Introduction
Epidermolysis bullosa simplex (EBS), a
mechanobullous genodermatosis characterized by skin fragility and
blisters on the skin and mucous membranes, results from external
trauma (1). EBS exhibits an
incidence rate of 7.87/one million live births (2). The severity of EBS varies, ranging
from mild blisters on the hands and feet to more generalized forms
with extracutaneous involvement. Occasionally EBS can have a fatal
outcome. The onset of EBS varies based on its subtype, typically
manifesting at birth or during infancy (3). For accurate classification of EBS, it
is imperative to consider the specific clinical features alongside
molecular findings. A previous reclassification identified numerous
clinical variants of EBS, including localized (previously referred
to as Weber-Cockayne), intermediate (formerly known as generalized
intermediate or Koebner) and severe EBS (previously termed as EBS
Dowling-Meara) (4). The mildest,
most common subtype is localized EBS, with a reported incidence of
3.67/one million live births (2).
However, since a notable percentage of mild cases may remain
undiagnosed, the actual incidence may be higher than that reported
(2). One of the typical
comorbidities associated with EBS is basal cell carcinoma, which
primarily occurs in patients with the most severe form of EBS. This
elevated risk of basal cell carcinoma in patients with most severe
EBS is attributed to recurrent and chronic basal keratinocyte
injury (5).
In EBS, gene aberrations have been implicated in
seven genes, with 75% of the patients harboring mutations in genes
encoding keratin 5 (KRT5) and KRT14, which are the
primary cytoskeletal components of basal keratinocytes (6). The predominant genetic alterations
are amino acid substitutions (missense variants). Inheritance
typically follows an autosomal dominant pattern, with some
exceptions. The clinical severity of EBS, in most cases, is
associated with the specific loci of these genetic changes
(7). Traditionally, diagnosis of
EBS necessitated procedures such as skin biopsies,
immunofluorescence microscopy and transmission electron microscopy.
However, modern diagnostic strategies prioritize the identification
of heterozygous pathogenic variants in KRT5 or KRT14
through molecular testing (8).
The present study describes two novel heterozygous
KRT5 gene variants from two patients with the sporadic form
