Introduction
Parkinson's disease (PD) has a prevalence of 0.5-1%
after 65 years of age and 1-3% after 80 years of age worldwide
(1). The main clinical symptoms of
PD include heterogeneous motor (including tremor at rest,
bradykinesia, rigidity and postural instability) and non-motor
signs (including cognitive decline, depression, anxiety,
dysautonomia, sleep disturbances and anosmia) with heterogeneous
pathological characteristics (including mild-motor predominant PD,
diffuse malignancies PD and intermediate PD) (2,3).
Essential tremor (ET) is defined as an action tremor lasting for ≥3
years, which primarily involves both upper limbs and can be with or
without tremors in other locations. ET mainly occurs in the absence
of other neurological disorders, such as dystonia, ataxia or
parkinsonism (4). Systematic
review and meta-analysis revealed that the prevalence of ET
increased with advancing age, and the global prevalence of ET was
2.87% in people aged ≥80 years (5,6).
Notably, according to the Consensus Statement on the Classification
of Tremors, individuals with long-term ET may eventually develop
other neurological disorders, such as dystonia or PD (4).
Although PD is considered to be a sporadic disorder,
10-30% patients with PD report having a first-degree family members
with PD (7). Previous linkage and
sequence analyses performed in patients with familial PD have
identified SNCA, LRRK2, GIGYF2, VPS35,
eukaryotic translation initiation factor 4γ1 (EIF4G1),
DNAJC13, CHCHD2 and TMEM230 as autosomal
dominant pathogenic genes, and PARK2, PINK1,
DJ-1, ATP13A2, PLA2G6, FBXO7,
DNAJC6, SYNJ1 and VPS13C as autosomal
recessive pathogenic genes (8-10).
Additionally, other genes in which mutations have been shown to be
associated with PD include UBTF, GRN,
FAM171A2, PODXL and PTRHD1, and mutations in
RAB39B have been reported to cause X-linked PD (11,12).
A systematic analysis performed in patients with sporadic PD
identified autosomal dominant deleterious mutations in the
SNCA, LRRK2, GIGYF2, VPS35,
EIF4G1, DNAJC13 and CHCHD2 genes (13). In particular, although mutations in
the EIF4G1 gene have been identified in both patients with
familial and sporadic PD, the role of EIF4G1 in PD etiology
remains elusive.
The EIF4G1 gene is located on chromosome
3q27.1 and covers an ~20.8-kb genomic region with 31 coding exons
(14). It encodes a 1,599-amino
acid eIF4G1 protein, which is abundantly and ubiquitously expressed
as a subunit of the translation initiation complex eIF4F. eIF4G1
serves as a scaffold protein that interacts with poly(A)-binding
protein, eIF3, eIF4E, the RNA helicase eIF4A and the 40S ribosomal
subunit (15). It serves an
important role in signal transduction (16), cell growth and mortality (17) and the translation of mRNAs
associated with these aforementioned processes (18). Overexpression of the eIF4G1 protein
has been associated with several malignant disorders in humans
(such as breast cancer, lung cancer, multiple myeloma, pancreatic
ductal adenocarcinoma and chronic lymphocytic leukemia), whereas
decreased eIF4G1 protein expression results in the reduction of
overall protein and reduced ATP production (19). Furthermore, the degradation of
eIF4G1, triggered by the activation of calpain, has been reported
to result in decreased protein synthesis and increased neuronal
cell death after ischemic injury in vitro (20).
Clinically, ET is defined by the presence of
isolated action tremors, whereas PD is characterized by the
presence of bradykinesia with either resting tremor or rigidity
(21). Patients with longstanding
ET who later develop a PD phenotype are thereby referred to as
having PD with ‘antecedent ET’ (22). Previous studies have been conducted
on patients with familial PD harboring EIF4G1 mutations;
these patients are clinically characterized by a relatively long,
mild course and retain a high level of cognition (23,24).
In the present study, a linkage and sequence
analysis was performed on a Chinese family with members exhibiting
ET or PD with antecedent ET. A novel mutation in EIF4G1 was
discovered in familial cases of ET and PD, thereby broadening the
range of pathogenic mutations associated with these conditions.
Materials and methods
Clinical characteristics
The ethical review board of the Affiliated Hospital
of Jining Medical University (Jining, China) approved the study
protocol (approval no. 2023-09-C031), and written informed consent
was obtained from all participating subjects. Between April and
June 2023, data on the family history of all the members comprising
this family was gathered. A total of 29 members were in this family
(two members are deceased), including 15 males and 14 females. All
living members ranged in age from 4-75, with a median ages of
37±21.25. In this investigation, a cohort of 19 individuals who
underwent exome sequencing were ultimately enrolled, while
individuals who were deceased or did not undergo genetic sequencing
were excluded from the analysis. Neurological examinations were
performed by three neurologists specializing in movement disorders
without knowledge of the participants' genotype. The assessment of
motor function in the nervous system encompassed evaluations of
muscle strength, tone, involuntary movements, coordination
(including finger-nose tests, rapid alternating movements,
heel-knee-tibia tests, Romberg's sign and gait), as well as
examinations of nerve reflexes (such as abdominal, biceps, triceps,
radioperiosteal, knee, Achilles tendon, Babinski, Gordon,
Oppenheim, Hoffmann, Kernig, Brudzinski and nuchal rigidity
reflexes) and sensory function (including superficial, deep, and
cortical sensations). The laboratory tests conducted encompassed
blood routine analysis, electrolyte levels, liver and kidney
function assessments, blood glucose monitoring, and ceruloplasmin
evaluation.
Patients were diagnosed with ET according to the
published Classification of Tremors by the Task Force on Tremor of
the International Parkinson and Movement Disorder Society (4). Patients were diagnosed with PD based
on the United Kingdom PD Society Brain Bank clinical diagnostic
criteria (25), where severity was
assessed using the Hoehn and Yahr scale (26) and the Movement Disorder
Society-sponsored revision of the Unified PD Rating Scale Part III
(UPDRS III) (27). Cognitive
impairment was assessed using the Mini-Mental State Examination
(MMSE) (28).
Whole-exome sequencing (WES)
To identify the gene responsible for ET and PD in
the family, 5 ml peripheral blood was collected from the most
severely affected patient, Case II-2 and sent to Beijing Kangso
Medical Inspection Co., Ltd. for WES. A FlexiGene DNA kit (cat. no.
51206; Qiagen China Co., Ltd.) was used to extract genomic DNA from
the peripheral blood samples. Quality testing of the extracted DNA,
library construction, hybrid capture and sequencing were performed
as previously described (29).
Agarose gel electrophoresis was used to assess the extent of DNA
degradation, the presence of RNA, and protein contamination (data
not shown). The DNA concentration was precisely quantified
utilizing a Qubit 2.0 fluorometer (cat. no. 32866; Thermo Fisher
Scientific, Inc.). The quantified genomic DNA was randomly sheared
into 180-280 bp segments followed by adaptor ligation and cohesive
end trimming. Next, the DNA library was amplified using the
TransNGS® Index Primers (384) Kit (cat. no.
