Introduction
Major depressive disorder (MDD) is a common
psychiatric condition, and it is predicted that MDD affects ~264
million individuals globally (1,2). In
China, the incidence of MDD is ~3.4%, and in the USA, the incidence
is up to 16.2%, causing a heavy burden for the families of patients
with MDD and the nation as a whole (3). At present, traditional
anti-depressants, such as monoamine oxidase inhibitors, tricyclic
agents and selective serotonin reuptake inhibitors, require up to
six weeks to induce obvious anti-depressive effects (4,5). As
patients with MDD carry a high risk of death by suicide, it is
urgent that solutions are found to reduce suicidal tendencies
amongst these individuals (6,7).
Therefore, investigations into the development of a fast-acting
anti-depressants are of great importance. Previously, a number of
studies have shown that ketamine (KTM), an intravenous anesthetic
agent, is effective in treating MDD, and notably has been revealed
to reduce suicidal tendencies in patients with MDD in a short
period of time (several days to 1-2 weeks) (6,8).
Although KTM is effective in treating MDD, the anti-depressive
mechanism of KTM remains unknown. Traditionally, it has been
hypothesized that KTM induces pharmacological effects via
inhibition of the cellular N-methyl-D-aspartic acid receptor.
However, recent studies have demonstrated that cannabinoid (CB)
receptors, including CB1 and CB2 receptors, are involved in
KTM-induced anti-depressive effects (9,10).
In the pathological processes of MDD, oxidative
injury-induced mitochondrial dysfunction plays a critical role
(11,12). In cells, mitochondria produce
energy for cellular activities, however in the leukocytes of
patients with MDD, a lower mitochondrial DNA copy number was
recorded compared with individuals without MDD (13,14).
In addition, serum antioxidant levels in patients with MDD are
notably lower than that of healthy individuals (15). Moreover, the cerebral glutamate
level in patients with MDD is higher than healthy individuals
(15). Due to the aforementioned
reasons, glutamate-induced cell injury is used widely to mimic
oxidative stress injury in neurons (16).
Therefore, in the present study, HT22 neuronal cells
were treated with glutamate to imitate oxidative injury in MDD, and
it was hypothesized that the CB1 receptor mediates KTM-induced
protection against glutamate via ameliorating mitochondrial
function in HT22 cells.
Materials and methods
Cells and reagents
HT22 cells, a murine hippocampal cell line, were
obtained from The Affiliated Hospital of The Xuzhou Medical
University (Jiangsu, China). KTM was purchased from Fujian Gutian
Pharmaceutical Co., Ltd. (Fujian, China). Dulbecco's Modified
Eagle's Medium (DMEM), fetal bovine serum (FBS),
penicillin-streptomycin mixed solution and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
powder were purchased from MilliporeSigma. Lactate dehydrogenase
(LDH, cat. no. A020-1-2), glutathione (GSH, cat. no. A006-1-1),
catalase (CAT, cat. no. A007-1-1) and oxidized GSH (GSSG, cat. no.
A061-2-1) reagent kits were obtained from Nanjing Jiancheng
Bioengineering Institute (Jiangsu, China).
Cell culture and treatments
HT22 cells were cultured in DMEM containing 10% FBS
(v:v) and 1% penicillin-streptomycin mixed solution
(1x104 IU penicillin and 1x104 µg
streptomycin/ml). The medium was changed 3 times/week, and the
cells were cultured at 37˚C in 95% oxygen and 5% carbon dioxide
with 100% humidity. The cells were passaged 2-3 times/week.
The cells were divided into five groups to determine
an ideal concentration of KTM. The groups were as follows: i)
Control group (cultured in the medium without any drug); ii)
glutamate exposure group (exposed to the medium with 15 mM
glutamate); iii) 1 µg/ml KTM treatment group (exposed to the medium
with 1 µg/ml KTM plus 15 mM glutamate); iv) 10 µg/ml KTM treatment
group (exposed to the medium with 10 µg/ml KTM plus 15 mM
glutamate); and v) 20 µg/ml KTM group (exposed to the medium with
20 µg/ml KTM plus 15 mM glutamate). After incubation for 24 h, the
degree of cell injury was assessed by determining the cell
viability and LDH release.
Next, the role of the CB1 receptor in the
KTM-treated cells was investigated and the cells were divided into
five groups, including: i) Control group; ii) 15 mM glutamate
group; iii) 10 µg/ml KTM + 15 mM glutamate group; iv) 10 µM CB1
antagonist AM251 + 10 µg/ml KTM + 15 mM glutamate group; and v) 10
µM AM251 + 15 mM glutamate group. After incubation for 24 h, the
degree of cell injury and intracellular oxidant/antioxidant levels
were assessed. Moreover, to explore the KTM-induced effects on the
intracellular superoxide dismutase (SOD) 1 and SOD2 levels, the
cells were divided into the control and 10 µg/ml KTM exposure
groups. After treatment for 24 h, intracellular SOD1/SOD2 activity
and expression levels were measured.
MTT and LDH assay
A 96-well cell culture plate was used to culture the
cells with a cell intensity of 1x105 cells/well. As the
treatments were completed, 20 µl of MTT solution with a
concentration of 5 mg/ml was added to each plate well, and the
plate was sent back into the incubator. After incubation for 4 h at
37˚C, the medium was removed and discarded, and the generated
formazan of the plate was dissolved by adding 150 µl DMSO into each
well. After shaking for 15 min using an agitator, once the formazan
was dissolved completely, an enzyme-labeled instrument (Tecan
Group, Ltd.) was used to measure the absorbance. An empty well
containing 150 µl distilled water was added to serve as a blank
control.
A 96-well cell culture plate was used to culture the
cells and upon treatment completion, 50 µl of supernatant of the
cell culture medium was collected and centrifuged at a speed of 400
x g for 30 min at room temperature. After centrifugation for 30
min, as previously described (17), 40 µl of the medium from each well
was harvested to measure the LDH release level.
Cell apoptosis
A 6-well plate was used to culture the cells with a
cell density of 1x106 cells/well. Upon treatment
completion, the medium of the plate was removed and discarded, and
the cells were collected through centrifugation at a speed of 400 x
g for 30 min at room temperature. A total of 2 ml
phosphate-buffered saline (PBS) at 4˚C was used to wash the cells
twice, and the cells were resuspended in binding buffer. Next, the
anti-annexin-V staining antibody (1:1,000; cat. no. SAB5702648;
MilliporeSigma), which was labelled with fluorescein isothiocyanate
isomer plus propidium iodide solution, was added into the buffer.