of EBS: c.1399A>T (p.Ile467Phe) and c.1412G>A (p.Arg471His).
These individuals exhibited typical clinical features of
intermediate and localized EBS, respectively.
Case report
Two patients consulted the Department of Dermatology
in Suining Central Hospital (Suining, China) on August 2022.
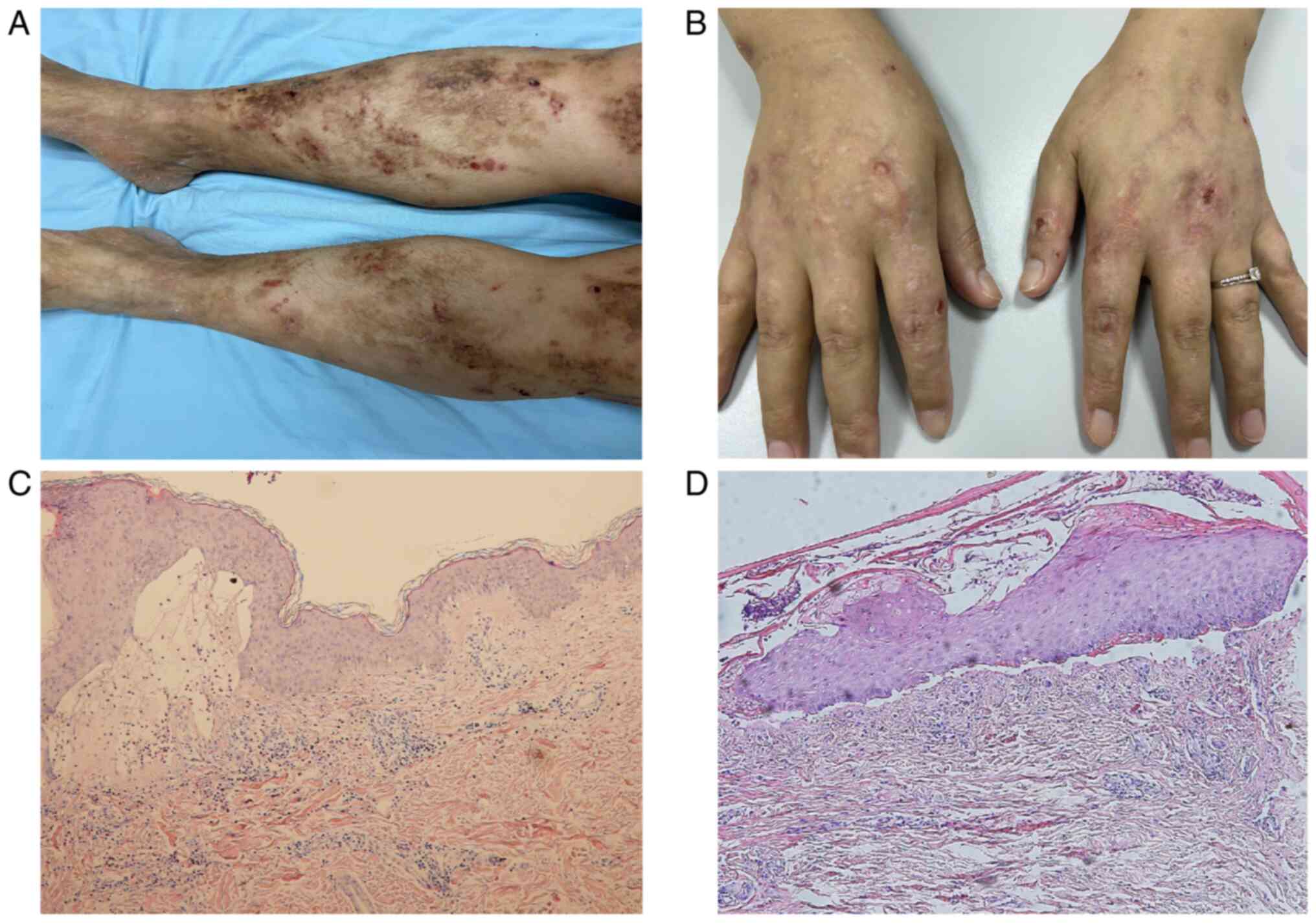
Patient 1 was a 43-year-old man with progressive trauma-induced
blisters on the arms and legs since birth. Initially, the blisters
appeared on legs at birth, followed by blister formation and skin
erosion on arms and joints after a minor trauma. There were no
signs of nail dystrophy, nail lysis or oral mucosal involvement.
The patient presented with pruritus, although he had no apparent
history or family history of similar disease (Fig. 1A). Patient 2 was a 31-year-old
woman with erosions and blisters on the hands from 6 months of age.
Typically, lesions healed without scarring or miliary formation.
Clinical observation showed stable blistering on the palms and
soles, with remission of symptoms in autumn and winter. Patient 2
had no family history of blister disease (Fig. 1B). Skin lesions measuring
1.2x1.0x0.6 cm from patient 1 and 1.0x1.0x0.8 cm from patient 2
were excised for histopathological examination. The tissues
underwent fixation in 4% neutral formalin at room temperature for
48 h, followed by dehydration with alcohol and xylene.
Subsequently, they were embedded in paraffin at 62˚C and cooled.
Serial sections of a 4-µm thickness were prepared and stained with
hematoxylin for 5 min and eosin for 2 min at room temperature.
Imaging was performed using light microscopy (Olympus BX51; Olympus
Corporation). The histology report of patient 1 showed subepidermal
blisters surrounded by eosinophils and lymph (Fig. 1C) while that of patient 2 showed
subepidermal blisters surrounded by neutrophils (Fig. 1D). Informed consent was obtained
from both patients and their parents, and ethics approval was
granted by the Ethics Committee of the Suining Central Hospital.
The study was conducted in accordance with the principles outlined
in the Declaration of Helsinki.
DNA was isolated from a 200-µl blood sample from
each patient using the Qiagen DNA Blood Midi/Mini kit (Qiagen
GmbH,). Genomic DNA fragments were generated using a Covaris
Ultrasonicator (Covaris, Inc.) and the DNA library was prepared.
