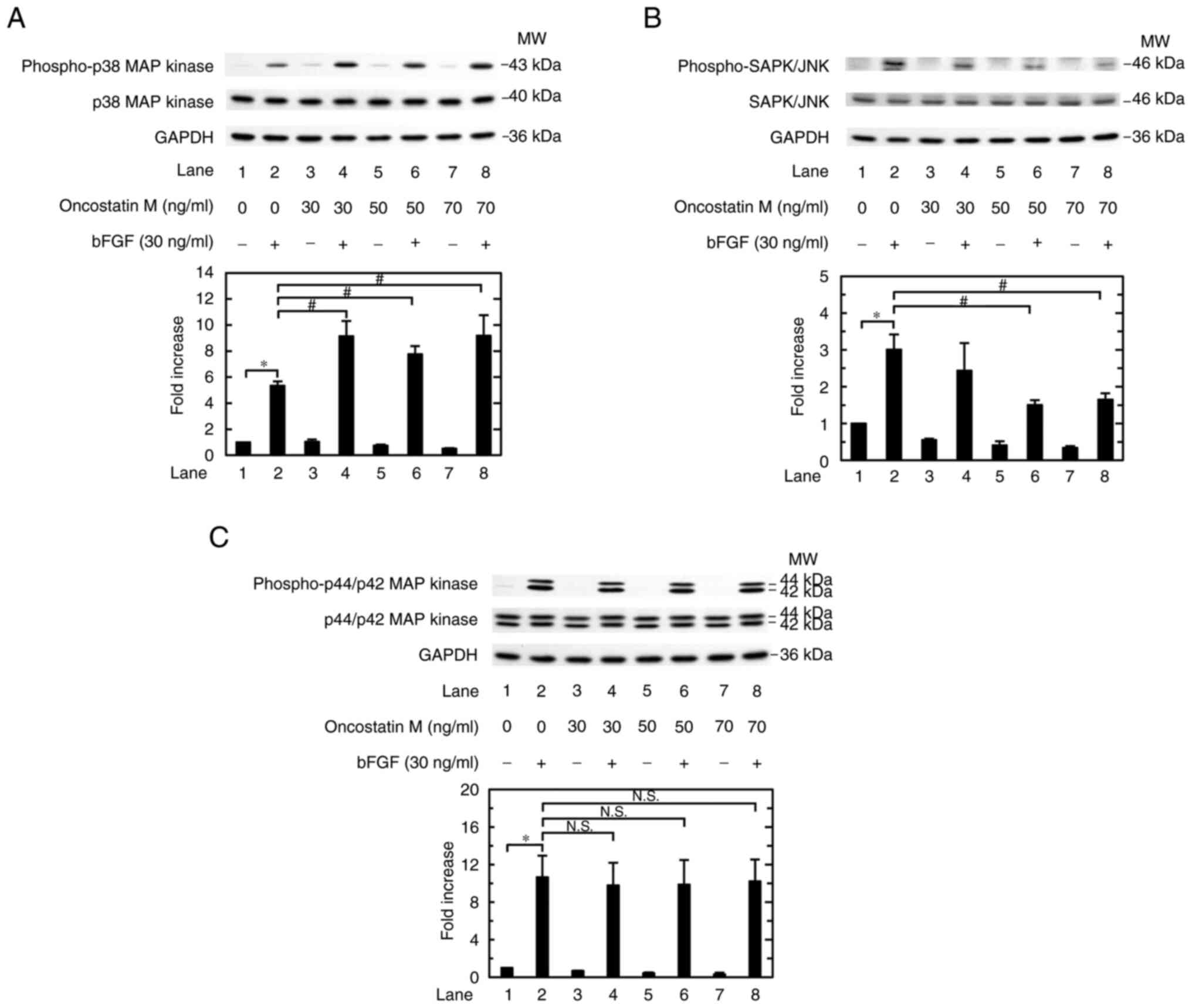 | Figure 3Effects of OSM on the bFGF-induced
phosphorylation of p38 MAPK, SAPK/JNK and p44/p42 MAPK in MC3T3-E1
cells. The cultured cells were pretreated with 0, 30, 50 or 70
ng/ml OSM for 60 min, and then stimulated with 30 ng/ml bFGF or
vehicle for (A) 10 or (B and C) 20 min. The cell extracts were then
subjected to SDS-PAGE and western blot analysis with antibodies
against (A) p-p38 MAPK, p38 MAPK and GAPDH; (B) p-SAPK/JNK,
SAPK/JNK and GAPDH; or (C) p-p44/p42 MAPK, p44/p42 MAPK and GAPDH.