After which, the cells were mixed with the buffer slowly. In
darkness at room temperature, the cells were incubated for 15 min.
Finally, cell apoptosis was measured by flow cytometry (FACSCanto
II; BD Biosciences), which was operated via BD FACSCanto System II
Software v2.4 (BD Biosciences).
Western blot analysis
A 6-well plate was used to culture the cells with
1x106 cells seeded into each well. Following treatment
completion, the cells were washed with PBS three times for 5 min.
After which, the cells were harvested using a cell scraper and the
protein level of the cells was measured using the Bradford method.
The western blotting procedure was conducted as previously
described (18). The subsequent
antibodies were used including anti-cleaved caspase-3 antibody
(cat. no. ab231289; Abcam), anti-CB1 primary antibody (cat. no.
ab259323, Abcam) and anti-SOD1/SOD2 antibody (cat. no.
ab13498/ab12533; Abcam) at a dilution of 1:200. β-tubulin (1:500;
cat. no. ab179511; Abcam) and β-actin (1:500; cat. no. ab8226;
Abcam) served as the loading control. Image Lab Software 6.0.1
(Bio-Rad Laboratories, Inc.) was taken to analyze the images from
the western blotting results.
Immunocytochemistry
A confocal microscope-specific cell culture plate
was taken to culture the cells, and the cells were seeded at a
density of 1x105 cells/well. Upon treatment completion,
the medium in the plate was discarded and PBS solution was used to
wash the cells three times for 5 min. After which, the cells were
incubated with 1 ml of 4% paraformaldehyde solution for 20 min, and
PBS was used to wash the cells three times for 5 min. Next, the
cells were incubated with 50 µl anti-CB1 primary antibody (1:50) at
4˚C for 12 h. After incubation, the primary antibody was discarded
and PBS was used to wash the plate three times. Next, 100 µl
Cy3-labelled goat-anti-rabbit antibody (red; 1:100; cat. no. A0516;
Beyotime Institute of Biotechnology) was added into each plate;
after 30-min incubation in darkness at room temperature, 50 µl DAPI
solution was added into each plate. After an additional incubation
in darkness for 10 min, the plate was washed again with PBS.
Finally, a laser confocal fluorescence microscope (FV10i; Olympus
Corporation) was used to observe the cell culture plate, and the
images were captured at random.
Intracellular GSH, CAT, GSSG and SOD
measurements
A 6-well plate was used to culture the cells, and
1x106 cells was seeded into each well. Following cell
lysis induction, 1 ml PBS (0.1 M; pH 7.4) was used. After which the
cells were centrifuged at a speed of 400 x g for 20 min at 4˚C. The
supernatants were harvested to measure the intracellular GSH, CAT
and GSSG levels, and a spectrophotometer (Tecan Group, Ltd.) was
used to detect the absorbance.
The SOD1 and SOD2 activities were measured according
to an investigation described previously (19). Briefly, total SOD activity was
measured first and then SOD2 activity was evaluated. As the total
SOD activity is equal to the SOD1 activity plus the SOD2 activity,
the SOD1 activity can be calculated according to the total SOD and
SOD2 values.
Intracellular ROS assay
A confocal microscope-specific cell culture plate
was used to culture the cells, and the cells were seeded at a
density of 2x104 cells/well. Upon treatment completion,
1 ml FBS-free cell culture medium containing 100 µM
dichlorofluorescein (DCF)-DA was added into each well, after
incubation for 20 min at 37˚C. After which, PBS was used to wash
the cells three times for 5 min. A confocal microscope was used to
observe and capture images, and the excitation and emission
wavelengths were 480 and 535 nm, respectively. Non-fluorescence
DCF-DA can be oxidized into fluorescence DCF (green) by
intracellular ROS, with more ROS producing more DCF. Image Pro-Plus
Software 6.0 (Media Cybernetics, Inc.) was used to assess the
fluorescence intensity of images.
Mitochondrial complex activity
assay
A 6-well plate was used to culture the cells, and
1x106 cells was seeded into each well. Upon treatment
completion, the cells were collected using a cell scraper. After
which, the cells were treated with trypsin for 1-2 min at room
temperature. Next, the cells were centrifuged at a speed of 400 x g
for 30 min at room temperature for mitochondrial isolation. A
mitochondrial isolation reagent kit (cat. no. 37612; Qiagen GmbH)
was taken to collect and isolate the mitochondria from the cells. A
spectrophotometer (Tecan Group, Ltd.) was used to measure the
mitochondrial complex I and complex IV activities at 30˚C.
Mitochondrial superoxide
measurement
Mitochondrial superoxide levels were evaluated using
a MitoSOX Red Reagent Kit (cat. no. M36008; Invitrogen; Thermo
Fisher Scientific, Inc.). Briefly, a confocal microscope-specific
plate was used to culture the cells, and the cell density was
2x104 cells/plate. Upon treatment completion, 5 µl
MitoSOX reagent was added into each plate. In darkness, the cells
were incubated for 20 min at 37˚C, and the nuclei of the cells were
marked by adding 20 µl DAPI staining solution into each plate. The
fluorescent images were captured at random following incubation for
15 min in darkness at room temperature. The excitation and emission
wavelengths for mitochondrial superoxide (red) were 510 and 580 nm,
respectively. The two wavelengths for the nuclei (blue) were 340
and 488 nm, respectively. An Image Pro-Plus Software 6.0 (Media
Cybernetics, Inc.) was used to assess the fluorescence intensity of
the images.
