Introduction
Cardiac hypertrophy constitutes an adaptive response
to pressure or volume overload, but prolonged cardiac hypertrophy
may cause heart failure, which is implicated in a large number of
cardiac disorders, including myocardial ischemia, myocardial
infarction and hypertrophic cardiomyopathy (1). Therefore, studying the molecular
mechanism of cardiac hypertrophy is beneficial in order for
effective measures to be taken to reduce the morbidity and
mortality of heart disease. There are numerous studies concerning
the therapeutic strategy for cardiac hypertrophy (2,3);
however, the practical therapeutic regimen remains to be explored.
Cardiac hypertrophy has a complex pathophysiology, which involves
numerous signaling pathways and cell factors, including
inflammation, apoptosis and redox reactions (4). Autophagy, an intracellular
self-degradative mechanism to maintain cellular homoeostasis by
degrading and recycling cytoplasmic proteins and organelles, serves
an important role in cardiac remodeling (5). However, the role of autophagy in
cardiac remodeling remains controversial. Numerous reports have
reported that autophagy serves a dual role, with basal autophagy
being adaptive and beneficial, and excessive or insufficient
autophagy being maladaptive and detrimental (6,7).
Therefore, autophagy regulation in cardiac remodeling needs to be
further studied and could provide novel treatment options for heart
failure.
Semaphorin-3A (Sema3A), a neurochemical inhibitor,
serves an important role in the distribution of sympathetic nerves,
and is involved in multiple signaling pathways through interactions
with neuropilin-1 (NP-1), a Semaphorin-3A receptor (8-10).
Sema3A is associated with many diseases, including cancer,
dermatitis, arthritis and kidney disease (11,12).
Our previous study reported that Sema3A is also expressed in
cardiomyocytes and serves a potential role in the regulation of the
induction of ventricular arrhythmia after infarction (13). Recently, it was reported that
Sema3A reduces cardiac inflammation and improves cardiac function
after myocardial infarction (14).
However, the roles of Sema3A in cardiac hypertrophy have not been
thoroughly studied to date.
The present study examined whether Sema3A
overexpression affected cardiac hypertrophy in the context of
isoproterenol (ISO) treatment. The possible mechanisms of Sema3A
involvement in cardiac hypertrophy were also assessed.
Materials and methods
Cell culture and treatment
The H9c2 rat embryonic ventricular myocytes cell
line was purchased from the Shanghai Institute of Biochemistry and
Cell Biology. Cells were cultured in high-glucose DMEM containing
10% fetal bovine serum (FBS) (both Shanghai ExCell Biology, Inc.)
at 37˚C in an incubator with 5% CO2. The ISO-induced
H9c2 cell model was established to study cardiomyocyte hypertrophy.
Cells were pretreated with 10 µM ISO (MilliporeSigma) and 50 nM
rapamycin (Cell Signaling Technology, Inc.) for 24 h before
collection.
Cell transfection
Sema3A overexpression was performed using lentivirus
transfection according to the manufacturer's instructions using a
fourth-generation self-inactivating lentivirus vector (Shanghai
GeneChem Co. Ltd.). H9c2 cells at 20-30% confluence
(~2x105 cells per well) in a 6-well plate, were
transfected with Sema3A-LV or Mock-LV (MOI, 20) containing a
cytomegalovirus-driven green-fluorescent protein (GFP) reporter
gene in 1 ml of a mixture of culture medium (10% FBS in DMEM) and
HiTransG P (1:25). After 15 h of transfection, cardiomyocytes were
cultured in 2 ml DMEM without FBS at 37˚C in an incubator with 5%
CO2. After treatment with ISO and rapamycin according to
the aforementioned method, cells were divided into six groups
including the Control (Mock lentivirus + PBS), Sema3A over (Sema3A
overexpression lentivirus + PBS), Control + ISO (Mock lentivirus +
ISO), Sema3A over + ISO (Sema3A overexpression lentivirus + ISO),
Control + ISO + Rap (Mock lentivirus group + ISO + rapamycin) and
Sema3A over + ISO + Rap (Sema3A overexpression lentivirus + ISO +
rapamycin) groups. Cells treated with ISO and rapamycin were
harvested after 72 h of transfection.
Cell surface area measurement using
rhodamine-phalloidin staining
H9c2 cells grown on coverslips were fixed with 4%
paraformaldehyde in phosphate buffer solution (PBS) for 15 min at
room temperature. After a PBS wash, cells were further incubated
with 0.1% Triton X-100 (Sangon Biotech Co., Ltd.) for 10 min. Cells
were then washed with PBS and incubated with 100 nM
rhodamine-phalloidin for 30 min (Sigma-Aldrich; Merck KGaA) at room
temperature, shielded from light. After three PBS washes, cells
were further stained with 100 nM DAPI (Sigma Aldrich; Merck KGaA)
at 4˚C. An Olympus FV3000 laser scanning confocal microscope
(Olympus Corporation) was used for imaging. Cell surface areas in
randomly selected fields (50 for each group) were assessed using
ImageJ (version 180; National Institutes of Health).
Reverse transcription-quantitative PCR
(RT-qPCR)
Sema3A, brain natriuretic factor (BNF) and β-myosin
heavy chain (β-MHC) mRNA expression levels were quantified using
RT-qPCR. Total RNA from H9c2 cells was extracted using SevenFast
Total RNA Extraction Kit for Cells [Seven Innovation (Beijing)
Biotechnology Co., Ltd.] according to the manufacturer's protocol.
cDNA was synthesized in a 20 µl reaction using the PrimeScript™ RT
reagent Kit containing gDNA Eraser (Perfect Real Time) (Takara Bio,
Inc.). Amplification with TB Green® Premix Ex Taq™ II
(Tli RNaseH Plus) (Takara Bio, Inc.) was performed on an Applied
Biosystems Prism 7500 Fast Sequence Detection System (Applied
Biosystems; Thermo Fisher Scientific, Inc.) with thermocycling at
95˚C for 30 sec followed by 40 cycles of 95˚C for 5 sec and 60˚C
for 34 sec. The primers used were as follows: Sema3A forward (F),
5'-AGACGACAGGATATAAGGAATGG-3' and reverse (R),
5'-GCAGACTACGAAGCAGGAG-3'; BNF forward, 5'-GTGCTGCCCCAG ATGATTCT-3'
and R, 5'-GCAGCTTCTGCATCGTGGAT-3'; β-MHC F,
5'-TGCTCTACAATCTCAAGGAGAGGT-3' and R, 5'-TGTTGACGGTCTTACCAGCTC-3';
and GAPDH F, 5'-GTCGGTGTGAACGGATTTGG-3' and R,
5'-GCTCCTGGAAGATGGTGATGG-3'. All primers were synthesized by
Shanghai GenePharma Co., Ltd. Triplicate reactions were analyzed
using the 2-ΔΔCq method (15), with GAPDH used for
normalization.
