Introduction
Acute myeloid leukemia (AML) is a common malignancy
of the blood system and has characteristics of genetic diversity,
aggression and high heterogeneity (1). Most AML patients have gene mutations
associated with the occurrence, development and prognosis of
leukemia (2-5),
including the internal tandem duplication (ITD) mutations of the
FLT3 tyrosine kinase (FLT3-ITD) gene. Of AML patients ~30% carry
these mutations (6,7) with adverse effects on treatment
outcomes (8,9). The FLT3-ITD mutation causes
autophosphorylation of the FLT3 receptor in the absence of its
ligand (10), resulting in
activation of downstream PI3K/AKT, RAS/MAPK/ERK and STAT5 signaling
pathways (11,12). Ultimately, proliferation and
survival of leukemia cells are both enhanced (13-15).
Therefore, the FLT3-ITD has been considered a therapeutic target
(16) and several FLT3 inhibitors
explored (4). However, inhibitory
effects are limited (17,18) by acquired resistance caused by a
secondary point mutation in the activation loop of the FLT3,
producing FLT3-TKD, which often causes patients to relapse after
remission (16,19,20).
Thus, the search continues for new drugs and treatment strategies
for AML patients.
The anti-tumor drug, paclitaxel (PTX) (21,22),
promotes tubulin aggregation, stabilizes microtubule structure,
inhibits cell mitosis and arrests cells in the G2/M
phase of the cell cycle (23). The
drug also regulates Bcl-2 and Caspase-3 to promote apoptosis of
cancer cells (24,25). Increasing evidence suggests that
PTX mediates the PI3K/AKT signaling pathway to inhibit
proliferation of solid tumor cells and induce apoptosis. Such
actions have been reported for nasopharyngeal carcinoma (26), cervical cancer (27) and lung cancer (28). Paclitaxel also enhances the
sensitivity of cancer cells to other antitumor drugs through
regulation of this pathway (28,29).
Inhibitory effects have also been found for PTX with leukemia cell
lines, such as HL-60(30) and K562
(31,32). Our previous study (33) showed that PTX combined with
quizartinib synergistically inhibited proliferation and induced
apoptosis of the FLT3-ITD+ AML cell-line, MV4-11, but
how PTX acts on the MV4-11 cells remains to be elucidated.
The current study sought to clarify the effect of
PTX on proliferation and apoptosis of MV4-11 cells and to
investigate the underlying mechanism. Paclitaxel was found to have
anti-proliferative and apoptosis-inducing effects on MV4-11 cells
via an underlying mechanism connected with the PI3K/AKT/mTOR
pathway.
Materials and methods
Cell culture
MV4-11 (FLT3-ITD+ AML cell-line) cells
were obtained from Nanjing Kebai Biotechnology Co., Ltd. and THP-1
and K562 (FLT3-ITD- leukemia cell-lines) cells from
Guangzhou Saiku Biotechnology Co., Ltd. and Procell Life Science
& Technology Co., Ltd., respectively. MV4-11 cells were
cultured in Iscove's Modified Dulbecco's Medium (Corning, Inc.) and
THP-1 and K562 cells in RPMI-1640 medium (Corning, Inc.) both
containing penicillin-streptomycin solution and 10% fetal bovine
serum (Gibco; Thermo Fisher Scientific, Inc.) 37˚C, 5%
CO2 in a humidified incubator.
Reagents
Paclitaxel and sorafenib were obtained from Shanghai
Yuanye Biotechnology Co., Ltd. and PKI-587 from Shanghai Macklin
Biochemical Co., Ltd. Cell Counting Kit-8 (CCK8) was from Dojindo
Laboratories, Inc. and Annexin V-FLTC/propidium iodide (PI)
Apoptosis Assay Kit (cat. no. 56584) from BD Biosciences. Reverse
transcription and PCR kits were from Takara Biotechnology Co., Ltd.
Anti-FLT3 antibody (1:1,000; cat. no. 3462), anti-AKT antibody
(1:1,000; cat. no. 4691), anti-phosphorylated (p-)AKT antibody
(1:2,000; cat. no. 4060), anti-S6K antibody (1:1,000; cat. no.
2708) and anti-p- S6K antibody (1:1,000; cat. no. 9234) were all
purchased from Cell Signaling Technology, Inc. and anti-GAPDH
antibody (1:5,000; cat. no. 10494-1-AP) was from Proteintech Group,
Inc. HRP-conjugated secondary antibodies goat anti-rabbit (1:2,000;
cat. no. M21002S) was purchased from Abmart Pharmaceutical
Technology Co., Ltd.
CCK8 assay
MV4-11, THP-1 and K562 cells were seeded into
96-well plates and 10 µl per well CCK-8 added at time-points
indicated for 1-4 h before the absorbance (OD) value was measured
at 450 nm by microplate reader. Mean OD values at each
concentration were used to calculate the proliferation inhibition
rate=[1-(experimental group OD value-blank group OD
value)/(negative control group OD value-blank group OD value)]
x100%. Experiments were performed in triplicate. Half-maximal
inhibitory concentration (IC50) values were calculated
by GraphPad Prism version 5.0 software (Dotmatics).
Effects of Paclitaxel plus PKI-587 on
proliferation of MV4-11 cells
The synergistic index, q, was calculated using the
Kingsdale formula (34) to assess
the combined effect of the two drugs. The formula was: q=E
(D1+2)/(D1+
D2-D1xD2), where D1 and
D2 are individual rates of inhibition by the two drugs
and D1+2 is the combined rate of inhibition. Values of
q>1.15, 0.85≤q≤1.15 and q<0.85 indicate synergistic, additive
and antagonistic effects, respectively.
Flow cytometry assay
Aliquots of 1x106 cells/ml of MV4-11,
THP-1 and K562 cells were treated for 48 h before harvesting and
washing once with PBS precooled at 4˚C. Cells were resuspended with
1X Binding Buffer and 5 µl Annexin V added with incubation at room
temperature for 15 min in the dark. 5 µl PI and 300 µl 1X Binding
Buffer were added, at room temperature for 5 min. FACSCalibur (BD
Biosciences) was used to perform flow cytometry within 5 min.
FlowJo 10.0.7 software (FlowJo LLC) was used for analysis.
Apoptotic rate=the percentage of early + late apoptotic cells.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted by TRIzol®
reagent (Thermo Fisher Scientific, Inc.) from 1x106
cells/ml of MV4-11 cells which had been exposed to various
concentrations of PTX for 48 h. Samples were dried and RNA
dissolved in DEPC water. The concentration was adjusted to give an
OD260/OD280 ratio between 1.8-2.0 and Prime
Script RT Master Mix reverse transcription kit used to perform the
reverse transcription into DNA at 37˚C for 15 min followed by
inactivation at 85˚C for 5 min, according to the manufacturer's
protocol. RT-qPCR was performed using TB Green Premix Ex Taq II kit
with GAPDH as internal reference, according to the manufacturer's
instructions. Fold change in expression levels was calculated using
the 2-IICq method (35). The Fluorescent PCR instrument,
Bio-Rad CFX Manager 2.1 was purchased from Bio-Rad Laboratories,
Inc. All RT-qPCR primers were designed by Sangon Biotech Co., Ltd.
and the primer sequences were as follows: FLT3, forward,
5'-GCAATCATAAGCACCAGCCAGGA-3' and reverse,
5'-TTCTGCGAGCACTTGAGGTTTCC-3'; PI3K, forward,
5'-CTTTGCGACAAGACTGCCGAGAG-3' and reverse,
5'-CGCCTGAAGCTGAGCAACATCC-3'; AKT, forward,
5'-ATGGAGTATGCCAACGGGGG-3' and reverse, 5'-TGTCGCGGTATACCACGTC-3';
mTOR, forward, 5'-CTTGCTGAACTGGAGGCTGATGG-3' and reverse,
5'-CCGTTTTCTTATGGGCTGGCTCT-3'; S6K, forward,
5'-TGCTGTGGATTGGTGGAGTTTGG-3' and reverse,
5'-TCTGGCTTCTTGTGTGAGGTAGGG-3'; GAPDH, forward,
5'-CTCTGCTCCTCCTGTTCGAC-3' and reverse, 5'-TAAAAAGCAGCCCTGGTGAC-3'.
The thermocycling conditions were: Initial denaturation at 95˚C for
30 sec, denaturation at 95˚C for 5 sec, annealing and elongation at
60˚C for 30 sec for a total of 40 cycles.
Western blotting
Total protein was extracted from 1x106
cells/ml MV4-11 cells which had been exposed to various
concentration of PTX for 48 h. In brief, cells were washed twice
with pre-cooled PBS and radioimmunoprecipitation assay lysis buffer
(RIPA; Beyotime Institute of Biotechnology) added before protein
concentrations were determined by Enhanced Bicinchoninic acid
Protein Assay Kit (BCA; Beyotime Institute of Biotechnology).