3KI241; TransGen Biotech Co., Ltd.) according to the manufacturer's
protocol: Initial denaturation was at 98˚C for 3 min; followed by 5
cycles of 30 sec at 98˚C, 35 sec at 60˚C and 30 sec at 72˚C, with a
final extension at 72˚C for 3 min. The subsequent adaptor-specific
primers were employed for the amplification of the DNA library:
Forward,
5'-AATGATACGGCGACCACCGAGATCTACACTAGCTGCCACACTCTTTCCCTACACGACCTCTTCCGATC-s-T-3'
and reverse
5'-CAAGCAGAAGACGGCATACGAGATTCCGCGAAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATC-s-T-3'
(-s-represents a phosphorothioate bond). The PCR products were
subsequently purified using MagicPure Size Selection DNA
Beads (cat. no. EC401; TransGen Biotech Co., Ltd.) in accordance
with the manufacturer's instructions. Subsequent DNA fragments were
subjected to liquid-phase hybridization with up to 500,000
biotin-labeled Agilent SureSelect Human All Exon V6 probes (cat.
no. 5190-8863; Agilent Technologies, Inc.), followed by capture
using streptomycin magnetic beads and amplification utilizing the
SureSelect Target Enrichment System (Agilent Technologies, Inc.)
under specific thermocycling conditions: Initial denaturation at
98˚C for 2 min; subsequent 15 cycles of 30 sec at 98˚C, 30 sec at
62˚C and 1 min at 72˚C, concluding with a final extension at 72˚C
for 10 min. Adaptor-specific primers were employed for the
amplification process: Forward, 5'-AATGATACGGCGACCACCGA-3' and
reverse primer, 5'-CAAGCAGAAGACGGCATACGA-3'. The products were
purified using MagicPure Size Selection DNA Beads as
aforementioned. The effective concentration (3 nM) of the library
was accurately quantified using the Qubit 2.0 fluorometer and an
Agilent Technologies 2200 TapeStation qPCR (7500 Fast Dx Real-Time
PCR Instrument; Thermo Fisher Scientific, Inc.). Subsequently,
single-read sequencing was performed on a NextSeq500 (Illumina,
Inc.).
Bioinformatics analysis
Data analysis was performed as described in previous
studies (29,30). Alignment between sequencing reads
and the human reference genome (version hg19) was performed using
the Burrows-Wheeler Alignment tool (version 0.7.15; https://github.com/lh3/bwa). Single-nucleotide
variants and small insertion or deletion variants were detected
using the GATK (v3.6; https://www.broadinstitute.org). CODEX (v1.14.1,
https://www.bioconductor.org/packages/release/bioc/html/CODEX.html),
XHMM (v1.0, https://zzz.bwh.harvard.edu/xhmm/index.shtml) and
Kangso Sequencing Copy Number Variation Detection Software v1.0
(Beijing Kangso Medical Inspection Co., Ltd.) were used to detect
the possible copy number variations (31,32).
The RefSeq (reference genome version, GRCH37/Hg19; https://www.ncbi.nlm.nih.gov/refseq), Ensembl
(April 2021 update; https://www.ensembl.org/index.html) and UCSC Genome
Browser (reference genome version, GRCH37/Hg19; https://genome.ucsc.edu) were employed for the
annotation of genes in the present study. Frequencies of annotated
variants in populations were investigated using the 1000G (2015
update; http://www.1000genomes.org), dbSNP
(v150; https://www.ncbi.nlm.nih.gov/SNP), and ExAC (v0.3;
ExAC is now in gnomAD; www.gnomad-sg.org) databases. Impacts of any mutations
on eIF4E function were investigated using the SIFT (version 2;
https://sift.bii.a-star.edu.sg),
PolyPhen2 (version 2; http://genetics.bwh.harvard.edu/pph2) and
MutationTaster (NCBI 37/Ensembl 69; http://www.mutationtaster.org) tools (33,34).
The Online Mendelian Inheritance in Man (https://www.omim.org/), Human Gene Mutation
(http://www.hgmd.org) and ClinVar databases
(https://submit.ncbi.nlm.nih.gov/clinvar/) were used to
perform disease-related annotations. SWISS-MODEL (http://swissmodel.expasy.org/interactive) was used to
predict the secondary and tertiary structures of the mutated and
wild-type eIF4F proteins. The American College of Medical Genetics
and Genomics (ACMG) Variation Interpretation Guidelines were used
to classify the variants (pathogenic, likely pathogenic, benign,
likely benign, and variants of uncertain significance) and conduct
clinical analyses (35).
Sanger sequencing verification
In total, 2 ml peripheral blood was collected from
19 participants and partners of Case II-1 and Case II-2, and sent
to Beijing Kangso Medical Inspection Co., Ltd. for Sanger
sequencing verification. The remaining family members (10/29)
declined to be tested. According to the WES results of a severely
affected patient, namely Case II-2, the c.1909A>T mutation in
the EIF4G1 gene was selected for further validation.
Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast) was
used for primer design (36)
according to the gene sequence from GenBank (accession no.
NM_198241). The primer sequences used were: Forward,
5'-CCGTGAGTTCCTGCTTGGTT-3' and reverse, 5'-CTTGCGTGGTTCTTTTCGGG-3'
(Tianyi Huiyuan Biotechnology Co., Ltd.). The c.1909A>T mutation
was amplified by PCR using a EasyTaq PCR SuperMix (cat. no. AS111;
Beijing TransGen Biotech Co., Ltd.), under the following
thermocycling conditions: Initial denaturation at 95˚C for 10 min;
followed by 35 cycles of 30 sec at 95˚C, 30 sec at 60˚C and 45 sec
at 72˚C; with a final extension at 72˚C for 5 min. The amplicons
were subsequently subjected to Sanger sequencing utilizing an ABI
3730xl DNA analyzer (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The resultant sequences were then compared with the findings
of WES, with false-positive variants identified through
next-generation sequencing being omitted.
Whole-exome sequence accession numbers
in ClinVar
The novel c.1909A>T (p.Ser637Cys) missense
mutation in EIF4G1 was deposited in ClinVar (https://submit.ncbi.nlm.nih.gov/clinvar;
accession no. SCV004039569).
Imaging examinations
MRI examinations were conducted using a 3.0T MRI
scanner (Ingenia CX; Philips Healthcare) with T1-weighted scan,
T2-weighted scan and 3D fluid-attenuated inversion recovery
performed in all 19 sequenced family members, with 8 symptomatic
cases undergoing additional high-resolution three-dimensional (3D)
susceptibility-weighted imaging (SWI) sequence. The presence or
absence of the swallow tail sign (STS) was assessed in the
cross-sectional images on the 3D SWI sequence in nigrosome-1, which
is located within the dorsolateral substantia nigra (SN). Previous
studies have shown that the absence of STS is both a highly
specific and sensitive indicator for the presence of PD (37,38).