Western blotting
The protein expression levels of the
autophagy-related proteins light-chain 3 (LC3), p62 and Beclin-1,
and the Akt/mTOR signaling associated proteins Akt, phosphorylated
(p)-Akt, mTOR, p-mTOR, 4E-binding protein 1 (4EBP1) and p-4EBP1
were assessed using western blotting. H9c2 cells were lysed using
RIPA lysis buffer (Beyotime Institute of Biotechnology) and the
protein concentration was assessed using a BCA Protein Assay Kit
(Thermo Fisher Scientific, Inc.). Equal amounts of total protein
(30 µg per lane) were separated by SDS-PAGE (5% stacking gel, 10%
separating gel) and transferred onto PVDF membranes. After blocking
with 5% nonfat milk at 37˚C for 2 h, the membranes were incubated
with primary antibodies against LC3 (1:2,000; cat. no. 12741S; Cell
Signaling Technology, Inc.), p62 (1:2,000; cat. no. 39749T; Cell
Signaling Technology, Inc.), Beclin-1 (1:2,000; cat. no. 3495S;
Cell Signaling Technology, Inc.), Akt (1:2,000; cat. no. 4691T;
Cell Signaling Technology, Inc.), p-Akt (1:2,000; cat. no. 9271S;
Cell Signaling Technology, Inc.), mTOR (1:2,000; cat. no. 2983T;
Cell Signaling Technology, Inc.), p-mTOR (1:2,000; cat. no. 5536S;
Cell Signaling Technology, Inc.), 4EBP1 (1:2,000; cat. no. 9644T;
Cell Signaling Technology, Inc.), p-4EBP1 (1:2,000; cat. no. 2855S;
Cell Signaling Technology, Inc.) and GAPDH (1:2,500; cat. no.
2118S; Cell Signaling Technology, Inc.) in 5% BSA at 4˚C overnight.
Membranes were then washed three times with TBST (0.1% Tween 20)
and incubated with HRP-Conjugated AffiniPure Goat Anti-rabbit
secondary antibodies (1:2,000; cat. no. BA1054; Wuhan Boster
Biological Technology Co., Ltd.) in 5% non-fat milk at 37˚C for 1
h. After washing, proteins were visualized using an ECL
luminescence kit (cat. no. AR1173; Wuhan Boster Biological
Technology Co., Ltd.). The ChemiDoc XRS System (Bio-Rad
Laboratories, Inc.) was utilized for imaging, and images were
analyzed with Image Lab™ Software 6.1 (Bio-Rad Laboratories, Inc.).
The protein expression levels of LC3, P62, Beclin-1, Akt, p-Akt,
mTOR, p-mTOR, 4EBP1 and p-4EBP1 were normalized to GAPDH
expression.
Statistical analysis
Data are presented as mean ± standard error of the
mean. GraphPad Prism 5.0 (Dotmatics) was used for data analysis.
Two groups and multiple groups were compared using unpaired
Student's t-test and one-way ANOVA followed by Tukey's post hoc
test, respectively. P<0.05 was considered to indicate a
statistically significant difference. Assays were performed in
triplicate.
Results
Sema3A alleviates ISO-induced
cardiomyocyte hypertrophy
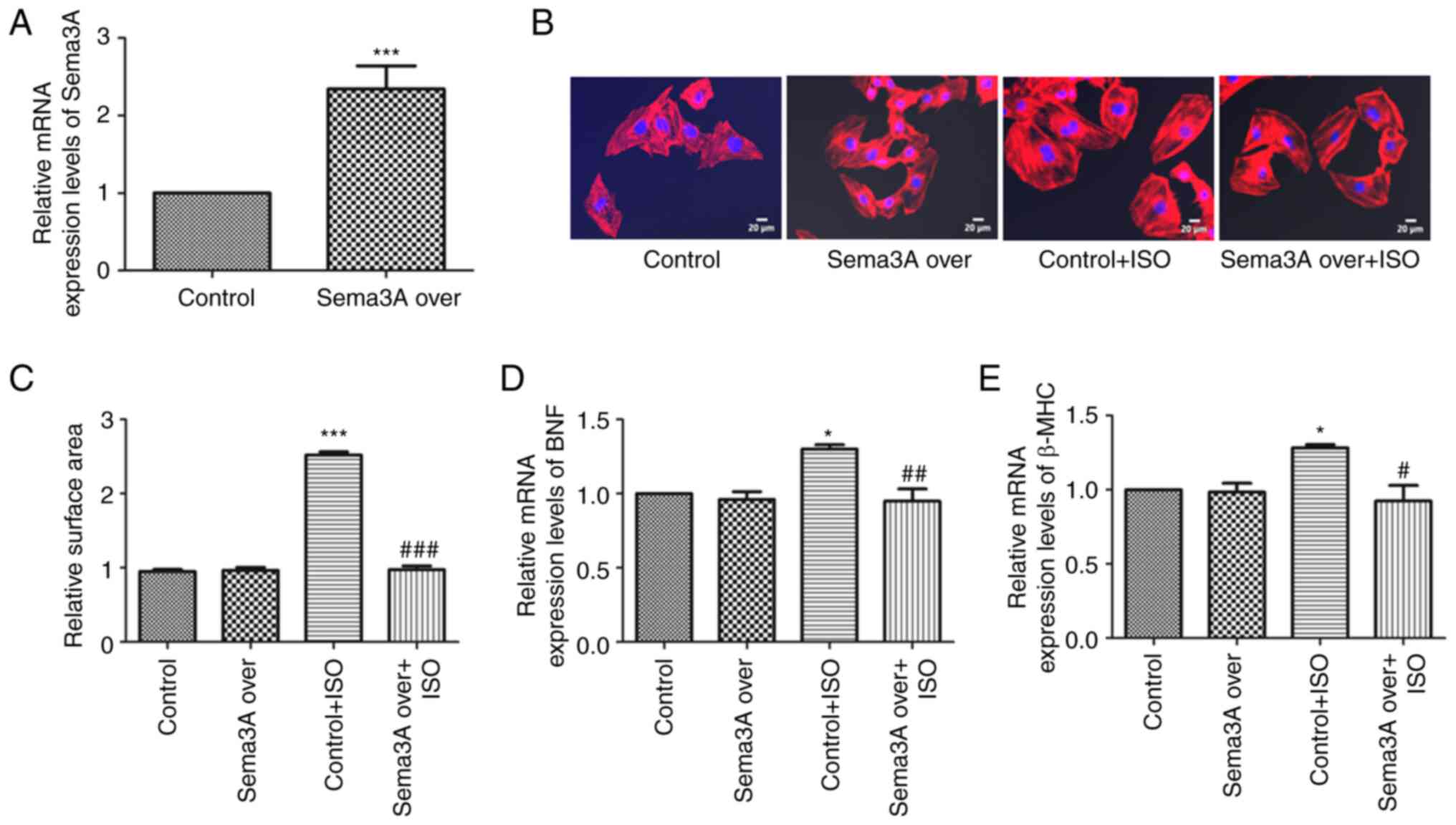
After transfection with Sema3A overexpression
lentivirus for 72 h, Sema3A mRNA expression levels were assessed
using RT-qPCR. Compared with the control group, Sema3A mRNA
expression levels were significantly increased in the Sema3A over
group (Fig. 1A), which indicated
that H9c2 cells transfected with the Sema3A lentivirus
significantly overexpressed Sema3A.