Protein samples (10 µl per lane) were separated with 10% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)
before transfer to PVDF membrane (Millipore, USA). Membranes were
blocked for 1 h at room temperature with 5% BSA blocking buffer
(Beijing Solarbio Science & Technology Co., Ltd.) and incubated
with primary antibodies overnight at 4˚C. Membranes were incubated
with secondary antibodies for 1 h at room temperature and ECL
luminescence substrate kit (Biosharp Life Sciences) and ImageJ
version 1.48 software (National Institutes of Health) were used to
visualize and quantify protein bands.
Statistical analysis
SPSS17.0 software (SPSS, Inc.) was used to perform
all statistical analyses. Data are presented as mean ± standard
deviation. When groups=3, differences were analyzed by one-way
ANOVA with the Least-Significant Difference (LSD) for post hoc
multiple comparisons. When groups >3, differences were analyzed
by one-way ANOVA with Tukey's for post hoc multiple comparisons.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effect of PTX on proliferation of
leukemia cells
To assess the effect of PTX on the
FLT3-ITD+ AML cells, MV4-11, the present study first
confirmed its effect on cell viability and compared it with that of
FLT3 inhibitor sorafenib, which has been widely used in treatment
of AML patients. It used various concentrations of PTX and
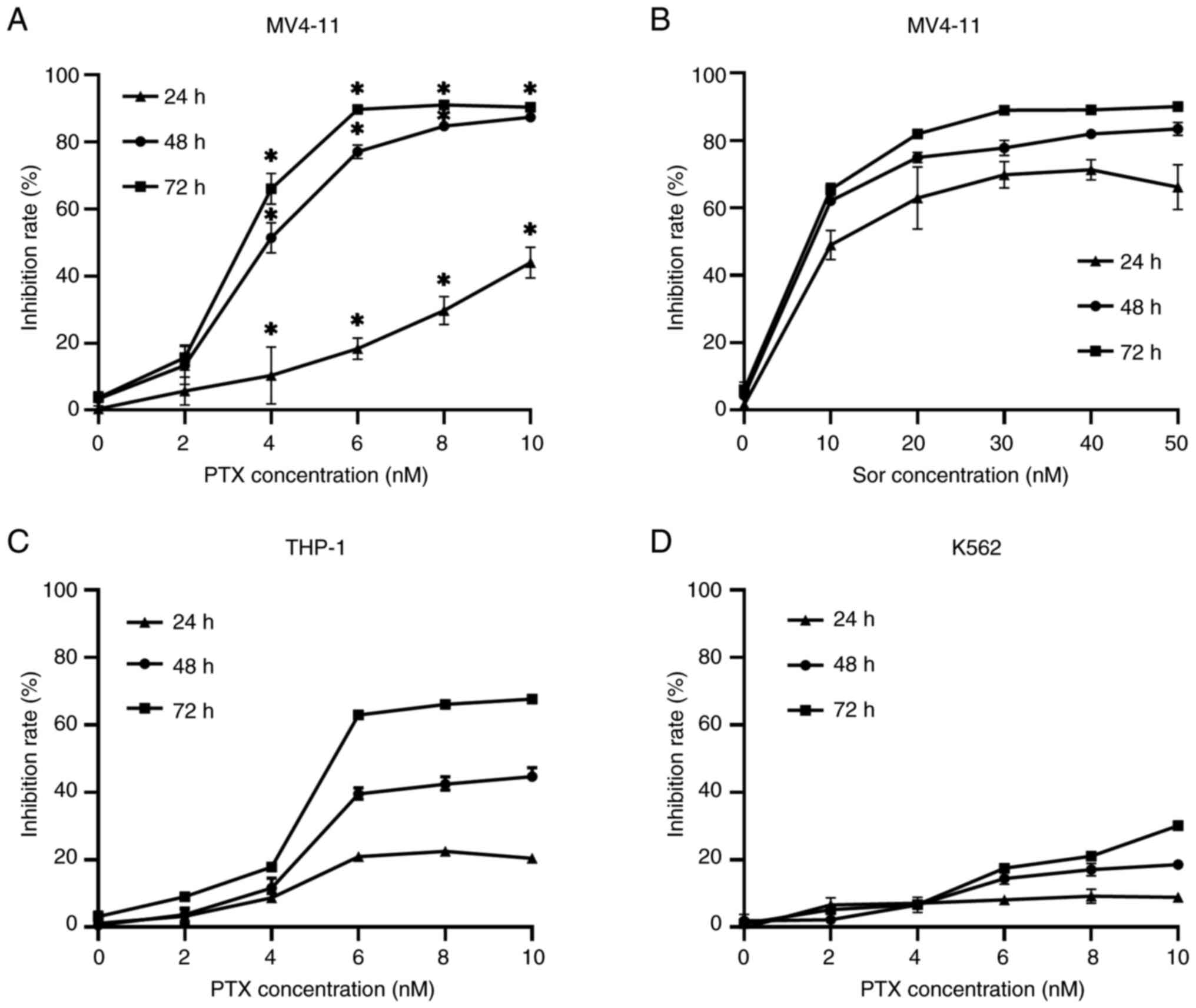
sorafenib to treat MV4-11 cells for 24, 48 and 72 h. The results
showed that PTX inhibited proliferation of MV4-11 cells in a
dose-dependent manner and with increased potency with prolonged
exposure (Fig. 1A). Sorafenib also
had a significant inhibitory effect on cell proliferation (Fig. 1B). By comparing the IC50
values of both drugs for inhibition of MV4-11 cell proliferation,
it was found that the inhibitory effect of PTX was stronger than
that of sorafenib (Table I). To
further test and compare the anti-proliferative effect of PTX on
the FLT3-ITD- leukemia cells, THP-1 and K562, the
present study used the same concentrations of PTX to treat THP-1
and K562 cells, respectively. It was observed that there was a more
moderate inhibitory effect on proliferation (Fig. 1C and D) and PTX IC50 values for
inhibition of THP-1 and K562 cell proliferation were larger than