Exon capturing was performed using Streptavidin-Coated Magnetic
Beads by NimbleGen (Roche NimbleGen, Inc.). The library quality was
assessed by linear PCR amplification. Subsequently, next-generation
sequencing (NGS), performed at Shanghai Anbailong Biotechnology,
was conducted on an Illumina HiSeq X Ten platform (Illumina, Inc.)
with a sequencing depth >200x and Q30>90% (9). The raw sequencing data was converted
to FASTQ format and the reads were aligned with the human genome
reference sequence (hg19) using Burrows-Wheeler Alignment to detect
the genetic variants (9). All the
identified variants were assessed by consulting various databases,
such as the NCBI database of Single Nucleotide Polymorphisms
(http://www.ncbi.nlm.nih.gov/SNP/),
Online Mendelian Inheritance in Man (http://www.omim.org/), The Human Gene Molecular
Biology Reports Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) and NCBI
ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
Sanger sequencing was performed for verification,
targeting the potential pathogenic variants identified by NGS.
In silico analysis tools, including PolyPhen2 (http://genetics.bwh.harvard.edu/pph2/)
and MutationTaster (https://www.mutationtaster.org/), were employed to
predict the impact of amino acid substitutions on KRT5 protein
structure and function. From January 2021 to September 2021, a
total of 100 unrelated healthy Chinese Han individuals (52% male,
48% female; aged 18-30 years) consisted of the control group in
Suining Central Hospital.
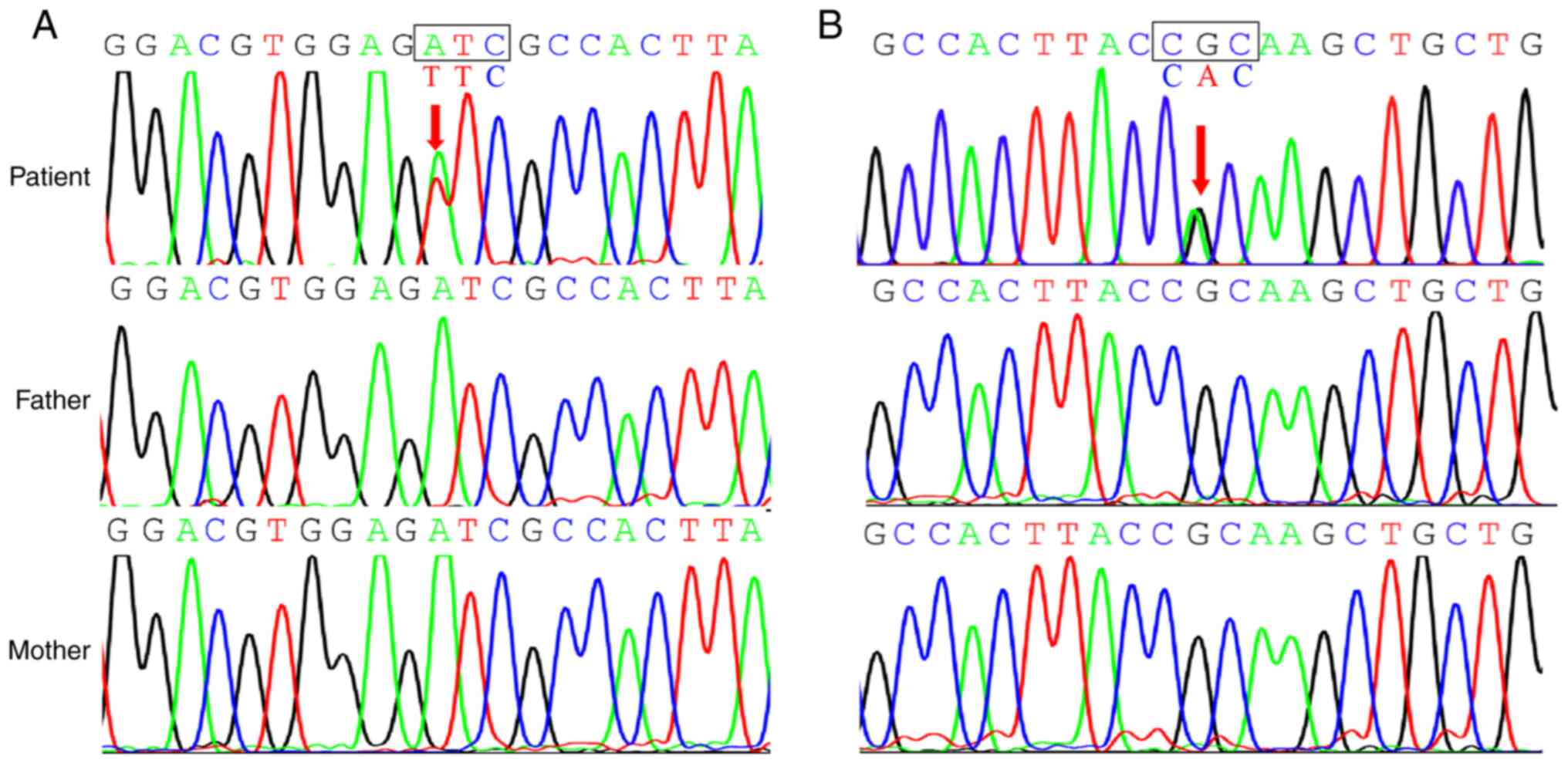
NGS analysis performed on the DNA isolated from the
blood sample of patient 1 revealed a novel KRT5 gene
variant, c.1399A>T (p.Ile467Phe; Fig. 2A) within the 2B-domain of exon 7.