H9c2 cells were incubated with 10 µM ISO for 24 h to
induce cardiomyocyte hypertrophy. Then, BNF and β-MHC mRNA
expression levels were assessed using RT-qPCR, and cardiomyocyte
surface area was assessed using rhodamine-phalloidin staining, to
validate the cardiomyocyte hypertrophic model. ISO significantly
increased the surface area of H9c2 cells, with cell surface areas
in the Control + ISO group being 2.65-fold greater than those of
the Control group. Sema3A overexpression significantly ameliorated
the change induced by ISO on the surface area of myocardial cells,
with cell surface area significantly decreased in the Sema3A over +
ISO group compared with the Control + ISO group (Fig. 1B and C). Similarly, BNF and β-MHC mRNA
expression levels were also significantly increased in cells
treated with ISO compared with the Control group; Sema3A
counteracted this effect, with the Sema3A over + ISO group showing
decreased BNF and β-MHC mRNA expression levels compared with the
Control + ISO group (Fig. 1D and
E). However, Sema3A overexpression
demonstrated no effect on BNF and β-MHC mRNA expression levels, or
cardiomyocyte surface area in H9c2 cells without ISO treatment.
Sema3A attenuates ISO-induced
autophagy in cardiomyocytes
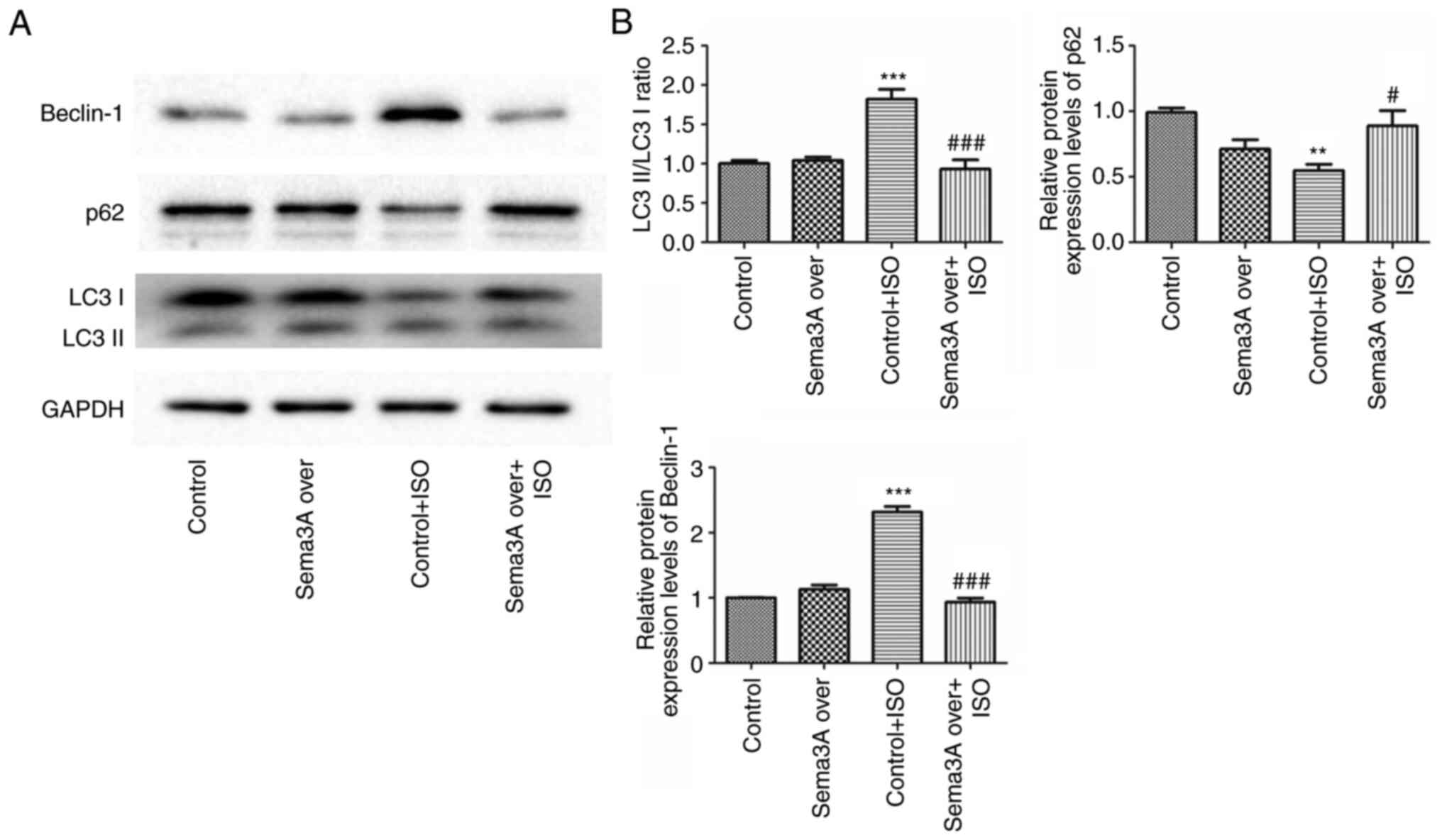
To assess the effect of Sema3A on ISO-induced
autophagy, the protein expression levels of the autophagy-related
proteins LC3, p62 and Beclin-1 were assessed using western
blotting. ISO-induced a significant increase in the LC3-II/I ratio
and Beclin-1 protein expression level and a significant decreased
in the p62 protein expression level in the Control + ISO group
compared with the Control, these effects were all significantly
attended by Sema3A (Fig. 2). In
agreement with the aforementioned results of the present study, the
Sema3A group and non-ISO H9c2 cells had similar autophagy levels.