that for MV4-11 cells (Table I).
These results indicated that PTX could clearly inhibit
proliferation of MV4-11 cells.
 | Table IIC50 values for growth
rate inhibition by paclitaxel and sorafenib for leukemia cells. |
Table I
IC50 values for growth
rate inhibition by paclitaxel and sorafenib for leukemia cells.
| | Paclitaxel
(nM) | Sorafenib (nM) |
|---|
| Drug action
duration | MV4-11 | THP-1 | K562 | MV4-11 |
|---|
| 24 h | 14.35±1.22 | 35.84±8.77 |
99.29±2.59a | 16.84±1.41 |
| 48 h | 5.65±0.35 |
11.53±1b |
33.13±3.85b |
11.41±0.9c |
| 72 h | 3.92±0.18 |
7.01±0.12a |
17.65±0.95a |
8.95±1.67c |
Effect of PTX on apoptosis in leukemia
cells
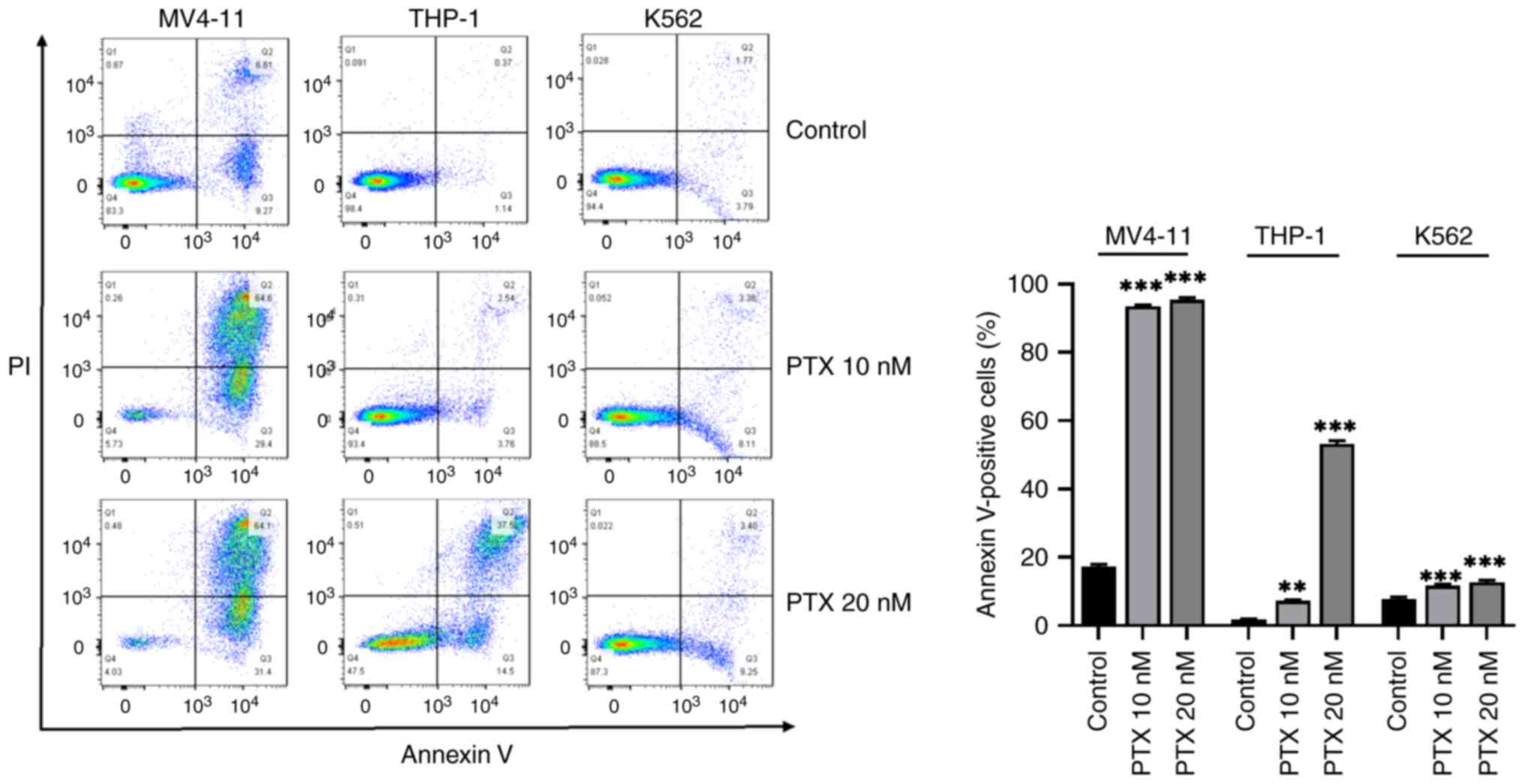
MV4-11, THP-1 and K562 cells were treated with a
range of PTX concentrations for 48 h before evaluation of apoptosis
by flow cytometry. Rates of apoptosis elicited by PTX in the
different cell-lines were: MV4-11, 10 nM PTX; 93.53±0.31% and 20 nM
PTX: 95.37±0.67%; THP-1 cells, 10 nM PTX: 7.31±0.23% and 20 nM PTX:
53.23±0.9%; K562 cells, 10 nM PTX: 11.67±0.43% and 20n M PTX:
12.68±0.58%. PTX induced apoptosis in leukemia cells at higher
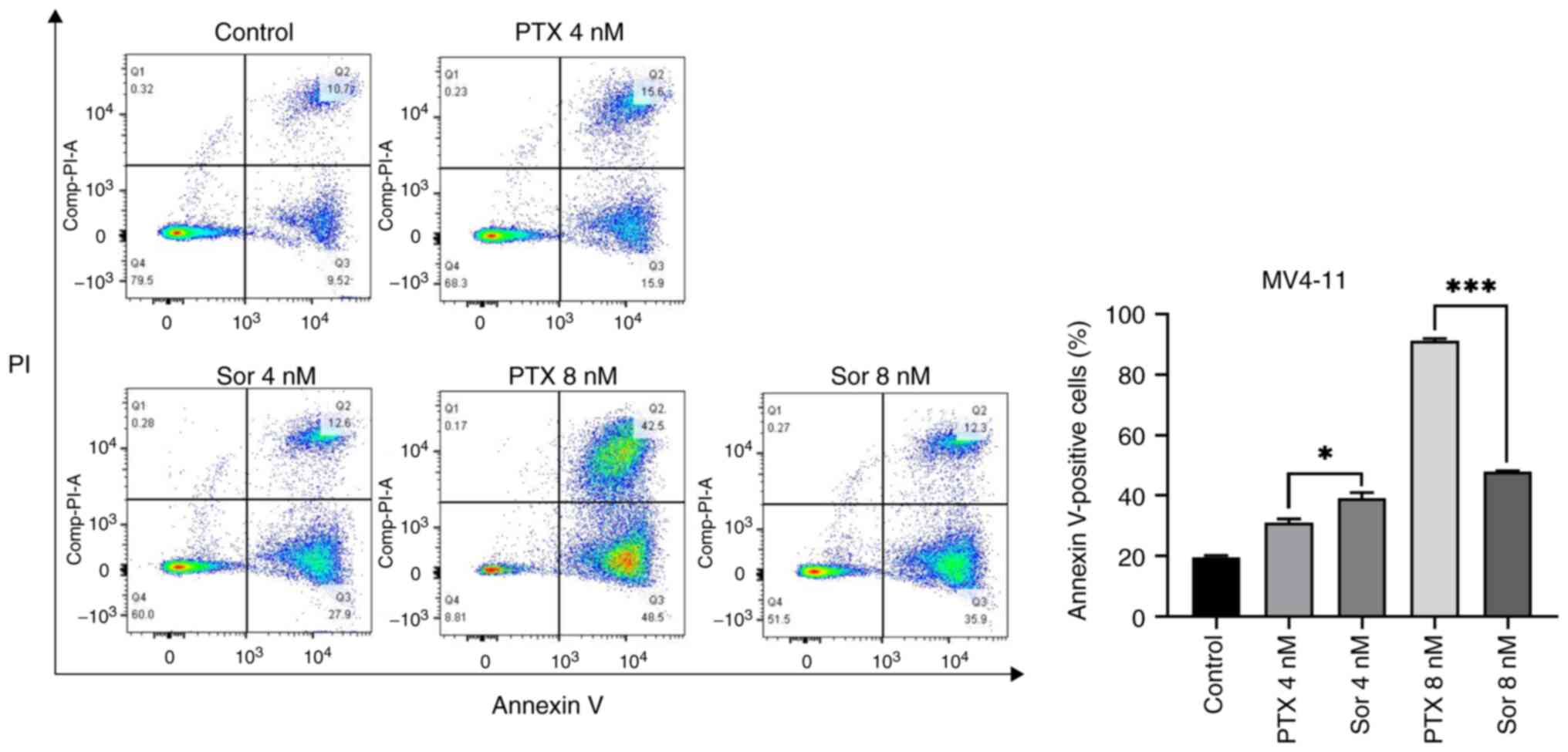
rates for MV4-11 cells compared with THP-1 and K562 cells (Fig. 2). Then, in order to compare the
pro-apoptotic effects of PTX and sorafenib on MV4-11 cells, the
same concentration of PTX and sorafenib was used to treat MV4-11
cells respectively for 48 h. It was found that the apoptosis rate
of sorafenib at 4 nM was slightly higher than that of PTX, but at 8
nM, the apoptosis rate of PTX, 91.2±0.82%, was significantly higher
compared with that of sorafenib, 48.03±0.15% (Fig. 3). Therefore, it was hypothesized
that the pro-apoptotic effect of PTX on MV4-11 cells was stronger
than sorafenib.
Effect of PTX on expression of FLT3
mRNA and protein in MV4-11 cells
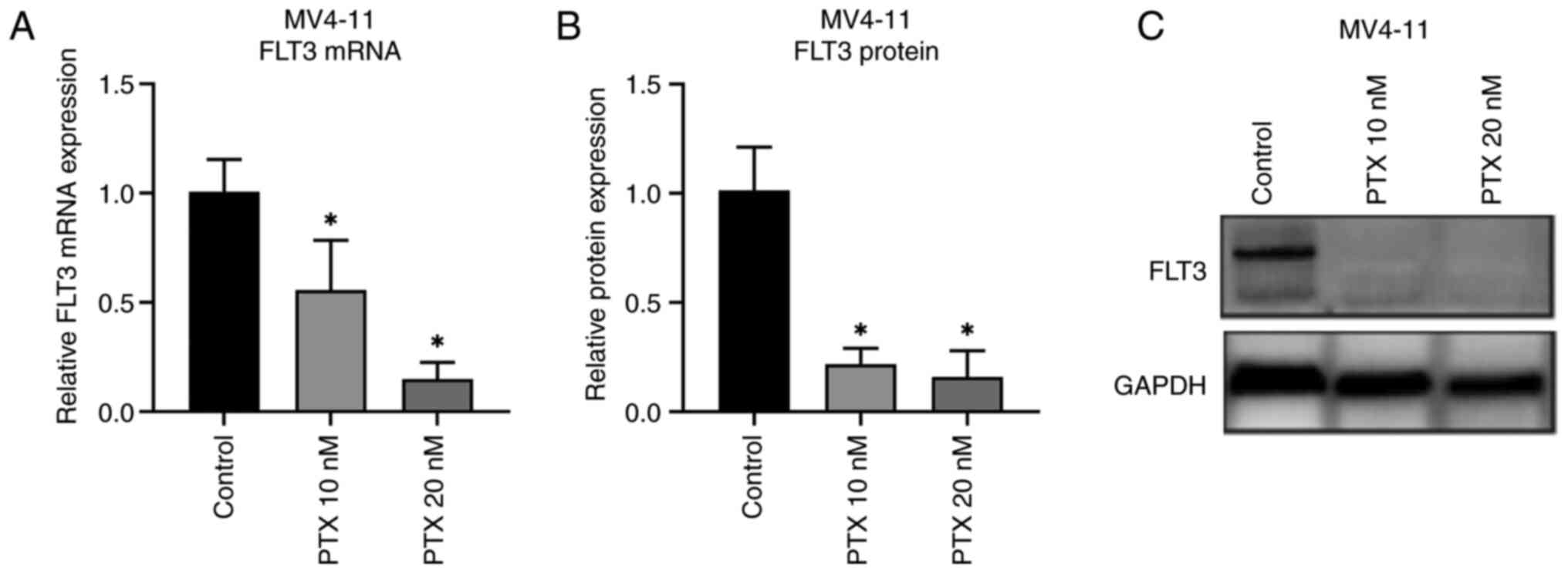
Given that PTX reduced proliferation and promoted
apoptosis in MV4-11 cells, the underlying mechanism was
investigated by treating MV4-11 cells with 10 or 20 nM PTX for 48
h, followed by PCR and western blotting. FLT3 mRNA (Fig. 4A) and protein (Fig. 4B and C) expression were significantly
downregulated compared with control cells and with the increase of
PTX concentration, FLT3 gene expression decreased significantly.