The histograms show the semi-quantitative representations of the
expression levels of (A) p-p38 MAPK after normalization to p38
MAPK, (B) p-SAPK/JNK after normalization to SAPK/JNK, and (C)
p-p44/p42 MAPK after normalization to p44/p42 MAPK obtained from
densitometric analysis. The levels were expressed as the fold
increase with respect to the basal levels presented in lane 1. Data
are presented as the mean ± SEM of (A) quadruplicate determinations
from four independent cell preparations or (B and C) triplicate
determinations from three independent cell preparations.
*P<0.05 vs. control; #P<0.05 vs. bFGF
alone. bFGF, basic fibroblast growth factor; MAPK,
mitogen-activated protein kinase; MW, molecular weight N.S., not
significant; p-, phosphorylated; SAPK/JNK, stress-activated protein
kinase/c-Jun N-terminal kinase. |
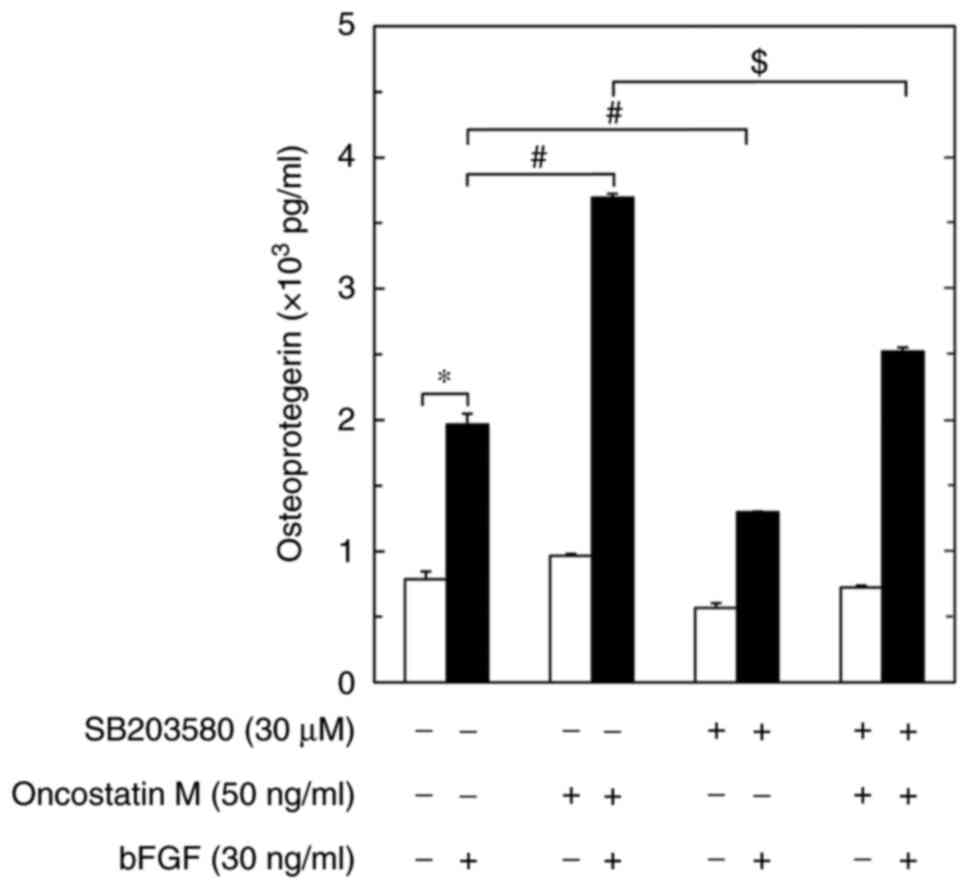
Effect of SB203580 on the
amplification by OSM of the bFGF-induced osteoprotegerin release
from MC3T3-E1 cells
To elucidate whether the enhancing effect of OSM on
the bFGF-stimulated osteoprotegerin release was truly mediated
through p38 MAPK, the present study examined the effect of
SB203580(30) on the amplification
by OSM of the osteoprotegerin release induced by bFGF in
osteoblast-like MC3T3-E1 cells. As previously reported (20), SB203580, which alone had little
effect on osteoprotegerin levels, significantly suppressed the
bFGF-induced osteoprotegerin release (Fig. 4). SB203580 also markedly reduced
the amplification of the bFGF-stimulated osteoprotegerin release
caused by OSM (Fig. 4).