These results suggested Sema3A alleviated excessive cardiac
autophagy caused by ISO.
Sema3A ameliorates cardiomyocyte
hypertrophy via suppression of autophagy
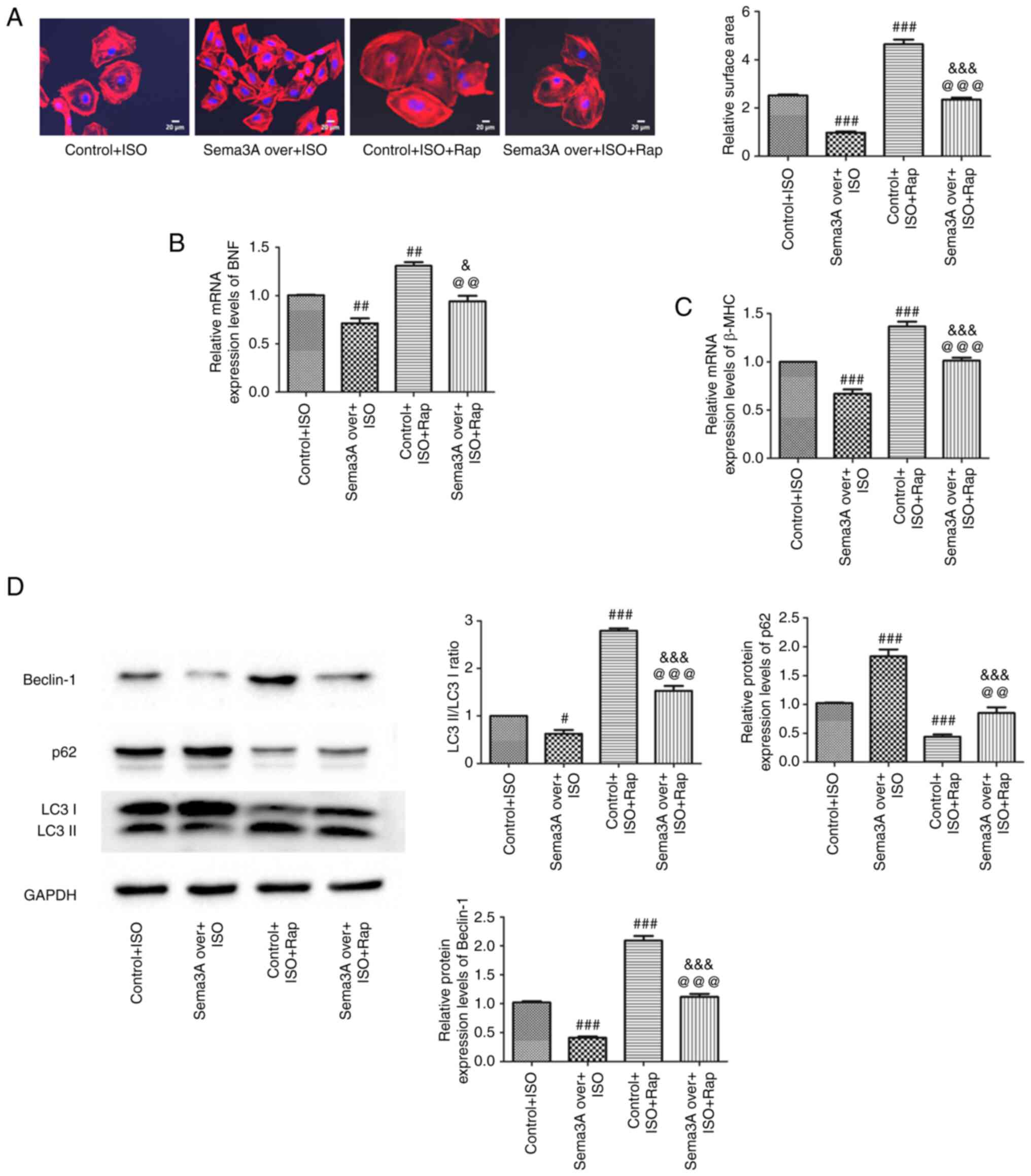
To further evaluate whether autophagy affected by
Sema3A was involved in cardiac hypertrophy, rapamycin, a potent
autophagy agonist, was used to induce cardiac autophagy. In
previous experiments, treatment of cells with ISO was demonstrated
to exacerbate autophagy in the process of cardiomyocyte
hypertrophy, which was repressed by Sema3A upregulation. Increases
in cell surface area, LC3-II/I ratio, and protein expression levels
of Beclin-1, BNF and β-MHC, as well as reduction in p62 protein
expression levels, were all significantly greater in the Control +
ISO + Rap group compared with the Control + ISO group. These data
suggested rapamycin aggravated autophagy induction to worsen
ISO-induced cardiac hypertrophy. Meanwhile, significant reductions
of cell surface area, LC3-II/I ratio and protein expression levels
of Beclin-1, BNF and β-MHC were demonstrated in the Sema3A over +
ISO + Rap group compared with the Control + ISO + Rap group.
Concomitantly, p62 expression was significantly upregulated in the
Sema3A over + ISO + Rap group compared with the Control + ISO + Rap
group. These findings indicated Sema3A upregulation still inhibited
autophagy in myocardial cells and improved cardiomyocyte
hypertrophy under treatment with ISO and rapamycin. Significant
increases were demonstrated for cell surface area and in the
LC3-II/I ratio and Beclin-1, BNF and β-MHC protein expression
levels, as well as a significant decrease in p62 protein expression
levels, in the Sema3A over + ISO + Rap group compared with the
Sema3A over + ISO group (Fig. 3).
Collectively, these results indicated rapamycin partly abolished
the anti-hypertrophy effects of Sema3a by regulating autophagy, and
further demonstrated that Sema3a improved ISO-induced cardiomyocyte
hypertrophy by inhibiting maladaptive cardiomyocyte autophagy.
