Introduction
Limb-girdle muscular dystrophies (LGMDs) are a major
group of muscular dystrophies with extreme genotypic and phenotypic
heterogeneity (1). LGMDs were
first classified as autosomal dominant (LGMDD4, MIM#618129) or
autosomal recessive (LGMD2A, MIM#253600) by the European
Neuromuscular Centre in 1995(2).
The incidence of LGMD is ~1 in 100,000 live births worldwide with a
variable age of onset (3). LGMD2A
is the most common type of LGMD and accounts for ~30% of all cases;
however, cases of LGMDD4 or calpainopathy are very rarely reported
(4,5). Patients with LGMDD4 usually manifest
with gradual, progressive weakness and atrophy of proximal muscles
more than distal muscles, leading to difficulties in walking and
running, a waddling gait, scapular winging and respiratory failure
at the advanced stages of the disease (6). Furthermore, thigh muscles, pelvic
girdle muscles, periscapular muscles and biceps are the most
affected areas, with effects on the facial muscles rare (6,7).
Patients with LGMDD4 are generally identified by elevated levels of
serum creatine kinase, dystrophic changes in muscle pathology, and
low or reduced expression of calpain 3 (CAPN3) (7). A germline heterozygous mutation in
the CAPN3 gene causes autosomal dominant calpainopathy
(LGMDD4) (8-10).
Among the reported variants of the CAPN3 gene, the most
common are missense mutations (70%) with 30% accounting for
loss-of-function variants, i.e., frameshift, nonsense and splice
site variants (11).
The CAPN3 gene is located on chromosome 15
and encodes the CAPN3 protein (9,10).
CAPN3 is a member of the calpain superfamily and is a
calcium-dependent non-lysosomal cysteine protease (11). CAPN3 is broadly distributed in
myocytes and sarcomeres, and has a significant role in maintaining
calcium homeostasis, regulating muscle contraction and stabilizing
the motility of sarcomere cells (12-14).
CAPN3 also plays a key role in the regulation of cell
differentiation, apoptosis and the cell cycle.
At present, very few LGMDD4 cases have been reported
worldwide (10,15-17).
Vissing et al (10)
reported a novel heterozygous 21-bp in-frame deletion
(c.643_663del21, p.Ser215_Gly221del) in the CAPN3 gene in
European families with LGMDD4. Martinez-Thompson et al
(15) also reported LGMDD4 due to
the same heterozygous 21-bp in-frame deletion (c.643_663del21,
p.Ser215_Gly221del) in the CAPN3 gene in American families.
This deletion is located in domain I (NS domain) of CAPN3 protein
and results in a reduction in the rigidity of domain I and
decreased inter-domain interactions with domain III, facilitating
CAPN3 inactivation. Furthermore, Cerino et al (16) reported a novel CAPN3 variant
(c.1333G>A; p.Gly445Arg) that caused LGMDD4 in patients from
four unrelated families. This variant is located in domain III
(C2-like domain) of CAPN3 protein and causes impairment of the
catalytic activity of mutated CAPN3 protein. This variant interacts
with domain IV (PEF domain), which has a major role in
calcium-binding and homodimerization of CAPN3. Finally,
González-Mera et al (17)
reported five heterozygous CAPN3 missense variants
(c.700G>A, p.Gly234Arg; c.1327T>C, p.Ser443Pro; c.1333G>A,
p.Gly445Arg; c.1661A>C, p.Tyr554Ser and c.1706T>C,
p.Phe569Ser) that caused LGMDD4 in seven unrelated families. The
first variant (p.Gly234Arg) is located at domain I (NS domain) and
induces the loss of the nuclear localization signal. However, all
other variants (p.Ser443Pro, p.Gly445Arg, p.Tyr554Ser and
p.Phe569Ser) are located in domain III and cause impairment of
CAPN3 activation by calmodulin. It is critical that more cases of
LGMDD4 are reported on in the future to gain an improved
understanding of the disease mechanism and inheritance patterns
(18,19).
In the present study, a 16-year-old male patient
with gradual and progressive weakness, and atrophy of the muscles
in both legs was investigated. The younger sister and mother of the
proband exhibited the same phenotypes as the proband. Whole exome
sequencing was performed to identify the disease-causing variant,
followed by functional characterization of this disease-causing
variant to demonstrate its pathogenicity.
Materials and methods
Subjects
A 16-year-old male patient clinically diagnosed with
LGMDD4 from a nonconsanguineous Han Chinese family was investigated
(Fig. 1A). The mother and younger
sister of the proband were also clinically diagnosed with LGMDD4
while the father was phenotypically normal.
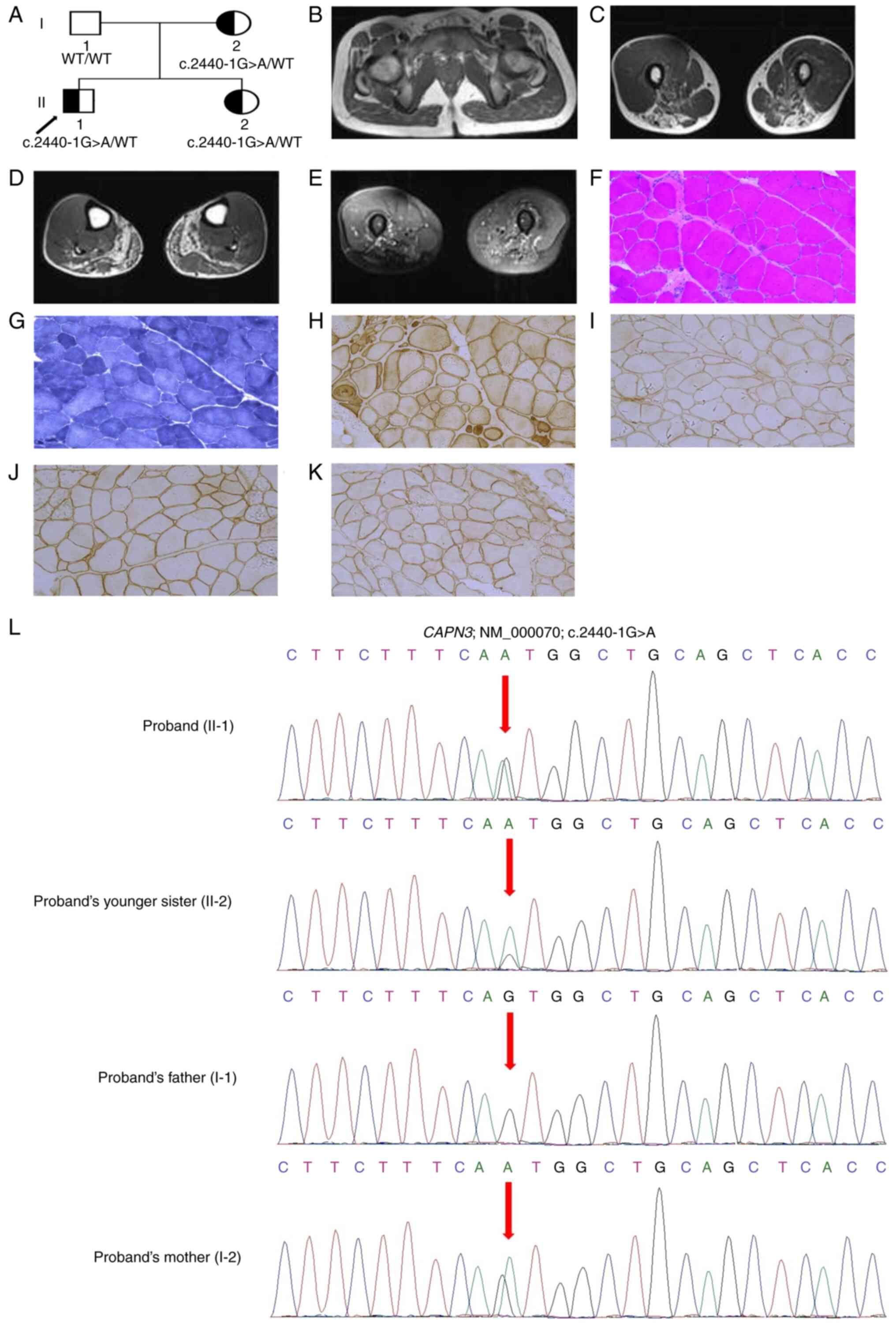 | Figure 1(A) Pedigree of the family. The
squares indicate male patients and the circles indicate female
patients. The half-filled symbols indicate affected patients
(including the proband) and the empty symbols indicate unaffected
healthy individuals. The arrow points to the proband. (B) Muscle
magnetic resonance imaging of the proband showed severe involvement
of adductor magnus, thigh muscles (biceps femoris, semimembranosus
and semitendinosus) and calf muscles (gastrocnemius and soleus).
(C) Hypertrophy of gracilis was identified. (D) Normal appearance
of the rectus femoris, quadriceps femoris and sartorius of thigh
muscles, was identified. (E) Normal appearance of tibialis
anterior, tibialis posterior and extensor digitorum longus of calf
muscles was found. (F-K) Muscle biopsy of the proband, (F) H&E,
(G) NADH, (H) caveolin-3, (I) Dys, (J) α-sarcoglycan and (K)
β-sarcoglycan staining. Total magnification, x200. (L) Partial DNA
sequences in the CAPN3 gene [NM_000070], as determined by
Sanger sequencing of the proband, their younger sister and parents.