Discussion
The present study used osteoblast-like MC3T3-E1
cells to explore the effects of OSM, a cytokine produced by osteal
macrophages (9), on the
bFGF-induced synthesis of osteoprotegerin and M-CSF, which are the
inhibitory factor and the promoting factor of osteoclastogenesis,
respectively (3,4,7). The
results revealed that the bFGF-induced release of osteoprotegerin
was clearly enhanced by OSM. In addition, the mRNA expression
levels of osteoprotegerin stimulated by bFGF were amplified by OSM,
indicating that the enhancing effect of OSM on the bFGF-induced
osteoprotegerin release may be mediated through transcriptional
events in these cells. Therefore, it is likely that OSM potentiates
the synthesis of osteoprotegerin stimulated by bFGF in osteoblasts.
The present study also demonstrated that the bFGF-stimulated M-CSF
release was suppressed by OSM. In addition, the mRNA expression
levels of M-CSF induced by bFGF were markedly reduced by OSM,
suggesting that the suppressive effect of OSM on the bFGF-induced
M-CSF release may be mediated through a reduction of
transcriptional events in these cells. It is likely that OSM could
diminish the synthesis of M-CSF stimulated by bFGF in osteoblasts.
Thus, OSM seems to regulate bFGF-stimulated osteoblast functions,
having diverse effects on the syntheses of osteoprotegerin and
M-CSF, amplifying the former and suppressing the latter. To the
best of our knowledge, this seems to be the first report that
clearly presents the effects of OSM on bFGF-induced osteoblast
activation. bFGF embedded in bone matrix is released by bone
resorption in the process of bone remodeling and affects osteoblast
lineage cells to promote osteogenesis (15,16),
and as such is considered a direct stimulator. OSM secreted by
osteal macrophages translocated for bone remodeling, can stimulate
the activation of osteoblasts and inhibit bone resorption (9), and as such is considered a modulator.
Notably, the present study revealed that OSM by itself hardly
affected the synthesis of osteoprotegerin or M-CSF in
osteoblast-like MC3T3-E1 cells. Thus, the reason the present study
performed pretreatment with OSM and stimulation with bFGF is that
OSM itself cannot stimulate but instead modulates for the
activation of osteoblast-like cells.
Regarding the intracellular signaling system
underlying the effects of bFGF in osteoblasts, our previous study
demonstrated that p38 MAPK and SAPK/JNK are involved in the
bFGF-stimulated osteoprotegerin synthesis in osteoblast-like
MC3T3-E1 cells (20). In addition,
it has been reported that p44/p42 MAPK is activated by bFGF
stimulation in these cells (21).
The present study revealed that both SB203580(30) and SP600125(31) significantly reduced the release of
M-CSF induced by bFGF, suggesting that activation of both p38 MAPK
and SAPK/JNK are involved in bFGF-induced M-CSF synthesis as
positive regulators in these cells. Furthermore, PD98059(29) was shown to hardly affect
bFGF-stimulated M-CSF release, thus it is unlikely that p44/p42
MAPK is involved in the M-CSF synthesis induced by bFGF in these
cells. Therefore, it is likely that activation of p38 MAPK and
SAPK/JNK, but not p44/p42 MAPK, is commonly involved in the
syntheses of osteoprotegerin and M-CSF in osteoblast-like MC3T3-E1
cells.