Statistical analysis
SPSS 12.0 (SPSS, Inc.) was used to conduct the
statistical assessments. All data were presented as the mean ±
standard deviation, and one-way ANOVA followed by Tukey's Multiple
Comparison Test were used to compare the differences between
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
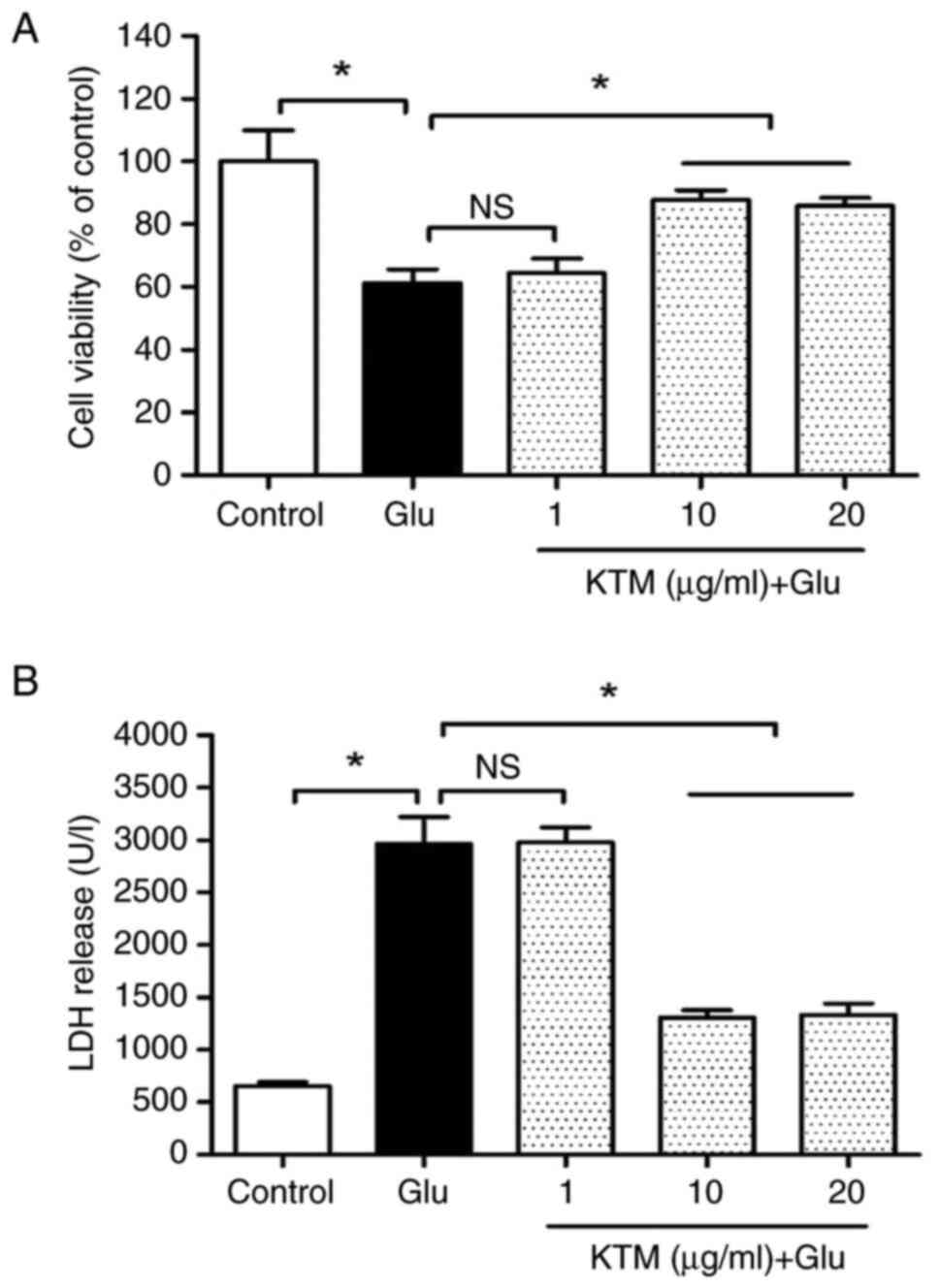
KTM decreases cell injury induced by
glutamate
Glutamate (15 mM) was used to imitate oxidative
injury in HT22 cells. To search for a suitable KTM dose, cell
viability and LDH release were used to assess the degree of cell
injury (Fig. 1). Compared with the
control (cultured in drug-free medium), 15 mM glutamate decreased
cell viability and enhanced the release of LDH significantly
(P<0.05), and 10 and 20 µg/ml KTM restored cell viability and
inhibited LDH release (P<0.05). However, 1 µg/ml KTM did not
induce marked protection against the cell injury caused by
glutamate (P>0.05). Therefore, 10 µg/ml KTM was the selected
dosage for subsequent experiments.
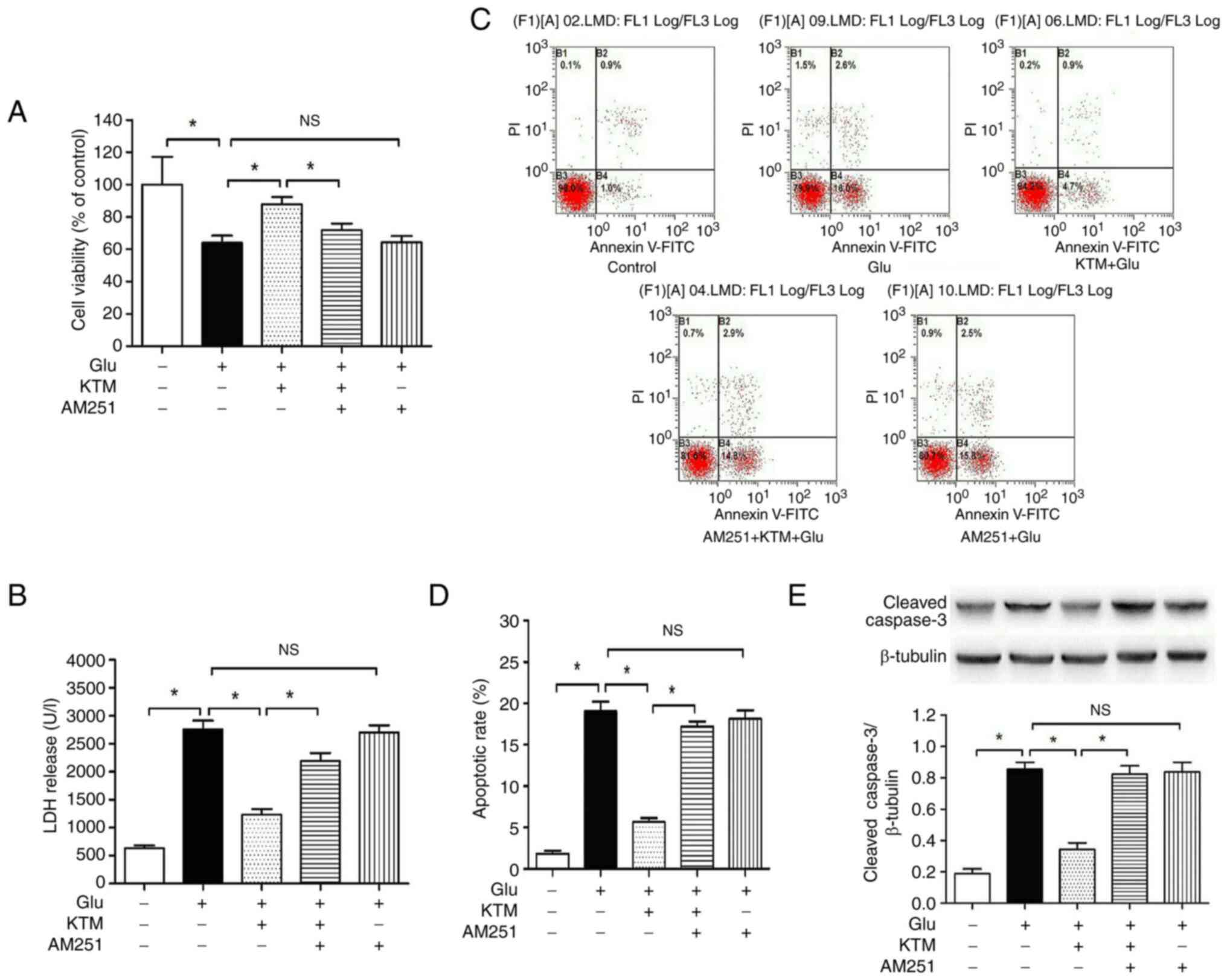
CB1 receptor antagonist AM251 reverses
KTM-induced neuroprotection against glutamate in neuronal
cells
To further explore the underlying cytoprotective
mechanism of KTM, AM251, a selective CB1 receptor antagonist, was
used to study the CB1 receptor in KTM-induced neuroprotection.
