Introduction
High-incidence neurodegenerative diseases, such as
Alzheimer's disease, Parkinson's disease (PD) and Huntington's
disease, predominantly affect elderly individuals (1). The United Nations' aging report has
predicted that the global population of individuals aged >60
years will reach 2.1 billion by 2050(2), posing significant challenges for
healthcare and social services in managing neurodegenerative
diseases. Accumulating evidence has suggested that impaired protein
turnover homeostasis and the subsequent accumulation of damaged or
aberrantly-modified proteins are common mechanistic causes of these
aforementioned neurological diseases (3). The autophagy-lysosome pathway is a
cellular degradation process that serves a crucial role in
maintaining protein homeostasis under metabolic stress or
pathological conditions (4).
Dysregulation of autophagy has been associated with the formation
of toxic proteins, such as α-synuclein, in patients with PD
(5,6), underscoring its potential importance
in disease development. Therefore, understanding the signals that
can regulate autophagy and intervening in this process may hold
promise for disease control.
Programmed cell death 4 (PDCD4) is a tumor
suppressor protein that was discovered in 2018, which has been
reported to be involved in cell cycle progression and apoptosis
(7). Initially studied in cancer,
PDCD4 was found to modulate protein synthesis by inhibiting
translation via reducing eukaryotic initiation factor 4A helicase
activity in HeLa cells (8). In
mitogen-stimulated cells, degradation of PDCD4 is required for
efficient protein translation, which is a prerequisite for
efficient cell proliferation (9).
The potential role of PDCD4 in other physiologies, such as vascular
remodeling and lipid metabolism, has only begun to be elucidated
over recent years (10,11). In addition, in the context of
protein synthesis, molecular pathways (such as mTOR, JAK/STAT and
PI3k/Akt) involved in tumor cell development tend to overlap
significantly with those associated with axonal growth and
regeneration processes in the nervous system (12,13).
Excluding nervous system malignancies, available research data on
the role of PDCD4 in other nervous system diseases remain limited.
Existing studies have revealed that PDCD4 knockout can reduce the
chronic stress-induced depression-like behavior in mice (14), whereas increased PDCD4 levels in
vitro can lead to significant reductions in axonal length
(15). In addition, PDCD4
knockdown has been observed to reduce infarct damage and cortical
neuronal apoptosis induced by brain ischemia/reperfusion injury
(16).
Previous studies have suggested that PDCD4 can
inhibit autophagy in certain conditions, such as diabetic
nephropathy (17), atherosclerosis
(18), and cardiac hypertrophy
(19). However, the involvement of
PDCD4 in neurodegenerative diseases and autophagy remains unclear.
In the context of PD, which is primarily associated with damage and
degeneration of the nigrostriatal dopaminergic pathway (20), it is conceivable that PDCD4
promotes dopaminergic neuron damage by inhibiting autophagy. To
explore this hypothesis, the present study established an in
vitro model of PD in SK-N-SH cells using
1-methyl-4-phenylpyridinium (MPP+) to investigate the
regulatory mechanism of PDCD4 in this context. The results from the
present study may offer novel insights and therapeutic targets for
future PD treatment.
Materials and methods
Cell culture and treatment
SK-N-SH cells (American Type Culture Collection)
were cultured in DMEM (Gibco; Thermo Fisher Scientific, Inc.)
supplemented with 10% FBS (HyClone; Cytiva) and treated with 1 mM
MPP+ (Sigma-Aldrich; Merck KGaA) for 12 h at 37˚C to
mimic PD in vitro (21).
Cells were transfected with either of two types of pLKO.1-Neo
short-hairpin RNA (shRNA; 100 nM) against PDCD4 or with scrambled
shRNA as a negative control (sh-NC; Changsha Aibiwei Biotechnology
Co., Ltd.) using Lipofectamine® 2000 (Invitrogen; Thermo
Fisher Scientific, Inc.) at 37˚C for 48 h. The target sequences for
shRNAs (5'-3') were as follows: sh-PDCD4-1, GCGGTTTGTAGAAGAATGTTT;
sh-PDCD4-2, CCTCCATTAACGAAGCTAGAA; and sh-NC, TTCTCCGAACGTGTCACGT.
The expression levels of PDCD4 were measured 48 h
post-transfection. For mechanistic studies, transfected cells were
pre-treated with 5 mM autophagy inhibitor 3-methyladenine (3-MA;
Sigma-Aldrich; Merck KGaA) for 30 min at 37˚C (22) and then treated with
MPP+.
Reverse transcription-quantitative PCR
(RT-qPCR)
SK-N-SH cells were homogenized with
TRIzol® reagent (Invitrogen; Thermo Fisher Scientific,
Inc.) on ice. Total RNA was quantified using NanoDrop®
equipment (NanoDrop; Thermo Fisher Scientific, Inc.). Total RNA was
reverse transcribed into cDNA using a SuperScript™ III
RT kit (Invitrogen; Thermo Fisher Scientific, Inc.) according to
the manufacturer's protocol, before 2X SYBR Green PCR Mastermix
(Beijing Solarbio Science & Technology Co., Ltd.) was applied
for qPCR. The thermocycling conditions used for qPCR were as
follows: 94˚C for 2 min, followed by 40 cycles of 94˚C for 15 sec
and 60˚C for 30 sec. The relative mRNA expression levels were
normalized to β-actin using the 2-IICq method (23). The primer sequences (5'-3') were as
follows: PDCD4, forward AACCCTGCAGAAAATGCTGG, reverse
CGCCTTTTTGCCTTGGCATT; and β-actin, forward CTTCGCGGGCGACGAT and
reverse CCACATAGGAATCCTTCTGACC.
Western blotting
SK-N-SH cells were lysed using RIPA buffer (Beyotime
Institute of Biotechnology) on ice, before total protein was
quantified using the BCA method. Proteins (25 µg/lane) were
separated by SDS-PAGE using 10 or 12% gels according to molecular
weight and electro-transferred onto PDVF membranes. The membranes
were blocked in 5% fat-free milk for 2 h at room temperature before
being incubated with primary antibodies against PDCD4 (cat. no.
9535S; 1:1,000; Cell Signaling Technology, Inc.), Bcl-2 (cat. no.
3498S; 1:1,000; Cell Signaling Technology, Inc.), Bax (cat. no.
50599-2-Ig; 1:10,000; ProteinTech Group, Inc.), cleaved-caspase3
(cat. no. 25128-1-AP; 1:1,000; ProteinTech Group, Inc.), LC3 (cat.
no. 14600-1-AP; 1:2,000; ProteinTech Group, Inc.), p62 (cat. no.