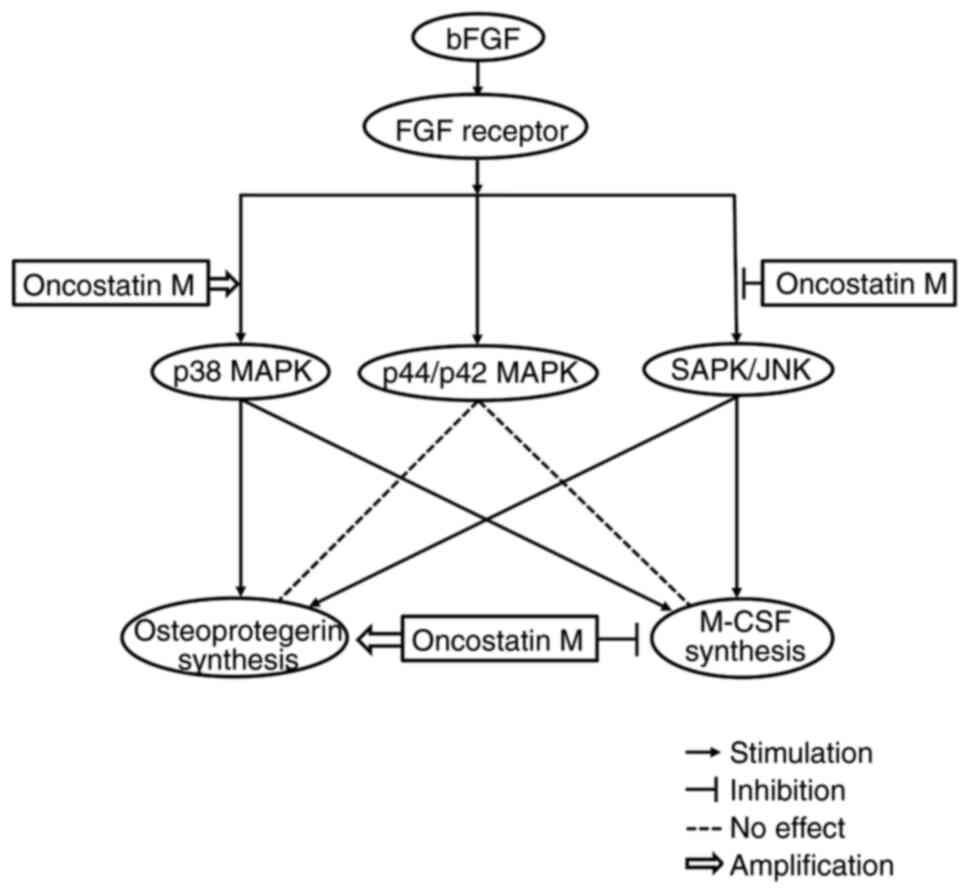
The present study demonstrated that OSM enhanced the
bFGF-induced phosphorylation of p38 MAPK, indicating that OSM may
upregulate activation of p38 MAPK induced by bFGF in MC3T3-E1
cells. By contrast, OSM did not enhance but suppressed the
bFGF-induced phosphorylation of SAPK/JNK, suggesting that OSM may
downregulate the activation of SAPK/JNK stimulated by bFGF in these
cells. In addition, it was confirmed that bFGF-stimulated
phosphorylation of p44/p42 MAPK was not affected by OSM, indicating
that OSM is not able to affect the p44/p42 MAPK activation by bFGF
in these cells. Thus, it is likely that the upregulation of p38
MAPK caused by OSM is involved in the enhancement of bFGF-induced
osteoprotegerin synthesis in osteoblast-like MC3T3-E1 cells. The
present study further examined the effect of SB203580(30) on the OSM-induced enhancement of
osteoprotegerin release stimulated by bFGF in these cells. The
results demonstrated that SB203580 markedly reduced the
amplification of bFGF-stimulated osteoprotegerin release caused by
OSM in these cells. Therefore, it seems that OSM may amplify
bFGF-induced osteoprotegerin synthesis, at least in part via the
upregulation of p38 MAPK activation in osteoblasts. As SB203580 is
a common selective inhibitor of p38α and p38β MAPK (32), it is not clear which subtype works
in the amplification of bFGF-induced osteoprotegerin synthesis by
OSM. However, it is recognized that p38α is the most highly
expressed isoform of p38 MAPK in osteoblasts (33), suggesting that p38α is a promising
candidate. In addition, OSM has been shown to downregulate the
activation of SAPK/JNK stimulated by bFGF in osteoblasts. Taking
into account this finding, it is possible that downregulating
SAPK/JNK activation could result in the suppression of
bFGF-stimulated M-CSF synthesis in osteoblasts. The schematic
illustration of the mechanism underlying osteoprotegerin and M-CSF
synthesis induced by bFGF, indicating where and how OSM affects
this, is presented as Fig. 5.