Compared with the cells of control group (Fig. 2), 15 mM glutamate decreased the
degree of cell viability (Fig.
2A), and enhanced the LDH level (Fig. 2B), cell apoptosis rate (Fig. 2C and D) and cleaved caspase-3 expression
(Fig. 2E) (P<0.05).
Co-administration of 10 µg/ml KTM significantly attenuated the
glutamate-induced cell injury (Fig.
2A-E); however the presence of the CB1 receptor antagonist
AM251 (10 µM), partially blocked the KTM-induced cytoprotection,
and AM251 did not bring about notable effects on the
glutamate-induced cell injury (P>0.05). According to the flow
cytometry results, the right two quadrants indicated early (lower
quadrant) and late (upper quadrant) apoptotic cells, respectively,
and the rate of apoptotic cells was the sum of the percentage of
the lower right and upper right quadrants (Fig. 2C and D).
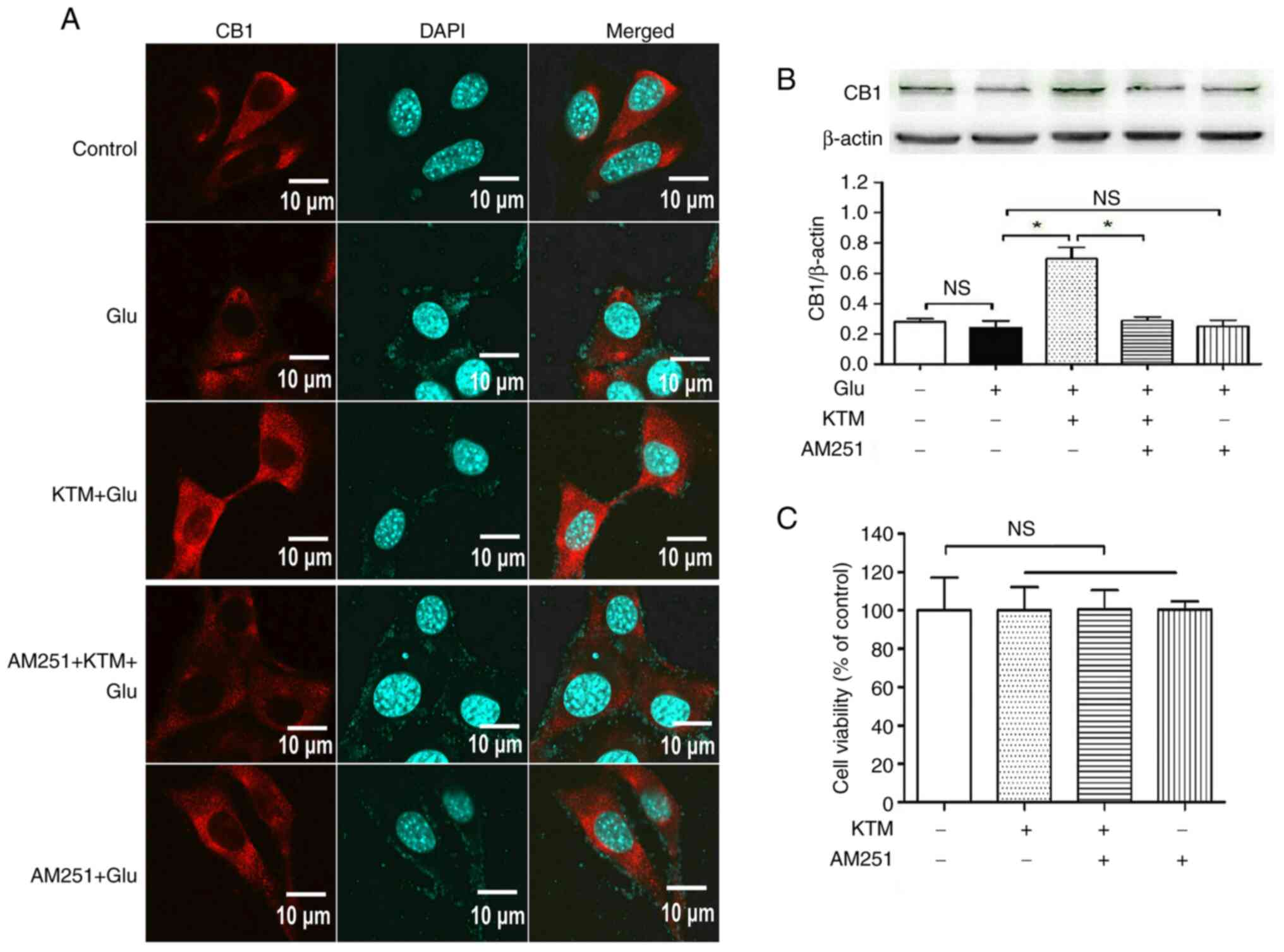
CB1 antagonist AM251 blocks
KTM-induced upregulation of the CB1 receptor in glutamate-treated
HT22 cells
Compared with the cells cultured in drug-free medium
(Fig. 3A and B), 15 mM glutamate did not cause marked
changes in CB1 receptor expression in HT22 cells (P>0.05), and
10 µg/ml KTM significantly upregulated CB1 expression. However, 10
µM CB1 antagonist AM251 (P<0.05) partially reversed the
KTM-induced CB1 upregulation (P<0.05); and AM251 did not induce
notable effects on CB1 expression, compared with the
glutamate-treated cells (P>0.05).