Sema3A suppresses autophagy by
activating the Akt/mTOR signaling pathway
To evaluate the molecular mechanisms involved in the
Sema3A-induced autophagic responses, protein expression levels of
Akt, p-Akt, mTOR, p-mTOR, 4EBP1 and p-4EBP1 were assessed using
western blotting. Compared with the control group, ISO
significantly decreased p-Akt, p-mTOR and p-4EBP1 levels in H9c2
cells. Sema3A overexpression resulted in a significant increase in
the p-Akt, p-mTOR and p-4EBP1 levels in ISO-treated H9c2 cells
(Fig. 4A). To further assess
whether Sema3A inhibited ISO-induced excessive autophagy via
activation of the Akt/mTOR pathway, the mTOR activator rapamycin
was used. In the present study, the levels of p-Akt, p-mTOR and
p-4EBP1 were significantly decreased in the Control + ISO + Rap
group compared with the Control + ISO group (Fig. 4B). Moreover, p-Akt, p-mTOR and
p-4EBP1 levels were significantly increased in the Sema3A over +
ISO + Rap group compared with the Control + ISO + Rap group, which
indicated that Sema3A overexpression activated the Akt/mTOR
signaling pathway in H9c2 cells after treatment with rapamycin and
ISO. Furthermore, the Sema3A over + ISO + Rap group had
significantly lower levels of p-Akt, p-mTOR and p-4EBP1 compared
with the Sema3A over + ISO group, which suggested that rapamycin
partially abolished the autophagic level change induced by Sem3A
overexpression via inhibition of Akt/mTOR signaling. These results
further demonstrated that Sema3A suppressed ISO‐induced autophagy
through regulation of the Akt/mTOR signaling pathway.
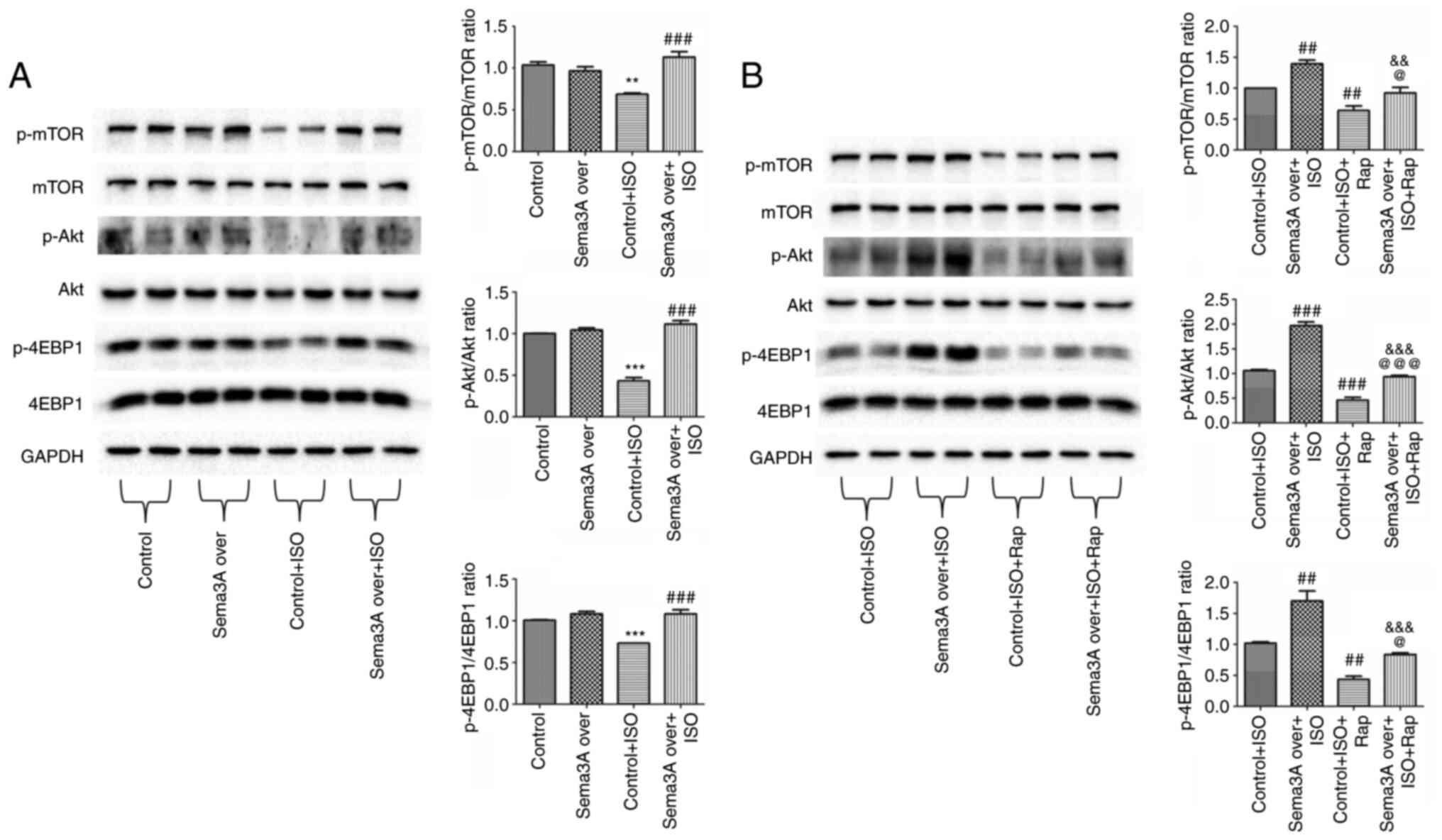 | Figure 4Sema3A inhibits autophagy by
activating the Akt/mTOR signaling pathway. H9c2 cells transfected
with Sema3A overexpression lentivirus were incubated with 10 µM ISO
for 24 h in the absence of (A) rapamycin or (B) after pretreatment
with 50 nM rapamycin. Protein expression levels of p-mTOR, mTOR,
p-Akt, Akt, p-4EBP1 and 4EBP1 were semi-quantified using western
blotting. Protein bands were semi-quantified by densitometry, with
GAPDH used for normalization. Data are presented as mean ± standard
error of the mean. **P<0.01 and
***P<0.001 vs. control; ##P<0.01 and
###P<0.001 vs. control + ISO;
&&P<0.01 and
&&&P<0.001 vs. Sema3A over + ISO;
@P<0.05 and @@@P<0.001 vs. Control +
ISO + Rap. Sema3A over, Semaphorin-3A overexpression lentivirus;
ISO, isoproterenol; Rap, rapamycin; p, phosphorylated; 4EBP1,
eukaryotic translation initiation factor 4E-binding protein 1. |
Discussion
Cardiac hypertrophy, a major pathological feature of
certain cardiovascular disorders, including myocardial ischemia,
diabetic cardiomyopathy and hypertrophic cardiomyopathy, is closely
related to heart failure (16,17).