18420-1-AP; 1:20,000; ProteinTech Group, Inc.), Beclin1 (cat. no.
11306-1-AP; 1:5,000; ProteinTech Group, Inc.) and GAPDH (cat. no.
10494-1-AP; 1:20,000; ProteinTech Group, Inc.) at 4˚C overnight.
The next day, the membranes were incubated with HRP-conjugated goat
anti-rabbit secondary antibodies (cat. no. ab6721; 1:2,000; Abcam)
for 2 h at room temperature. The membranes were then developed with
a BeyoECL Plus reagent (Beyotime Institute of Biotechnology) before
the results were analyzed using ImageJ software (version 1.52;
National Institutes of Health).
Lactate dehydrogenase (LDH)
activity
SK-N-SH cells were seeded into 96-well plates and
grown to 80% confluence. Cells received MPP+
stimulation, before the LDH release reagent (cat. no. C0016;
Beyotime Institute of Biotechnology) diluted 10-fold was added to
the wells and incubated at 37˚C for an additional 1 h. Cells
without MPP+ stimulation were also supplemented with LDH
release reagent and served as a maximum enzyme release control.
After centrifugation at 400 x g for 5 min at room temperature, the
supernatant of each well was collected and the absorbance was
measured at 490 nm using a microplate reader (Molecular Devices,
LLC). LDH release (%)=absorbance of the experimental
group/absorbance of the maximum release control group x100, and the
result was normalized to the control group.
Flow cytometry
SK-N-SH cells were subjected to MPP+
stimulation, washed twice with PBS and suspended in 1X binding
buffer at a density of 1x106/ml. Cells were incubated
with 5 µl Annexin V-FITC working solution for 5 min and 5 µl
propidium iodide working solution for 5 min both at room
temperature in the dark. These solutions were contained within the
Annexin V-FITC/PI apoptosis detection kit (cat. no. CA1020; Beijing
Solarbio Science & Technology Co., Ltd.). Cell apoptosis was
then immediately analyzed using a flow cytometer (BD
FACSCanto™; BD Biosciences) and FlowJo software (version
10.0.7; FlowJo LLC).
ELISA
The levels of inflammatory factors TNF-α (cat. no.
PT518), IL-1β (cat. no. PI305) and IL-6 (cat. no. PI330) were
assessed using ELISA kits (Beyotime Institute of Biotechnology)
according to the manufacturer's protocols. Cell supernatant was
gathered after centrifugation at 300 x g for 5 min at 4˚C, before
the absorbance was measured using a microplate reader (Molecular
Devices, LLC).
Cellular reactive oxygen species (ROS)
levels
SK-N-SH cells were incubated with diluted DCFH-DA
probe (Nanjing KeyGen Biotech Co., Ltd.) at a final concentration
of 10 µmol/l in the dark at 37˚C for 30 min. The cells were then
washed twice with PBS and imaged from five fields of view using a
fluorescence microscope (Olympus Corporation). The fluorescence
intensity was analyzed using ImageJ software (version 1.52;
National Institutes of Health).
Oxidative stress parameters
Malondialdehyde (MDA; cat. no. D799761; Sangon
Biotech Co., Ltd.) and 4-hydroxynonenal (4-HNE; cat. no. D751041;