To exclude the potential cytotoxicity caused by KTM
or AM251, the cell grouping was as follows (Fig. 3C): i) Control; ii) 10 µg/ml KTM
group; iii) KTM + AM251 group and iv) 10 µM AM251 group. After
incubation for 24 h, no notable cell injury was observed in the
other three groups (P>0.05), compared with that of the control
group, indicating that the KTM-induced cytoprotection was via
pharmacological effects rather than cytotoxic effects.
KTM blocks glutamate-induced oxidative
injury in HT22 cells and cytoprotection is reversed by the CB1
antagonist AM251
A high concentration of glutamate brings about
oxidative injury to neuronal cells. In the present study, compared
with the control cells, 15 mM glutamate decreased intracellular
redox glutathione (Fig. 4A) and
CAT (Fig. 4E) levels (P<0.05),
and enhanced the GSSG (Fig. 4B)
and ROS (Fig. 4C and D) levels. In addition, 10 µg/ml KTM
notably blocked the glutamate-induced consumption of intracellular
GSH and CAT, and decreased the generation of ROS and GSSG
(P<0.05). The CB1 antagonist AM251, however, significantly
reversed the aforementioned KTM-induced effects on intracellular
antioxidants and oxidants, and AM251 alone did not cause marked
influence on the glutamate-induced effects on intracellular GSH,
GSSG, ROS and CAT (P>0.05). These findings supported the
hypothesis that the CB1 receptor mediates KTM-induced antioxidative
injury against glutamate in neurons.
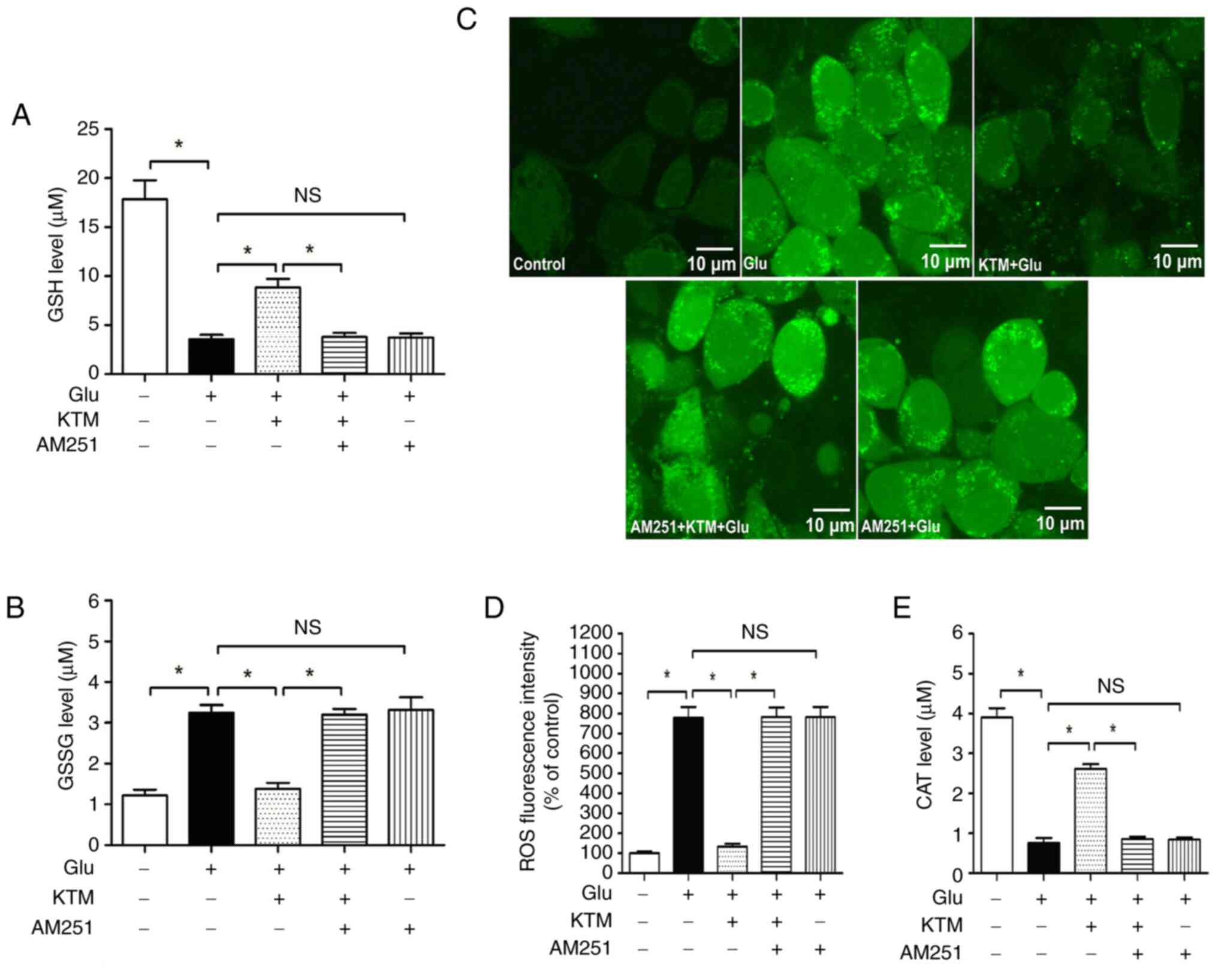 | Figure 4CB1 antagonist AM251 reverses
KTM-induced ameliorations of intracellular antioxidants/oxidants.
Cells were grouped as described in Fig. 2. Intracellular GSH, GSSG, ROS and
CAT levels were measured, and the fluorescence images were captured
using a fluorescence microscope. (A) AM251 reversed KTM-induced
ameliorations of intracellular GSH levels (n=6). (B) AM251 reversed
KTM-induced intracellular GSSG decrease (n=6). (C) Intracellular
ROS images (magnification, x400). (D) AM251 reversed KTM-induced
intracellular ROS decrease (n=6). (E) AM251 reversed KTM-induced
ameliorations of intracellular CAT level (n=6).