In bone metabolism, osteoprotegerin is a pivotal
regulator of osteoclastogenesis and osteoclastic bone resorption to
competitively interrupt RANK and RANKL binding as the decoy
receptor for RANKL (7,8). Thus, the enhancement of
osteoblast-derived osteoprotegerin synthesis by OSM seems to
suppress the accelerated bone resorption in metabolic bone diseases
such as osteoporosis. In addition, M-CSF, which plays a pivotal
role in osteoclastogenesis to promote the proliferation and
differentiation of osteoclast progenitor cells (3,4), is
recognized to activate osteoclastic bone resorption in cooperation
with RANK and RANKL binding (5,6). It
can be hypothesized that OSM-induced downregulation of M-CSF
synthesis by osteoblasts would reduce both the number and activity
of osteoclasts. It is probable that OSM is a potent functional
modulator of osteoblasts to suppress osteoclastic bone resorption
in bone remodeling. In addition, osteocytes, differentiated from
osteoblasts, which are recognized as the most abundant cell type in
bone, also produce the cytokines M-CSF, RANKL and osteoprotegerin
(34). Taking this into account,
the effects of OSM on the synthesis of osteoprotegerin and M-CSF by
bFGF-stimulated osteocytes in addition to osteoblasts needs to be
elucidated to clarify the detailed regulatory mechanism of bone
remodeling. Regarding the effect of OSM on M-CSF synthesis, we
recently reported that TGF-β-stimulated M-CSF synthesis is reduced
by OSM via suppression of p44/p42 MAPK and SAPK/JNK in
osteoblast-like cells (23). As
aforementioned, it is probable that bFGF-elicited activation of
p44/p42 MAPK is not affected by OSM in these cells. Moreover,
SAPK/JNK is likely involved in the M-CSF synthesis induced by bFGF
in these cells. Thus, the mechanisms underlying M-CSF synthesis and
the outcome of OSM effect on the synthesis are dependent on what
stimulates the osteoblasts. Such a precisely regulated signaling
mechanism of M-CSF synthesis and OSM effect might indicate the
importance of both M-CSF synthesis and OSM action in the functions
of osteoblasts in bone remodeling. Osteal macrophages may play a
pivotal role as functional cells in regulating bone remodeling
through OSM, which promotes bone formation. Thus, the present
findings may provide new insights into the mechanism underlying
physiological bone metabolism.
There are several limitations in the present study.
First, the present study could not show that the effects of
SB203580 were not detected in the suppressive effect of OSM on
bFGF-induced M-CSF release. Furthermore, the effects of PD98059 and
SP600125 on the release of osteoprotegerin and M-CSF stimulated by
bFGF and OSM were not detected. However, these experiments may be
unnecessary, because it is unlikely that p44/p42 MAPK is involved
in the effects of OSM, or that SAPK/JNK would be involved in the
down regulation by OSM of bFGF-stimulated M-CSF release.
Furthermore, the findings were not confirmed in other cell lines.
In addition, the spontaneous differentiation of MC3T3-E1 cells
during culture could affect the levels of response to OSM. Further
investigations, including those using primary cultured cells, or
disease and development-related animal models, would be necessary
to clarify the details. The effect of OSM via bFGF-stimulated
osteoblasts on bone remodeling could also be strengthened if the
conditioned medium from osteoblast-like cells treated with OSM and
bFGF had a considerable impact on osteoclastogenesis.
In conclusion, the findings of the present study
strongly suggested that OSM may possess diverse effects on
bFGF-induced osteoblast activation via p38 MAPK and SAPK/JNK,
leading to the amplification of osteoprotegerin synthesis and the
attenuation of M-CSF synthesis. These results may provide novel
insights for bone remodeling.
Acknowledgements
The authors would like to thank Mrs. Yumiko Kurokawa
(Department of Pharmacology, Gifu University Graduate School of
Medicine, Gifu, Japan) for their skillful technical assistance. The
authors would also like to thank Dr. Masayoshi Kumegawa (Graduate
School of Dentistry, Department of Dentistry, Meikai University,
Sakado, Japan) for donating cloned MC3T3-E1 osteoblast-like
cells.
Funding
Funding: The present study was supported in part by
Grants-in-Aid for Scientific Research (grant nos. 19K09370, 19K1847
and 22K09438) from the Ministry of Education, Culture, Sports,
Science and Technology of Japan, and the Research Funding for
Longevity Sciences (grant nos. 20-12 and 21-1) from the National
Center for Geriatrics and Gerontology, Japan.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
OK and HT conceived and designed the experiments.