Thus, downregulation of the FLT3 gene may be linked to decreased
growth of MV4-11 cells.
Effect of PTX on PI3K/AKT/mTOR pathway
in MV4-11 cells
The FLT3 receptor activates the downstream
PI3K/AKT/mTOR signaling pathway which is known to be involved in
growth of leukemia cells (36).
According to the results of the previous experiment, PTX can
inhibit the expression of FLT3 gene so, to test whether PTX have
any effect on the PI3K/AKT/mTOR pathway, MV4-11 cells were treated
with 0, 10 or 20 nM PTX for 48 h and expression of genes dependent
on PI3K/AKT/mTOR signaling measured by PCR and western blotting.
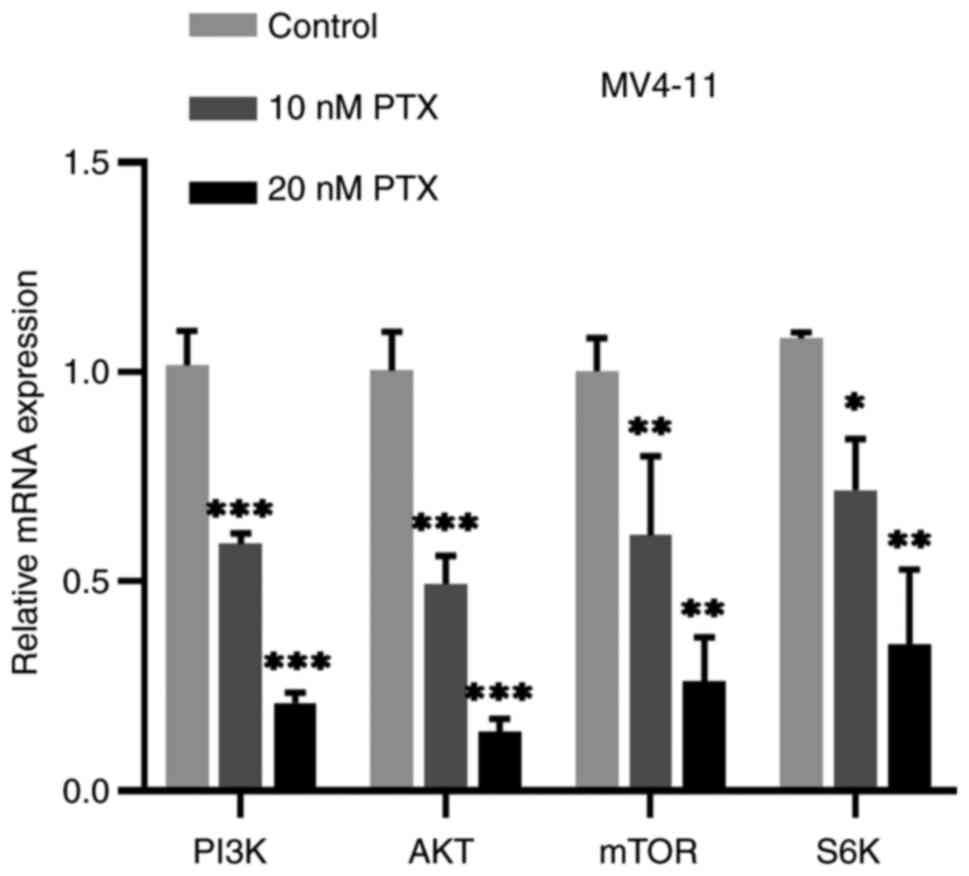
Production of PI3K, AKT, mTOR and S6K mRNA were all downregulated
compared with the control cells (Fig.
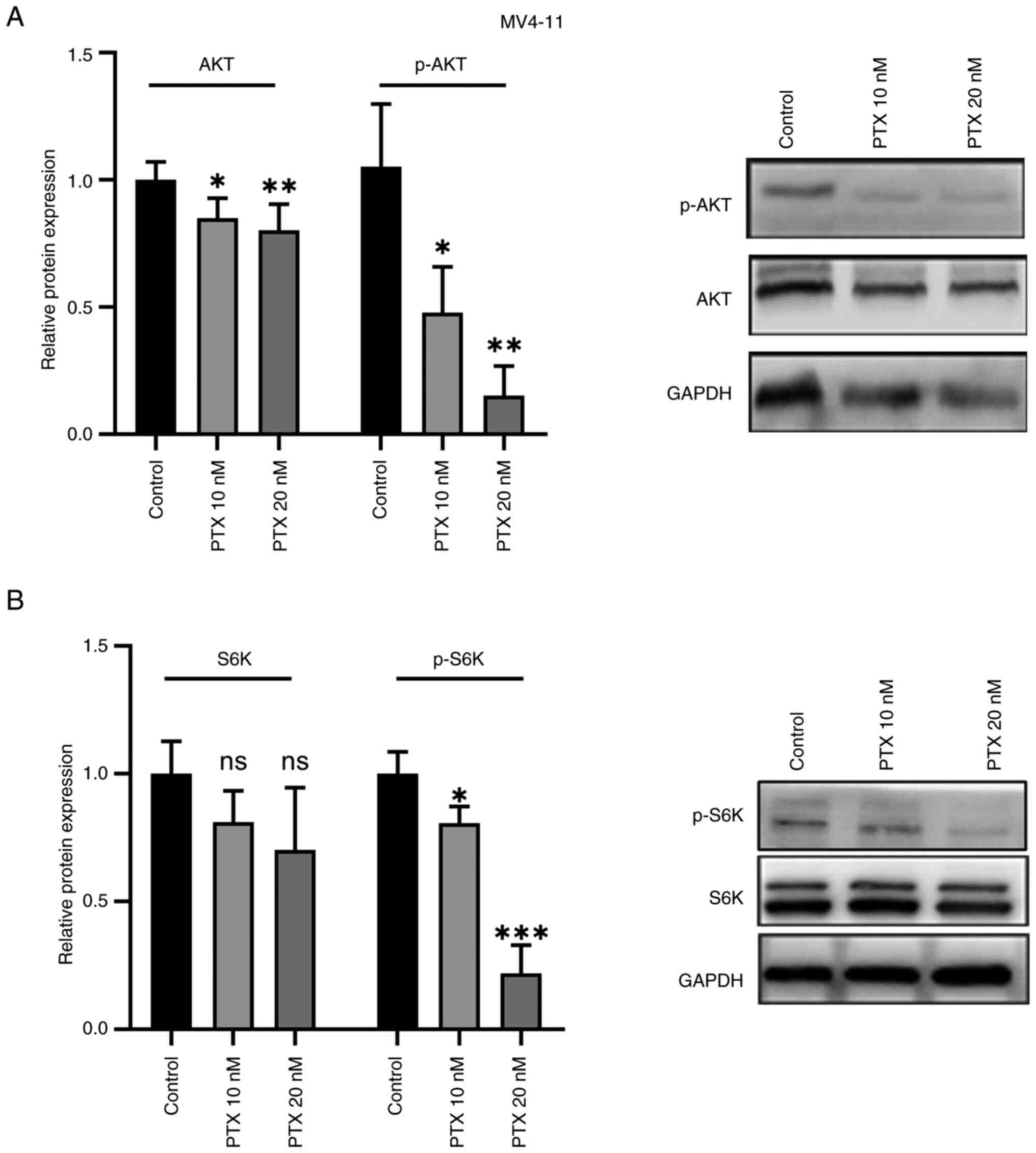
5). Compared with the control group, the expression of p-AKT
(Fig. 6A) and p-S6K (Fig. 6B) protein were also decreased
significantly, but the changes of AKT, S6K protein were not evident
(Fig. 6B). Based on this
experiment, it was hypothesized that PTX might mainly inhibit the
phosphorylation of AKT and S6K. Thus, the inhibitory effect of PTX
on MV4-11 cell growth may be linked to inhibition of the
PI3K/AKT/mTOR pathway.