The mechanisms of cardiac hypertrophy are complex and could be
regulated by numerous factors, including inflammation, apoptosis
and redox reactions (4). Sema3A, a
secreted axon guidance molecule, serves as a chemorepellent during
axonal guidance (11,12). Previous studies have reported that
Sema3A was associated with myocardial injury and malignant
arrhythmia after myocardial injury (18-20).
However, the role of Sema3A in cardiac hypertrophy remains unknown.
In the present study, the regulatory function of Sema3A in
ISO-induced cardiac hypertrophy in cardiomyocytes was
investigated.
The results demonstrated that ISO induced
cardiomyocyte hypertrophy, with markedly increased cardiomyocyte
area. Meanwhile, the protein expression levels of BNF and β-MHC
were significantly higher compared with controls. These data
indicated that Sema3A overexpression significantly decreased
cardiomyocyte size, as well as BNF and β-MHC protein expression
levels in cardiomyocytes, which suggested that Sema3A could protect
cardiomyocytes from ISO-induced hypertrophy. Multiple studies have
assessed Sema3A in cardiovascular disease (13,14,19,21),
but the role of Sema3A remains unclear. Our previous study reported
that Sema3A overexpression after myocardial infarction reduced the
inducibility of ventricular arrhythmia as a result of attenuated
sympathetic reinnervation (13).
Similarly, Wen et al (21)
reported that Sema3A ameliorated electrical remodeling at infarct
border zones after myocardial infarction. Rienks et al
(14) reported that Sema3A
promoted the resolution of cardiac inflammation after myocardial
infarction, to maintain cardiac function. All the previous studies
noted above indicated a protective role for Sema3A in
cardiovascular disease. Another study reported that Sema3A
deficiency improved hypoxia-induced myocardial injury by
alleviating inflammation and cardiomyocytes apoptosis (19). However, little is known about the
function of Sema3A in cardiac hypertrophy. In the present study,
Sema3A inhibited ISO-induced myocardial hypertrophic remodeling.
Cardiomyocyte area and, BNF and β-MHC protein expression levels
demonstrated no significant changes in the Sema3A over group
without ISO treatment, which indicated Sem3A overexpression had no
effect on cardiac function under normal conditions.
Autophagy, a conserved homeostatic process for
recycling cellular waste and biologically active monomers, serves
an important role in pathophysiological changes in numerous
diseases, including cancer, neurodegeneration, aging and heart
disease (22-24).
However, the roles of autophagy in cardiac remodeling and heart
failure remain controversial. Studies assessing the role of
autophagy in ISO-induced cardiac hypertrophy have reported
different outcomes, and whether autophagy activation or inhibition
is protective may depend on the model, stimuli, signaling pathway,
and/or disease stage and severity (25-27).
A recent study suggested that excessive or uncontrolled autophagy
induced by pressure stress can be detrimental (28). In the present study, the autophagic
markers LC3-II/I ratio, Beclin-1 and p62 were assessed to evaluate
autophagic level. It is known that transformation from LC3-I to
LC3-II is important in autophagosome formation and autophagy
activation (29). Beclin-1, also
named Atg6, can positively regulate autophagy by combining with
PI3KCIII and other positive or negative co-factors, which are
required for the initiation of the formation of the autophagosome
in autophagy (30). p62, a linker
between LC3 and ubiquitinated substrates, may be degraded during
autophagy (31). These data
demonstrated that autophagy was significantly enhanced in the ISO
group, indicated by significantly increased LC3II/I ratio and
Beclin-1 protein expression levels and significantly decreased p62
protein expression levels, while Sem3A overexpression significantly
reduced the LC3II/I ratio and Beclin-1 protein expression levels
and significantly increased p62 protein expression levels. As shown
previously, ISO-induced cardiomyocyte hypertrophy and excessive
cardiomyocyte autophagy are greatly attenuated by Sema3A
overexpression. These results indicated a potential protective role
for Sema3A in the development of cardiomyocyte hypertrophy by
normalizing the autophagic process. However, autophagy in the
Sema3A over group without ISO treatment remained unchanged compared
with the control group, which suggested that Sema3A overexpression
did not regulate autophagy under non-diseased conditions. It was
considered that Sema3A may serve as a signaling molecule, which may
not be initiated or activated in the normal myocardial cell
environment, which indicated that Sema3A could affect autophagy and
myocardial hypertrophy in H9c2 cells under pathological conditions,
such as after induction by ISO. Therefore, it could be hypothesized
that Sema3A could exert similar effects in myocardial hypertrophy
induced by stress load in animal models. The role of Sema3A in
cardiac hypertrophy required further, in vivo, study.
The roles of autophagy in cardiac remodeling and
heart failure remain controversial, and autophagy seems to have a
dual effect in cardiac hypertrophy (6), which requires further research. It
has been suggested autophagy is an adaptive response to myocardial
hypertrophy (32,33), and that excessive autophagy
exacerbates stress-induced myocardial hypertrophy (34). A previous study reported that
excessive or uncontrolled autophagy induced by ISO may be harmful.
In cardiac hypertrophy induced by transverse aortic constriction
and angiotensin II (Ang II), Beclin-1 and LC3II/I ratio increased
significantly, and p62 protein levels significantly decreased
compared with the control group. The significant increase in
autophagy suggested that excessive autophagy exacerbated myocardial
hypertrophy (28). Qi et al
(35) reported that the use of
abdominal aortic coarctation (AAC) and Ang II also induced
myocardial hypertrophy and excessive autophagy, and that inhibition
of excessive autophagy could significantly alleviate heart failure
and myocardial hypertrophy. Another study reported that the LC3II/I
ratio increased over time within a 48 h period, in cardiac
hypertrophy induced by ISO (36).
The results of the present study indicated that Sema3A inhibited
excessive autophagy induced by ISO to ameliorate cardiac
hypertrophy. Rapamycin, an autophagy inducer, was used to assess
whether autophagy participated in cardiac hypertrophy alleviated by
Sema3A. In the present study, rapamycin treatment markedly enhanced
autophagy, with rapamycin treatment significantly increasing the
LC3II/I ratio and Beclin-1 protein expression levels, and
significantly downregulating the protein expression levels of p62.