TH, JT, KU and RMN performed the experiments. TH, RMN, HI, OK and
HT analyzed the data. TH, OK and HT wrote the paper. All authors
read and approved the final manuscript. OK and HT confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Kular J, Tickner J, Chim SM and Xu J: An
overview of the regulation of bone remodeling at the cellular
level. Clin Biochem. 45:863–873. 2012.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Kim JM, Lin C, Stavre Z, Greenblatt MB and
Shim JH: Osteoblast-osteoclast communication and bone homeostasis.
Cells. 9(2073)2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Fuller K, Owens JM, Jagger CJ, Wilson A,
Moss R and Chambers TJ: Macrophage colony-stimulating factor
stimulates survival and chemotactic behavior in isolated
osteoclasts. J Exp Med. 178:1733–1744. 1993.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Fixe P and Praloran V: M-CSF:
Haematopoietic growth factor or inflammatory cytokine? Cytokine.
10:32–37. 1998.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Walsh MC, Kim N, Kadono Y, Rho J, Lee SY,
Lorenzo J and Choi Y: Osteoimmunology: Interplay between the immune
system and bone metabolism. Annu Rev Immunol. 24:33–63.
2006.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Amarasekara DS, Yun H, Kim S, Lee N, Kim H
and Rho J: Regulation of osteoclast differentiation by cytokine
networks. Immune Netw. 18(e8)2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Simonet WS, Lacey DL, Dunstan CR, Kelley
M, Chang MS, Lüthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et
al: Osteoprotegerin: A novel secreted protein involved in the
regulation of bone density. Cell. 89:309–319. 1997.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Theoleyre S, Wittrant Y, Tat SK, Fortun Y,
Redini F and Heymann D: The molecular triad OPG/RANK/RANKL:
Involvement in the orchestration of pathophysiological bone
remodeling. Cytokine Growth Factor Rev. 15:457–475. 2004.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Sinder BP, Pettit AR and McCauley LK:
Macrophages: Their emerging roles in bone. J Bone Miner Res.
30:2140–2149. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zarling JM, Shoyab M, Marquardt H, Hanson
MB, Lioubin MN and Todaro GJ: Oncostatin M: A growth regulator
produced by differentiated histiocytic lymphoma cells. Proc Natl
Acad Sci USA. 83:9739–9743. 1986.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Sims NA and Quinn JMW: Osteoimmunology:
Oncostatin M as a pleiotropic regulator of bone formation and
resorption in health and disease. Bonekey Rep.
3(527)2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Bellido T, Stahl N, Farruggella TJ, Borba
V, Yancopoulos GD and Manolagas SC: Detection of receptors for
interleukin-6, interleukin-11, leukemia inhibitory factor,
oncostatin M, and ciliary neurotrophic factor in bone marrow
stromal/osteoblastic cells. J Clin Invest. 97:431–437.
1996.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Walker EC, McGregor NE, Poulton IJ, Solano
M, Pompolo S, Fernandes TJ, Constable MJ, Nicholson GC, Zhang JG,
Nicola NA, et al: Oncostatin M promotes bone formation
independently of resorption when signaling through leukemia
inhibitory factor receptor in mice. J Clin Invest. 120:582–592.
2010.PubMed/NCBI View
Article : Google Scholar
|
|
14
|
Guihard P, Boutet MA, Brounais-Le Royer B,
Gamblin AL, Amiaud J, Renaud A, Berreur M, Rédini F, Heymann D,
Layrolle P and Blanchard F: Oncostatin m, an inflammatory cytokine
produced by macrophages, supports intramembranous bone healing in a
mouse model of tibia injury. Am J Pathol. 185:765–775.
2015.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Aguirre JI, Leal ME, Rivera MF, Vanegas
SM, Jorgensen M and Wronski TJ: Effects of basic fibroblast growth
factor and a prostaglandin E2 receptor subtype 4 agonist on
osteoblastogenesis and adipogenesis in aged ovariectomized rats. J
Bone Miner Res. 22:877–888. 2007.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Charoenlarp P, Rajendran AK and Iseki S:
Role of fibroblast growth factors in bone regeneration. Inflamm
Regen. 37(10)2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Abboud SL and Pinzani M: Peptide growth
factors stimulate macrophage colony-stimulating factor in murine
stromal cells. Blood. 78:103–109. 1991.PubMed/NCBI
|
|
18
|
Xie Y, Su N, Yang J, Tan Q, Huang S, Jin
M, Ni Z, Zhang B, Zhang D, Luo F, et al: FGF/FGFR signaling in
health and disease. Signal Transduct Target Ther.