*P<0.05. NS, no significance; GSH, glutathione; GSSG,
oxidized GSH; ROS, reactive oxygen species; CAT, catalase; KTM,
ketamine; CB1, cannabinoid receptor 1; Glu, glutamate. |
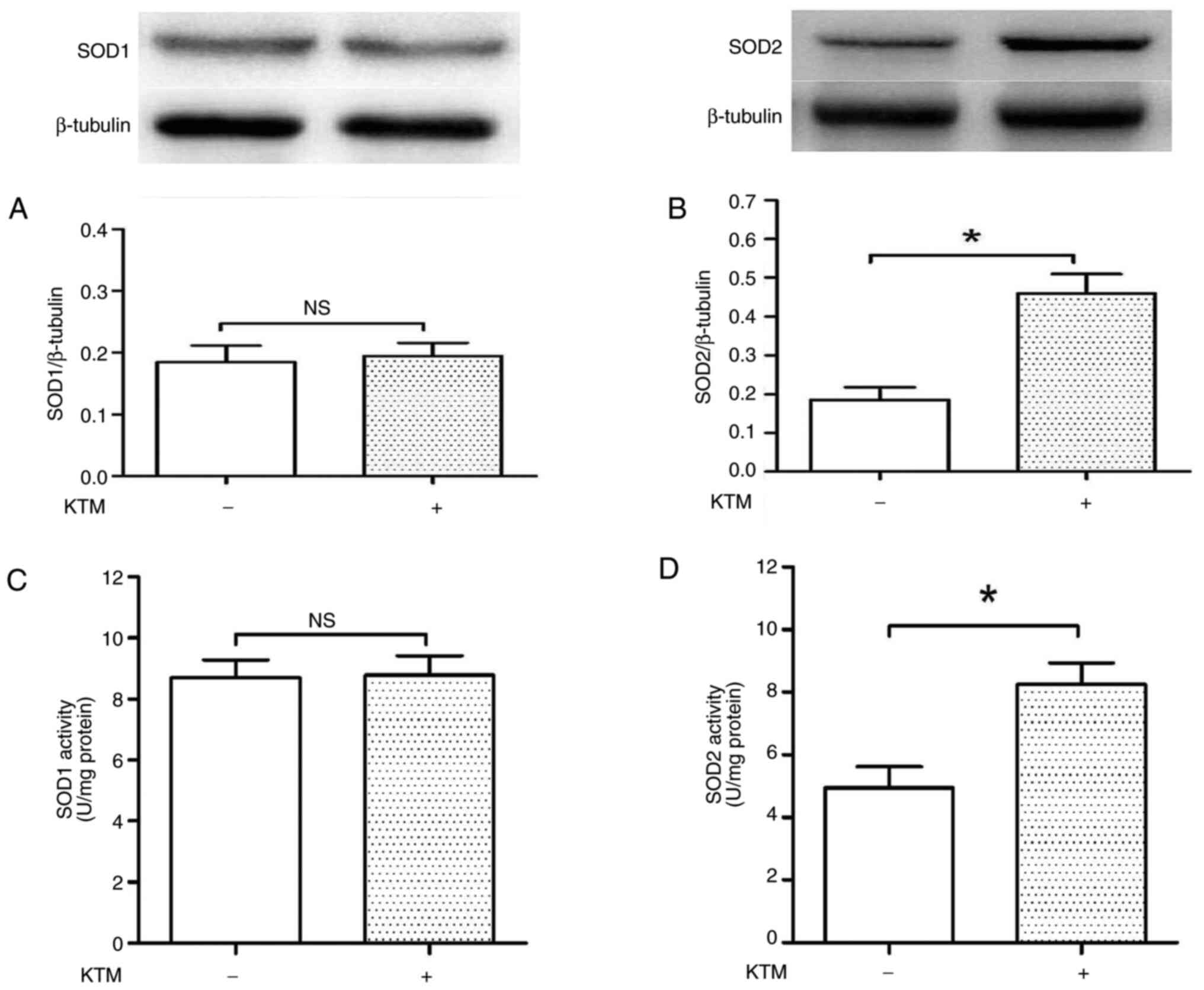
KTM increases intracellular SOD2
levels, but not SOD1 levels
To further explore the KTM-induced antioxidative
injury mechanism, intracellular SOD1 and SOD2 levels were measured.
A concentration of 10 µg/ml KTM was used to treat the cells. After
treatment for 24 h, western blotting and SOD reagent kits were used
to evaluate intracellular SOD1 and SOD2 expression levels and
activities (Fig. 5A-D). KTM
exposure did not alter SOD1 expression and activity (P>0.05);
however, it increased SOD2 expression and activity in HT22 cells
(P<0.05), compared with the cells of the control group. As SOD1
is expressed in the cytoplasm, and mitochondria contain SOD2
protein, according to the findings of the present study, it can be
inferred that the KTM-induced cytoprotection against glutamate may
be via maintaining mitochondrial function and SOD2 levels.