Arrows point to the mutation. CAPN3, calpain 3; H&E,
hematoxylin and eosin; NADH, nicotinamide adenine dinucleotide
dehydrogenase; Dys, dystrophin. |
Muscle biopsy
A muscle biopsy was performed for the proband.
Frozen sections of skeletal muscle were evaluated at The
Reproductive Medicine Centre, The First Hospital of Lanzhou
University (Lanzhou, China).
Fresh frozen sections of skeletal
muscle
Excess moisture was first removed from the skeletal
tissue sample simply by blotting thoroughly with a paper towel.
Optimal cutting temperature compound was placed in the bottom of a
shallow cryomold to provide a foundation for the section of muscle.
The skeletal muscle section was carefully placed into the mold.
Isopentane (2-methylbutane), which has a high thermal conductivity,
does not form a vapor halo. Therefore, isopentane chilled with
liquid nitrogen freezes muscle tissues more effectively and evenly
than putting the tissues directly into liquid nitrogen. Therefore,
prechilled isopentane was then chilled and maintained at -140˚C in
a flask by adding liquid nitrogen. Then, the skeletal muscle
section was kept in isopentane for 10-20 sec. Fresh frozen sections
of skeletal muscle were stored at -80˚C. Then, cryo-sectioning of
the frozen sections of skeletal muscles was performed and 10-µm
sections were produced (20).
These slides were processed further for staining.
Hematoxylin and eosin (H&E)
staining
Slides were incubated at room temperature with
hematoxylin solution for 10 min and washed with distilled water.
Next, the slides were stained with eosin solution for 5 min at room
temperature, followed by treatment with ethanol [70% ethanol for 30
sec, 90% ethanol for 30 sec and absolute (100%) ethanol for 1 min]
at room temperature and finally xylene for 3 min at room
temperature (21). Then, the
slides were air-dried, mounted with xylene and stored at room
temperature. Digital microscopy (variation of a traditional
optical/light microscope; total magnification, x200) was used to
capture the H&E-stained images.
Nicotinamide adenine dinucleotide
dehydrogenase (NADH) staining
Firstly, TRIS buffer (0.05 M, pH 7.6), NADH
solution, Nitro blue tetrazolium (NBT) solution and acetone
de-staining solutions (30, 60 and 90%) were prepared. The NADH
solution was gently thawed and 5 ml NBT solution was added to it.
The slides were incubated in this solution for 30 min at 37˚C and
washed three times with deionized water. The slides were washed
again with acetone de-staining solutions first in increasing and
then in decreasing concentrations. Finally, the slides were rinsed
three times with deionized water and mounted with glycerol and
phenol (22). A light microscope
(total magnification, x200) was used to capture the images.
Immunohistochemistry analysis
The frozen skeletal tissue block was transferred to
a cryotome cryostat at -20˚C prior to sectioning and the
temperature of the frozen skeletal muscle tissue block was allowed
to equilibrate to the temperature of the cryotome cryostat.
Sections of the frozen skeletal muscle tissue block of the desired
thickness (3, 5 and 8 µm) were prepared. Then, the skeletal muscle
tissue sections were placed onto glass slides suitable for
immunohistochemistry. Next, these sections were dipped in distilled
water and treated with 5% hydrogen peroxidase for endogenous
peroxidase blocking and incubated for 30 min at room temperature.
Then, these sections were incubated with BondTM Primary Antibody
Diluent 0.5L (cat. no. AR9352; Leica Microsystems, Inc.) at room
temperature for 30 min for non-specific background blocking.
Immunohistochemical analysis of skeletal muscle samples was
performed with four primary antibodies (dystrophin, α-sarcoglycan,
β-sarcoglycan and caveolin-3) and Chromogenic Multiplex IHC for
BOND RX/RXm (Leica Microsystems, Inc.) was used as
detection agent (23,24). Anti-Mouse UltraPolymer HRP
(2MH-050: Anti-Mouse UltraPolymer HRP 50 ml; Cell IDx; Leica
Microsystems, Inc.) was used to increase the sensitivity and signal
amplification. The following primary antibodies were used:
Dystrophin (dilution, 1:100; cat. no. ab275391; Abcam; 60 min at
25˚C), α-sarcoglycan (dilution, 1:1,000; cat. no. ab189254; Abcam;
60 min at 25˚C), β-sarcoglycan (dilution, 1:100; cat. no. ab135954;
Abcam; 60 min at 25˚C) and caveolin-3 (dilution, 1:1,000; cat. no.
ab289544; Abcam; 60 min at 25˚C). The following secondary antibody
was used: Anti-Rabbit IgG H&L (HRP) (dilution, 1:2,000; cat.
no. ab205718; Abcam; 10 h at 4˚C). A light microscope (total
magnification, x200) was used to capture the images.
Whole exome sequencing
A blood sample was collected from the proband and
genomic DNA was extracted (QIAamp DNA Blood Mini Kit; Qiagen GmbH)
according to the manufacturer's instructions. The genomic DNA of
the proband was subjected to whole exome sequencing (25). Illumina® DNA Prep with
Exome 2.5 Enrichment (cat. No. 20025524; Illumina, Inc.) was used
to prepare DNA samples for whole exome sequencing. DNA was
quantified using Qubit Fluorometric Quantitation (Thermo Fisher
Scientific, Inc.). The quality of DNA was assessed by 1.5% agarose
gel electrophoresis and visualized by ethidium bromide with using a
GelDoc Go Gel Imaging System (Bio-Rad Laboratories, Inc.). The
recommended read length of whole exome sequencing was 2x150 bp.
SureSelect Human All Exon v6 (cat. No. 5190-8864, Agilent
Technologies, Inc.) was used to capture the sequences. The
sequencing library was prepared and the enriched library was
subjected to whole exome sequencing on an Illumina HighSeq4000
(HiSeq 3000/4000 SBS Kit; cat. no. FC-410-1003; Illumina, Inc.).
The loading concentration of the final sequencing library was
250-300 pM as determined using the Kapa Library quantification kit
(part no. KK4824; Roche Diagnostics) and size-corrected using
fragment analysis. Sequencing of both the forward and reverse
strands was performed (paired end). After sequencing, the alignment
of sequencing reads was performed using Burrows-Wheeler Aligner
software (v0.59; https://bio-bwa.sourceforge.net/). Then, local
realignment of the Burrows-Wheeler-aligned reads was performed by
Genome Analysis Toolkit (GATK) IndelRealigner (26). Next, base quality recalibration of
the Burrows-Wheeler-aligned reads was carried out using GATK
BaseRecalibrator (26). Next,
single-nucleotide variants (SNV) and insertions or deletions were
identified using GATK Unified Genotyper (26). Then, these variants were annotated
with the Consensus Coding Sequences Database (https://www.ncbi.nlm.nih.gov/CCDS/CcdsBrowse.cgi)
at the National Centre for Biotechnology Information (NCBI).
Illumina pipeline was used for image analysis and base calling. In
addition, indexed primers were designed and used for data fidelity
surveillance. SOAP aligner (soap2.21) software (27) was used to align the clean
sequencing reads with human reference genome (hg19). Lastly, to
assemble the consensus sequence and call genotypes in target
regions, SOAPsnp (v1.05) software (28) was used.
Bioinformatics data analysis and
interpretation
Variants obtained from whole exome sequencing were
collected. Variants with minor allele frequency (MAF) of <0.01
in dbSNP, HapMap, 1000 Genomes Project and our in-house database of
50,000 Chinese Han samples were selected. Public databases, namely,
dbSNP (https://www.ncbi.nlm.nih.gov), HapMap
(https://www.genome.gov), 1000 Genome Database
(http://www.internationalgenome.org)
and our in-house database for 50,000 Chinese Han samples were used
(29-31).
All of these databases were used to identify the MAFs of genetic
variations in different populations. Using the MAF of a genetic
variant in a specific population, the possible pathogenicity of
this genetic variant for a specific case can be interpreted. The
Human Gene Mutation database (HGMD, www.hgmd.cf.ac.uk/) contains all the reported genetic
variants associated with monogenic disorders. Hence, by using this
database, it can be interpreted whether the identified genetic
variant is a novel or previously reported cause of any disease
(32). Online Mendelian
Inheritance in Man (OMIM, https://www.omim.org) is a highly comprehensive and
freely available database, comprised of >16,000 human genes
associated with all known Mendelian disorders (33). This database also provides
information regarding genotype-phenotype correlation. Exome
Aggregation Consortium (ExAC, http://exac.broadinstitute.org) contains exome
sequencing data from largescale sequencing projects from different
populations worldwide (34). Thus,
the possible pathogenicity of identified genetic variants was
confirmed by comparing their frequencies in different populations.
Genome Aggregation Database (gnomAD, https://gnomad.broadinstitute.org) also comprises both
genome and exome sequencing data from large-scale genome or exome
sequencing projects from different populations around the world
(35). Therefore, by comparing the
frequencies of any reported genetic variant in different
populations, the pathogenicity of the identified genetic variants
in the present study was interpreted.