Sangon Biotech Co., Ltd.) levels, and catalase (CAT; cat. no.
BC0200; Beijing Solarbio Science & Technology Co., Ltd.) and
superoxidase dismutase (SOD; cat. no. D799593; Sangon Biotech Co.,
Ltd.) activities were quantified using the corresponding commercial
assay kits according to the manufacturers' instructions. Optical
density was measured using a microplate reader.
Immunofluorescence (IF)
SK-N-SH cells were subjected to MPP+
stimulation followed by 4% paraformaldehyde fixation at room
temperature for 15 min. These cells were blocked with 5% goat serum
(Gibco; Thermo Fisher Scientific, Inc.) at room temperature for 1
h, and then incubated with a primary antibody against LC3B (cat.
no. ab51520; 1:2,000; Abcam) at 4˚C overnight, followed by
incubation with FITC-labelled goat anti-rabbit secondary antibody
(cat. no. SA00003-2; 1:500; ProteinTech Group, Inc.) for 1 h at
room temperature. The nuclei were counterstained with 0.5 µg/ml
DAPI for 5 min at room temperature. Cells were finally imaged from
five fields of view under a fluorescence microscope (Olympus
Corporation). The fluorescence intensity was analyzed using ImageJ
software (version 1.52; National Institutes of Health).
Statistical analysis
All experiments were performed at least three times
independently. GraphPad Prism 8.0 software (Dotmatics) was used for
statistical analysis and data are presented as the mean ± standard
deviation. Statistical differences between two groups were assessed
using the unpaired Student's t-test, whereas one-way ANOVA followed
by Tukey's post hoc test was applied for comparisons among multiple
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Role of PDCD4 in cytotoxic injury
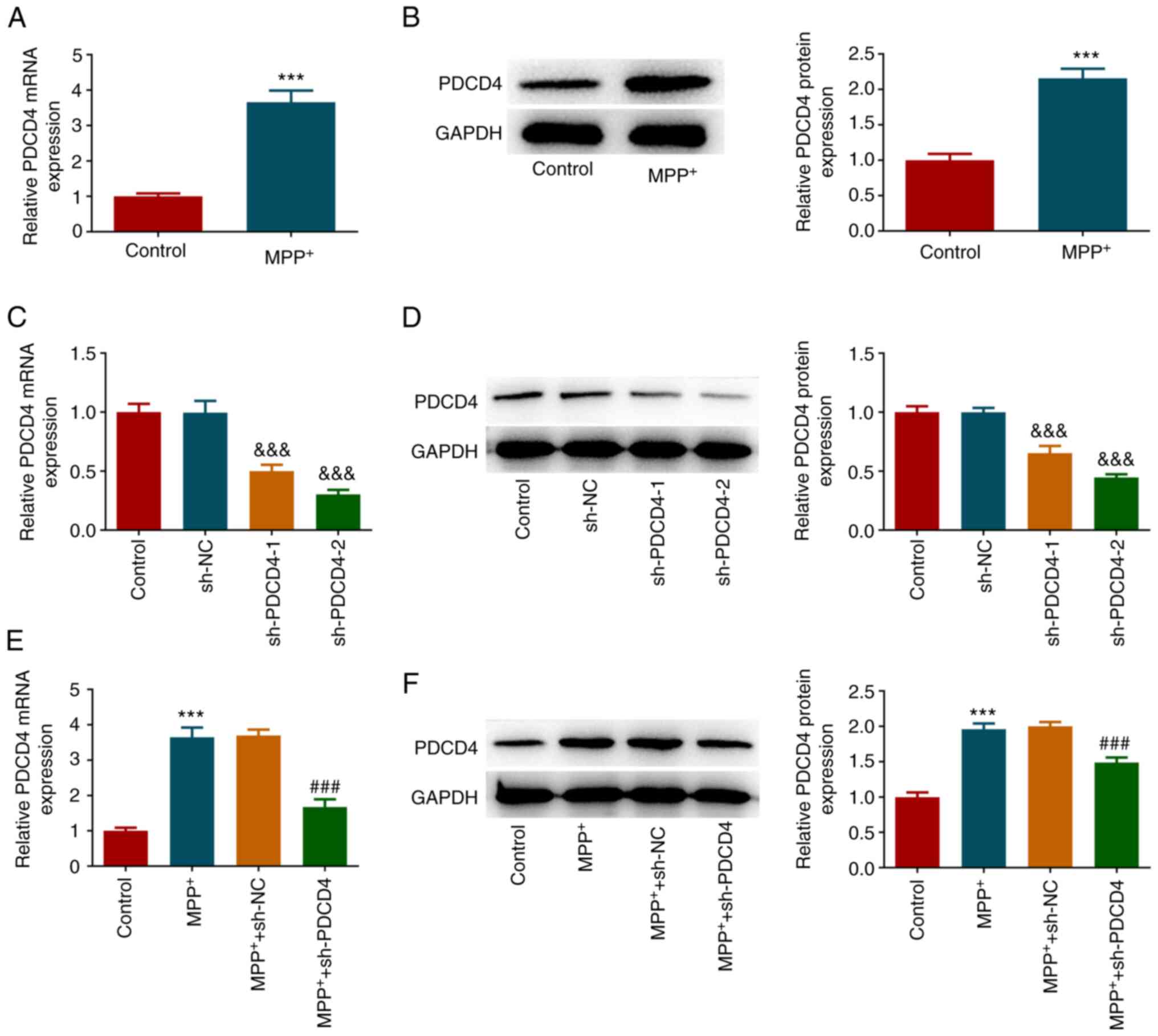
After SK-N-SH cells were treated with
MPP+, the mRNA and protein expression levels of PDCD4
were significantly increased (Fig.
1A and B). To explore the role
of PDCD4, cells were subjected to PDCD4 knockdown by shRNA
transfection. Transfection efficiency was then verified by RT-qPCR
and western blotting (Fig. 1C and
D). PDCD4 mRNA and protein levels
were significantly reduced in the sh-PDCD4-1 and sh-PDCD4-2 groups
compared with the sh-NC group. Since transfection efficiency in the
sh-PDCD4-2 group was superior, this transfection group was selected
for subsequent experiments. This group of transfected cells and
cells in the sh-NC group were then treated with MPP+,
and PDCD4 expression was significantly decreased in the
MPP+ + sh-PDCD4 group compared with that in the
MPP+ + sh-NC group (Fig.
1E and F). Subsequently, the
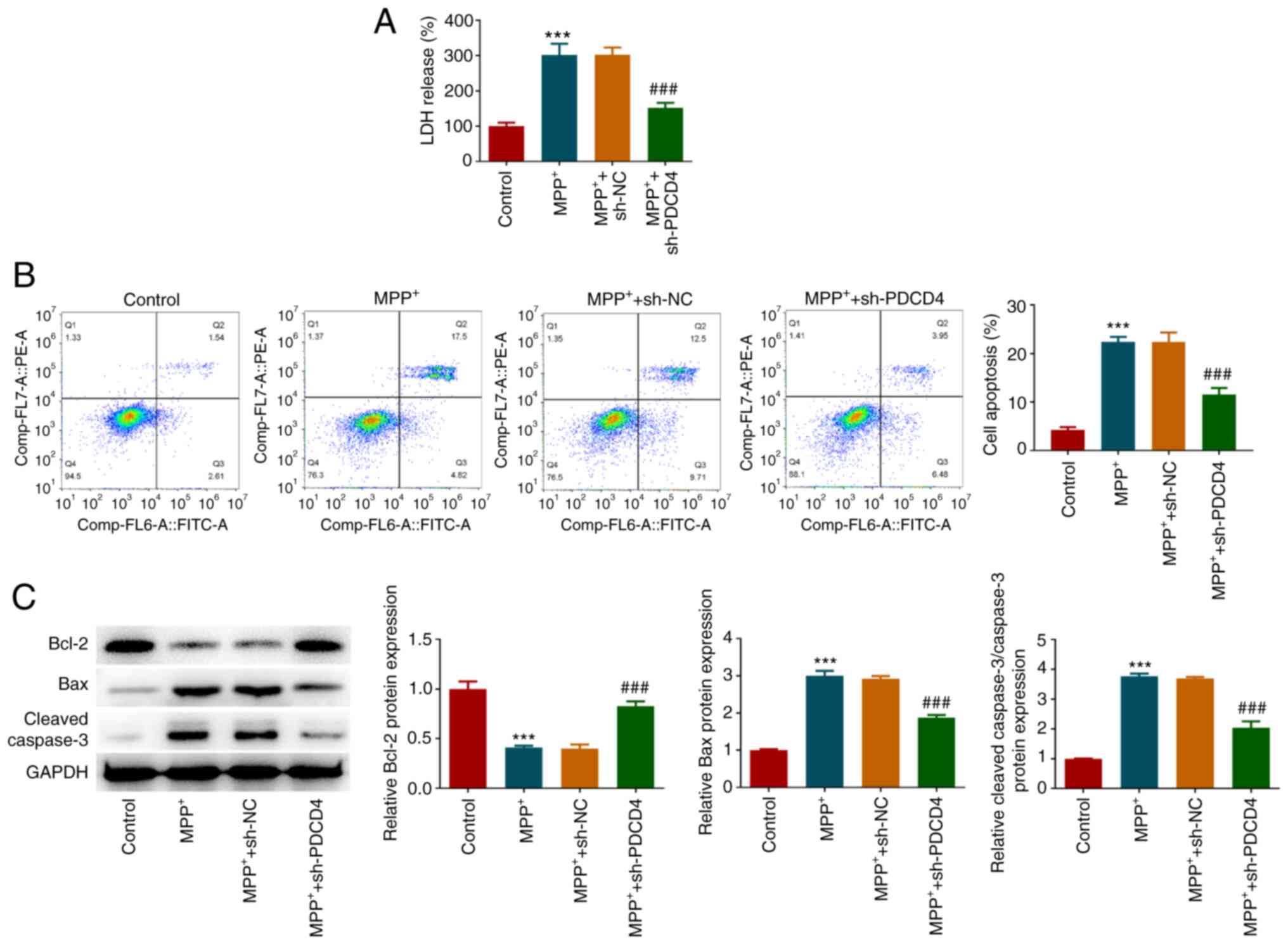
effects of MPP+ and PDCD4 on cytotoxic injury in each
group were evaluated. MPP+ caused a significant increase
in LDH activity, which was significantly reversed by PDCD4
knockdown (Fig. 2A). The results
of flow cytometry (Fig. 2B) showed
that MPP+ promoted cell apoptosis, which was supported
by the western blotting results (Fig.