Results
Clinical features
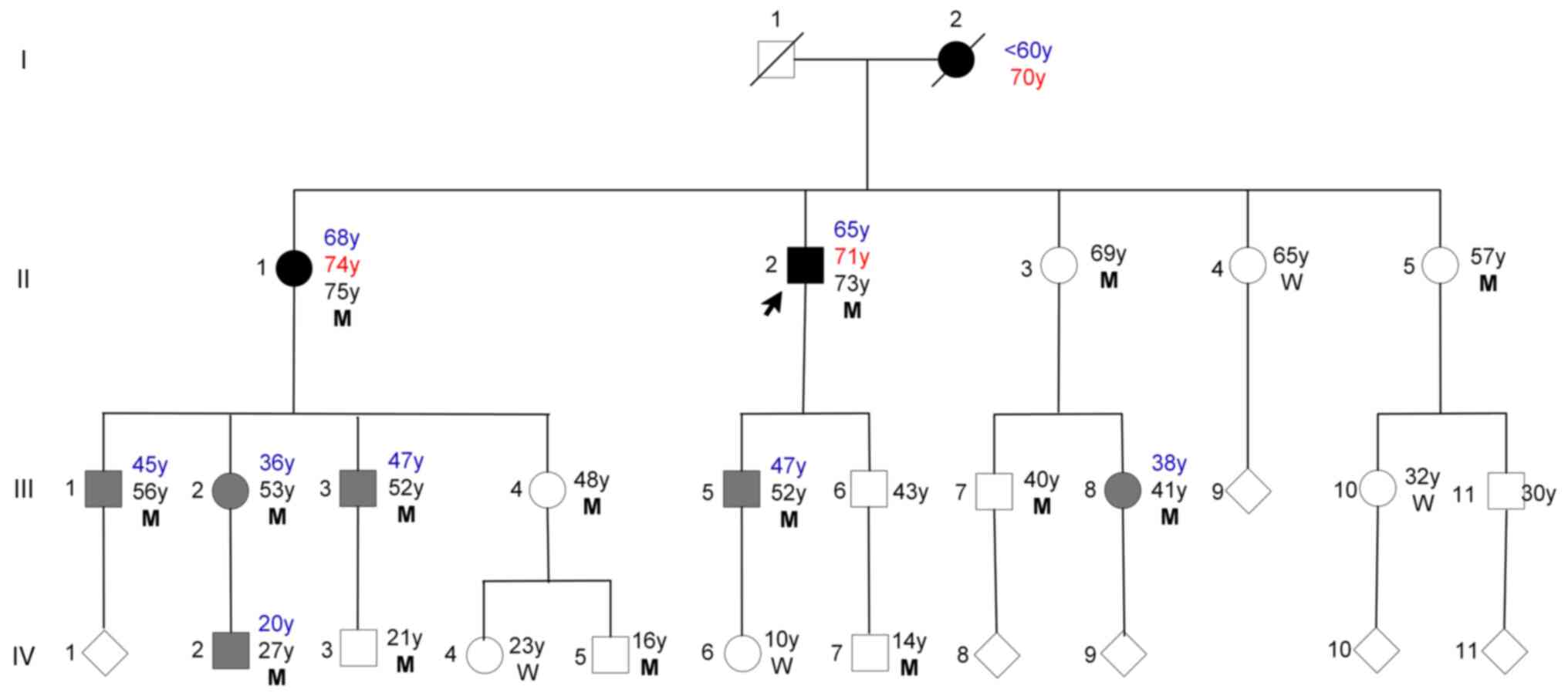
The family lived in eastern China, and there were no
consanguineous marriages in the family. There were a total of 29
individuals in this family, comprising 15 males and 14 females. The
age range of living family members in the study varied from 4 to 75
years, with a median age of 37±21.25. Case II-1 and Case II-2 were
admitted to the Affiliated Hospital of Jining Medical University
for the first time in April 2023. In total, 19 individuals from
this family spanning three generations were clinically assessed in
May 2023 at the Affiliated Hospital of Jining Medical University
and were genetically sequenced (Fig.
1). The first generation, consisting of two members, has passed
away. A total of eight family members (III-6,9,11, IV-1,8-11) chose
not to participate in the study. Based on the aforementioned
diagnostic criteria, three patients were diagnosed with PD [I-2
(deceased), II-1,2] and six were diagnosed with ET (III-1-3,5,8,
IV-2) in June 2023. All three patients with PD reported having a
multiple-year history of tremors before the clinical appearance of
PD signs.
Case I-2 died at the age of 75 years. According to
the patient's grandchildren, they had trembling of the hands in
their 60s. These symptoms then worsened as they advanced to >70
years of age, manifesting as involuntary shaking of limbs at rest,
movement retardation and a tendency to fall. The patient was unable
to take care of themself and was almost completely bedridden before
death. Neither the individual's parents nor their three siblings
had a similar disease history. Based on the patient's disease
history, a diagnosis of clinically possible PD may be considered in
accordance with the criteria established by the Movement Disorder
Society (39).
Case II-1 is a 75-year-old woman who had been
otherwise healthy until aged 68 years, when they first experienced
action tremor in their hands, which has persisted thereafter (for 7
years). The past medical history of this patient included
hypertension. The patient presented with postural tremor in the
hands and hyposmia. However, one year prior to the hospital visit,
at 74 years of age, the individual experienced the onset of resting
tremor in the upper left limb, followed by the gradual development
of resting tremor in the lower left limb and upper right limb.
These symptoms were accompanied by mild bradykinesia, decreased arm
swing during walking, a decline in short-term memory and loss of
smell. PD was initially diagnosed in the patient at the age of 75
on June 5, 2023 at the age of 75 with a UPDRS III score of 35 and
Hoehn-Yahr stage II when they were completely independent. The
patient was initially treated with oral levodopa (50 mg) and
benserazide hydrochloride (12.5 mg) three times daily in June 2023.
Due to the suboptimal control of resting tremor, the dosage of oral
medication was escalated to levodopa (100 mg) and benserazide
hydrochloride (25 mg) after a two-week period. The patient
underwent biweekly post-treatment monitoring. One month later, the
patient responded well to medication with an UPDRS III score of 18
on July 3, 2023 at the age of 75.
Case II-2 appears as the most severely affected
living patient within the present study. This 73-year-old male
individual initially presented with tremor in his hands eight years
ago, at the age of 65, prior to hospitalization. The medical
history included a diagnosis of type II diabetes. Initially
presented with intention tremor, the patient's symptoms exacerbated
during periods of nervousness and were less noticeable at rest.
Subsequently, two years prior to his hospitalization (at the age of
71), the patient developed resting tremor in his upper right limb,
which subsequently extended to the lower right limb and upper left
limb. These symptoms were accompanied by bradykinesia, a stiff
facial expression, reduced arm swing during walking, shuffling gait
and occasionally coughing when drinking water. PD was first
diagnosed on June 12, 2023 at the age of 73 with a UPDRS III score
of 45 and Hoehn-Yahr stage II when the patient was completely
independent. The treatment regimen was identical to that received
by his older sister, namely levodopa and benserazide hydrochloride.