Variant interpretation was performed based on the
variant interpretation guidelines of American College of Medical
Genetics and Genomics (ACMG; Bethesda, Maryland, USA) (36). All the heterozygous, homozygous and
compound heterozygous variants were selected based on gene function
and disease association by OMIM and the literature. The expression
of variants was confirmed using ‘Mutalyzer 2.0.35’ software
(https://v2.mutalyzer.nl/) which strongly follows
the Human Genome Variation Society guidelines (37). The whole exome sequencing quality
control data are described in Table
I.
 | Table IQuality control data of whole exome
sequencing. |
Table I
Quality control data of whole exome
sequencing.
| Sequencing quality
control data | Value |
|---|
| Raw reads (mapped
to hg19) | 9,487,685 |
| Raw data yield,
Mb | 850.26 |
| Reads mapped to
target region | 5,978,717 |
| Reads mapped to
flanked 100 bp region | 6,269,868 |
| Data mapped to
target region, Mb | 488.78 |
| Data mapped to
flanked 100 bp region, Mb | 498.98 |
| Length of target
region, bp | 887,989 |
| Length of flanked
100 bp region, bp | 979,880 |
| Number of covered
bases on target region | 804,587 |
| Coverage of target
region, % | 99.87 |
| Number of covered
bases on flanked 100 bp region | 987,890 |
| Coverage of flanked
100 bp region, % | 99.92 |
| Average (mean)
sequencing depth of target region (30X) | 590.89 |
| Average (mean)
sequencing depth (30X) of flanked 100 bp region | 486.98 |
Sanger sequencing
Sanger sequencing was performed to validate the
possible disease-causing mutations identified by whole exome
sequencing. Primers for polymerase chain reaction (PCR) were
designed based on the reference genomic sequences of the Human
Genome from GenBank (NCBI; https://www.ncbi.nlm.nih.gov/genbank/). PCR products
were subjected to Sanger sequencing. Sanger sequencing data were
compared and analyzed.
The heterozygous novel mutation identified through
whole exome sequencing was validated by Sanger sequencing. Sanger
sequencing was performed with the following primers: Forward
(F)'-5'-TGGTGGAGGGAAGGGATAGG-3'; reverse
(R)5'-TGGCAAAGGGACAAGGGTTT-3'; the reference sequence NM_000070 of
CAPN3 gene was used.
Reverse transcription-PCR (RT-PCR) and
Sanger sequencing
In order to understand the effect of the novel
splice-site mutation of the CAPN3 gene on the splicing event
of CAPN3 mRNA, RT-PCR followed by Sanger sequencing was
performed. Muscle samples were collected from the proband and their
family members, and total RNA was extracted using
RNAqueous™ Total RNA Isolation Kit (Thermo Fisher
Scientific, Inc.) and reverse transcribed to cDNA using PrimeScript
cDNA Synthesis Kit (cat. no. RR037A; Takara Biotechnology, Co.,
Ltd.) according to the manufacturer's recommendations. cDNA from
the proband and their family members were amplified with the use of
primers (F, 5'-CTGCTTCGTTAGGCTGGAGG-3'; R,
5'-GAAGCCTGTAGGGGTGTAGC-3') encompassing the coding sequence from
exon 22 to exon 24. Then, the amplified cDNAs were run on a 2%
agarose gel (2 g agarose powder was mixed with 100 ml 1xTAE in a
microwavable flask and the gel was made). purified using
DNAclear™ Purification Kit (Thermo Fisher Scientific,
Inc.). Finally, Sanger sequencing was performed as aforementioned
for the proband and family members and the data were analyzed.
Relative expression of CAPN3 mRNA
Reverse-transcribed cDNA was collected for
fluorescence quantitative detection by quantitative (qPCR). The
One-Step TB Green® PrimeScript™ RT-PCR Kit II
(Perfect Real Time; cat. no. RR086A; Takara Biotechnology Co.,
Ltd.) was used. The thermocycling conditions were as follows: 15
min at 95˚C to activate the chemically modified hot-start Taq DNA
polymerase, followed by 35-45 cycles of a 15-sec denaturation at
95˚C and 60 sec annealing and extension at 60˚C. The target gene
and housekeeping gene (GAPDH; F-5'-GTCTCCTCTGACTTCAACAGCG-3' and
R-5'-ACCACCCTGTTGCTGTAGCCAA-3') of each sample were subjected to
(q)PCR. The change in RNA expression based on the detected Cq value
was calculated. Primers used in qPCR were as follows: CAPN3
F, 5'-CTGCTTCGTTAGGCTGGAGG-3'; R, 5'-GAACTTACTTGAGGCATATG-3'. The
RNA sample from the normal healthy individual was collected from
our hospital's sample bank (The Reproductive Medicine Centre Sample
Bank, The First Hospital of Lanzhou University, Lanzhou, China).
The hospital's sample bank (The Reproductive Medicine Centre Sample
Bank, The First Hospital of Lanzhou University, Lanzhou, China)
collected the RNA sample from the normal healthy individual after
getting written informed consent. Data were analysed using the
comparative threshold cycle (2-ΔΔCq) method (38).
Results
Clinical description of proband
(II-1)
The proband, a 16-year-old male patient from China,
manifested with gradual and progressive weakness of upper and lower
extremities. The clinical symptoms first appeared at 8 years old
with slow, progressive weakness of muscles and an abnormal gait. At
10 years old, the proband presented with slow, progressive weakness
of lower limbs with frequent falls, difficulty in standing, walking
and climbing stairs. At the age of 16 years, physical testing
revealed that the proband could only walk unaided for <15 min
and that the proband was unable to raise their arms above their
head. No sensory, ocular or bulbar abnormalities were identified,
and neurological examination showed severe weakness of proximal
muscles in all limbs, pelvic and shoulder girdles. At the age of 16
years, the proband had a normal mental status with no oculomotor or
facial abnormalities with sensory and coordination examinations
also finding no abnormalities. In the present study, bilateral
atrophy of the biceps, shoulder muscles, hip adductors, posterior
thigh muscles and knee flexors, as well as moderate hypertrophy on
both sides of the calves and scapular winging, was reported. In
order to assess muscle strength, the Medical Research Council (MRC)
Scale was used (39). The MRC
scale is divided into five grades (grade 5, normal muscle; grade 4,
movement of the limb against gravity and resistance; grade 3,
movement of the limb against gravity over the full range; grade 2,
movement of the limb but not against gravity; grade 1, visible
contraction without movement of the limb; grade 0, no visible
contraction). There were six muscles examined (shoulder abductors,
elbow flexors, wrist extensors, hip flexors, knee extensors and
foot dorsiflexors) in both the upper and lower limbs on both left
and right sides, each with a score from 0 to 5. Hence, the total
score ranged from 60 (normal) to 0 (quadriplegic) (39). The muscle power of proximal upper
and lower extremities was grade 3, while distal upper and lower
extremities was 4+ according to the MRC scale. Moreover, deep
tendon reflexes were normal in the proband's arm while no deep
tendon reflexes were found in their lower extremities. Gowers' sign
was used to ascertain the weakness of the muscle in the pelvic
girdle or proximal muscle in lower limbs (40). This sign is a medical term that
describes the strength of hip and thigh muscle, and is often
identified amongst patients in the advanced stages of muscular
dystrophies. The presence of Gowers' sign with decreasing plantar
reflexes was identified in the proband. No abnormalities were found
in the nerve conduction study; however, an electromyographic study
revealed chronic myopathy. Thyroid tests were in the normal range;
however, laboratory tests identified highly elevated levels (4,754
IU/l; normal, 35-232 IU/l) of serum creatine kinase. Normal levels
of serum lactate and transaminases were observed in the proband. No
abnormalities were identified in the electrocardiogram and
echocardiogram, and the pulmonary function test (PFT) was
normal.
According to these aforementioned clinical
presentations, the proband was clinically diagnosed with LGMD.
Proband's younger sister (II-2)
The younger sister of the proband, a 14-year-old
girl from China, manifested with progressive weakness of muscles
since childhood. They presented with similar clinical symptoms,
disease onset and progression as the proband. At 7 years old, they
demonstrated difficulty standing, walking or running with frequent
falls. These clinical symptoms gradually and progressively
developed, and at 10 years old, they had exercise intolerance and
difficulty standing from the sitting position independently. They
developed weakness in both of the upper and lower extremities and
finally lost the ability to ambulate independently at 11 years old.
They also showed atrophy of both bilateral biceps and quadriceps
with neurological examination showing weakness in the limb-girdle
muscles. Their muscle strength was gradually and progressively
reduced in the upper extremities of the proximal muscle, and lower
extremities of both the proximal and distal muscles. Absence or
diffusely reduced deep tendon reflexes were identified in the lower
extremities; however, no abnormalities were found in extraocular
movements and cognition. Diffuse atrophy in all upper and lower leg
muscles was identified, PFT found no abnormalities, and physical
and neurological examinations revealed that the muscle power of the
proximal upper and lower extremities was grade 3 and 2,
respectively, according to the MRC scale. The presence of Gowers'
sign, as well as pelvic girdle and leg muscle atrophy, was
observed; however, no abnormalities in facial muscles were
identified. Laboratory tests found elevated levels (1,050 IU/l;
normal, 35-232 IU/l) of serum creatine kinase; however, the levels
of serum lactate and transaminases were normal. Electrocardiogram
and echocardiogram showed no cardiac abnormalities, and no
abnormalities were found in nerve conduction velocity.