2C). This was indicated by the significant increase in Bax and
cleaved-caspase3 protein expression levels, and the significant
decrease in Bcl-2 expression levels (Fig. 2C). After cells with PDCD4 knockdown
were treated with MPP+, the level of apoptosis was
significantly lower compared with that in the MPP+ +
sh-NC group, according to both flow cytometry and western blotting
results (Fig. 2C and D).
Role of PDCD4 in the inflammation and
oxidative stress
The effects of MPP+ and PDCD4 on the
inflammatory response and oxidative stress of cells in each
treatment group were next evaluated. After measuring the levels of
inflammatory factors in the cell supernatant, it was found that
MPP+ significantly increased the levels of TNF-α, IL-1β
and IL-6, which was significantly reversed by PDCD4 knockdown
compared with those in the MPP+ + sh-NC group (Fig. 3A). The results of DCFH-DA probe
imaging revealed that MPP+ induced a significant
increase in ROS levels in the cells, which was also significantly
reversed by PDCD4 knockdown (Fig.
3B). In addition, MPP+ significantly increased MDA
and 4-HNE levels, an effect that was significantly reversed by
PDCD4 knockdown. The activities of CAT and SOD were significantly
weakened by MPP+, but were significantly restored after
PDCD4 knockdown (Fig. 3C).
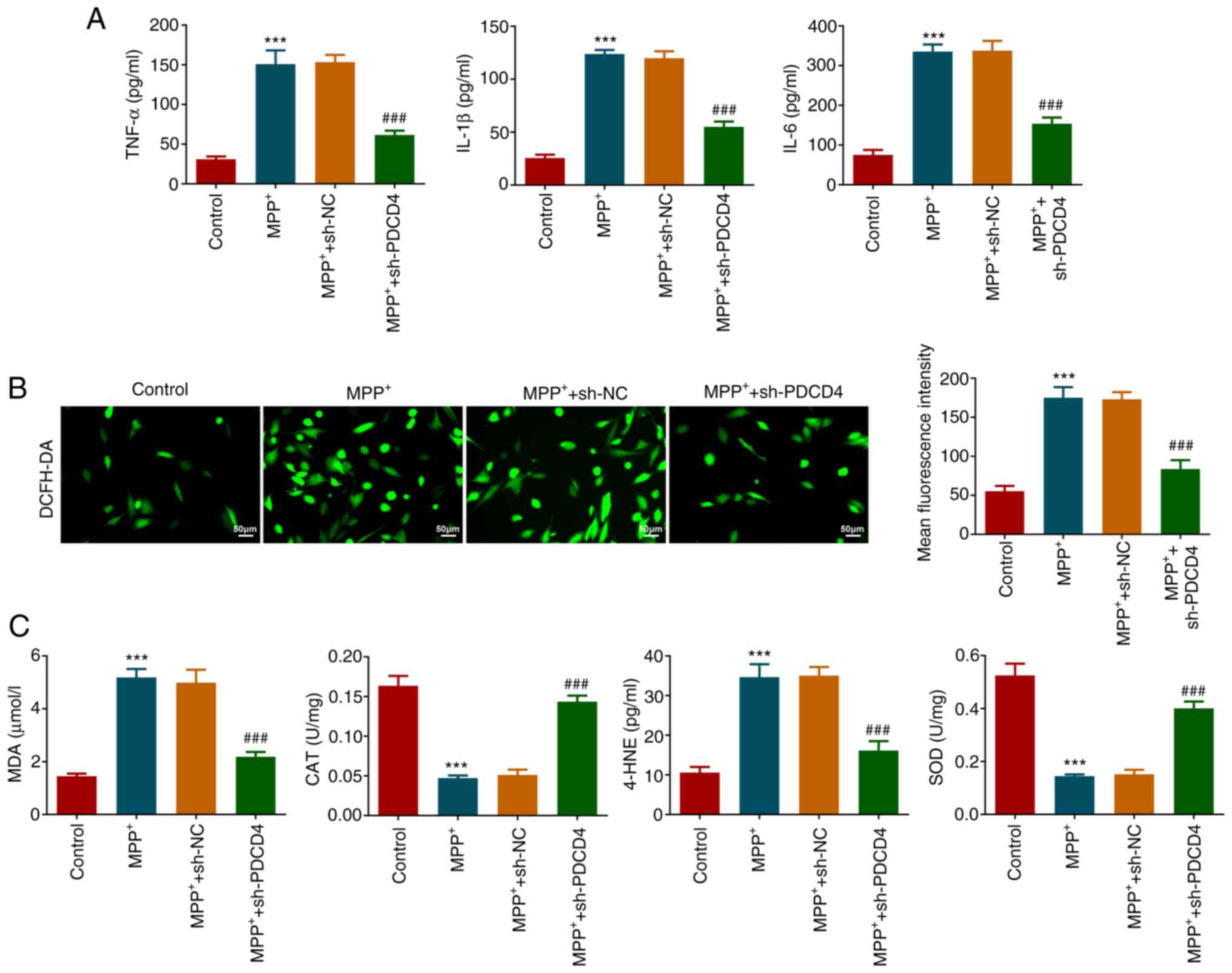 | Figure 3Role of PDCD4 in inflammation and
oxidative stress. (A) Levels of inflammatory factors in the cell
supernatant were measured by ELISA. (B) DCFH-DA probe was used to
measure reactive oxygen series content in cells. Magnification,
x200. (C) Levels of MDA and 4-HNE, and activities of CAT and SOD,
were used to measure the degree of oxidative stress in cells.