However, during the two subsequent follow-up visits, which occurred
every two weeks, the patient exhibited poor response to the
prescribed drug therapy with a UPDRS III score of 45 on July 9,
2023 at the age of 73. Subsequently, the patient was recommended
undergoing a levodopa challenge test (LCT) or be treated with
arotinolol to control the tremors. The patient refused LCT due to
concerns about drug withdrawal reactions and adverse effects of
LCT, but accepted the addition of arotinolol. Therefore, arotinolol
(10 mg) was given once daily, which alleviated the tremors
substantially according to a telephone follow-up after 2 weeks. At
the 2-month follow-up after starting the treatment, the patient was
satisfied with their symptom improvement with a UPDRS III score of
21 on August 6, 2023 at the age of 73.
Case III-1-3, 5, 8 and IV-2 all had action (postural
and kinetic) tremors in their hands, with or without head tremors,
which worsened with nervousness and disappeared with drinking
alcohol, but did not show clinical manifestations of PD. The time
to action tremor onset in these patients was earlier (at 40-50
years of age) compared with their parents and grandparents. The
other individuals (II-3, 4, 5; III-4, 7, 10; and IV-3, 4, 5, 6, 7)
did not show any clinical manifestations of ET and/or PD. The
remaining 8 members (III-6, 9, 11, IV-1 and 8-11) declined
participation in the study and consequently did not undergo WES,
laboratory tests and MRI examination, precluding an analysis of
their respective conditions.
Neurological assessments of all participants
indicated that Case II-1 exhibited mild bradykinesia, diminished
arm swing during ambulation and cognitive decline in short-term
memory. Case II-2 displayed bradykinesia, facial rigidity,
decreased arm swing during walking and a shuffling gait, while
Cases III-1-3, 5, 8 and IV-2 demonstrated action tremors in the
hands, with or without accompanying head tremors and tremors could
exacerbated by anxiety. All patients had normal tendon reflexes,
negative pathological signs and normal limb sensations. Examination
of the cranial nerves revealed that Case II-1 exhibited anosmia and
the other patients had no abnormalities. General physical
examinations and routine laboratory tests did not reveal any
obvious pathognomonic alterations. The MMSE results showed that
none of the patients had cognitive impairment. The information of 8
symptomatic patients (2 patients with PD and 6 patients with ET)
and 1 deceased patient with PD are listed in Table I, whereas those of the other
asymptomatic patients are not listed.
 | Table IClinicopathological features of
affected individuals within the family. |
Table I
Clinicopathological features of
affected individuals within the family.
| Individual ID | I-2 | II-1 | II-2 | III-1 | III-2 | III-3 | III-5 | III-8 | IV-2 |
|---|
| Sex | F | F | M | M | F | M | M | F | M |
| Age at ET onset,
years | <60 | 68 | 65 | 45 | 36 | 47 | 47 | 38 | 20 |
| Age at PD onset,
years | 70 | 74 | 71 | - | - | - | - | - | - |
| Age at examination,
years | / | 75 | 73 | 56 | 53 | 52 | 52 | 41 | 27 |
| Disease duration,
years | >15 | 7 | 8 | 11 | 17 | 5 | 5 | 3 | 7 |
| Resting tremor | + | + | + | - | - | - | - | - | - |
| Action tremor | + | + | + | + | + | + | + | + | + |
| MMMSE scoring | / | 22 | 23 | 26 | 28 | 24 | 30 | 30 | 30 |
| UPDRS III
scoring | / | 35 | 45 | - | - | - | - | - | - |
| Hoehn-Yahr
stage | / | II | II | - | - | - | - | | - |
| Concomitant
diseases | / | Hypertension | Type II diabetes
mellitus | - | - | - | - | - | - |
Imaging features
The results of cranial 3T MRI of 19 included family
members are displayed in Fig. 2.
Both Case II-1and II-2 showed multiple ischemic degeneration foci,
old infarct foci with softening foci in the bilateral cerebral
hemispheres and basal ganglia. There was also evidence of bilateral
hemispheric atrophy and lateral periventricular white matter
degeneration. Specifically, Case II-1 displayed a significant
decrease in signal intensity in both SN regions, along with absence
of the bilateral STS. In contrast, Case II-2 showed a notable
reduction in signal intensity in the right substantia nigra and a
slight decrease in signal intensity in the left substantia nigra,
accompanied by bilateral disappearance of the STS.
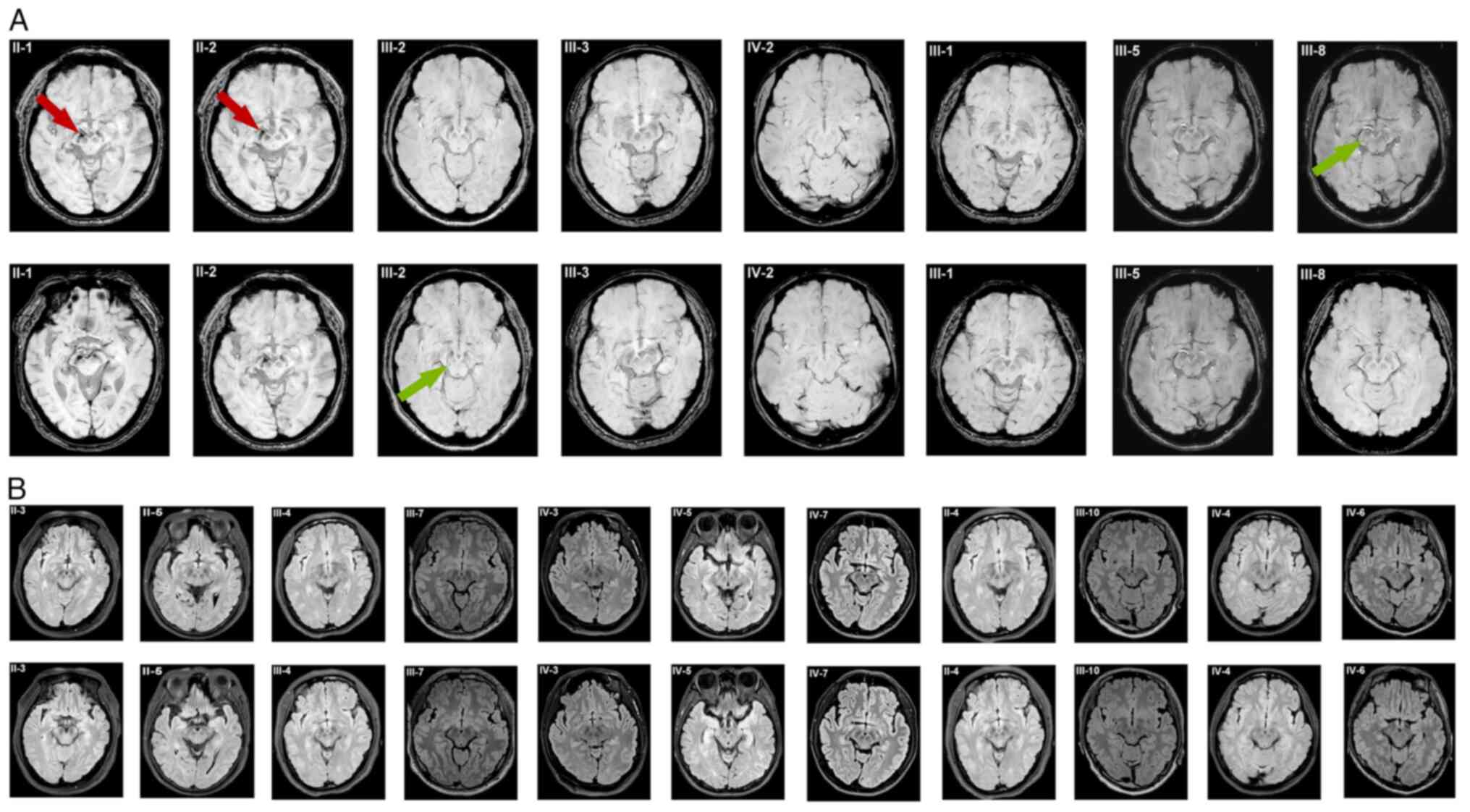 | Figure 2The cranial 3T MRI of 19 included
family members. (A) Cranial 3T MRI with high-resolution 3D
susceptibility-weighted imaging findings for Cases II-1, -2,
III-1-3, -5, -8, and IV-2 in the same family. Cases II-1 and Case
II-2 showed reduced signals in the SN region and a typical loss of
the swallow tail sign. Case III-2 showed slightly reduced signal in
the SN with an identified swallow tail morphology. Case III-3
showed slightly reduced signal in the SN with a faint swallow tail
morphology. Cases III-1, III-5, -8 and IV-2 showed normal
hyperintense signals in the SN region and a clear swallow tail
morphology. (B) Cranial 3T MRI with high-resolution 3D
fluid-attenuated inversion recovery findings for other family
members (II-3-5; III-4, -6, -7 and -9-11; IV-1 and -3-7) did not
show any obvious pathognomonic alterations. SN, substantia nigra.