Proband's mother (I-2)
The mother of the proband was a 42-year-old woman
from China that presented with gradual, progressive weakness, and
atrophy of both proximal and distal muscles. At 14 years old, they
presented with difficulty running, climbing stairs, standing up
from a sitting position and a waddling gait. At 18 years old, they
struggled to walk independently and lift weights, and by their
early twenties, they had lost the ability to walk independently,
showing a complete loss of ambulation. They had an unremarkable
medical history with normal sensation, no intellectual
disabilities, and had never been reported to have fasciculations or
pseudo-hypertrophy of calf muscles or joint contractures. However,
neurological examination showed weakness and atrophy of limb-girdle
muscles and an absence of all deep tendon reflexes. Laboratory
tests found elevated levels (2,000 IU/l; normal, 35-232 IU/l) of
serum creatine kinase. No abnormalities were identified in the
levels of serum lactate and transaminases. The muscle power of both
proximal upper and lower extremities, and distal upper and lower
extremities were grade 2 according to the MRC scale, and the
presence of Gowers' sign with atrophy of limb-girdle muscles was
identified. No cardiac, motor or sensory nerve conduction
abnormalities were identified; however, typical myopathic right
biceps, severe atrophy of lower limbs and atrophy of paraspinal
muscle were identified. At 30 years old, they developed back pain,
hyperlordosis and myalgia, and both their proximal limb and
abdominal wall muscles became weak. At present, they have
manifested with paraspinal muscle atrophy.
Magnetic resonance imaging (MRI) of
the muscle
MRI of the muscles of the proband was performed. The
MRI revealed significant involvement of adductor magnus, thigh
muscles (biceps femoris, semimembranosus and semitendinosus) and
calf muscles (gastrocnemius and soleus) (Fig. 1B-E). Hypertrophy of the gracilis
was detected; however, normal appearance of the rectus femoris,
quadriceps femoris and sartorius of the thigh muscle was
identified. Moreover, normal appearance of the tibialis anterior,
tibialis posterior and extensor digitorum longus of the calf muscle
was reported.
Clinical descriptions of the proband, their younger
sister and mother have been comprehensively summarized in Table II.
| Table IIClinical characteristics of the
proband, their younger sister and mother. |
Table II
Clinical characteristics of the
proband, their younger sister and mother.
| Clinical
characteristics | Proband (II-1) | Proband's younger
sister (II-2) | Proband's mother
(I-2) |
|---|
| Age, years | 16 | 14 | 42 |
| Sex | Male | Female | Female |
| Age of onset,
years | 8 | 7 | 14 |
| Clinical
symptoms | Difficulty in
standing, walking, climbing stairs, walking unaided for <15 min
and inability to raise arms above head. | Difficulty in
standing, walking, climbing stairs or running with frequent falls,
exercise intolerance, and difficulty rising from the floor or
standing from the sitting position independently. | Difficulty in
running, climbing stairs, walking independently, lifting weights,
standing up from sitting position and a waddling gait. |
| Neurological
examination | Severe weakness of
the proximal muscles of all limbs, pelvic and shoulder
girdles. | Weakness and
atrophy of limb-girdle muscles. | Weakness and
atrophy of limb-girdle muscles. |
| Muscle weakness and
atrophy | Bilateral atrophy
of the biceps, shoulder muscles, hip adductors, posterior thigh
muscles and knee flexors and moderate hypertrophy on both sides of
the calves and scapular winging. | Diffuse atrophy in
all upper and lower leg muscles. | Typical myopathic
right biceps, severe atrophy of lower limbs and atrophy of
paraspinal muscle, back pain, hyperlordosis and myalgia. |
| Muscle power (MRC
Scale) | Proximal upper and
lower extremities were grade 3, while distal upper and lower
extremities were 4+. | Proximal upper and
lower extremities and proximal lower extremities were grade 3 and
2, respectively. | Proximal upper and
lower extremities were grade 2, and distal upper and lower
extremities were grade 2. |
| Deep tendon
reflexes | Normal in arms;
absent in lower extremities | Absent or diffusely
reduced in lower extremities. | Absent of all. |
| Gowers' sign | Present with
decreasing plantar reflexes. | Present with pelvic
girdle and leg muscle atrophy. | Present with
limb-girdle muscle atrophy. |
| Creatine kinase
levels, IU/l | 4,754 | 1,050 | 2,000 |
Muscle biopsy pathology
Muscle biopsies of the proband showed mild to
moderate dystrophic changes with H&E staining (Fig. 1F). These changes included increased
fiber size variation with scattered atrophic and hypertrophic
fibers, necrotic fibers undergoing myophagocytosis, grouped
regeneration, endomysial fibrosis and fatty replacement (Fig. 1F). NADH staining revealed the
mildly disorganized structure of the intermyofibrillar network
(Fig. 1G) The immunostaining of
caveolin-3, dystrophin, α-sarcoglycan and β-sarcoglycan indicated
normal expression (positive staining) (Fig. 1H-K).
According to the aforementioned clinical symptoms
and test results, the clinical diagnosis, therapeutic interventions
and disease management were confirmed for the studied family.
Identification of a novel mutation in
the CAPN3 gene
Whole exome sequencing and Sanger sequencing
identified a novel heterozygous splice-acceptor site mutation
(c.2440-1G>A) in intron 23 of the CAPN3 gene in the
proband. Sanger sequencing confirmed that this heterozygous novel
mutation was also present in the mother and younger sister of the
proband, while the father did not harbor the mutation (Fig. 1L).
The mutation was not present in 100
ethnically-matched normal healthy controls from our in-house
database. This previously generated in-house control database
collected genomic DNA samples from 100 normal healthy individuals,
after getting their written informed consent, and performed whole
exome sequencing to obtain raw sequencing data, followed by
bioinformatics data analysis and interpretation. This normal
control database has been used as a reference database, thus if any
novel variant is identified in any gene, we can compare whether it
is already present in the in-house database in order to understand
its pathogenicity. This mutation was also absent in the HGMD, OMIM,
ExAC, dbSNP, gnomAD and 1000 Genome Database, as well as our
in-house database, which consists of ~50,000 Chinese Han
samples.
The mutation (c.2440-1G>A) is classified as
‘likely pathogenic’ [absent in population database (PM2) +
predicted null variant in a gene where loss-of-function is a known
mechanism of disease (PVS1) + co-segregation of disease in multiple
affected family members (PP1)] according to the variant
interpretation guidelines of ACMG (36). The mutation (c.2440-1G>A) is
co-segregated with the disease phenotype in this family with an
autosomal dominant mode of inheritance.
Functional characterization of the
novel splice-acceptor site mutation
Individuals with wild-type (father of the proband)
and mutated (proband, their mother and younger sister) CAPN3
underwent PCR for amplification of CAPN3 cDNA encompassing
the coding sequence (exon 22-24), which was run on a 2% agarose
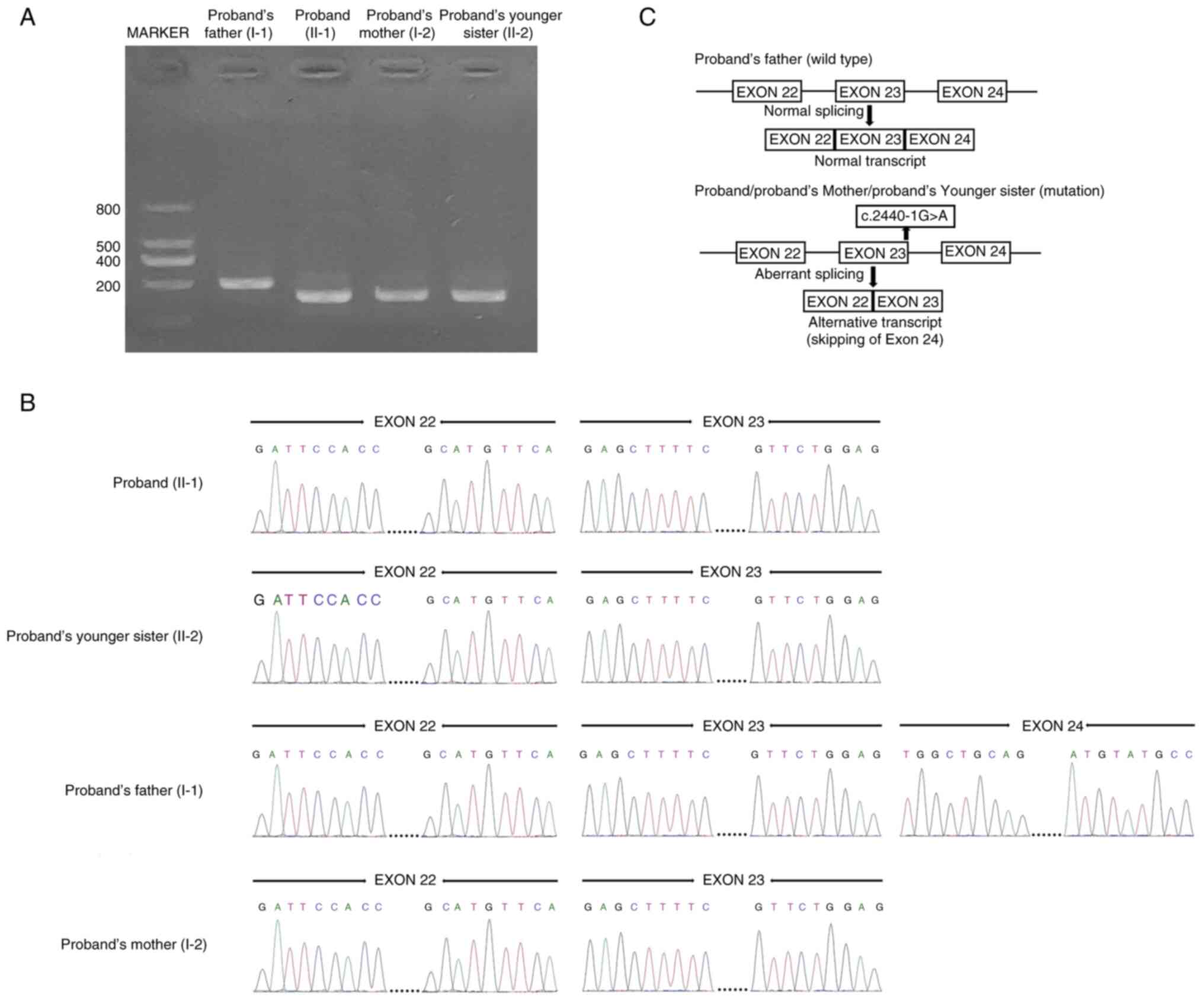
gel. A 200-bp band for wild-type CAPN3 cDNA was found,
whereas a 176-bp band for mutated CAPN3 cDNA was identified
(Fig. 2A).