This gene variant resulted in an amino acid substitution from
isoleucine to phenylalanine at position 467 in the KRT5 protein.
Notably, this variant was absent in both the unaffected parents and
a control group of 100 healthy individuals. Patient 1 was diagnosed
with intermediate EBS.
Similarly, patient 2, who was diagnosed with
localized EBS, harbored a novel point mutation, c.1412G>A
(p.Arg471His), in exon 7 of KRT5 gene. This mutation was
also within the 2B-domain (Fig.
2B) and resulted in amino acid substitution from arginine to
histidine at position 471 in the KRT5 protein. PolyPhen2 prediction
of pathogenicity revealed that both p.Ile467Phe and p.Arg471His
variants were harmful. MutationTaster predicted that these two
variants were ‘disease-causing’. In addition, these variants were
considered novel since they were not found in the ExAC (http://exac.broadinstitute.org/dbsnp),
ESP (https://esp.gs.washington.edu/drupal/), 1000G
(http://www.1000genomes.org/) and HGMD
(http://www.hgmd.org) databases. In line with the
American College of Medical Genetics and Genomics guidelines
(10), both variants were
classified as ‘likely pathogenic’, further supporting their
potential role in the development of EBS and confirming the
diagnosis.
For the two patients, the treatment approach
primarily targeted symptom relief to enhance functionality and
improve quality of life. Given the significant role of inflammation
in EBS and its exacerbating effects on phenotypes, antibiotics and
corticosteroids were administered to patient 1 while botulinum
toxin was administered to patient 2 to decrease blistering in the
affected surface area.
Discussion
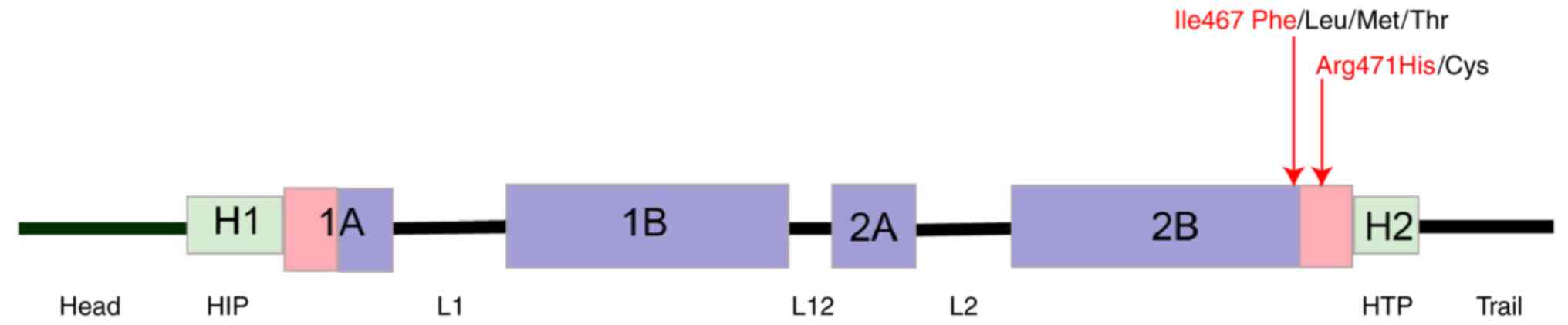
KRT5 gene, located on chromosome 12qx,
comprises nine exons spanning ~6.1 kilobases (11). The rod domain of this keratin
protein, which is encoded by the KRT5 gene, comprises four
α-helical regions (1A, 1B, 2A and 2B) interspersed with three short
linker sequences (L1, L12 and L2) at conserved regions (12). At the C-terminus of the 2B domain
is a distinct sequence motif, TYRKLLEGE, which exhibits
near-perfect conservation across all intermediate filament proteins
(13).