The red arrow points to the SN region and the green arrow points to
the STS region. |
Case III-2 showed small degenerative foci of the
right frontal lobe, slightly reduced signal in the bilateral SN and
a visually identified STS. Case III-3 showed ischemic degeneration
foci in the center of the right semiovale and slightly reduced
signal in the bilateral SN, with a faintly present STS. Case IV-3
showed no abnormalities and a positive bilateral STS. Cranial 3T
MRI (Fig. 2) did not reveal any
obvious pathognomonic alterations in the other family members.
Gene discovery
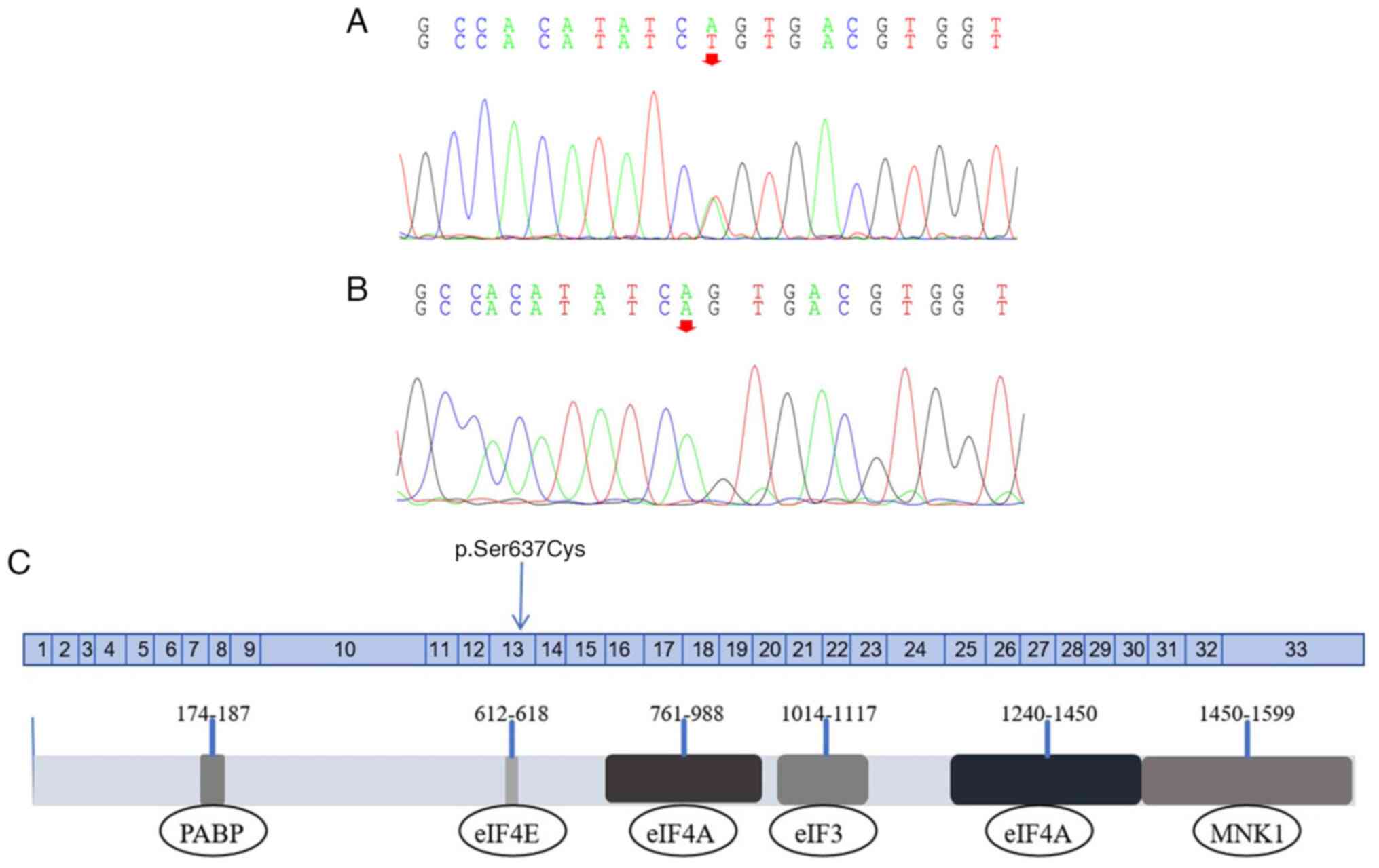
WES was performed in Case II-2. A missense mutation
was identified in the EIF4G1 gene, specifically c.1909A>T
in exon 13 on chr3:184040722 (Fig.
3). Using Sanger sequencing, a total of 15 family members,
namely II-1-3 and -5; III-1-5,-7 and -8; and IV-2,-3,-5 and-7, were
found to carry the same heterozygous missense mutation in
EIF4G1. No other family members, nor the partners of Case
II-1 and Case II-2, had this c.1909A>T variant. This mutation
resulted in a serine-to-cysteine substitution at position 637
(Ser637Cys), near the eIF4E binding region (Fig. 3C). To the best of our knowledge,
this variant has not been previously reported in the Online
Mendelian Inheritance in Man, Human Gene Mutation or ClinVar
databases. However, no changes in the secondary or tertiary
structure were predicted at the protein level according to
SWISS-MODEL.