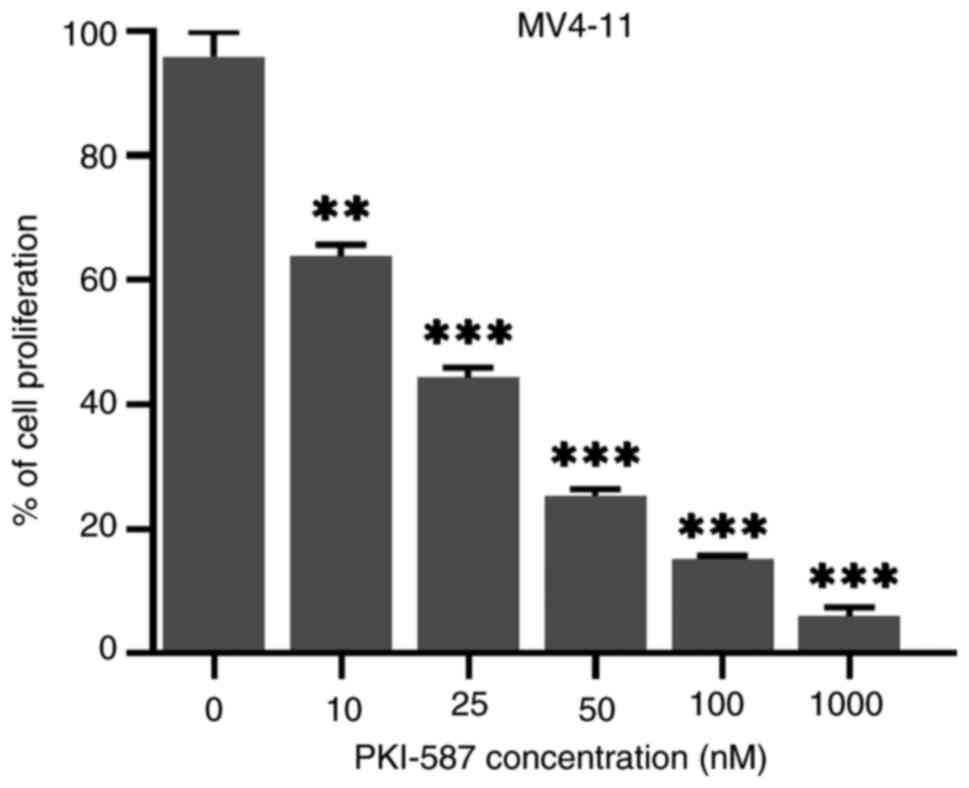
Effect of the PI3K/AKT/mTOR pathway
inhibitor, PKI-587, in inhibiting growth of MV4-11 cells
The above experiments showed that PTX could not only
have anti-proliferative and pro-apoptotic effect on MV4-11 cells,
but also inhibited PI3K/AKT/mTOR pathway in the cells. To test
whether the effect of PTX on MV4-11 cells depend on this signaling
pathway, cells were treated with PI3K/AKT/mTOR pathway inhibitor
PKI-587, which also known as gedatolisib or PF 05212384. PKI-587
could inhibit cell viability (Fig.
7). Taken together, it was hypothesized that the inhibitory
effect of PTX on MV4-11 cells proliferation was related to the
PI3K/AKT/mTOR pathway.
Effect of PTX combined with PKI-587 on
cell proliferation and apoptosis of MV4-11 cells
Abnormal activation of PI3K/AKT/mTOR pathway is not
only related to the occurrence and development of AML, but also
drug resistance (37). As a dual
PI3K/AKT/mTOR inhibitor, PKI-587 has been shown to efficiently
inhibit cell proliferation, block colony formation and induce
apoptosis of sorafenib-resistant AML cells (38). The results of the present study
confirmed that PKI-587 also had clear anti-proliferative on MV4-11
cells. As it was observed that PTX could inhibit the PI3K/AKT/mTOR
pathway, it was hypothesized that PKI-587 might enhance the effect
of PTX in MV4-11 cells, or the two drugs might have synergistic
effect. To verify this hypothesis, first, 2 nM PTX combined with
10, 25 or 50 nM PKI-587 was used to treated MV4-11 cells, followed
by CCK-8 assay to detect cell viability. As shown in Table II, comparing with PTX or PKI-587
monotherapy, the cell proliferation inhibition rate of the
combination group was significantly increased. The synergistic
index q value calculated by Kingsdale formula suggested that the
combination of PTX and PKI-587 had synergistic or additive
effect.
| Table IIEffect of the combination of
paclitaxel and PKI-587 on MV4-11 cell proliferation. |
Table II
Effect of the combination of
paclitaxel and PKI-587 on MV4-11 cell proliferation.
| Group | Inhibition rate
(%) | q-value | Interaction |
|---|
| Control | 3.11±0.63 | | |
| 2 nM PTX | 10.5±3.3 | | |
| 10 nM PKI-587 | 14.63±5.93 | | |
| 25 nM PKI-587 | 66.68±3.44 | | |
| 50 nM PKI-587 | 69.65±5.63 | | |
| 2 nM PTX + 10 nM
PKI-587 |
35.41±2.63a | 1.50 | Synergy |
| 2 nM PTX + 25 nM
PKI-587 |
83.09±0.56a | 1.18 | Synergy |
| 2 nM PTX + 50 nM
PKI-587 |
82.07±5.87a | 1.13 | Additive |
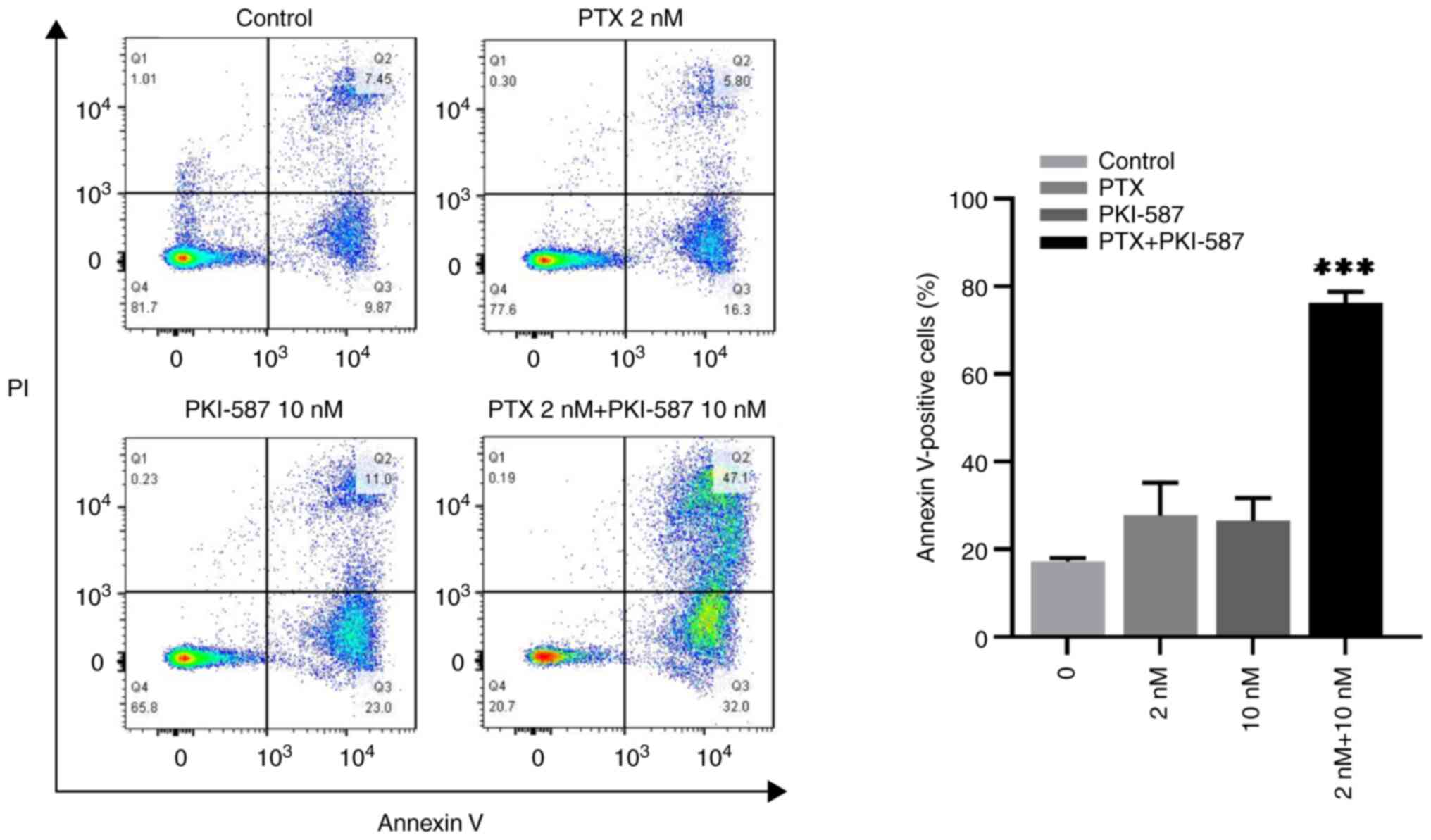
Second, its effect on cells apoptosis was confirmed.