***P<0.001 vs. control; ###P<0.001 vs.
MPP+ + sh-NC. MPP+,
1-methyl-4-phenylpyridinium; PDCD4, programmed cell death 4; sh,
short hairpin; NC, negative control; MDA, malondialdehyde; CAT,
catalase; 4-HNE, 4-hydroxynonenal; SOD, superoxide dismutase. |
Role of autophagy in PDCD4
regulation
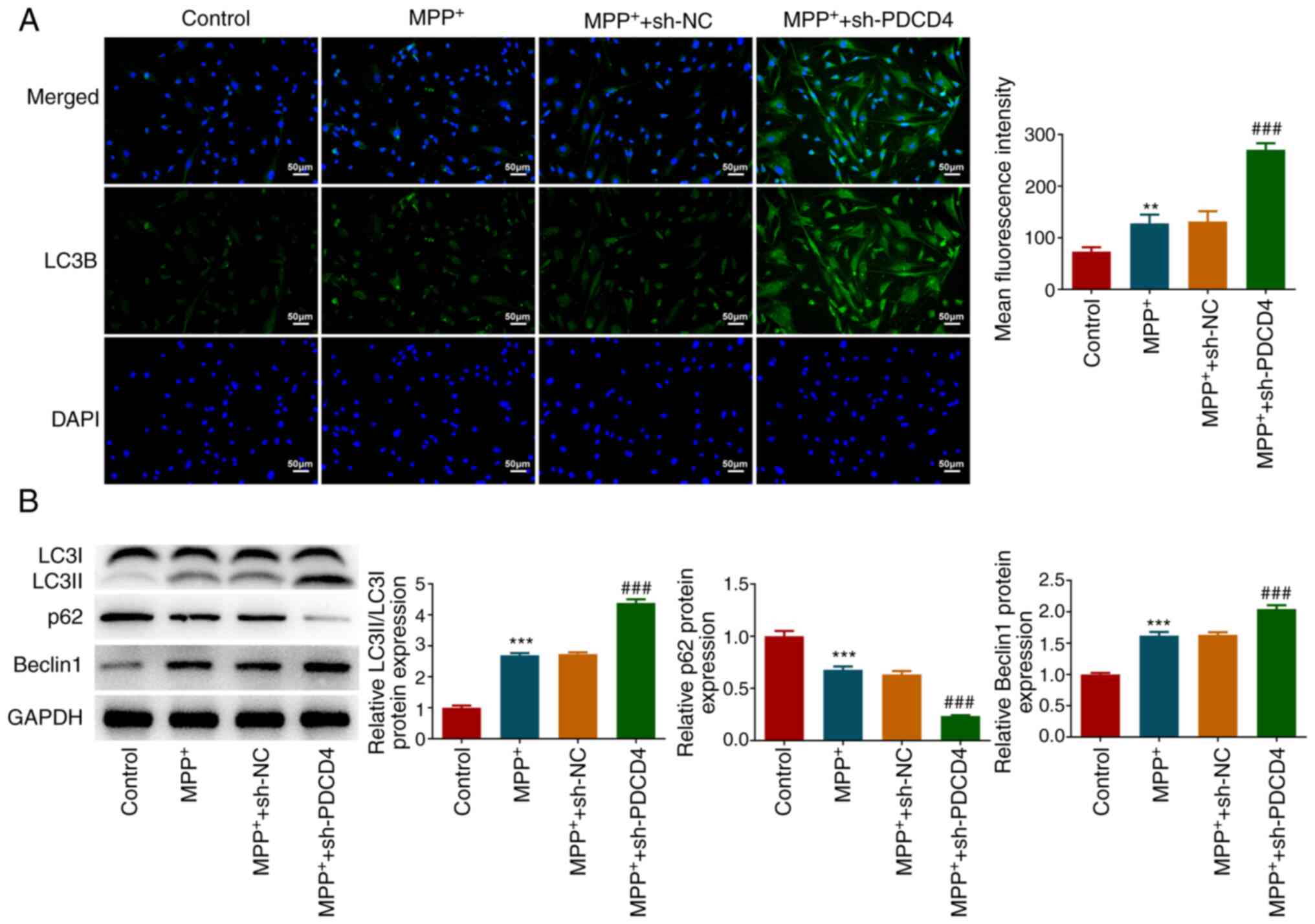
Immunofluorescence revealed that MPP+
slightly but significantly promoted the autophagic behavior of
cells, which was significantly potentiated by PDCD4 knockdown
(Fig. 4A). Similar findings were
also reflected in the results of western blotting (Fig. 4B). Specifically, LC3II/LC3I and
Beclin1 expression levels were significantly increased, whereas p62
expression was decreased, after MPP+ treatment. In
addition, these aforementioned alterations were significantly
potentiated in the MPP+ + sh-PDCD4 group (Fig. 4B).
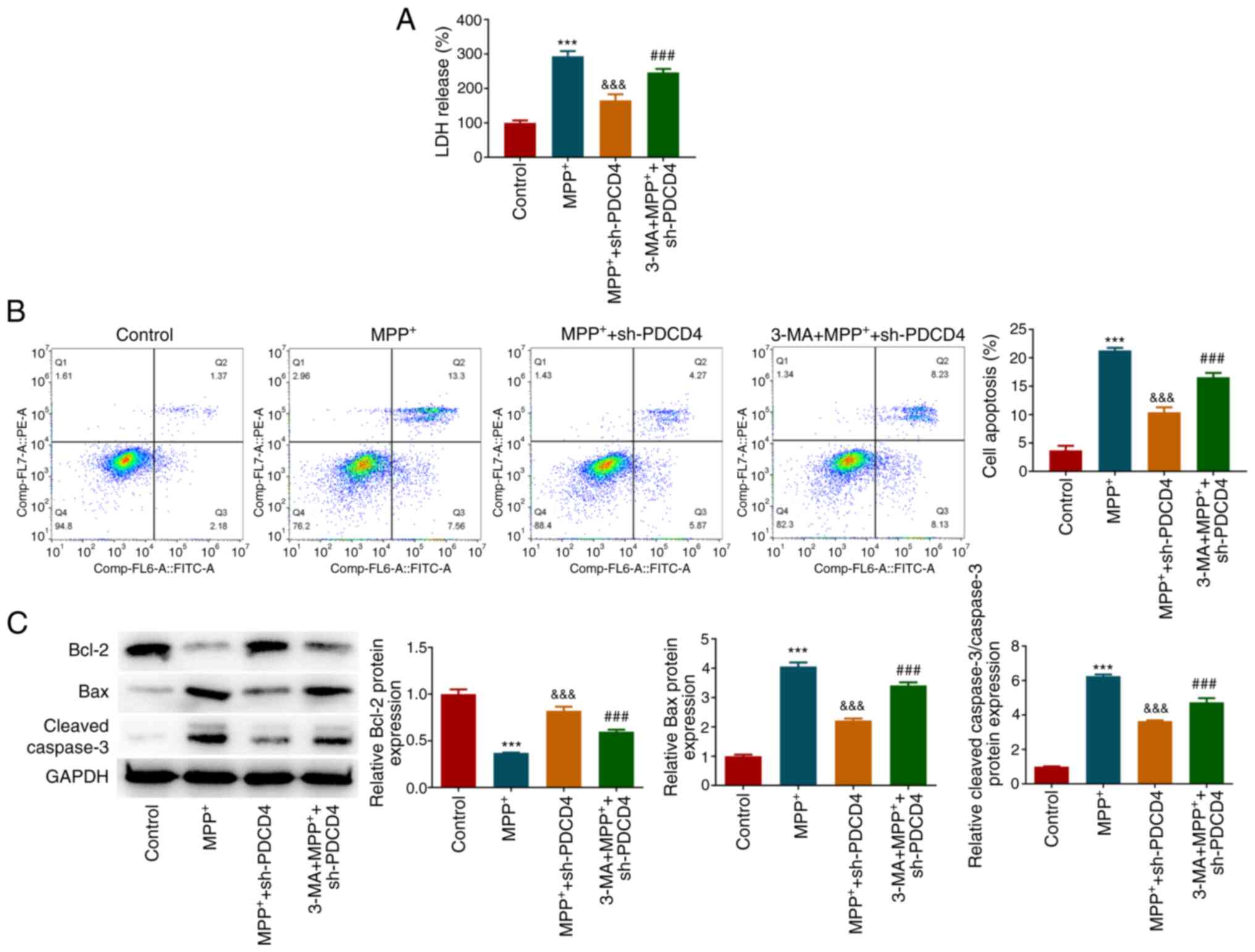
To explore whether autophagy mediated the regulatory
mechanism of PDCD4, cells were treated with 3-MA. Under conditions
of MPP+ stimulation and PDCD4 knockdown, 3-MA
significantly increased the activity of cellular LDH (Fig. 5A) and significantly promoted cell
apoptosis compared with those in the MPP+ + sh-PDCD4