Subsequent SIFT and PolyPhen2 analyses indicated
that the p.Ser637Cys mutation is potentially ‘Deleterious’ and
‘Possibly damaging’, respectively. MutationTaster software
identified the Ser637Cys mutation as a ‘disease causing’ mutation.
According to the ACMG criteria (35), this variant was considered as
having uncertain significance. In particular, it was categorized as
pathogenic moderate (PM1; located in a mutational hot spot and/or
critical and well-established functional domain; e.g., active site
of an enzyme, without benign variation), as it was located in a
mutational hot spot and/or critical and well-established functional
domain, such as the active site of an enzyme, without benign
variation. Furthermore, it was categorized as benign supporting 4,
since there were multiple lines of computational evidence
suggesting no impact on the gene or gene product in terms of
conservation, evolutionary and splicing impact.
Discussion
In the present study, the clinical details of a
family of patients with ET or PD with antecedent ET are described.
There are 29 members in this family, where the 2 members in the
first generation have died. A total of 19 participants underwent a
systematic physical examination, laboratory tests, imaging tests
and genetic sequencing. At first, one of the deceased patients
(Case I-2) was diagnosed with PD according to the description
provided by their grandchildren. In addition, 2 members of the
family were diagnosed with PD, both of whom had a ≥6-year tremor
history before the PD signs appeared. By contrast, 6 family members
were diagnosed with ET, with the other 11 family members currently
asymptomatic. Consistent with the previously reported clinical
characteristics of patients with EIF4G1 variants, the
patients in the present family showed late-onset PD with mild
progression, cognitive preservation and a good response to
L-levodopa treatment (23,40). However, unlike such previously
reported cases, the patients with PD in the present family all had
ET prior to PD onset.
Case II-1 and II-2 both showed reduced signals in
the SN region and a typical loss of the STS in cranial 3T MRI with
high-resolution 3D SWI. Nigrosome-1 is the largest cluster of
dopaminergic neurons and is located in the dorsolateral SN
(41). It is considered to be a
hyperintense structure on high-resolution 3D SWI sequences on 3T
MRI and is surrounded by hypointense portions in the SN and medial
lemniscus (41). Such imaging
features result in the appearance of an STS (42). Degeneration of nigrosome-1 in
patients with PD results in the loss of normal hyperintense signals
within the dorsolateral SN, which appears as an absence of the STS
(43). However, the
third-generation patients with ET in the present study showed
slightly reduced signals in the SN with a faint or identifiable STS
morphology. In the fourth-generation patients with ET, normal
hyperintense signals in the SN region and the STS morphology were
both clearly observed. Therefore, in the family in the present
study, there is a gradual decrease in signal within the SN region
and disappearance of the STS with increasing age and disease
progression.
Further genetic testing revealed that all family
members with confirmed PD or ET had the c.1909A>T (p.Ser637Cys)
variant in the EIF4G1 gene. The inheritance mode was
consistent with it being autosomal dominant. Previously,
mutations/variants in the EIF4G1 gene were deemed to be
associated with autosomal dominant PD (ADPD) (22,23).
The missense mutation p.Arg1205His in EIF4G1 was found to
segregate with disease in a large French family with ADPD. This
variant was found to perturb eIF4G1-eIF3E binding, which is a
crucial step in recruiting the 40S ribosomal subunit (23,44).
In another study, the p.Ala502Val variant has been shown to disrupt
eIF4G1-eIF4E binding, which may affect the binding of mRNA to
ribosomes (22,45). Subsequently, p.E462delInsGK was
identified in two affected siblings as a possible novel
disease-causing agent of ADPD (46). The discovery of variants
p.Gly686Cys in sporadic PD patients and p.Arg1197Trp in healthy
individuals challenges the assumed pathogenicity characteristic of
these variants (47). In another
study with 975 patients with PD and 1,014 healthy controls, novel
non-synonymous variants p.Thr318IIe, p.Val541Gly and p.Gly698Ala
were predicted to be ‘probably or possibly damaging’ by Polyphen2,
whereas p.Gly698Ala was predicted to be ‘disease causing’ by
MutationTaster (47). In the
family of the present study, although the p.Ser637Cys mutation was
considered as having uncertain significance according to the ACMG
criteria, it was predicted to be ‘possibly damaging’ by Polyphen2
and ‘disease causing’ by MutationTaster.
In addition, the p.Arg1205His mutation was also
identified in three healthy controls (47), which raises questions regarding the
potential of the EIF4G1 variant to cause PD. Subsequent
studies in patients with sporadic PD from different ethnic
backgrounds, including Asian (48,49),
African (50) and European
(51) showed mutations in the
EIF4G1 gene are not a significant or frequent risk factor
for Parkinson's disease. However, the incomplete penetrance of
p.Arg1205His in the EIF4G1 gene was similar to the
p.Gly2019Ser mutation in the LRRK2 gene for late-onset PD,
which should be considered (52).
Therefore, members of the family in the present study who were
harboring the p.Ser637Cys variant but did not show any clinical
manifestations of PD may have incomplete penetrance of the
EIF4G1 variant. In addition, the onset of PD is
age-dependent (53) and most
family members in the present study had not reached the average age
of onset (age at onset 61.7; standard deviation ±8.57) for late
onset PD in 26 Italian families (24). Therefore, members in this family
will require regular annual follow-ups for the next 20 years.
A variant of the PARK1 gene (p.Ala53Thr) was
previously identified in members of a family afflicted with PD,
which showed Mendelian segregation. This suggests that PD may have
a strong genetic component (54).
To date, ~30 genes have been reported to be associated with PD
(55). Genome-wide association
studies and candidate gene association studies have collectively
identified ≥90 common variants of independent loci that modify
disease risk (56). In addition,
accumulating evidence has suggested that exposure to various toxins
and changes in dietary habits may affect the occurrence and
progression of PD (57). Studying
this complex interplay between genetic and environmental factors
will improve the understanding of the pathophysiology of PD. The
present study reported the clinical features and sequencing
analysis results of a family with ET or PD with ‘antecedent ET’.
The emergence of a new mutation in EIF4G1 in a family with
tremors suggested that the role of EIF4G1 variants in
familial PD should be considered.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Postdoctoral
Program of the Affiliated Hospital of Jining Medical University
(grant no. JYFY303573), the Health Commission of Shandong Province
(grant no. 202006010928), Academician Lin He New Medicine in Jining
Medical University (grant no. JYHL2018FMS05) and the Affiliated
Hospital of Jining Medical University (grant no. 2018-BS-004).
Availability of data and materials
The c.1909A>T (p.Ser637Cys) missense mutation in
EIF4G1 generated in the present study may be found in
ClinVar under accession no. SCV004039569 or at the following URL:
(https://submit.ncbi.nlm.nih.gov/clinvar/). The other
data generated in the present study may be requested from the
corresponding author.