From Table II, it can be seen
that the synergistic index q value was the largest at 2 nM PTX
combined with 10 nM. Therefore, 2 nM PTX combined with 10 nM
PKI-587 was selected to treat cells. The results showed that the
apoptosis rate of PTX or PKI-587 alone was 27.69±7.45% and
26.42±5.24%, respectively, but at the combination group, the
apoptosis rate was up to 76.23±2.55% (Fig. 8). Taken together, it was concluded
that PKI-587 might enhance effect of PTX on MV4-11 cells, the two
drugs having a synergistic or additive effect. It was also further
proved that the proliferation inhibition and pro-apoptosis effect
of PTX on MV4-11 cells were related to the PI3K/AKT/mTOR signaling
pathway.
Discussion
The prognosis of AML patients carrying the FLT3-ITD
mutation remains poor and its 5-year survival rate is only ~20%,
despite advances in treatments (39). Thus, there is a pressing need to
identify new and effective treatment strategies for these patients
(39). PTX is considered an
effective antitumor treatment for a variety of malignancies. The
present study found that PTX had a time- and dose-dependent
anti-proliferative and apoptosis-inducing effect on MV4-11 cells
carrying the FLT3-ITD mutation. PTX also inhibited expression of
FLT3 mRNA and protein and exerted a regulatory effect on the
PI3K/AKT/mTOR pathway.
Leukemia is a common malignant tumor derived from
the hematopoietic system during which bone marrow, peripheral blood
and other organs are filled with uncontrollably proliferating
primitive white blood cells (40,41).
Previous studies have demonstrated anti-proliferative effects of
PTX on the leukemia cells, HL-60(30) and K562 (31,32)
and a similar inhibitory effect on MV4-11 cells was predicted. The
results showed that PTX had a significant anti-proliferative and
apoptosis-inducing effect on MV4-11 cells. To observe and compare
its effect on the FLT3-ITD negative leukemia cell lines, THP-1 and
K562, both cell lines were treated under the same conditions. PTX
could also inhibit cell proliferation and induce apoptosis of THP-1
or K562 cells; however, compared with MV4-11 cells, its effect was
weaker. Therefore, it was hypothesized that the anti-proliferative
and pro-apoptosis effect of PTX might depend on the leukemia cell
type and that it had a more potent effect on FLT3-ITD+
AML cells.
As the FLT3-ITD mutation often indicates high risk
in AML patients, several FLT3 inhibitors have been developed and
used in clinics for AML patients, such as sorafenib, midostaurin
and gilteritinib (42,43). The most widely used is sorafenib,
which has been used to treat relapsed or refractory AML patients,
or as an adjunct to transplantation, although it is merely a
multi-kinase inhibitor (44). To
further identify the effect of PTX on MV4-11 cells, its inhibitory
and pro-apoptotic effect were compared with sorafenib. The results
showed that the IC50 values of sorafenib were greater
than PTX, indicating the anti-proliferative effect of PTX was
stronger than sorafenib. Moreover, the apoptosis rate of sorafenib
was lower compared with PTX. However, sorafenib is a multi-kinase
inhibitor; in order to improve our understanding of the
anti-leukemia effect of PTX, it is necessary to further compare the
efficacy of PTX with gilteritinib, a new FLT3/AXL dual inhibitor
which has a relatively definite efficacy in relapsed or refractory
AML patients (45). Taken
together, it was suggested that PTX had distinct effect on MV4-11
cells.
FLT3 is often overexpressed in hematopoietic tumors
(6), affecting the development,
growth and differentiation of blood cells and promoting leukemia
progression. The FLT3-ITD mutation can decrease treatment efficacy
and result in adverse outcomes (46) by activating downstream signaling
pathways (11,12), such as the PI3K/AKT signaling
pathway, which has a great influence on survival, proliferation and
differentiation of hematopoietic cells (36). Abnormal activation of the PI3K/AKT
signaling pathway associated with reduced overall survival, has
been identified in ~60% of AML patients (47,48).
A number of studies have shown that mutations or activation of
receptor tyrosine kinases, for example, the FLT3-ITD mutation, can
stimulate the PI3K/AKT signaling pathway in most human
malignancies, including AML (13,49,50).
PTX is a potent anticancer drug and several studies have shown that
it can inhibit cell proliferation and induce apoptosis by
regulating the PI3K/AKT pathway in various solid tumors, such as
nasopharyngeal carcinoma (26,51),
cervical cancer (27) and lung
cancer. Furthermore, the pathway inhibitors or other drugs
inactivating the PI3K/AKT pathway can enhance the chemosensitivity
of PTX in various cancer cells (52-54).
The present study observed that PTX could downregulate the
expression of FLT3 mRNA and protein in MV4-11 cells. In addition,
upon PTX treatment, the mRNA expression level of PI3K, AKT, mTOR
and S6K and the proteins of p-AKT, p-S6K all declined distinctly.
However, the changes of AKT, S6K protein were not clear and it was
hypothesized that PTX might mainly influence the phosphorylation of
AKT and S6K, perhaps through inactivating the PI3K/AKT/mTOR
pathway, to affect cell proliferation and apoptosis. It has been
reported that PI3K/mTOR pathway inhibitors can inhibit cell
proliferation and induce apoptosis of MV4-11 cells and another
FLT3-ITD+ AML cell line, MOLM-13 cells. The present
study also demonstrated that the resistance of AML cells to
sorafenib might have resulted from overexpression of p-AKT and
p-S6K proteins of the PI3K/mTOR pathway. Inhibition of this pathway
enhanced the anti-leukemia effect of sorafenib, lengthening the
life of mice with transplanted tumors (38). The present study found that
PKI-587, a dual PI3K/mTOR inhibitor did have cytotoxic effect on
MV4-11 cells, which was consistent with a previous study (38). It was also combined with PTX to
treat the cells and it was found that this combination showed a
significant synergistic or additive effect in inhibiting cell
proliferation and inducing apoptosis. These findings further
demonstrated that the effect of PTX on MV4-11 cells might be
associated with the PI3K/AKT/mTOR pathway. Future studies will
further detect whether the effect of PTX increases when the
expression of p-AKT and p-S6K are downregulated. However, there is
an important question; how PTX regulates the FLT3 is unclear. On
the basis of previous studies, it is hypothesized that PTX might
inhibit heat shock protein 90 (Hsp90) to downregulate the
expression of FLT3.
FLT3 as an important therapeutic target and its
expression can be inhibited by a variety of small molecule
inhibitors, such as Hsp90, proteasome, RET and other inhibitors
(55). Hsp90 is a molecular
chaperone that is overexpressed in leukemia cells and its
overexpression is associated with indefinite cells proliferation
and drug resistance, mainly through forming multiprotein complexes
to stabilize its client proteins (55,56).
It was hypothesized that PTX might, through inhibition of Hsp90,
downregulate the expression of FLT3 for the following reasons.
First, several studies have confirmed that FLT3 is one of the Hsp90
client proteins; Hsp90 inhibitors, such as 17-AAG, can reduce its
expression and inhibit proliferation of leukemia cells and induce
apoptosis (57-59).
Second, AKT is also an Hsp90 client protein; Hsp90 inhibitors can
inactivate AKT and downregulate the expression of p-AKT. The
present study verified that PTX could reduce the expression of both
FLT3 and p-AKT. Third, triptolide and tripterine, isolated from
Tripeterygium wilfordii Hook f, both are terpenoid
compounds. Previous studies have demonstrated that tripterine
demonstrates anti-proliferation and pro-apoptosis effect on various
leukemia cells by inhibiting the expression of Hsp90 (60-62).
It has been reported that triptolide can also inhibit the
expression of Hsp90(63).
Considering that PTX is also a terpenoid compound, such as
triptolide and tripterine, it was hypothesized that it might also
inhibit the expression of Hsp90. For these reasons, it was further
hypothesized that PTX might inhibit the expression of FLT3 and
p-AKT by reducing the expression of Hsp90. This remains a
hypothesis and further work will be conducted to identify its
veracity.
In conclusion, the present study revealed the
anti-proliferative and apoptosis-inducing effect of PTX on MV4-11
cells and its molecular mechanism, which was connected to the
inhibition of PI3K/AKT/mTOR pathway. The aim of the present study
was to establish the potential utility of PTX as a therapy for
FLT3-ITD+ AML patients, supported by a theoretical
basis. However, the present study was limited by its in
vitro nature and did not establish the efficacy of PTX in
patients. In the future, we will further explore the effects of PTX
on primary cells and further animal experiments will be planned
which may pave the way for clinical trials.