Sanger sequencing of mutated cDNA from the proband,
their mother and younger sister revealed that the mutation
(c.2440-1G>A) at the last nucleotide of intron 23 of the
CAPN3 gene disrupts the wild-type CAPN3 exon 24
splice acceptor-site and leads to aberrant splicing of CAPN3
mRNA, finally resulting in the skipping of exon 24 (24 bp).
Complete loss of exon 24 causes the formation of a truncated CAPN3
protein (p.Trp814*) with the removal of eight amino acids from the
C-terminal (Fig. 2B). However,
Sanger sequencing of the wild-type CAPN3 cDNA showed normal
splicing of exon 22 to exon 24 (Fig.
2B). The splicing mechanism of both wild-type and mutated cDNA
is schematically presented (Fig.
2C).
Relative expression of CAPN3 mRNA
determined by RT-qPCR
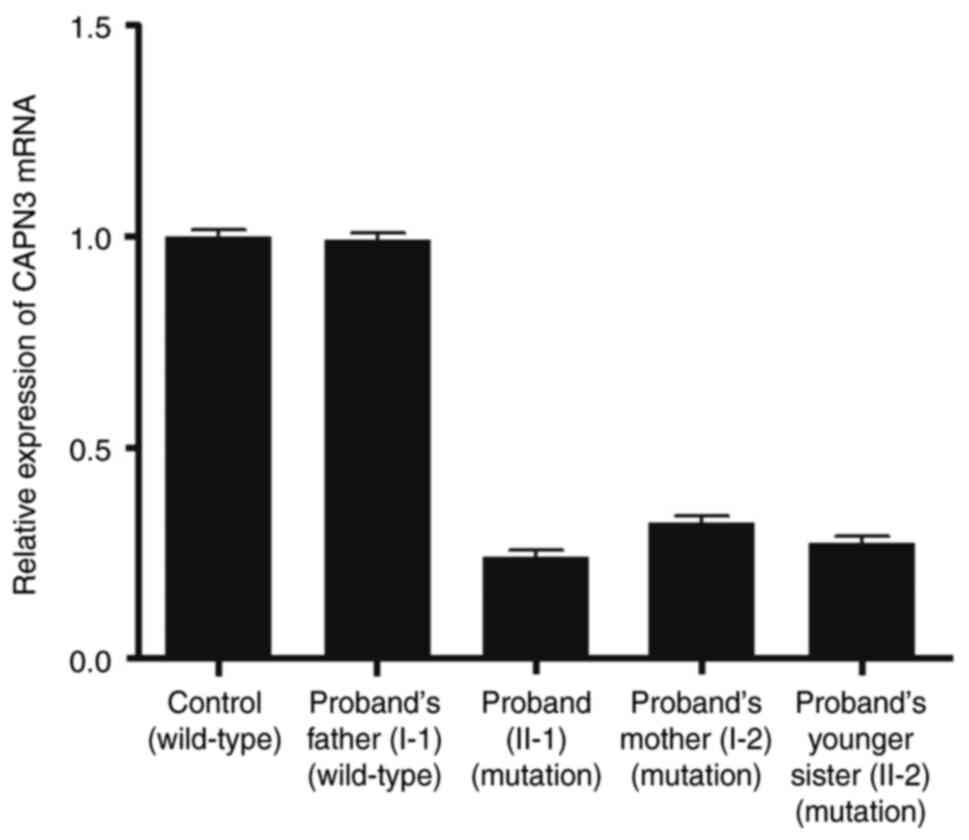
Relative expression levels of CAPN3 mRNA
revealed a significantly decreased mutated CAPN3 transcript
level in the proband, their younger sister and mother while the
father of the proband showed normal expression (same as the normal
healthy control individual) of wild-type CAPN3 mRNA (Fig. 3). These results also suggested that
the mutant CAPN3 transcript (proband, proband's mother and younger
sister) was present at detectable levels. Therefore, the mutated
CAPN3 transcript was not degraded by the nonsense-mediated mRNA
decay pathway.
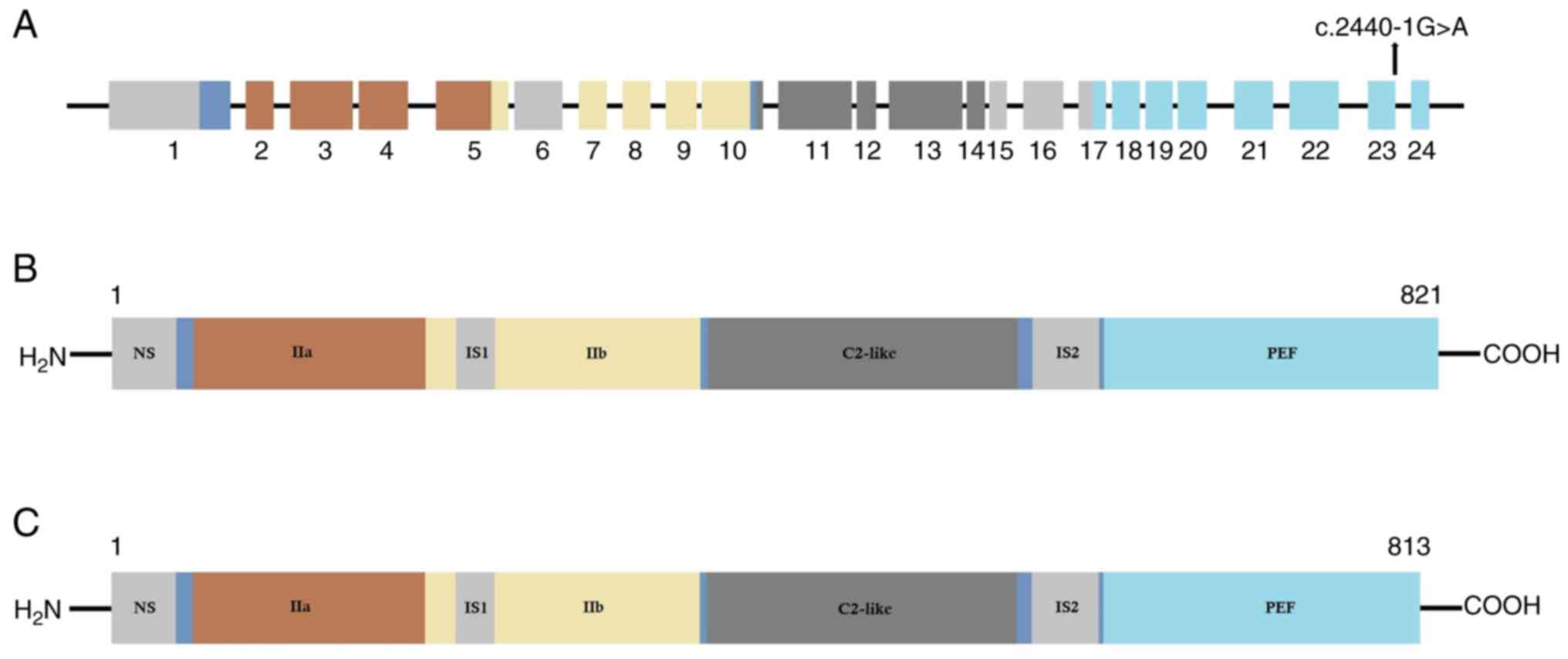
All 24 exons of the CAPN3 gene are
schematically presented in Fig.
4A, and the mutation is located at the last nucleotide of exon
23. The wild type CAPN3 protein structure is shown in Fig. 4B. The mutated CAPN3 protein (with
complete loss of exon 24) is presented in Fig. 4C. According to the structural point
of view, this mutation causes partial loss of domain IV of the
CAPN3 protein, which is involved in calcium binding and
homodimerization (41).