group, which was reflected in the significant increase in the
proportion of apoptotic cells (Fig.
5B). Additionally, 3-MA increased the expression levels of Bax
and cleaved-caspase 3 and reduced Bcl-2 levels, corroborating the
increase in apoptosis (Fig. 5C).
3-MA treatment was also found to significantly increase the levels
of inflammatory factors TNF-α, IL-1β and IL-6 in cells (Fig. 6A). Furthermore, the levels of
cellular ROS, as reflected by fluorescence imaging (Fig. 6B), and the levels of oxidative
stress indicators MDA and 4-HNE, as detected using respective kits,
were all significantly increased in response to 3-MA compared with
those in the MPP+ + sh-PDCD4 group. By contrast, CAT and
SOD activities were significantly decreased after 3-MA treatment
compared with those in the MPP+ + sh-PDCD4 group
(Fig. 6C). These results suggested
that 3-MA largely reversed the effects of PDCD4 knockdown.
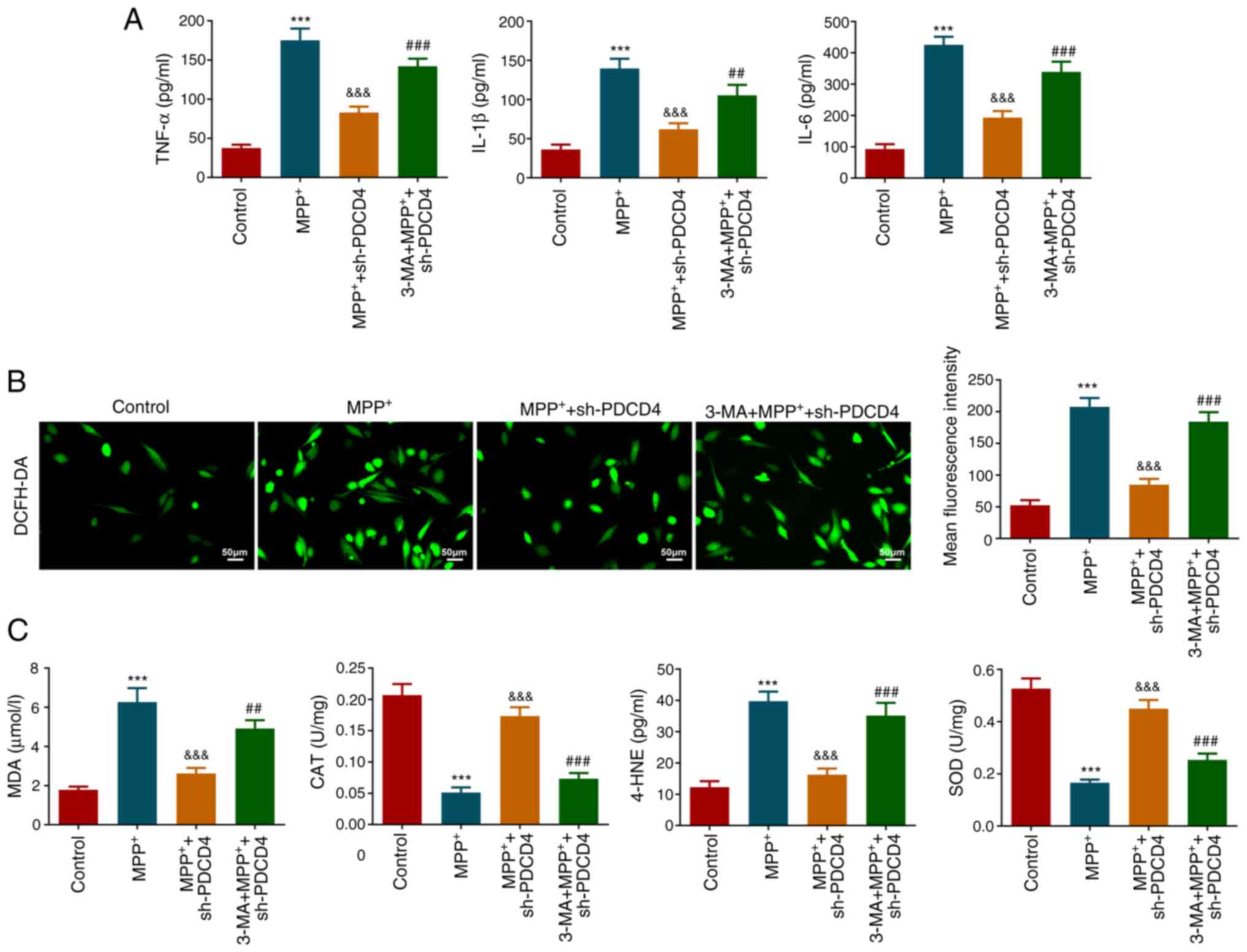 | Figure 6Role of autophagy in inflammation and
oxidative stress. (A) Effect of 3-MA on the levels of inflammatory
factors was measured. (B) Effect of 3-MA on the level of reactive
oxygen species production was assessed using DCFH-DA probe.