Authors' contributions
RHL, YK and QXK designed the study. XYX, LY, YYJ,
XCW and JG collected the data. RHL, YK and XYX contributed to the
data analysis and interpretation. RHL drafted the manuscript. YK
and QXK contributed to the revision of the manuscript. XYX and LY
confirm the authenticity of all the raw data. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by The Ethics
Committee of The Affiliated Hospital of Jining Medical University
(approval no. 2023-09-C031; Jining, China). All participants
provided written informed consent to participate.
Patient consent for publication
All participants provided written informed consent
for the publication of any associated data as well as any
accompanying images.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Nussbaum RL and Ellis CE: Alzheimer's
disease and Parkinson's disease. N Engl J Med. 348:1356–1364.
2003.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Hayes MT: Parkinson's disease and
Parkinsonism. Am J Med. 132:802–807. 2019.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Fereshtehnejad SM, Zeighami Y, Dagher A
and Postuma RB: Clinical criteria for subtyping Parkinson's
disease: Biomarkers and longitudinal progression. Brain.
140:1959–1976. 2017.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Bhatia KP, Bain P, Bajaj N, Elble RJ,
Hallett M, Louis ED, Raethjen J, Stamelou M, Testa CM and Deuschl
G: Tremor Task Force of the International Parkinson and Movement
Disorder Society. Consensus Statement on the classification of
tremors. From the task force on tremor of the International
Parkinson and Movement Disorder Society. Mov Disord. 33:75–87.
2018.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Song P, Zhang Y, Zha M, Yang Q, Ye X, Yi Q
and Rudan I: The global prevalence of essential tremor, with
emphasis on age and sex: A meta-analysis. J Glob Health.
11(04028)2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Louis ED and Ferreira JJ: How common is
the most common adult movement disorder? Update on the worldwide
prevalence of essential tremor. Mov Disord. 25:534–541.
2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
de Lau LM and Breteler MM: Epidemiology of
Parkinson's disease. Lancet Neurol. 5:525–535. 2006.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Clarimón J and Kulisevsky J: Parkinson's
disease: From genetics to clinical practice. Curr Genomics.
14:560–567. 2013.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Funayama M, Ohe K, Amo T, Furuya N,
Yamaguchi J, Saiki S, Li Y, Ogaki K, Ando M, Yoshino H, et al:
CHCHD2 mutations in autosomal dominant late-onset Parkinson's
disease: A genome-wide linkage and sequencing study. Lancet Neurol.
14:274–282. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Lesage S, Drouet V, Majounie E,
Deramecourt V, Jacoupy M, Nicolas A, Cormier-Dequaire F, Hassoun
SM, Pujol C, Ciura S, et al: Loss of VPS13C function in
autosomal-recessive Parkinsonism causes mitochondrial dysfunction
and increases PINK1/Parkin-Dependent mitophagy. Am J Hum Genet.
98:500–513. 2016.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Nalls MA, Blauwendraat C, Vallerga CL,
Heilbron K, Bandres-Ciga S, Chang D, Tan M, Kia DA, Noyce AJ, Xue
A, et al: Identification of novel risk loci, causal insights, and
heritable risk for Parkinson's disease: A meta-analysis of
genome-wide association studies. Lancet Neurol. 18:1091–1102.
2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Puschmann A: New genes causing hereditary
Parkinson's disease or Parkinsonism. Curr Neurol Neurosci Rep.
17(66)2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Yang N, Zhao Y, Liu Z, Zhang R, He Y, Zhou
Y, Xu Q, Sun Q, Yan X, Guo J and Tang B: Systematically analyzing
rare variants of autosomal-dominant genes for sporadic Parkinson's
disease in a Chinese cohort. Neurobiol Aging. 76:215.e1–215.e7.
2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Yan R and Rhoads RE: Human protein
synthesis initiation factor eIF-4 gamma is encoded by a single gene
(EIF4G) that maps to chromosome 3q27-qter. Genomics. 26:394–398.
1995.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Jackson RJ, Hellen CU and Pestova TV: The
mechanism of eukaryotic translation initiation and principles of
its regulation. Nat Rev Mol Cell Biol. 11:113–127. 2010.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Raught B, Gingras AC, Gygi SP, Imataka H,
Morino S, Gradi A, Aebersold R and Sonenberg N: Serum-stimulated,
Rapamycin-sensitive phosphorylation sites in the eukaryotic
translation initiation factor 4GI. EMBO J. 19:434–444.
2000.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ramírez-Valle F, Braunstein S, Zavadil J,
Formenti SC and Schneider RJ: eIF4GI links nutrient sensing by mTOR
to cell proliferation and inhibition of autophagy. J Cell Biol.
181:293–307. 2008.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Haimov O, Sehrawat U, Tamarkin-Ben Harush
A, Bahat A, Uzonyi A, Will A, Hiraishi H, Asano K and Dikstein R:
Dynamic interaction of eukaryotic initiation factor 4G1 (eIF4G1)
with eIF4E and eIF1 Underlies Scanning-Dependent and -independent
translation. Mol Cell Biol. 38:e00139–18. 2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Howard A and Rogers AN: Role of
translation initiation factor 4G in lifespan regulation and
age-related health. Ageing Res Rev. 13:115–124. 2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Vosler PS, Gao Y, Brennan CS, Yanagiya A,
Gan Y, Cao G, Zhang F, Morley SJ, Sonenberg N, Bennett MV and Chen
J: Ischemia-induced calpain activation causes eukaryotic
(translation) initiation factor 4G1 (eIF4GI) degradation, protein
synthesis inhibition, and neuronal death. Proc Natl Acad Sci USA.
110:18102–18107. 2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Algarni M and Fasano A: The overlap
between Essential tremor and Parkinson disease. Parkinsonism Relat
Disord. 46 (Suppl 1):S101–S104. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Tarakad A and Jankovic J: Essential Tremor
and Parkinson's disease: Exploring the relationship. Tremor Other
Hyperkinet Mov (NY). 8(589)2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chartier-Harlin MC, Dachsel JC,
Vilariño-Güell C, Lincoln SJ, Leprêtre F, Hulihan MM, Kachergus J,
Milnerwood AJ, Tapia L, Song MS, et al: Translation initiator
EIF4G1 mutations in familial Parkinson disease. Am J Hum Genet.
89:398–406. 2011.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Gialluisi A, Reccia MG, Modugno N, Nutile
T, Lombardi A, Di Giovannantonio LG, Pietracupa S, Ruggiero D,
Scala S, Gambardella S, et al: Identification of sixteen novel
candidate genes for late onset Parkinson's disease. Mol
Neurodegener. 16(35)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Gibb WR and Lees AJ: The relevance of the
Lewy body to the pathogenesis of idiopathic Parkinson's disease. J
Neurol Neurosurg Psychiatry. 51:745–752. 1988.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Hoehn MM and Yahr MD: Parkinsonism: Onset,
progression and mortality. Neurology. 17:427–442. 1967.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Goetz CG, Tilley BC, Shaftman SR, Stebbins
GT, Fahn S, Martinez-Martin P, Poewe W, Sampaio C, Stern MB, Dodel
R, et al: Movement disorder Society-sponsored revision of the
Unified Parkinson's Disease Rating Scale (MDS-UPDRS): Scale
presentation and clinimetric testing results. Mov Disord.