The results showed that Sema3A overexpression reduced cardiomyocyte
area, BNF, β-MHC and Beclin-1 protein expression levels and the
LC3II/I ratio and significantly increased p62 protein expression
levels in cardiomyocytes after ISO treatment. Similar to previous
reports, in the present study Sema3A overexpression exhibited the
same protective effects in cardiomyocytes treated with both ISO and
rapamycin, including the prevention of cardiomyocyte hypertrophy
progression and the inhibition of excessive cardiomyocyte
autophagy. The present study also demonstrated that rapamycin
significantly attenuated p62 upregulation and impeded decreases in
cardiomyocyte area, LC3II/I ratio and Beclin-1, as well as BNF and
β-MHC downregulation induced by Sema3A. In other words, the effects
of Sema3A on cardiomyocyte hypertrophy were partly abolished by
rapamycin via autophagy regulation. These results indicated that
Sema3A ameliorated cardiomyocyte hypertrophy by inhibiting
autophagy.
Autophagy in cardiomyocytes is a complex process
involving numerous signaling pathways. To elucidate the mechanism
by which Sema3A suppressed autophagy, the present study evaluated
the Akt/mTOR signaling pathway causing autophagy inhibition
(37). Activation of Akt/mTOR
signaling, demonstrated by enhanced phosphorylation of Akt, mTOR
and its downstream effector 4EBP1, downregulates autophagy. The
Akt/mTOR signaling pathway serves a vital role in most cellular
processes, while recent studies have reported Akt/mTOR signaling
pathway suppression contributes to autophagy and apoptotic cell
death (38,39). Wang et al (40) also reported that ISO downregulated
p-mTOR, which led to excessive autophagy. Fan et al
(36) reported that ISO could
inhibit activation of the Akt/mTOR signaling pathway, and that the
Traditional Chinese Medicine Qili Qiangxin significantly increased
the levels of p-Akt and p-mTOR to inhibit excessive autophagy and
alleviate ISO induced myocardial hypertrophy. In the present study,
decreased phosphorylation of Akt, mTOR and 4EBP1 was demonstrated
in the Control + ISO group, whereas Sema3A overexpression increased
levels of p-Akt, p-mTOR and p-4EBP1. Rapamycin, an mTOR specific
inhibitor, blunted the effects of Sema3A on the Akt/mTOR signaling
pathway. These results indicated that Sema3A inhibited excessive
autophagy, which may have been via activation of the Akt/mTOR
signaling pathway. In the present study, the effect of Sema3A on
myocardial hypertrophy was only demonstrated in vitro and
further work is required to demonstrate the role of Sema3A in
cardiac hypertrophy in animals. Moreover, numerous signaling
pathways are known to regulate autophagy, so further study of the
molecular mechanisms related to autophagy is also required.
In conclusion, Sema3A alleviated ISO-induced cardiac
hypertrophy, at least in part, by inhibition of excessive cardiac
autophagy via the Akt/mTOR signaling pathway. These findings
suggested Sema3A may be a potential target for the prevention and
treatment of cardiac hypertrophy and heart failure.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YS and JY designed the study. YS, XC and JY
performed the experiments and analyzed the data. YS drafted the
manuscript and interpreted the data. JY and JD revised the
manuscript for important intellectual content. JD, JW and BL
supervised the project and contributed to conception and design. JD
and JY confirm the authenticity of all the raw data. All authors
have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Haque Z and Wang D: How cardiomyocytes
sense pathophysiological stresses for cardiac remodeling. Cell Mol
Life Sci. 4:983–1000. 2017.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Qin W, Du N, Zhang L, Wu X, Hu Y, Li X,
Shen N, Li Y, Yang B, Xu C, et al: Genistein alleviates pressure
overload-induced cardiac dysfunction and interstitial fibrosis in
mice. Br J Pharmacol. 172:5559–5572. 2015.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ma ZG, Dai J, Zhang WB, Yuan Y, Liao HH,
Zhang N, Bian ZY and Tang QZ: Protection against cardiac
hypertrophy by geniposide involves the GLP-1 receptor/AMPKα
signalling pathway. Br J Pharmacol. 173:1502–1516. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Fan W, Zhang B, Wu C, Wu H, Wu J, Wu S,
Zhang J, Yang X, Yang L, Hu Z and Wu X: Plantago asiatica L. seeds
extract protects against cardiomyocyte injury in
isoproterenol-induced cardiac hypertrophy by inhibiting excessive
autophagy and apoptosis in mice. Phytomedicine.
91(153681)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wu QQ, Xiao Y, Yuan Y, Ma ZG, Liao HH, Liu
C, Zhu JX, Yang Z, Deng W and Tang QZ: Mechanisms contributing to
cardiac remodelling. Clin Sci (Lond). 131:2319–2345.
2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Wang EY, Biala AK, Gordon JW and
Kirshenbaum LA: Autophagy in the heart: Too much of a good thing? J
Cardiovasc Pharmacol. 60:110–117. 2012.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Rifki OF and Hill JA: Cardiac autophagy:
Good with the bad. J Cardiovasc Pharmacol. 60:248–252.
2012.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Tamagnone L, Artigiani S, Chen H, He Z,
Ming GI, Song H, Chedotal A, Winberg ML, Goodman CS, Poo M, et al:
Plexins are a large family of receptors for transmembrane,
secreted, and GPI-anchored semaphorins in vertebrates. Cell.
99:71–80. 1999.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Soker S, Miao HQ, Nomi M, Takashima S and
Klagsbrun M: VEGF165 mediates formation of complexes containing
VEGFR-2 and neuropilin-1 that enhance VEGF165-receptor binding. J
Cell Biochem. 85:357–368. 2002.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Bagnard D, Vaillant C, Khuth ST, Dufay N,
Lohrum M, Puschel AW, Belin MF, Bolz J and Thomasset N: Semaphorin
3A-vascular endothelial growth factor165 balance mediates migration
and apoptosis of neural progenitor cells by the recruitment of
shared receptor. Neurosci. 21:3332–3341. 2001.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Neufeld G, Cohen T, Shraga N, Lange T,
Kessler O and Herzog Y: The neuropilins: Multifunctional semaphorin
and VEGF receptors that modulate axon guidance and angiogenesis.
Trends Cardiovasc Med. 12:13–79. 2002.PubMed/NCBI View Article : Google Scholar
|
|
12
|
He Z and Tessier-Lavigne M: Neuropilin is
a receptor for the axonal chemorepellent Semaphorin III. Cell.