Discussion
In the present study, a nonconsanguineous family
from China clinically diagnosed with LGMDD4 was investigated and
analyzed. The proband (II-1), their mother (I-2) and younger sister
(II-2) were clinically diagnosed with LGMDD4, while the father of
the proband (I-1) was phenotypically normal. Whole exome sequencing
identified a novel heterozygous splice-acceptor site mutation
(c.2440-1G>A) in the CAPN3 gene in the proband. Sanger
sequencing confirmed that the mother and younger sister of the
proband were also carriers of this mutation, while the father was
devoid of it. This splice-site mutation causes aberrant splicing of
CAPN3 mRNA, leading to the complete loss of exon 24, finally
resulting in the formation of a truncated (p.Trp814*) CAPN3 protein
of 813 amino acids, hence, it is a loss-of-function mutation. The
proband, the mother and the younger sister had mutations in the
CAPN3 gene (as established by whole exome sequencing and
Sanger sequencing). Thus, the CAPN3 mRNA expression in the proband,
sister and mother was expression of the mutated CAPN3. The mutated
CAPN3 mRNA showed significantly reduced expression compared with
the wild-type CAPN3 mRNA. This mutation causes both structural and
functional changes in CAPN3 protein, which lead to LGMDD4 in the
proband and all affected family members. In the present study,
other pathogenic or likely pathogenic variants in other genes
associated with muscular dystrophies were not identified. In
addition, no other variants in genes (titin, dysferlin, filamin C
and ATP2A2) that interact with CAPN3 were identified;
therefore, a very rare form of LGMDD4 in a Chinese family was
reported in the present study.
The patients in the present study showed
comparatively milder phenotypes than that of previously reported
patients with LGMDD4 (15-17).
Additionally, intrafamilial phenotypic variability was identified
in this family with the level of serum creatine kinase not showing
an association with disease severity. This is in line with the
literature, as some patients with LGMDD4 are reported to have
normal serum creatine kinase during their clinical course, while
others have elevated levels of serum creatine kinase (42).
It has previously been reported that CAPN3 interacts
with tropomyosin, α-actinin-3 and LIM-domain binding protein 3
(14,43). CAPN3 strongly interacts with
ryanodine receptor type 1 (RyR1), calsequestrin and
sarco/endoplasmic reticulum calcium ATPase proteins to maintain
calcium homeostasis (43-45).
In addition, CAPN3 increases the activity of NCX3, which also
regulates calcium homeostasis (46). Maintenance and remodeling of
sarcomeres is regulated by CAPN3 and titin, and CAPN3 is also
involved in modulating the function of mitochondria (45,46).
Germline mutations in the CAPN3 gene lead to the formation
of non-functional CAPN3 protein, which in turn causes reduced
expression of RyR1 and reduced release of calcium from the
sarcoplasmic reticulum, thus finally resulting in the dysregulation
of calcium homeostasis and manifesting as LGMDD4 (45-47).
Additionally, reduced expression of RyR1 and
calcium/calmodulin-dependent kinase II signaling has already been
reported in muscles among patients with LGMDD4(48). Hence, this is the
pathophysiological mechanism underlying the disease phenotype among
patients with LGMDD4. The pathophysiological mechanism of
CAPN3-associated LGMDD4 is associated with the NF-κB pathway
(49). CAPN3 activates NF-κB by
calcium-dependent degradation of IκBα, a NF-κB inhibitor. Activated
NF-κB causes degradation of protein, inflammation and fibrosis of
skeletal muscle (49). Therefore,
mutations in the CAPN3 gene lead to dysregulation of the
NF-κB pathway and could be the pathophysiological mechanism for
LGMDD4. Additionally, the ubiquitin-proteasome system (UPS) and the
autophagy-lysosome pathway are, reportedly, involved in the
proteolytic processes of cell regulatory turnover of protein in
muscles. It has been reported that the UPS is a main pathway
leading to muscle atrophy (49,50).
At present, ~20 different types of LGMD have been
reported with extreme intra- and inter-familial phenotypic
heterogeneity, even among patients with the same LGMD subtype
(51,52). Assessing the serum creatine kinase
level or analyzing electrical activity of the muscle is the key
point of clinical diagnosis of muscular disorders; however,
diagnosing the exact type of muscular disorder is the biggest
challenge at present (53).
Clinical diagnosis of calpainopathies is usually achieved through
muscle biopsy, to measure the presence of CAPN3 in the muscle
(54). Immunohistochemical
staining is not a simple or confirmatory diagnostic procedure for
patients with calpainopathies because of the rapid autolysis of
CAPN3(41). Several types of LGMD
have been identified with overlapping clinical symptoms, and
genetic testing is the most efficient way to ensure accurate and
timely clinical diagnosis. Hence, genetic molecular diagnosis
through whole exome sequencing is the most easy, useful, accurate,
and least time-consuming method for clinical diagnosis of this
disease (41).
Among all the reported mutations of the CAPN3
gene associated with LGMD, the majority are missense mutations
(60-70%) (55,56). Most of the mutations in the
CAPN3 gene are located in exons 1, 4, 5, 8, 10, 11 and 21
(57,58). At present, ~300 mutations of
CAPN3 have been reported and according to the location of
these mutations, CAPN3 gene consists of two hotspots, one on
exon 11 and the other on exon 21. Among these reported mutations,
some are founder but the majority are private variants (53,59,60).
Duguez et al (61)
described the first study, which involved 548 patients with
myopathy, including LGMD, from 181 families and 19 countries, and
reported 97 pathogenic mutations of the CAPN3 gene (62).
Structurally, the CAPN3 protein is comprised of four
domains. Domain I is an N-terminal domain containing the nuclear
localization signal. Germline mutations located in domain I cause a
loss of the nuclear transport function of CAPN3(41). Domain II (IIa, IS1 and IIb) is an
evolutionarily conserved cysteine protease domain, which is
involved in protease activity. Mutations in domain II causes a loss
of the protease activity of CAPN3(49). Domain III is directly responsible
for structural changes of activated CAPN3. Mutations occurring in
domain III lead to no structural changes in CAPN3 upon its
activation (43). Lastly, domain
IV is majorly involved in calcium ion (Ca2+) binding and
homodimerization of CAPN3. Germline mutations in domain IV cause
formation of non-functional CAPN3, which is unable to bind with
Ca2+ and lacks the ability to homodimerize (46). Hence, these mutations located in
different domains of CAPN3 gene can cause LGMDD4(50).
At present, several gene therapy-based strategies
have been used to treat patients with LGMDD4 that harbor a
CAPN3 mutation. Moreover, cellular therapies, drug therapies
(glucocorticoid treatment) or gene therapies (AAV-mediated therapy
and CRISPR-Cas9 gene editing) have been performed either in
preclinical or clinical phases; however, there is no cure (63). Endoplasmic reticulum stress
factor-targeting inhibitors and small molecules
(tauroursodeoxycholic acid, salubrinal and rapamycin) have also
been considered as potential therapeutic strategies (11). However, at present, no therapeutic
strategies have been developed to treat the progressive muscle loss
and premature death of patients with LGMD (64). In the present study, a single
interesting case of CAPN3-associated LGMDD4 was reported. A
limitation of the study is that due to the unavailability of fresh
muscle samples, western blotting to confirm the relative expression
of CAPN3 protein among the proband and all affected family members
could not be performed. In the future, more cases of
CAPN3-associated LGMDD4 should be analyzed and reported on
in order to understand the disease mechanism, genotype-phenotype
correlation and possible clinical management for the patients.
In conclusion, in the present study, a 16-year-old
male patient from China with gradual and progressive weakness, and
atrophy of the muscles in both the legs was reported on. The
younger sister and mother of the proband displayed similar clinical
symptoms as the proband. Whole exome sequencing identified a
heterozygous novel splice-site mutation (c.2440-1G>A) in intron
23 of the CAPN3 gene in the proband. This mutation causes
aberrant splicing of CAPN3 mRNA and leads to the skipping of
exon 24 and, finally, results in the formation of a truncated
(p.Trp814*) CAPN3 protein. To the best of our knowledge, the
present study is the first to report on a case of CAPN3
gene-associated LGMDD4 in the Chinese population.
Acknowledgements
Not applicable.
Funding
Funding: This study is supported by the Key Research and
Development Plan of Gansu Province (grant no. 21YF1FA115), Key
Research and Development Plan of Gansu Province in 2022 (grant no.
22YF7FA084) and Natural Science Foundation of Gansu Province in
2022 (grant no. 22JR5RA911).
Availability of data and materials
The datasets generated and/or analyzed during the
current study are not available to patient privacy but are
available from the corresponding author on reasonable request.
Authors' contributions
BM, JY, XJ, LihZ and XM were responsible for the
clinical investigation (radiology, histology and
immunohistochemistry) for the proband and his family members. XS
and LilZ performed genetic analysis. BM, JY, XZ and XM performed
clinical investigation. SB, LilZ and XM were responsible for
project administration. BM, JY, XJ, LihZ and XM acquired materials.