Acknowledgements
The authors thank Dr Luo Bin from Guangxi Medical
University (Guangxi Zhuang Autonomous Region, China) for technical
assistances with the CCK-8 and PCR.
Funding
Funding: The present study was funded by National Natural
Science Foundation of China (grant no. 81700172).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
YS and WZ conceived and designed the study. YS, BZ
and ZB performed the experiment. RP was responsible for cultivating
cells. MW and ZL provided technical guidance. YS wrote the
manuscript. WZ revised and finalized the manuscript. YS and WZ
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Estey E and Döhner H: Acute myeloid
leukaemia. Lancet. 368:1894–1907. 2006.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Yang F, Anekpuritanang T and Press RD:
Clinical utility of next-generation sequencing in acute myeloid
leukemia. Mol Diagn Ther. 24:1–13. 2020.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Ley TJ, Miller C, Ding L, Raphael BJ,
Mungall AJ, Robertson A, Hoadley K, Triche TJ Jr, Laird PW, Baty
JD, et al: Genomic and epigenomic landscapes of adult de novo acute
myeloid leukemia. N Engl J Med. 368:2059–2074. 2013.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Daver N, Schlenk RF, Russell NH and Levis
MJ: Targeting FLT3 mutations in AML: Review of current knowledge
and evidence. Leukemia. 33:299–312. 2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Döhner H, Estey E, Grimwade D, Amadori S,
Appelbaum FR, Büchner T, Dombret H, Ebert BL, Fenaux P, Larson RA,
et al: Diagnosis and management of AML in adults: 2017 ELN
recommendations from an international expert panel. Blood.
129:424–447. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Sun YM, Wang WT, Zeng ZC, Chen TQ, Han C,
Pan Q, Huang W, Fang K, Sun LY, Zhou YF, et al: CircMYBL2, a
circRNA from MYBL2, regulates FLT3 translation by recruiting PTBP1
to promote FLT3-ITD AML progression. Blood. 134:1533–1546.
2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
De Kouchkovsky I and Abdul-Hay M: ‘Acute
myeloid leukemia: A comprehensive review and 2016 update’. Blood
Cancer J. 6(e441)2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Whitman SP, Ruppert AS, Radmacher MD,
Mrózek K, Paschka P, Langer C, Baldus CD, Wen J, Racke F, Powell
BL, et al: FLT3 D835/I836 mutations are associated with poor
disease-free survival and a distinct gene-expression signature
among younger adults with de novo cytogenetically normal acute
myeloid leukemia lacking FLT3 internal tandem duplications. Blood.
111:1552–1559. 2008.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Whitman SP, Archer KJ, Feng L, Baldus C,
Becknell B, Carlson BD, Carroll AJ, Mrózek K, Vardiman JW, George
SL, et al: Absence of the wild-type allele predicts poor prognosis
in adult de novo acute myeloid leukemia with normal cytogenetics
and the internal tandem duplication of FLT3: A cancer and leukemia
group b study. Cancer Res. 61:7233–7239. 2001.PubMed/NCBI
|
|
10
|
Almatani MF, Ali A, Onyemaechi S, Zhao Y,
Gutierrez L, Vaikari VP and Alachkar H: Strategies targeting FLT3
beyond the kinase inhibitors. Pharmacol Ther.
225(107844)2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Levis M and Small D: FLT3: It does matter
in leukemia. Leukemia. 17:1738–1752. 2003.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Takahashi S: Downstream molecular pathways
of FLT3 in the pathogenesis of acute myeloid leukemia: Biology and
therapeutic implications. J Hematol Oncol. 4(13)2011.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Brandts CH, Sargin B, Rode M, Biermann C,
Lindtner B, Schwäble J, Buerger H, Müller-Tidow C, Choudhary C,
McMahon M, et al: Constitutive activation of Akt by FLT3 internal
tandem duplications is necessary for increased survival,
proliferation, and myeloid transformation. Cancer Res.
65:9643–9650. 2005.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Rocnik JL, Okabe R, Yu JC, Lee BH, Giese
N, Schenkein DP and Gilliland DG: Roles of tyrosine 589 and 591 in
STAT5 activation and transformation mediated by FLT3-ITD. Blood.
108:1339–1345. 2006.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Döhner H, Weisdorf DJ and Bloomfield CD:
Acute myeloid leukemia. N Engl J Med. 373:1136–1152.
2015.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Smith CC, Wang Q, Chin CS, Salerno S,
Damon LE, Levis MJ, Perl AE, Travers KJ, Wang S, Hunt JP, et al:
Validation of ITD mutations in FLT3 as a therapeutic target in
human acute myeloid leukaemia. Nature. 485:260–263. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Piloto O, Levis M, Huso D, Li Y, Li H,
Wang MN, Bassi R, Balderes P, Ludwig DL, Witte L, et al: Inhibitory
anti-FLT3 antibodies are capable of mediating antibody-dependent
cell-mediated cytotoxicity and reducing engraftment of acute
myelogenous leukemia blasts in nonobese diabetic/severe combined
immunodeficient mice. Cancer Res. 65:1514–1522. 2005.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Alvarado Y, Kantarjian HM, Luthra R,
Ravandi F, Borthakur G, Garcia-Manero G, Konopleva M, Estrov Z,
Andreeff M and Cortes JE: Treatment with FLT3 inhibitor in patients
with FLT3-mutated acute myeloid leukemia is associated with
development of secondary FLT3-tyrosine kinase domain mutations.
Cancer. 120:2142–2149. 2014.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Weisberg E, Barrett R, Liu Q, Stone R,
Gray N and Griffin JD: FLT3 inhibition and mechanisms of drug
resistance in mutant FLT3-positive AML. Drug Resist Updat.
12:81–89. 2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Alotaibi AS, Yilmaz M, Kanagal-Shamanna R,
Loghavi S, Kadia TM, DiNardo CD, Borthakur G, Konopleva M, Pierce
SA, Wang SA, et al: Patterns of resistance differ in patients with
acute myeloid leukemia treated with type I versus type II FLT3
inhibitors. Blood Cancer Discov. 2:125–134. 2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Zhu L and Chen L: Progress in research on
paclitaxel and tumor immunotherapy. Cell Mol Biol Lett.
24(40)2019.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Tamburin S, Park SB, Alberti P, Demichelis
C, Schenone A and Argyriou AA: Taxane and epothilone-induced
peripheral neurotoxicity: From pathogenesis to treatment. J
Peripher Nerv Syst. 24 (Suppl 2):S40–S51. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Leung JC and Cassimeris L: Reorganization
of paclitaxel-stabilized microtubule arrays at mitotic entry: Roles
of depolymerizing kinesins and severing proteins. Cancer Biol Ther.
20:1337–1347. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Vassileva V, Allen CJ and Piquette-Miller
M: Effects of sustained and intermittent paclitaxel therapy on
tumor repopulation in ovarian cancer. Mol Cancer Ther. 7:630–637.
2008.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Al-Mahayri ZN, AlAhmad MM and Ali BR:
Current opinion on the pharmacogenomics of paclitaxel-induced
toxicity. Expert Opin Drug Metab Toxicol. 17:785–801.
2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Li T: Pacilitaxel induces human
nasopharyngeal carcinoma cell line CNE2 apoptosis and growth
inhibition by suppressing PI3K/AKT/P53 signaling pathway. Lin Chung
Er Bi Yan Hou Tou Jing Wai Ke Za Zhi. 29:2147–2150. 2015.PubMed/NCBI(In Chinese).
|
|
27
|
Guertin DA and Sabatini DM: Defining the
role of mTOR in cancer. Cancer Cell. 12:9–22. 2007.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Ma D, Li S, Cui Y, Li L, Liu H, Chen Y and
Zhou X: Paclitaxel increases the sensitivity of lung cancer cells
to lobaplatin via PI3K/AKT pathway. Oncol Lett. 15:6211–6216.
2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Moschetta M, Pretto F, Berndt A, Galler K,
Richter P, Bassi A, Oliva P, Micotti E, Valbusa G, Schwager K, et
al: Paclitaxel enhances therapeutic efficacy of the F8-IL2
immunocytokine to EDA-fibronectin-positive metastatic human
melanoma xenografts. Cancer Res. 72:1814–1824. 2012.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Ying J, Yang W, Xie CY, Ni QC, Pan XD,
Dong JH, Liu ZM and Wang XS: Induction of caspase-3-dependent
apoptosis in human leukemia HL-60 cells by δ-elemene. Yakugaku
Zasshi. 131:1383–1394. 2011.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Xia RL, Lu Y, Zhu LN, Zhang SF, Zhao FK
and Fu CY: Different regulatory pathways are involved in the
proliferative inhibition of two types of leukemia cell lines
induced by paclitaxel. Oncol Rep. 30:1853–1859. 2013.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Meshkini A and Yazdanparast R: Involvement
of oxidative stress in taxol-induced apoptosis in chronic
myelogenous leukemia K562 cells. Exp Toxicol Pathol. 64:357–365.