Magnification, x200. (C) Levels of MDA and 4-HNE, and activities of
CAT and SOD, were measured following 3-MA treatment.
***P<0.001 vs. control;
&&&P<0.001 vs. MPP+;
##P<0.01 and ###P<0.001 vs.
MPP+ + sh-PDCD4. 3-MA, 3-methyladenine; MPP+,
1-methyl-4-phenylpyridinium; PDCD4, programmed cell death 4; sh,
short hairpin; MDA, malondialdehyde; CAT, catalase; 4-HNE,
4-Hydroxynonenal; SOD, superoxide dismutase. |
Discussion
PD is the second most common neurodegenerative
disease, which pathologically manifests as the damage and
progressive loss of dopaminergic neurons (24). Although current treatment methods,
such as medication (Levodopa, monoamine oxidase type B inhibitors,
dopamine receptor agonists), deep brain stimulation surgery and
rehabilitation, can alleviate symptoms in some patients, they
cannot halt disease progression (3). Therefore, elucidating the etiology of
this disease is critical for the development of effective
diagnostics and therapies. Neurodegenerative diseases are marked by
the presence of deleterious protein aggregates in the cytoplasm and
nucleus, leading to cytotoxicity and neuronal cell death (25). Autophagy is a process that can
engulf long-lived proteins and protein aggregates, which are then
targeted for lysosomal degradation (26,27).
Therefore, autophagy serves a key role in maintaining cytoplasmic
homeostasis by clearing damaged proteins and/or organelles
(28). Although advances have been
made in elucidating the role of the autophagy machinery and how its
components are regulated, the mechanism underlying its dysfunction
and how it influences PD pathology remains unclear. The present
study found that MPP+ increased PDCD4 expression and
enhanced the extent of autophagy in cells, suggesting that PDCD4
may be an upstream regulator of the autophagy process in this cell
type. Knockdown of PDCD4 expression was found to promote autophagy
further, and to reduce cell apoptosis and mitigate oxidative
stress. These findings suggested that PDCD4 could be a potential
target for autophagy regulation in PD.
In the context of PD, the relationship between PDCD4
and autophagy was assessed in the present study. Similar
associations have been previously documented. During
atherosclerosis, macrophages exposed to low-density fatty acids
have been reported to trigger autophagy at the initial stages of
foam cell formation (18). PDCD4
deficiency was also found to enhance lipophagy in macrophages, but
to suppress apoptosis and mitochondrial dysfunction. Another
previous study demonstrated that PDCD4 can reduce the expression of
autophagy-related (ATG) 5 and hence formation of the ATG12/ATG5
complex in human ovarian cancer Skov3 cells, thereby exerting a
negative regulatory function on autophagy (29). In addition, in mouse
neuro-inflammation models, PDCD4 has been found to potentially
serve as a hub regulatory molecule, simultaneously boosting
microglial inflammatory activation and neuronal apoptosis in the
central nervous system (30).
To assess the effects of PDCD4 regulatory signaling
on autophagy, the present study used 3-MA to inhibit autophagy in
cells. Previous studies have used 3-MA to investigate the
regulation of autophagy in neurological disease models. Autophagy
has been observed to be impaired in the hippocampus of rats with
doxorubicin-induced neuronal injury, but 3-MA treatment resulted in
persistent oxidative stress and neuronal apoptosis (31). In a
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced PD mouse
model, 3-MA was found to reverse the therapeutic effects of
kaempferol on tyrosine hydroxylase-positive neuron injury and
neuroinflammation (32).
Furthermore, dopaminergic neurons are particularly metabolically
active with high mitochondrial energy demands (33). Therefore, they are particularly
vulnerable to the insufficient clearance of damaged mitochondria
(33). A recent study found that
PDCD4 knockdown was able to maintain mitochondrial membrane
potential in mouse dopaminergic neuronal MN9D cells (34).
In summary, to the best of our knowledge, the
present study was the first to reveal the regulatory effects of
PDCD4 on autophagy in MPP+-induced SK-N-SH cells,
offering potential targets for PD therapy. The results of the
present study may be used to potentially advance the development of
targeted interventions to mitigate the pathophysiology of PD.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
GC and PL contributed to the project design and
experiments. TK and NL contributed to experiments and formal
analysis. All authors read and approved the final manuscript, and
confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Hou Y, Dan X, Babbar M, Wei Y, Hasselbalch
SG, Croteau DL and Bohr VA: Ageing as a risk factor for
neurodegenerative disease. Nat Rev Neurol. 15:565–581.
2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Amuthavalli Thiyagarajan J, Mikton C,
Harwood RH, Gichu M, Gaigbe-Togbe V, Jhamba T, Pokorna D, Stoevska
V, Hada R, Steffan GS, et al: The UN Decade of healthy ageing:
Strengthening measurement for monitoring health and wellbeing of
older people. Age Ageing. 51(afac147)2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Hou X, Watzlawik JO, Fiesel FC and
Springer W: Autophagy in Parkinson's disease. J Mol Biol.
432:2651–2672. 2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Li C, Wang X, Li X, Qiu K, Jiao F, Liu Y,
Kong Q, Liu Y and Wu Y: Proteasome inhibition activates
autophagy-lysosome pathway associated with TFEB dephosphorylation
and nuclear translocation. Front Cell Dev Biol.
7(170)2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Park H, Kang JH and Lee S: Autophagy in
neurodegenerative diseases: A hunter for aggregates. Int J Mol Sci.
21(3369)2020.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Martini-Stoica H, Xu Y, Ballabio A and
Zheng H: The autophagy-lysosomal pathway in neurodegeneration: A
TFEB perspective. Trends Neurosci. 39:221–234. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wang Q and Yang HS: The role of Pdcd4 in
tumour suppression and protein translation. Biol Cell:
10.1111/boc.201800014, 2018 (Epub ahead of print).
|
|
8
|
Moustafa-Kamal M, Kucharski TJ, El-Assaad
W, Abbas YM, Gandin V, Nagar B, Pelletier J, Topisirovic I and
Teodoro JG: The mTORC1/S6K/PDCD4/eIF4A axis determines outcome of
mitotic arrest. Cell Rep. 33(108230)2020.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Dorrello NV, Peschiaroli A, Guardavaccaro
D, Colburn NH, Sherman NE and Pagano M: S6K1- and betaTRCP-mediated
degradation of PDCD4 promotes protein translation and cell growth.