SB, LilZ and XM supervised the present study; BM, JY, XJ, SB, LihZ
and XM wrote the original draft. SB, LilZ and XM wrote, reviewed
and edited the manuscript. LilZ and XM confirm the authenticity of
all the raw data. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The ethics committee of The Reproductive Medicine
Centre, The First Hospital of Lanzhou University approved the
present study (approval no. 2021-A-8900920; Lanzhou, China) in
accordance with the recommendations of The Declaration of Helsinki.
Written informed consent was obtained from all family members and
the parents of the minor patients for their participation in this
study. The genomic DNA samples from the 100 normal healthy
individuals were collected from our hospital's sample bank (The
Reproductive Medicine Centre Sample Bank, The First Hospital of
Lanzhou University, Lanzhou, China). The hospital's sample bank
(The Reproductive Medicine Centre Sample Bank, The First Hospital
of Lanzhou University, Lanzhou, China) collected the genomic DNA
samples from the 100 normal healthy individuals after obtaining
written informed consent. The ethics committee of The Reproductive
Medicine Centre, The First Hospital of Lanzhou University (approval
no. 2021-C-723149; Lanzhou, China) approved the use of the genomic
DNA samples from 100 normal healthy individuals from the hospital's
sample bank (The Reproductive Medicine Centre Sample Bank, The
First Hospital of Lanzhou University, Lanzhou, China) for the
present study. The RNA sample from the normal healthy individual
was collected from our hospital's sample bank (The Reproductive
Medicine Centre Sample Bank, The First Hospital of Lanzhou
University, Lanzhou, China). The hospital's sample bank (The
Reproductive Medicine Centre Sample Bank, The First Hospital of
Lanzhou University, Lanzhou, China) collected the RNA sample from
the normal healthy individual after getting written informed
consent. The ethics committee of The Reproductive Medicine Centre,
The First Hospital of Lanzhou University (approval no.
2021-C-934572; Lanzhou, China) approved the use of the RNA sample
from a normal healthy individual from the hospital's sample bank
(The Reproductive Medicine Centre Sample Bank, The First Hospital
of Lanzhou University, Lanzhou, China) for the present study.
Patient consent for publication
Written informed consent was obtained from all
family members and the parents of the minor patients for the
publication of this study.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Allamand V, Broux O, Richard I,
Fougerousse F, Chiannilkulchai N, Bourg N, Brenguier L, Devaud C,
Pasturaud P, Pereira de Souza A, et al: Preferential localization
of the limb-girdle muscular dystrophy type 2A gene in the proximal
part of a 1-cM 15q15.1-q15.3 interval. Am J Hum Genet.
56:1417–1430. 1995.PubMed/NCBI
|
|
2
|
Bushby KM: Diagnostic criteria for the
limb-girdle muscular dystrophies: Report of the ENMC consortium on
limb-girdle dystrophies. Neuromuscul Disord. 5:71–74.
1995.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wang CH, Liang WC, Minami N, Nishino I and
Jong YJ: Limb-girdle muscular dystrophy type 2A with mutation in
CAPN3: The first report in Taiwan. Pediatr Neonatol. 56:62–65.
2015.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Zheng J, Xu X, Zhang X, Wang X, Shu J and
Cai C: Variants of CAPN3 cause limb-girdle muscular dystrophy type
2A in two Chinese families. Exp Ther Med. 21(104)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Richard I, Broux O, Allamand V,
Fougerousse F, Chiannilkulchai N, Bourg N, Brenguier L, Devaud C,
Pasturaud P, Roudaut C, et al: Mutations in the proteolytic enzyme
calpain 3 cause limb-girdle muscular dystrophy type 2A. Cell.
81:27–40. 1995.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Feng X, Luo S, Li J, Yue D, Xi J, Zhu W,
Gao X, Guan X, Lu J, Liang Z and Zhao C: Fatty infiltration
evaluation and selective pattern characterization of lower limbs in
limb-girdle muscular dystrophy type 2A by muscle magnetic resonance
imaging. Muscle Nerve. 58:536–541. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Mercuri E, Bushby K, Ricci E, Birchall D,
Pane M, Kinali M, Allsop J, Nigro V, Sáenz A, Nascimbeni A, et al:
Muscle MRI findings in patients with limb girdle muscular dystrophy
with calpain 3 deficiency (LGMD2A) and early contractures.
Neuromuscul Disord. 15:164–171. 2005.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Richard I, Brenguier L, Dinçer P, Roudaut
C, Bady B, Burgunder JM, Chemaly R, Garcia CA, Halaby G, Jackson
CE, et al: Multiple independent molecular etiology for limb-girdle
muscular dystrophy type 2A patients from various geographical
origins. Am J Hum Genet. 60:1128–1138. 1997.PubMed/NCBI
|
|
9
|
Groen EJ, Charlton R, Barresi R, Anderson
LV, Eagle M, Hudson J, Koref MS, Straub V and Bushby KM: Analysis
of the UK diagnostic strategy for limb girdle muscular dystrophy
2A. Brain. 130:3237–3249. 2007.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Vissing J, Barresi R, Witting N, Van
Ghelue M, Gammelgaard L, Bindoff LA, Straub V, Lochmüller H, Hudson
J, Wahl CM, et al: A heterozygous 21-bp deletion in CAPN3 causes
dominantly inherited limb girdle muscular dystrophy. Brain.
139:2154–2163. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Richard I, Roudaut C, Saenz A, Pogue R,
Grimbergen JE, Anderson LV, Beley C, Cobo AM, de Diego C, Eymard B,
et al: Calpainopathy-a survey of mutations and polymorphisms. Am J
Hum Genet. 64:1524–1540. 1999.PubMed/NCBI View
Article : Google Scholar
|
|
12
|
Kramerova I, Kudryashova E, Venkatraman G
and Spencer MJ: Calpain 3 participates in sarcomere remodeling by
acting upstream of the ubiquitin-proteasome pathway. Hum Mol Genet.
14:2125–2134. 2005.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Ermolova N, Kudryashova E, DiFranco M,
Vergara J, Kramerova I and Spencer MJ: Pathogenity of some limb
girdle muscular dystrophy mutations can result from reduced
anchorage to myofibrils and altered stability of calpain 3. Hum Mol
Genet. 20:3331–3345. 2011.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Taveau M, Bourg N, Sillon G, Roudaut C,
Bartoli M and Richard I: Calpain 3 is activated through autolysis
within the active site and lyses sarcomeric and sarcolemmal
components. Mol Cell Biol. 23:9127–9135. 2003.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Martinez-Thompson JM, Niu Z, Tracy JA,
Moore SA, Swenson A, Wieben ED and Milone M: Autosomal dominant
calpainopathy due to heterozygous CAPN3 C.643_663del21. Muscle
Nerve. 57:679–683. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Cerino M, Campana-Salort E, Salvi A,
Cintas P, Renard D, Morales RJ, Tard C, Leturcq F, Stojkovic T,
Bonello-Palot N, et al: Novel CAPN3 variant associated with an
autosomal dominant calpainopathy. Neuropathol Appl Neurobiol.
46:564–578. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
González-Mera L, Ravenscroft G,
Cabrera-Serrano M, Ermolova N, Domínguez-González C, Arteche-López
A, Soltanzadeh P, Evesson F, Navas C, Mavillard F, et al:
Heterozygous CAPN3 missense variants causing autosomal-dominant
calpainopathy in seven unrelated families. Neuropathol Appl
Neurobiol. 47:283–296. 2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Sáenz A and López de Munain A: Dominant
LGMD2A: Alternative diagnosis or hidden digenism? Brain.
140(e7)2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Vissing J and Duno M: Reply: Dominant
LGMD2A: Alternative diagnosis or hidden digenism? Brain.
140(e8)2017.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Cotta A, Carvalho E, da-Cunha-Júnior AL,
Valicek J, Navarro MM, Junior SB, da Silveira EB, Lima MI, Cordeiro
BA, Cauhi AF, et al: Muscle biopsy essential diagnostic advice for
pathologists. Surg Exp Pathol. 4(3)2021.
|
|
21
|
Wang C, Yue F and Kuang S: Muscle
histology characterization using H&E staining and muscle fiber
type classification using immunofluorescence staining. Bio Protoc.
7(e2279)2017.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Nix JS and Moore SA: What every
neuropathologist needs to know: The muscle biopsy. J Neuropathol
Exp Neurol. 79:719–733. 2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Suriyonplengsaeng C, Dejthevaporn C,
Khongkhatithum C, Sanpapant S, Tubthong N, Pinpradap K, Srinark N
and Waisayarat J: Immunohistochemistry of sarcolemmal
membrane-associated proteins in formalin-fixed and
paraffin-embedded skeletal muscle tissue: A promising tool for the
diagnostic evaluation of common muscular dystrophies. Diagn Pathol.
12(19)2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Danielsson O and Häggqvist B: Skeletal
muscle immunohistochemistry of acquired and hereditary myopathies.
Curr Opin Rheumatol. 33:529–536. 2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhang R, Chen S, Han P, Chen F, Kuang S,
Meng Z, Liu J, Sun R, Wang Z, He X, et al: Whole exome sequencing
identified a homozygous novel variant in CEP290 gene causes Meckel
syndrome. J Cell Mol Med. 24:1906–1916. 2020.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Van der Auwera GA, Carneiro M, Hartl C,
Poplin R, del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen
D, Thibault J, et al: From FastQ data to high confidence variant
calls: The genome analysis toolkit best practices pipeline. Curr
Protoc Bioinformatics: Mar 15, 2018 (Epub ahead of print). doi:
10.1002/0471250953.bi1110s43.
|
|
27
|
Gu S, Fang L and Xu X: Using SOAPaligner
for short reads alignment. Curr Protoc Bioinformatics.