2012.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Bai ZW, Wu MQ, Zhou BW, Shi ZY, Yao YB,
Liu ZF, Pang RL and Zhao WH: Effects of paclitaxel and quizartinib
alone and in combination on aml cell line MV4-11 and Its STAT5
signal pathway. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 30:671–676.
2022.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
34
|
Jin ZJ and Zhang XW: Equal probability and
curve with ‘q50’-A new method to estimate the effect of drug
combination. Journal of Shanghai Second Medical College.
(01)(15-18+86)1981.(In Chinese).
|
|
35
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-delta delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Nepstad I, Hatfield KJ, Grønningsæter IS
and Reikvam H: The PI3K-AKT-mTOR signaling pathway in human acute
myeloid leukemia (AML) cells. Int J Mol Sci.
21(2907)2020.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Martelli AM, Evangelisti C, Chappell W,
Abrams SL, Bäsecke J, Stivala F, Donia M, Fagone P, Nicoletti F,
Libra M, et al: Targeting the translational apparatus to improve
leukemia therapy: Roles of the PI3K/PTEN/Akt/mTOR pathway.
Leukemia. 25:1064–1079. 2011.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Lindblad O, Cordero E, Puissant A,
Macaulay L, Ramos A, Kabir NN, Sun J, Vallon-Christersson J,
Haraldsson K, Hemann MT, et al: Aberrant activation of the
PI3K/mTOR pathway promotes resistance to sorafenib in AML.
Oncogene. 35:5119–5131. 2016.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Port M, Böttcher M, Thol F, Ganser A,
Schlenk R, Wasem J, Neumann A and Pouryamout L: Prognostic
significance of FLT3 internal tandem duplication, nucleophosmin 1,
and cebpa gene mutations for acute myeloid leukemia patients with
normal karyotype and younger than 60 years: A systematic review and
meta-analysis. Ann Hematol. 93:1279–1286. 2014.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Almond LM, Charalampakis M, Ford SJ,
Gourevitch D and Desai A: Myeloid sarcoma: Presentation, diagnosis,
and treatment. Clin Lymphoma Myeloma Leuk. 17:263–267.
2017.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Ganzel C and Douer D: Extramedullary
disease in APL: A real phenomenon to contend with or not? Best
Pract Res Clin Haematol. 27:63–68. 2014.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Levis M: Midostaurin approved for
FLT3-mutated AML. Blood. 129:3403–3406. 2017.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Pulte ED, Norsworthy KJ, Wang Y, Xu Q,
Qosa H, Gudi R, Przepiorka D, Fu W, Okusanya OO, Goldberg KB, et
al: FDA approval summary: Gilteritinib for relapsed or refractory
acute myeloid leukemia with a FLT3 mutation. Clin Cancer Res.
27:3515–3521. 2021.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Antar A, Otrock ZK, El-Cheikh J,
Kharfan-Dabaja MA, Battipaglia G, Mahfouz R, Mohty M and Bazarbachi
A: Inhibition of FLT3 in AML: A focus on sorafenib. Bone Marrow
Transplant. 52:344–351. 2017.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Cucchi DGJ, Denys B, Kaspers GJL, Janssen
J, Ossenkoppele GJ, de Haas V, Zwaan CM, van den Heuvel-Eibrink MM,
Philippé J, Csikós T, et al: RNA-based FLT3-ITD allelic ratio is
associated with outcome and ex vivo response to FLT3 inhibitors in
pediatric AML. Blood. 131:2485–2489. 2018.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Mrózek K, Marcucci G, Paschka P, Whitman
SP and Bloomfield CD: Clinical relevance of mutations and
gene-expression changes in adult acute myeloid leukemia with normal
cytogenetics: Are we ready for a prognostically prioritized
molecular classification? Blood. 109:431–448. 2007.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Min YH, Eom JI, Cheong JW, Maeng HO, Kim
JY, Jeung HK, Lee ST, Lee MH, Hahn JS and Ko YW: Constitutive
phosphorylation of Akt/PKB protein in acute myeloid leukemia: Its
significance as a prognostic variable. Leukemia. 17:995–997.
2003.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Chen W, Drakos E, Grammatikakis I,
Schlette EJ, Li J, Leventaki V, Staikou-Drakopoulou E, Patsouris E,
Panayiotidis P, Medeiros LJ and Rassidakis HZ: Mtor signaling is
activated by FLT3 kinase and promotes survival of FLT3-mutated
acute myeloid leukemia cells. Mol Cancer. 9(292)2010.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Nepstad I, Hatfield KJ, Grønningsæter IS,
Aasebø E, Hernandez-Valladares M, Hagen KM, Rye KP, Berven FS,
Selheim F, Reikvam H and Bruserud Ø: Effects of insulin and pathway
inhibitors on the PI3K-Akt-mTOR phosphorylation profile in acute
myeloid leukemia cells. Signal Transduct Target Ther.
4(20)2019.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Watanabe D, Nogami A, Okada K, Akiyama H,
Umezawa Y and Miura O: FLT3-ITD activates RSK1 to enhance
proliferation and survival of AML cells by activating mTORC1 and
eIF4B cooperatively with PIM or PI3K and by inhibiting BAD and BIM.
Cancers (Basel). 11(1827)2019.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Dong P, Hao F, Dai S and Tian L:
Combination therapy eve and pac to induce apoptosis in cervical
cancer cells by targeting PI3K/AKT/mTOR pathways. J Recept Signal
Transduct Res. 38:83–88. 2018.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Ding Z, Xu F, Li G, Tang J, Tang Z, Jiang
P and Wu H: Knockdown of Akt2 expression by shRNA inhibits
proliferation, enhances apoptosis, and increases chemosensitivity
to paclitaxel in human colorectal cancer cells. Cell Biochem
Biophys. 71:383–388. 2015.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Lin YH, Chen BY, Lai WT, Wu SF, Guh JH,
Cheng AL and Hsu LC: The Akt inhibitor MK-2206 enhances the
cytotoxicity of paclitaxel (Taxol) and cisplatin in ovarian cancer
cells. Naunyn Schmiedebergs Arch Pharmacol. 388:19–31.
2015.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Liu X, Xie C, Li A, Zhang Y, Liu X, Zhou
S, Shen J, Huo Z, Cao W, Ma Y, et al: BEZ235 enhances
chemosensitivity of paclitaxel in hepatocellular carcinoma through
inhibiting the PI3K/Akt/mTOR pathway. Am J Transl Res.
11:7255–7271. 2019.PubMed/NCBI
|
|
55
|
Hacıhanefioglu A, Gonullu E, Mehtap O,
Keski H, Yavuz M and Ercin C: Effect of heat shock protein-90
(HSP90) and vascular endothelial growth factor (VEGF) on survival
in acute lymphoblastic leukemia: An immunohistochemical study. Med
Oncol. 28:846–851. 2011.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Han SY: Small molecule induced FLT3
degradation. Pharmaceuticals (Basel). 15(320)2022.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Yao Q, Nishiuchi R, Li Q, Kumar AR, Hudson
WA and Kersey JH: FLT3 expressing leukemias are selectively
sensitive to inhibitors of the molecular chaperone heat shock
protein 90 through destabilization of signal
transduction-associated kinases. Clin Cancer Res. 9:4483–4493.
2003.PubMed/NCBI
|
|
58
|
Ly BT, Chi HT, Yamagishi M, Kano Y, Hara
Y, Nakano K, Sato Y and Watanabe T: Inhibition of FLT3 expression
by green tea catechins in FLT3 mutated-AML cells. PLoS One.
8(e66378)2013.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Al Shaer L, Walsby E, Gilkes A, Tonks A,
Walsh V, Mills K, Burnett A and Rowntree C: Heat shock protein 90
inhibition is cytotoxic to primary AML cells expressing mutant FLT3
and results in altered downstream signalling. Br J Haematol.
141:483–493. 2008.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Hieronymus H, Lamb J, Ross KN, Peng XP,
Clement C, Rodina A, Nieto M, Du J, Stegmaier K, Raj SM, et al:
Gene expression signature-based chemical genomic prediction
identifies a novel class of HSP90 pathway modulators. Cancer Cell.
10:321–330. 2006.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Peng B, Xu L, Cao F, Wei T, Yang C, Uzan G
and Zhang D: HSP90 inhibitor, celastrol, arrests human monocytic
leukemia cell U937 at G0/G1 in thiol-containing agents reversible
way. Mol Cancer. 9(79)2010.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Lu Z, Jin Y, Qiu L, Lai Y and Pan J:
Celastrol, a novel HSP90 inhibitor, depletes Bcr-Abl and induces
apoptosis in imatinib-resistant chronic myelogenous leukemia cells
harboring T315I mutation. Cancer Lett. 290:182–191. 2010.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Zhang FZ, Ho DH and Wong RH: Triptolide, a
HSP90 middle domain inhibitor, induces apoptosis in triple manner.
Oncotarget. 9:22301–22315. 2018.PubMed/NCBI View Article : Google Scholar
|