A previous study revealed a genotype-phenotype
correlation exists in patients with EBS (14). Disease severity is associated with
the physicochemical properties of the substituted amino acids and
their location within the protein structure (15). Specifically, substitutions of
highly conserved amino acids in the helix initiation or termination
motifs disrupt heterodimerization of keratin 5 and 14 polypeptides
resulting in severe EBS (4).
Conversely, substitutions in other regions tend to result in milder
clinical phenotypes of EBS (16).
The present study identified two novel variants in
the KRT5 gene: c.1399A>T (p.I467F) in patient 1 resulting
in the amino acid substitution p.Ile467Phe and c.1412G>A
(p.R471H) in patient 2 resulting in the amino acid substitution
p.Arg471His. These two KRT5 variants have not been reported
in the ExAC, ESP, 1000G and HGMD databases. The in silico
pathogenicity predictions for these variants using Polyphen-2
revealed that they were harmful. Furthermore, pathogenicity
prediction using MutationTaster revealed that these two variants
were pathogenic.
The p.I467F variant in patient 1 resulted in
intermediate EBS while p.R471H variant in patient 2 led to
localized EBS. Notably, these variants were situated within the 2B
domain of the KRT5 gene (Fig.
3), a well-established hotspot for variants (17). Similar substitutions, namely,
p.I467L, p.I467M, and p.I467T, have been previously identified as
pathogenic variants resulting in EBS-localized, EBS-generalized and
EBS Dowling-Meara, respectively (15). Additionally, another similar
substitution, p.R471C, was previously reported as a pathogenic
variant resulting in EBS-generalized (15). The various clinical phenotypes
associated with these substitutions may be attributed to their
occurrence within the TYRKLLEGE motif of the KRT5 protein, which
may cause the surface-exposed residues to be affected. This motif
exhibits low tolerance for side chain modifications such that even
minor alterations in side chain chemistry can lead to significant
differences in EBS severity (13).
A comprehensive analysis of phenotypical changes introduced by
amino acid substitutions may reveal the genotype-phenotype
association in EBS (18).
Early stage molecular genetic analysis is essential
in patients suspected of having EBS as the results obtained can
contribute to personalized treatment, thereby resulting in improved
prognosis. A multidisciplinary approach, with recent advancements
in medicine focusing on wound care, pain management, pruritus
relief and nutritional support, is pivotal for the management of
EBS (6). Restoration of skin
integrity has become the primary goal of targeted therapy,
including protein-, cell- and gene-based interventions for EBS
(19). Topical calcipotriol and
diacerein are potential drugs to enhance the healing of skin
lesions in patients with EBS (20).
The present KRT5 gene variants contribute to
the expanding mutation spectrum of EBS. The present study
identified two novel de novo heterozygous missense variants
of KRT5, c.1399A>T (p.Ile467Phe) and c.1412G>A
(p.Arg471His), within the 2B domain of KRT5. These findings
not only broaden understanding of the underlying pathophysiology of
EBS but also hold potential significance for genetic diagnosis,
counseling and therapy. Furthermore, they also provide valuable
insights into the phenotype-genotype association observed in
EBS.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Sichuan Medical
Youth Innovation Research Project (grant no. Q23014).
Availability of data and materials
The data generated in the present study may be found
in the NCBI database under accession number (PRJNA1054300) or at
the following URL: http://www.ncbi.nlm.nih.gov/bioproject/1054300.
Authors' contributions
LL and CY designed the study and confirm the
authenticity of all the raw data. LL conducted the genetic and
bioinformatics analysis. HL and QL collected the clinical data. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Suining Central Hospital (Suining, China; approval no.
LLSNCH20200042), and written informed consent was obtained from all
subjects.
Patient consent for publication
The patients provided consent for publication of
their data and images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hon KL, Chu S and Leung AKC: Epidermolysis
bullosa: Pediatric perspectives. Curr Pediatr Rev. 18:182–190.
2022.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Jo-David and dermatology FJJ. Epidemiology
of inherited epidermolysis bullosa based on incidence and
prevalence estimates from the national epidermolysis bullosa
registry. JAMA Dermatol. 152:1231–1238. 2016.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Pânzaru M-C, Caba L, Florea L, Braha EE
and Gorduza EV: Epidermolysis bullosa-a different genetic approach
in correlation with genetic heterogeneity. Diagnostics (Basel).