44(11.11.1-17)2013.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Li R, Yu C, Li Y, Lam TW, Yiu SM,
Kristiansen K and Wang J: SOAP2: An improved ultrafast tool for
short read alignment. Bioinformatics. 25:1966–1967. 2009.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Sherry ST, Ward MH, Kholodov M, Baker J,
Phan L, Smigielski EM and Sirotkin K: dbSNP: The NCBI database of
genetic variation. Nucleic Acids Res. 29:308–311. 2001.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Clark AG, Hubisz MJ, Bustamante CD,
Williamson SH and Nielsen R: Ascertainment bias in studies of human
genome-wide polymorphism. Genome Res. 15:1496–1502. 2005.PubMed/NCBI View Article : Google Scholar
|
|
31
|
1000 Genomes Project Consortium. Auton A,
Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL,
McCarthy S, McVean GA and Abecasis GR: A global reference for human
genetic variation. Nature. 526:68–74. 2015.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Stenson PD, Ball EV, Mort M, Phillips AD,
Shiel JA, Thomas NST, Abeysinghe S, Krawczak M and Cooper DN: Human
gene mutation database (HGMD): 2003 Update. Hum Mutat. 21:577–581.
2003.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Hamosh A, Scott AF, Amberger JS, Bocchini
CA and McKusick VA: Online mendelian inheritance in man (OMIM), a
knowledgebase of human genes and genetic disorders. Nucleic Acids
Res. 33:D514–D517. 2005.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Karczewski KJ, Weisburd B, Thomas B,
Solomonson M, Ruderfer DM, Kavanagh D, Hamamsy T, Lek M, Samocha
KE, Cummings BB, et al: The ExAC browser: Displaying reference data
information from over 60 000 exomes. Nucleic Acids Res. 45
(D1):D840–D845. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Karczewski KJ, Francioli LC, Tiao G,
Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A,
Birnbaum DP, et al: The mutational constraint spectrum quantified
from variation in 141,456 humans. Nature. 581:434–443.
2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Lefter M, Vis JK, Vermaat M, den Dunnen
JT, Taschner PEM and Laros JFJ: Mutalyzer 2: Next generation HGVS
nomenclature checker. Bioinformatics. 37:2811–2817. 2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Vanhoutte EK, Faber CG, van Nes SI, Jacobs
BC, van Doorn PA, van Koningsveld R, Cornblath DR, van der Kooi AJ,
Cats EA, van den Berg LH, et al: Modifying the medical research
council grading system through Rasch analyses. Brain.
135:1639–1649. 2012.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Chang RF and Mubarak SJ: Pathomechanics of
Gowers' sign: A video analysis of a spectrum of Gowers' maneuvers.
Clin Orthop Relat Res. 470:1987–1991. 2012.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Partha SK, Ravulapalli R, Allingham JS,
Campbell RL and Davies PL: Crystal structure of calpain-3
penta-EF-hand (PEF) domain-a homodimerized PEF family member with
calcium bound at the fifth EF-hand. FEBS J. 281:3138–3149.
2014.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Gallardo E, Saenz A and Illa I:
Limb-girdle muscular dystrophy 2A. Handb Clin Neurol. 101:97–110.
2011.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Huang Y, de Morrée A, van Remoortere A,
Bushby K, Frants RR, den Dunnen JT and van der Maarel SM: Calpain 3
is a modulator of the dysferlin protein complex in skeletal muscle.
Hum Mol Genet. 17:1855–1866. 2008.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Toral-Ojeda I, Aldanondo G,
Lasa-Elgarresta J, Lasa-Fernández H, Fernández-Torrón R, López de
Munain A and Vallejo-Illarramendi A: Calpain 3 deficiency affects
SERCA expression and function in the skeletal muscle. Expert Rev
Mol Med. 18(e7)2016.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Jahnke VE, Peterson JM, Van Der Meulen JH,
Boehler J, Uaesoontrachoon K, Johnston HK, Defour A, Phadke A, Yu
Q, Jaiswal JK and Nagaraju K: Mitochondrial dysfunction and
consequences in calpain-3-deficient muscle. Skelet Muscle.
10(37)2020.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Chen L, Tang F, Gao H, Zhang X, Li X and
Xiao D: CAPN3: A muscle-specific calpain with an important role in
the pathogenesis of diseases (review). Int J Mol Med.
48(203)2021.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Lasa-Elgarresta J, Mosqueira-Martín L,
González-Imaz K, Marco-Moreno P, Gerenu G, Mamchaoui K, Mouly V,
López de Munain A and Vallejo-Illarramendi A: Targeting the
ubiquitin-proteasome system in limb-girdle muscular dystrophy with
CAPN3 mutations. Front Cell Dev Biol. 10(822563)2022.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Kramerova I, Torres JA, Eskin A, Nelson SF
and Spencer MJ: Calpain 3 and CaMKIIβ signaling are required to
induce HSP70 necessary for adaptive muscle growth after atrophy.
Hum Mol Genet. 27:1642–1653. 2018.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Lasa-Elgarresta J, Mosqueira-Martín L,
Naldaiz-Gastesi N, Sáenz A, López de Munain A and
Vallejo-Illarramendi A: Calcium mechanisms in limb-girdle muscular
dystrophy with CAPN3 mutations. Int J Mol Sci.
20(4548)2019.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Place N, Ivarsson N, Venckunas T, Neyroud
D, Brazaitis M, Cheng AJ, Ochala J, Kamandulis S, Girard S,
Volungevičius G, et al: Ryanodine receptor fragmentation and
sarcoplasmic reticulum Ca2+ leak after one session of
high-intensity interval exercise. Proc Natl Acad Sci USA.
112:15492–15497. 2015.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Mitsuhashi S and Kang PB: Update on the
genetics of limb girdle muscular dystrophy. Semin Pediatr Neurol.
19:211–218. 2012.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Guglieri M, Magri F, D'Angelo MG, Prelle
A, Morandi L, Rodolico C, Cagliani R, Mora M, Fortunato F, Bordoni
A, et al: Clinical, molecular, and protein correlations in a large
sample of genetically diagnosed Italian limb girdle muscular
dystrophy patients. Hum Mutat. 29:258–266. 2008.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Balci B, Aurino S, Haliloglu G, Talim B,
Erdem S, Akcören Z, Tan E, Caglar M, Richard I, Nigro V, et al:
Calpain-3 mutations in Turkey. Eur J Pediatr. 165:293–298.
2006.PubMed/NCBI View Article : Google Scholar
|
|
54
|
de Paula F, Vainzof M, Passos-Bueno MR, de
Cássia M, Pavanello R, Matioli SR, V B Anderson L, Nigro V and Zatz
M: Clinical variability in calpainopathy: What makes the
difference? Eur J Hum Genet. 10:825–832. 2002.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Fanin M, Nascimbeni AC, Fulizio L and
Angelini C: The frequency of limb girdle muscular dystrophy 2A in
northeastern Italy. Neuromuscul Disord. 15:218–224. 2005.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Fanin M, Nascimbeni AC, Tasca E and
Angelini C: How to tackle the diagnosis of limb-girdle muscular
dystrophy 2A. Eur J Hum Genet. 17:598–603. 2009.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Dorobek M, Ryniewicz B, Kabzińska D,
Fidziańska A, Styczyńska M and Hausmanowa-Petrusewicz I: The
Frequency of c.550delA mutation of the CANP3 gene in the Polish
LGMD2A population. Genet Test Mol Biomarkers. 19:637–640.
2015.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Piluso G, Politano L, Aurino S, Fanin M,
Ricci E, Ventriglia VM, Belsito A, Totaro A, Saccone V, Topaloglu
H, et al: Extensive scanning of the calpain-3 gene broadens the
spectrum of LGMD2A phenotypes. J Med Genet. 42:686–693.
2005.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Pogoda TV, Krakhmaleva IN, Lipatova NA,
Shakhovskaya NI, Shishkin SS and Limborska SA: High incidence of
550delA mutation of CAPN3 in LGMD2 patients from Russia. Hum Mutat.
15(295)2000.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Milic A and Canki-Klain N: Calpainopathy
(LGMD2A) in Croatia: Molecular and haplotype analysis. Croat Med J.
46:657–663. 2005.PubMed/NCBI
|
|
61
|
Duguez S, Bartoli M and Richard I: Calpain
3: A key regulator of the sarcomere? FEBS J. 273:3427–3436.
2006.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Foley AR, Donkervoort S and Bönnemann CG:
Next-generation sequencing still needs our generation's clinicians.
Neurol Genet. 1(e13)2015.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Şahin İO, Özkul Y and Dündar M: Current
and future therapeutic strategies for limb girdle muscular
dystrophy type R1: Clinical and experimental approaches.
Pathophysiology. 28:238–249. 2021.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Li C and Samulski RJ: Engineering
adeno-associated virus vectors for gene therapy. Nat Rev Genet.
21:255–272. 2020.PubMed/NCBI View Article : Google Scholar
|