5(181)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Suzuki A, Shinoda J, Kanda S, Oiso Y and
Kozawa O: Basic fibroblast growth factor stimulates
phosphatidylcholine-hydrolyzing phospholipase D in osteoblast-like
cells. J Cell Biochem. 63:491–499. 1996.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Kuroyanagi G, Otsuka T, Yamamoto N,
Matsushima-Nishiwaki R, Nakakami A, Mizutani J, Kozawa O and Tokuda
H: Down-regulation by resveratrol of basic fibroblast growth
factor-stimulated osteoprotegerin synthesis through suppression of
Akt in osteoblasts. Int J Mol Sci. 15:17886–17900. 2014.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Tokuda H, Kozawa O and Uematsu T: Basic
fibroblast growth factor stimulates vascular endothelial growth
factor release in osteoblasts: Divergent regulation by p42/p44
mitogen-activated protein kinase and p38 mitogen-activated protein
kinase. J Bone Miner Res. 15:2371–2379. 2000.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Tokuda H, Hirade K, Wang X, Oiso Y and
Kozawa O: Involvement of SAPK/JNK in basic fibroblast growth
factor-induced vascular endothelial growth factor release in
osteoblasts. J Endocrinol. 177:101–107. 2003.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Doi T, Hioki T, Tachi J, Ueda K,
Matsushima-Nishiwaki R, Iida H, Ogura S, Kozawa O and Tokuda H:
Oncostatin M reduces the synthesis of macrophage-colony stimulating
factor stimulated by TGF-β via suppression of p44/p42 MAP kinase
and JNK in osteoblasts. Biomed Res. 43:41–51. 2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Sudo H, Kodama HA, Amagai Y, Yamamoto S
and Kasai S: In vitro differentiation and calcification in a new
clonal osteogenic cell line derived from newborn mouse calvaria. J
Cell Biol. 96:191–198. 1983.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zambelli A, Mongiardini E, Villegas SN,
Carri NG, Boot-Handford RP and Wallis GA: Transcription factor
XBP-1 is expressed during osteoblast differentiation and is
transcriptionally regulated by parathyroid hormone (PTH). Cell Biol
Int. 29:647–653. 2005.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Simpson DA, Feeney S, Boyle C and Stitt
AW: Retinal VEGF mRNA measured by SYBR green I fluorescence: A
versatile approach to quantitative PCR. Mol Vis. 6:178–183.
2000.PubMed/NCBI
|
|
27
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Laemmli UK: Cleavage of structural
proteins during the assembly of the head of bacteriophage T4.
Nature. 227:680–685. 1970.PubMed/NCBI View
Article : Google Scholar
|
|
29
|
Alessi DR, Cuenda A, Cohen P, Dudley DT
and Saltiel AR: PD 098059 is a specific inhibitor of the activation
of mitogen-activated protein kinase kinase in vitro and in vivo. J
Biol Chem. 270:27489–27494. 1995.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Cuenda A, Rouse J, Doza YN, Meier R, Cohen
P, Gallagher TF, Young PR and Lee JC: SB 203580 is a specific
inhibitor of a MAP kinase homologue which is stimulated by cellular
stresses and interleukin-1. FEBS Lett. 364:229–233. 1995.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Bennett BL, Sasaki DT, Murray BW, O'Leary
EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, et
al: SP600125, an anthrapyrazolone inhibitor of Jun N-terminal
kinase. Proc Natl Acad Sci USA. 98:13681–13686. 2001.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Cuenda A and Rousseau S: p38 MAP-kinases
pathway regulation, function and role in human diseases. Biochim
Biophys Acta. 1773:1358–1375. 2007.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Rodríguez-Carballo E, Gámez B and Ventura
F: p38 MAPK signaling in osteoblast differentiation. Front Cell Dev
Biol. 4(40)2016.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Han Y, You X, Xing W, Zhang Z and Zou W:
Paracrine and endocrine actions of bone-the functions of secretory
proteins from osteoblasts, osteocytes, and osteoclasts. Bone Res.
6(16)2018.PubMed/NCBI View Article : Google Scholar
|