12(1325)2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Has C, Bauer JW, Bodemer C, Bolling MC,
Bruckner-Tuderman L, Diem A, Fine JD, Heagerty A, Hovnanian A,
Marinkovich MP, et al: Consensus reclassification of inherited
epidermolysis bullosa and other disorders with skin fragility. Br J
Dermatol. 183:614–627. 2020.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Fine JD, Johnson LB, Weiner M, Li KP and
Suchindran C: Epidermolysis bullosa and the risk of
life-threatening cancers: The national EB registry experience,
1986-2006. J Am Acad Dermatol. 60:203–211. 2009.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Bardhan A, Bruckner-Tuderman L, Chapple
ILC, Fine JD, Harper N, Has C, Magin TM, Marinkovich MP, Marshall
JF, McGrath JA, et al: Epidermolysis bullosa. Nat Rev Dis Primers.
6(78)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Mariath L, Santin JT, Schuler-Faccini L
and Kiszewski AE: Inherited epidermolysis bullosa: Update on the
clinical and genetic aspects. An Bras Dermatol. 95:551–569.
2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Lucky AW, Dagaonkar N, Lammers K, Husami A
and Zhang K: A comprehensive next-generation sequencing assay for
the diagnosis of epidermolysis bullosa. Pediatr Dermatol.
35:188–197. 2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wang W, Qin W, Ge H, Kong X, Xie C, Tang Y
and Li M: Clinical and molecular characteristics of thirty NF1
variants in Chinese patients with neurofibromatosis type 1. Mol
Biol Rep. 46:4349–4359. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Whittock NV, Eady RA and Mcgrath JA:
Genomic organization and amplification of the human epidermal type
II keratin genes K1 and K5. Biochem Biophys Res Commun.
274:149–152. 2000.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Hanukoglu I and Fuchs EJC: The cDNA
sequence of a type II cytoskeletal keratin reveals constant and
variable structural domains among keratins. Cell. 33:915–924.
1983.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Lee CH, Kim MS, Chung BM, Leahy DJ and
Coulombe PA: Structural basis for heteromeric assembly and
perinuclear organization of keratin filaments. Nat Struct Mol Biol.
19:707–715. 2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Sørensen CB, Ladekjaer-Mikkelsen AS,
Andresen BS, Brandrup F, Veien NK, Buus SK, Anton-Lamprecht I,
Kruse TA, Jensen PK, Eiberg H, et al: Identification of novel and
known mutations in the genes for keratin 5 and 14 in Danish
patients with epidermolysis bullosa simplex: Correlation between
genotype and phenotype. J Invest Dermatol. 112:184–190.
1999.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Banerjee S, Wu Q, Yu P, Qi M and Li C: In
silico analysis of all point mutations on the 2B domain of K5/K14
causing epidermolysis bullosa simplex: A genotype-phenotype
correlation. Mol Biosyst. 10:2567–2577. 2014.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhang J, Yan M, Liang J, Li M and Yao Z: A
novel KRT5 mutation associated with generalized severe
epidermolysis bullosa simplex in a 2-year-old Chinese boy. Exp Ther
Med. 12:2823–2826. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Schuilenga-Hut PH, Vlies Pv, Jonkman MF,
Waanders E, Buys CH and Scheffer H: Mutation analysis of the entire
keratin 5 and 14 genes in patients with epidermolysis bullosa
simplex and identification of novel mutations. Hum Mutat. 21:447.
2003.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Stawczyk-Macieja M, Wertheim-Tysarowska K,
Jakubowski R, Szczerkowska-Dobosz A, Krygier M, Wilkowska A,
Sawicka J, Nowak W, Bal J and Nowicki R: A novel de novo mutation
p.Ala428Asp in KRT5 gene as a cause of localized epidermolysis
bullosa simplex. Exp Dermatol. 28:1131–1134. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Cohn HI and Teng JM: Advancement in
management of epidermolysis bullosa. Curr Opin Pediatr. 28:507–516.
2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Prodinger C, Reichelt J, Bauer JW and
Laimer M: Epidermolysis bullosa: Advances in research and
treatment. Exp Dermatol. 28:1176–1189. 2019.PubMed/NCBI View Article : Google Scholar
|