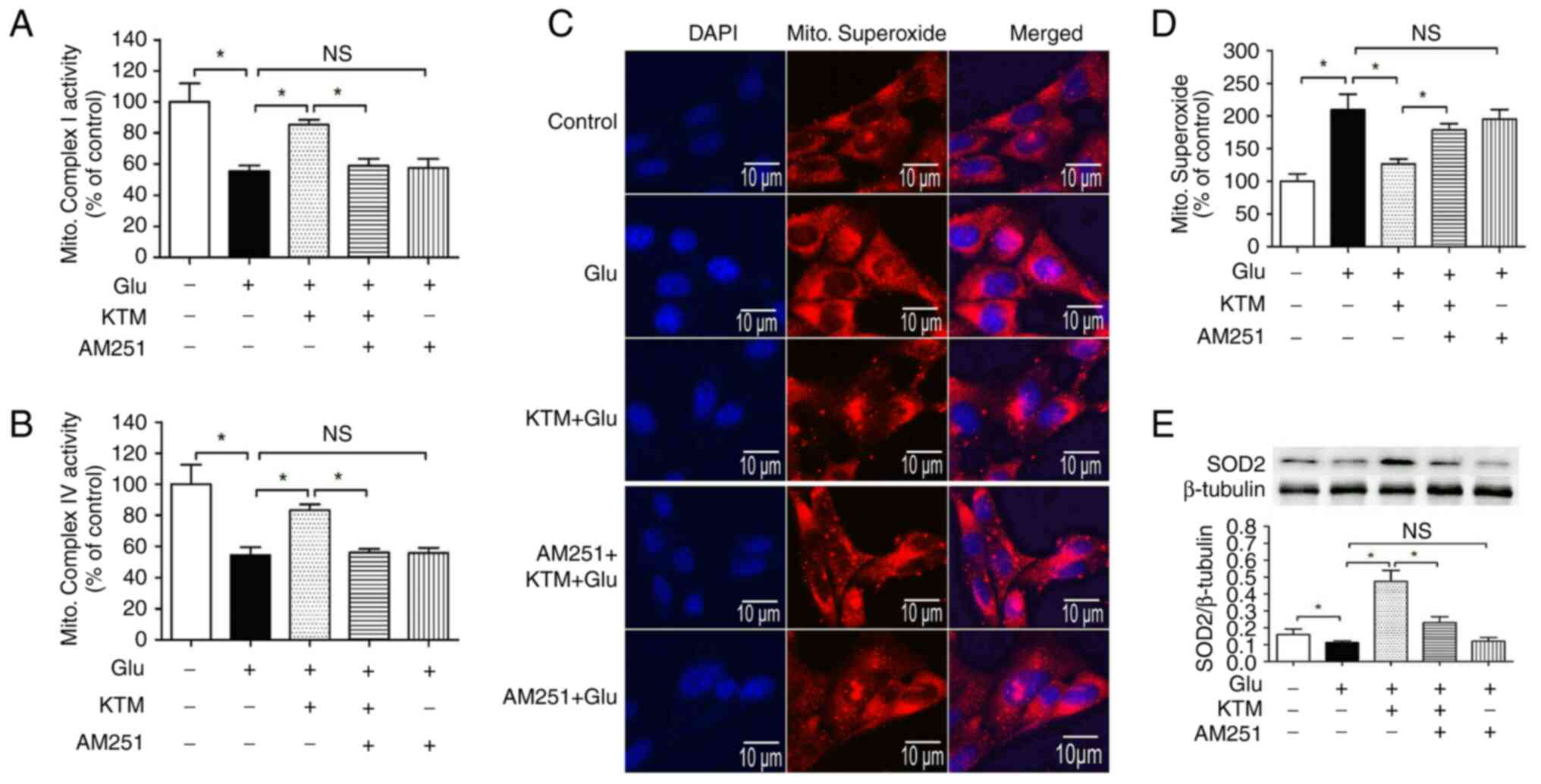
CB1 antagonist AM251 reverses
KTM-induced mitochondrial function amelioration, superoxide
decrease and SOD2 upregulation
To explore the role of CB1 in KTM-induced
cytoprotection, the mitochondrial function, superoxide and SOD2
protein levels were measured. Compared with the control, the
mitochondrial complex activities (complex I and IV) and SOD2
expression levels (Fig. 6A,
B and E) were significantly reduced in the
presence of 15 mM glutamate, and mitochondrial superoxide levels
(Fig. 6C and D) were increased. In addition, 10 µg/ml
KTM significantly increased the mitochondrial complex activities
and SOD2 expression levels, and decreased mitochondrial superoxide
levels in the glutamate-treated neuronal cells. AM251 significantly
blocked the KTM-induced effects on complex I and IV levels, and
mitochondrial superoxide and SOD2 expression levels (P<0.05).
AM251 did not cause notable changes in the aforementioned single
glutamate-induced oxidative injuries. These findings suggested that
KTM-induced cytoprotection against glutamate is mediated by the CB1
receptor through maintaining mitochondrial function.
Discussion
In the present study, HT22 neuronal cells were
treated with glutamate to imitate oxidative stress injury in MDD,
and it was found that glutamate decreased cell viability and
intracellular antioxidants levels, including GSH, CAT and SOD2, and
inhibited mitochondrial complex levels. In addition, LDH release,
intracellular ROS, GSSG, cell apoptosis, cleaved caspase-3
expression and mitochondrial superoxide levels were increased.
However, the presence of KTM significantly decreased the
glutamate-induced cell oxidative injury and the effects on the
intracellular antioxidant/oxidant levels, and upregulated
mitochondrial function and CB1 receptor expression simultaneously.
Co-administration with the CB1 antagonist AM251 notably blocked the
KTM-induced cytoprotective effects against glutamate, and
downregulated CB1 upregulation. AM251 did not induce marked effects
on glutamate-induced oxidative injury. These findings indicated
that KTM can decrease glutamate-induced oxidative injury in
neuronal cells, and the CB1 receptor may mediate the aforementioned
protective mechanisms.
MDD is a common psychiatric condition, with symptoms
including prolonged low mood, insomnia and pessimism, and a high
risk of suicide (20,21). In the United States, MDD costs the
economy $210 billion USD (45% direct losses, 50% workplace losses
and 5% suicide-associated losses), causing a great economic load to
families of patients and to society as a whole (21). Previous investigations have
indicated that a sub-clinical dose of KTM can bring about notable
therapeutic effects to treat MDD, and the administration of KTM may
also decrease the risk of suicide in patients with MDD (6,8).
However, the therapeutic mechanisms of KTM in the treatment of MDD
remain unknown. Previous studies have indicated that administering
KTM can increase CB1 and CB2 receptor expressions in vivo,
and that CB receptors may be involved in the anti-MDD effects of
KTM (9,10). Moreover, a previous study showed
that CB receptors can modulate the psychostimulant effects of KTM
in mice (22). At present, two CB
receptors, CB1 and CB2, have been discovered in the brain tissue.
The CB1 receptor is located in neurons and astrocytes, and the CB2
receptor is expressed in microglial cells and astrocytes (23). Therefore, in the present study, the
role of the CB1 receptor in KTM-induced anti-depressive effects was
explored.
Previous studies have identified that intracellular
antioxidants could be reduced in the pathological mechanisms of
MDD, meanwhile, neuronal mitochondrial function may also be
decreased (24,25). In patients with MDD, it is reported
that mitochondrial ATP production may be inhibited. Moreover,
chronic stress could inhibit the activities of mitochondrial
complex I, III and IV. This inhibition on mitochondrial complex
activity could be reversed by KTM, thus supporting the hypothesis
that KTM-induced anti-MDD therapeutic effects may act via
attenuating oxidative stress injury (15). Glutamate is an excitatory
neurotransmitter in the central nervous system, and high levels of
glutamate are commonly used to imitate oxidative stress injury
in vitro (16). In the
pathophysiological processes of MDD, cerebral glutamate
concentrations are elevated (26).