23:2129–2170. 2008.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Norris D, Clark MS and Shipley S: The
mental status examination. Am Fam Physician. 94:635–641.
2016.PubMed/NCBI
|
|
29
|
Wang XC, Liu RH, Wang T, Wang Y, Jiang Y,
Chen DD, Wang XY, Hou TS and Kong QX: A novel missense mutation in
SPAST causes hereditary spastic paraplegia in male members of a
family: A case report. Mol Med Rep. 27(79)2023.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Liu YD, Ma MY, Hu XB, Yan H, Zhang YK,
Yang HX, Feng JH, Wang L, Zhang H, Zhang B, et al: Brain proteomic
profiling in intractable epilepsy caused by TSC1 truncating
mutations: A small sample study. Front Neurol.
11(475)2020.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Fromer M, Moran JL, Chambert K, Banks E,
Bergen SE, Ruderfer DM, Handsaker RE, McCarroll SA, O'Donovan MC,
Owen MJ, et al: Discovery and statistical genotyping of copy-number
variation from whole-exome sequencing depth. Am J Hum Genet.
91:597–607. 2012.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Jiang Y, Oldridge DA, Diskin SJ and Zhang
NR: CODEX: A normalization and copy number variation detection
method for whole exome sequencing. Nucleic Acids.
43(e39)2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American College
of Medical Genetics and Genomics and the Association for Molecular
Pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Tsai FM, Lin YJ, Cheng YC, Lee KH, Huang
CC, Chen YT and Yao A: PrimerZ: Streamlined primer design for
promoters, exons and human SNPs. Nucleic Acids Res. 35:W63–W65.
2007.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Liu X, Wang N, Chen C, Wu PY, Piao S, Geng
D and Li Y: Swallow tail sign on susceptibility map-weighted
imaging (SMWI) for disease diagnosing and severity evaluating in
Parkinsonism. Acta Radiol. 62:234–242. 2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Kim DS, Tung GA, Akbar U and Friedman JH:
The evaluation of the swallow tail sign in patients with
Parkinsonism and gait disorders. J Neurol Sci.
428(117581)2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Postuma RB, Berg D, Stern M, Poewe W,
Olanow CW, Oertel W, Obeso J, Marek K, Litvan I, Lang AE, et al:
MDS clinical diagnostic criteria for Parkinson's disease. Mov
Disord. 30:1591–1601. 2015.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Deng H, Wu Y and Jankovic J: The EIF4G1
gene and Parkinson's disease. Acta Neurol Scand. 132:73–78.
2015.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Damier P, Hirsch EC, Agid Y and Graybiel
AM: The substantia nigra of the human brain. I. Nigrosomes and the
nigral matrix, a compartmental organization based on calbindin D
(28K) immunohistochemistry. Brain. 122:1421–1436. 1999.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Cosottini M, Frosini D, Pesaresi I,
Donatelli G, Cecchi P, Costagli M, Biagi L, Ceravolo R, Bonuccelli
U and Tosetti M: Comparison of 3T and 7T susceptibility-weighted
angiography of the substantia nigra in diagnosing Parkinson
disease. AJNR Am J Neuroradiol. 36:461–466. 2015.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Schwarz ST, Afzal M, Morgan PS, Bajaj N,
Gowland PA and Auer DP: The ‘swallow tail’ appearance of the
healthy nigrosome-a new accurate test of Parkinson's disease: A
case-control and retrospective cross-sectional MRI study at 3T.
PLoS One. 9(e93814)2014.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Villa N, Do A, Hershey JW and Fraser CS:
Human eukaryotic initiation factor 4G (eIF4G) protein binds to
eIF3c, -d, and -e to promote mRNA recruitment to the ribosome. J
Biol Chem. 288:32932–32940. 2013.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Sonenberg N and Hinnebusch AG: Regulation
of translation initiation in eukaryotes: Mechanisms and biological
targets. Cell. 136:731–745. 2009.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Lesage S, Condroyer C, Klebe S, Lohmann E,
Durif F, Damier P, Tison F, Anheim M, Honoré A, Viallet F, et al:
EIF4G1 in familial Parkinson's disease: Pathogenic mutations or
rare benign variants? Neurobiol Aging. 33:2233.e1–2233.e5.
2012.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Schulte EC, Mollenhauer B, Zimprich A,
Bereznai B, Lichtner P, Haubenberger D, Pirker W, Brücke T, Molnar
MJ, Peters A, et al: Variants in eukaryotic translation initiation
factor 4G1 in sporadic Parkinson's disease. Neurogenetics.
13:281–285. 2012.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Li K, Tang BS, Guo JF, Lou MX, Lv ZY, Liu
ZH, Tian Y, Song CY, Xia K and Yan XX: Analysis of EIF4G1 in ethnic
Chinese. BMC Neurol. 13(38)2013.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Chen Y, Chen K, Song W, Chen X, Cao B,
Huang R, Zhao B, Guo X, Burgunder J, Li J and Shang HF: VPS35
Asp620Asn and EIF4G1 Arg1205His mutations are rare in Parkinson
disease from southwest China. Neurobiol Aging. 34:1709.e7–e8.
2013.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Blanckenberg J, Ntsapi C, Carr JA and
Bardien S: EIF4G1 R1205H and VPS35 D620N mutations are rare in
Parkinson's disease from South Africa. Neurobiol Aging.
35:445.e1–e3. 2014.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Gagliardi M, Annesi G, Tarantino P,
Nicoletti G and Quattrone A: Frequency of the ASP620ASN mutation in
VPS35 and Arg1205His mutation in EIF4G1 in familial Parkinson's
disease from South Italy. Neurobiol Aging. 35:2422.e1–e2.
2014.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Kumari U and Tan EK: LRRK2 in Parkinson's
disease: Genetic and clinical studies from patients. FEBS J.
276:6455–6463. 2009.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Marchetti B, Tirolo C, L'Episcopo F,
Caniglia S, Testa N, Smith JA, Pluchino S and Serapide MF:
Parkinson's disease, aging and adult neurogenesis: Wnt/β-catenin
signalling as the key to unlock the mystery of endogenous brain
repair. Aging Cell. 19(e13101)2020.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Lunati A, Lesage S and Brice A: The
genetic landscape of Parkinson's disease. Rev Neurol (Paris).
174:628–643. 2018.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Bandres-Ciga S, Diez-Fairen M, Kim JJ and
Singleton AB: Genetics of Parkinson's disease: An introspection of
its journey towards precision medicine. Neurobiol Dis.
137(104782)2020.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Polymeropoulos MH, Higgins JJ, Golbe LI,
Johnson WG, Ide SE, Di Iorio G, Sanges G, Stenroos ES, Pho LT,
Schaffer AA, et al: Mapping of a gene for Parkinson's disease to
chromosome 4q21-q23. Science. 274:1197–1199. 1996.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Pirooznia SK, Rosenthal LS, Dawson VL and
Dawson TM: Parkinson disease: Translating insights from molecular
mechanisms to neuroprotection. Pharmacol Rev. 73:33–97.
2021.PubMed/NCBI View Article : Google Scholar
|