90:739–751. 1997.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Chen RH, Li YG, Jiao KL, Zhang PP, Sun Y,
Zhang LP, Fong XF, Li W and Yu Y: Overexpression of Sema3a in
myocardial infarction border zone decreases vulnerability of
ventricular tachycardia post-myocardial infarction in rats. J Cell
Mol Med. 17:608–616. 2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Rienks M, Carai P, Bitsch N, Schellings M,
Vanhaverbeke M, Verjans J, Cuijpers I, Heymans S and Papageorgiou
A: Sema3A promotes the resolution of cardiac inflammation after
myocardial infarction. Basic Res Cardiol. 112(42)2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Balakumar P and Jagadeesh G: Multifarious
molecular signaling cascades of cardiac hypertrophy: Can the muddy
waters be cleared? Pharmacol Res. 62:365–383. 2010.PubMed/NCBI View Article : Google Scholar
|
|
17
|
McKinsey TA and Kass DA: Small-molecule
therapies for cardiac hypertrophy: Moving beneath the cell surface.
Nat Rev Drug Discov. 6:617–635. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
18
|
Liu X, Zhou N, Sui X, Pei Y, Liang Z and
Hao S: Hrd1 induces cardiomyocyte apoptosis via regulating the
degradation of IGF-1R by sema3a. Biochim Biophys Acta Mol Basis
Dis. 1864:3615–3622. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Zhao C, Liu J, Zhang M and Wu Y:
Semaphorin 3A deficiency improves hypoxia-induced myocardial injury
via resisting inflammation and cardiomyocytes apoptosis. Cell Mol
Biol (Noisy-le-grand). 62:8–14. 2016.PubMed/NCBI
|
|
20
|
Ieda M, Kanazawa H, Kimura K, Hattori F,
Ieda Y, Taniguchi M, Lee JK, Matsumura K, Tomita Y, Miyoshi S, et
al: Sema3a maintains normal heart rhythm through sympathetic
innervation patterning. Nat Med. 13:604–612. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
21
|
Wen HZ, Jiang H, Li L, Xie P, Li JY, Lu ZB
and He B: Semaphorin 3A attenuates electrical remodeling at infarct
border zones in rats after myocardial infarction. Tohoku J Exp Med.
225:51–57. 2011.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sagrillo-Fagundes L, Bienvenue-Pariseault
J and Vaillancourt C: Melatonin: The smart molecule that
differentially modulates autophagy in tumor and normal placental
cells. PLoS One. 14(e0202458)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Esteves AR, Palma AM, Gomes R, Santos D,
Silva DF and Cardoso SM: Acetylation as a major determinant to
microtubule-dependent autophagy: Relevance to Alzheimer's and
Parkinson disease pathology. Biochim Biophys Acta Mol Basis Dis.
1865:2008–2023. 2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Nemchenko A, Chiong M, Turer A, Lavandero
S and Hill JA: Autophagy as a therapeutic target in cardiovascular
disease. J Mol Cell Cardiol. 51:584–593. 2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Nakai A, Yamaguchi O, Takeda T, Higuchi Y,
Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, et
al: The role of autophagy in cardiomyocytes in the basal state and
in response to hemodynamic stress. Nat Med. 13:619–624.
2007.PubMed/NCBI View
Article : Google Scholar
|
|
27
|
Xu X and Ren J: Unmasking the janus faces
of autophagy in obesity-associated insulin resistance and cardiac
dysfunction. Clin Exp Pharmacol Physiol. 39:200–208.
2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Liu L, Wang C, Sun D, Jiang S, Li H, Zhang
W, Zhao Y, Xi Y, Shi S, Lu F, et al: Calhex231
ameliorates cardiac hypertrophy by inhibiting cellular autophagy in
vivo and in vitro. Cell Physiol Biochem. 36:1597–1612.
2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Tukaj C: The significance of
macroautophagy in health and disease. Folia Morphol (Warsz).
72:87–93. 2013.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Pankiv S, Clausen TH, Lamark T, Brech A,
Bruun JA, Outzen H, Øvervatn A, Bjørkøy G and Johansen T:
p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of
ubiquitinated protein aggregates by autophagy. J Biol Chem.
282:24131–24145. 2007.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Xu X, Hua Y, Nair S, Bucala R and Ren J:
Macrophage migration inhibitory factor deletion exacerbates
pressure overload-induced cardiac hypertrophy through mitigating
autophagy. Hypertension. 63:490–499. 2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
McMullen JR, Sherwood MC, Tarnavski O,
Zhang L, Dorfman AL, Shioi T and Izumo S: Inhibition of mTOR
signaling with rapamycin regresses established cardiac hypertrophy
induced by pressure overload. Circulation. 109:3050–3055.
2004.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhu H, Tannous P, Johnstone JL, Kong Y,
Shelton JM, Richardson JA, Le V, Levine B, Rothermel BA and Hill
JA: Cardiac autophagy is a maladaptive response to hemodynamic
stress. J Clin Invest. 117:1782–1793. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
35
|
Qi H, Ren J, Ba L, Song C, Zhang Q, Cao Y,
Shi P, Fu B, Liu Y and Sun H: MSTN attenuates cardiac hypertrophy
through inhibition of excessive cardiac autophagy by blocking AMPK
/mTOR and miR-128/PPARγ/NF-κB. Mol Ther Nucleic Acids. 19:507–522.
2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Fan C, Tang X, Ye M, Zhu G, Dai Y, Yao Z
and Yao X: Qi-Li-Qiang-Xin alleviates isoproterenol-induced
myocardial injury by inhibiting excessive autophagy via
activating AKT/mTOR pathway. Front Pharmacol.
10(1329)2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Li L, Xu J, He L, Peng L, Zhong Q, Chen L
and Jiang Z: The role of autophagy in cardiac hypertrophy. Acta
Biochim Biophys Sin (Shanghai). 48:491–500. 2016.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Chen L, Liu P, Feng X and Ma C:
Salidroside suppressing LPS induced myocardial injury by inhibiting
ROS-mediated PI3K/Akt/mTOR pathway in vitro and in vivo. J Cell Mol
Med. 21:3178–3189. 2017.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Tang ZB, Lei Y and Zhang XS: Vitexin
mitigates myocardial ischemia reperfusion-induced damage by
inhibiting excessive autophagy to suppress apoptosis via the
PI3K/Akt/mTOR signaling cascade. RSC Adv. 7:56406–56416. 2017.
|
|
40
|
Wang H, Wang H, Liang EY, Zhou LX, Dong
ZL, Liang P, Weng QF and Yang M: Thrombopoietin protects H9C2 cells
from excessive autophagy and apoptosis in doxorubicin-induced
cardiotoxicity. Oncol Lett. 15:839–848. 2017.PubMed/NCBI View Article : Google Scholar
|