Science. 314:467–471. 2006.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Chai L, Wang Q, Wang Y, Li D, Zhang Q,
Chen Y, Liu J, Chen H, Qiu Y, Shen N, et al: Downregulation of
PDCD4 through STAT3/ATF6/autophagy mediates MIF-induced PASMCs
proliferation/migration and vascular remodeling. Eur J Pharmacol.
956(175968)2023.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Du X, Osoro EK, Chen Q, Yan X, Gao D, Wu
L, Ren J, Feng L, Wu N, Lu K, et al: Pdcd4 promotes lipid
deposition by attenuating PPARα-mediated fatty acid oxidation in
hepatocytes. Mol Cell Endocrinol. 545(111562)2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Li J, Kim SG and Blenis J: Rapamycin: One
drug, many effects. Cell Metab. 19:373–379. 2014.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Akram R, Anwar H, Javed MS, Rasul A, Imran
A, Malik SA, Raza C, Khan IU, Sajid F, Iman T, et al: Axonal
regeneration: underlying molecular mechanisms and potential
therapeutic targets. Biomedicines. 10(3186)2022.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Li Y, Jia Y, Wang D, Zhuang X, Li Y, Guo
C, Chu H, Zhu F, Wang J, Wang X, et al: Programmed cell death 4 as
an endogenous suppressor of BDNF translation is involved in
stress-induced depression. Mol Psychiatry. 26:2316–2333.
2021.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Di Paolo A, Eastman G, Mesquita-Ribeiro R,
Farias J, Macklin A, Kislinger T, Colburn N, Munroe D, Sotelo Sosa
JR, Dajas-Bailador F and Sotelo-Silveira JR: PDCD4 regulates axonal
growth by translational repression of neurite growth-related genes
and is modulated during nerve injury responses. RNA. 26:1637–1653.
2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Shan W, Ge H, Chen B, Huang L, Zhu S and
Zhou Y: Upregulation of miR-499a-5p decreases cerebral
ischemia/reperfusion injury by targeting PDCD4. Cell Mol Neurobiol.
42:2157–2170. 2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Osoro EK, Du X, Liang D, Lan X, Farooq R,
Huang F, Zhu W, Ren J, Sadiq M, Tian L, et al: Induction of PDCD4
by albumin in proximal tubule epithelial cells potentiates
proteinuria-induced dysfunctional autophagy by negatively targeting
Atg5. Biochem Cell Biol. 99:617–628. 2021.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Li S, Gao G, Wu F, Liu D, Zhao H, Ke J,
Liu Y, Li F, Li J, Chen Z, et al: Programmed cell death protein 4
deficiency suppresses foam cell formation by activating autophagy
in advanced glycation end-product low-density lipoprotein-induced
macrophages. J Cell Biochem. 120:7689–7700. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wang L, Ye N, Lian X, Peng F, Zhang H and
Gong H: MiR-208a-3p aggravates autophagy through the PDCD4-ATG5
pathway in Ang II-induced H9c2 cardiomyoblasts. Biomed
Pharmacother. 98:1–8. 2018.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Liu Z and Cheung HH: Stem cell-based
therapies for Parkinson disease. Int J Mol Sci.
21(8060)2020.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Liu W, Zhang F, Liang W, Huang K, Jia C,
Zhang J, Li X, Wei W, Gong R and Chen J: Integrated insight into
the molecular mechanisms of selenium-modulated,
MPP+-induced cytotoxicity in a Parkinson's disease
model. J Trace Elem Med Biol. 79(127208)2023.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhang L, Park JY, Zhao D, Kwon HC and Yang
HO: Neuroprotective effect of astersaponin i against Parkinson's
disease through autophagy induction. Biomol Ther (Seoul).
29:615–629. 2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Surmeier DJ: Determinants of dopaminergic
neuron loss in Parkinson's disease. FEBS J. 285:3657–3668.
2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Picca A, Guerra F, Calvani R, Romano R,
Coelho-Júnior HJ, Bucci C and Marzetti E: Mitochondrial
dysfunction, protein misfolding and neuroinflammation in
Parkinson's disease: Roads to biomarker discovery. Biomolecules.
11(1508)2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Liu J, Liu W and Yang H: Balancing
apoptosis and autophagy for Parkinson's disease therapy: Targeting
BCL-2. ACS Chem Neurosci. 10:792–802. 2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Corti O, Blomgren K, Poletti A and Beart
PM: Autophagy in neurodegeneration: New insights underpinning
therapy for neurological diseases. J Neurochem. 154:354–371.
2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Abdellatif M, Ljubojevic-Holzer S, Madeo F
and Sedej S: Autophagy in cardiovascular health and disease. Prog
Mol Biol Transl Sci. 172:87–106. 2020.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Song X, Zhang X, Wang X, Zhu F, Guo C,
Wang Q, Shi Y, Wang J, Chen Y and Zhang L: Tumor suppressor gene
PDCD4 negatively regulates autophagy by inhibiting the expression
of autophagy-related gene ATG5. Autophagy. 9:743–755.
2013.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Chen Q, Lu H, Duan C, Zhu X, Zhang Y, Li M
and Zhang D: PDCD4 simultaneously promotes microglia activation via
PDCD4-MAPK-NF-κB positive loop and facilitates neuron apoptosis
during neuroinflammation. Inflammation. 45:234–252. 2022.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Zhou X, Xu P, Dang R, Guo Y, Li G, Qiao Y,
Xie R, Liu Y and Jiang P: The involvement of autophagic flux in the
development and recovery of doxorubicin-induced neurotoxicity. Free
Radic Biol Med. 129:440–445. 2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Han X, Sun S, Sun Y, Song Q, Zhu J, Song
N, Chen M, Sun T, Xia M, Ding J, et al: Small molecule-driven NLRP3
inflammation inhibition via interplay between ubiquitination and
autophagy: Implications for Parkinson disease. Autophagy.
15:1860–1881. 2019.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Haddad D and Nakamura K: Understanding the
susceptibility of dopamine neurons to mitochondrial stressors in
Parkinson's disease. FEBS Lett. 589:3702–3713. 2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Li Y, Pang J, Wang J, Dai G, Bo Q, Wang X
and Wang W: Knockdown of PDCD4 ameliorates neural cell apoptosis
and mitochondrial injury through activating the PI3K/AKT/mTOR
signal in Parkinson's disease. J Chem Neuroanat.
129(102239)2023.PubMed/NCBI View Article : Google Scholar
|