In the present study, the glutamate exposure decreased
intracellular antioxidants, including GSH, CAT and SOD2, and
generated oxidants simultaneously, such as GSSH, ROS and
mitochondrial superoxide. These findings revealed that glutamate
can induce oxidative injury in HT22 cells. During oxidative injury,
intracellular GSH can be oxidized into GSSG, therefore the
intracellular GSH/GSSG levels are often measured to assess cellular
oxidative injury. As mitochondria generate energy for cell
metabolism, mitochondrial dysfunction can cause energy (ATP) supply
deficiency for cellular activities. In the serum of patients with
MDD, the level of the antioxidant vitamin E is lower than that of
healthy individuals. In addition, patients with MMD have less
mitochondrial DNA copy numbers than that of healthy individuals
(13,14). This may explain why mitochondria
play an important role in the progression of MDD. Moreover, SOD can
be divided into three types including SOD1, SOD2 and SOD3. In
animals, SOD1 and SOD3 are in the cytoplasm and intercellular
space, respectively, and SOD2 is expressed in the mitochondria
(27). As the present study was
in vitro, SOD1 and SOD2 levels were observed. KTM exposure
induced significant upregulations in SOD2 activity and expression
compared with SOD1. Therefore, the KTM-induced effects on the
mitochondrial functions of HT22 cells were observed. It was found
that KTM notably ameliorated the glutamate-induced decrease of
mitochondrial complex I and IV activities. Generally, high levels
of mitochondrial complex I and IV indicate healthy mitochondrial
function (28). In the present
study, it was observed that the CB1 receptor antagonist AM251
partially blocked KTM-induced neuroprotection, which supported the
hypothesis that the CB1 receptor mediates KTM-induced
neuroprotection against antioxidative injury. In fact, in neurons,
the CB1 receptor is expressed in the cell membrane and
mitochondria, and CB1 receptor upregulation protects neurons and
maintains mitochondrial function (28). In the present study, it was
observed that KTM restored the mitochondrial complex activities,
upregulated SOD2 expression and activity, and also decreased
mitochondrial superoxide levels. As SOD2 is located in the
mitochondria, the aforementioned results indicated that KTM-induced
neuroprotection may occur through amelioration of neuronal
mitochondrial function, which may be mediated by the mitochondrial
CB1 receptor. However, further experiments are required to verify
cell membrane or mitochondrial CB1-mediated KTM-induced
neuroprotection.
The present study investigated the possible
therapeutic mechanisms underlying the effects of KTM on MDD, and
also showed that the mitochondrial CB1 receptor may be a potential
therapeutic target in treating MDD. However, two limitations were
identified in the present study. Firstly, a neuronal cell line was
used in the present study and it is therefore not known whether
similar findings would be discovered when using primary cultured
neurons or in vivo experiments. Secondly, a CB1 antagonist
was used in the present study and this tool uncovered the
neuroprotective mechanism of KTM from the perspective of
pharmacology. However, whether similar results can be obtained
using small interfering RNA or adenoviruses remains unknown. In
future studies, the anti-MDD mechanism of KTM will be explored
using more tools, and the findings of the present study will be
verified in animal experiments.
Finally, in the present study it was found that KTM
decreased glutamate-induced oxidative injury in HT22 neuronal
cells, and the protection was mediated via the CB1 receptor.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by The National Science
Basic Research Program of Xi'an City (grant. no. 21YXYJ0117).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
HB and CW cultured the cells and performed the
western blotting and immunocytochemistry. BH and XX performed the
statistical analysis. QG designed the study and revised the final
version of the manuscript. HB and QG confirm the authenticity of
all the raw data. All authors read and approved the final version
of the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Carvalho S, Gonçalves ÓF, Brunoni AR,
Fernandes-Gonçalves A, Fregni F and Leite J: Transcranial direct
current stimulation as an add-on treatment to cognitive-behavior
therapy in first episode drug-naïve major depression patients: The
ESAP study protocol. Front Psychiatry. 11(563058)2020.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Cosker E, Moulard M, Schmitt S,
Angioi-Duprez K, Baumann C, Laprévote V, Schwan R and Schwitzer T:
Portable light therapy in the treatment of unipolar non-seasonal
major depressive disorder: Study protocol for the LUMIDEP
randomised controlled trial. BMJ Open. 11(e049331)2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Huang Y, Wang Y, Wang H, Liu Z, Yu X, Yan
J, Yu Y, Kou C, Xu X, Lu J, et al: Prevalence of mental disorders
in China: A cross-sectional epidemiological study. Lancet
Psychiatry. 6:211–224. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Doboszewska U, Wlaź P, Nowak G,
Radziwoń-Zaleska M, Cui R and Młyniec K: Zinc in the monoaminergic
theory of depression: Its relationship to neural plasticity. Neural
Plast. 2017(3682752)2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
McCall WV, Lisanby SH, Rosenquist PB,
Dooley M, Husain MM, Knapp RG, Petrides G, Rudorfer MV, Young RC,
McClintock SM, et al: Effects of continuation electroconvulsive
therapy on quality of life in elderly depressed patients: A
randomized clinical trial. J Psychiatr Res. 97:65–69.
2018.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Nikayin S and Sanacora G: Evaluating the
role of ketamine/esketamine in the management of major depressive
disorder with suicide risk. CNS Drugs. 35:1069–1079.
2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Roca M, Del Amo AR, Riera-Serra P,
Pérez-Ara MA, Castro A, Roman Juan J, García-Toro M, García-Pazo P
and Gili M: Suicidal risk and executive functions in major
depressive disorder: A study protocol. BMC Psychiatry.
19(253)2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Nowak W, Grendas LN, Sanmarco LM, Estecho
IG, Arena ÁR, Eberhardt N, Rodante DE, Aoki MP, Daray FM, Carrera
Silva EA and Errasti AE: Pro-inflammatory monocyte profile in
patients with major depressive disorder and suicide behaviour and
how ketamine induces anti-inflammatory M2 macrophages by NMDAR and
mTOR. EBioMedicine. 50:290–305. 2019.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Pacheco DDF, Romero TRL and Duarte IDG:
Ketamine induces central antinociception mediated by endogenous
cannabinoids and activation of CB1 receptors. Neurosci Lett.
699:140–144. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Khakpai F, Ebrahimi-Ghiri M, Alijanpour S
and Zarrindast MR: Ketamine-induced antidepressant like effects in
mice: A possible involvement of cannabinoid system. Biomed
Pharmacother. 112(108717)2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Pitsillou E, Bresnehan SM, Kagarakis EA,
Wijoyo SJ, Liang J, Hung A and Karagiannis TC: The cellular and
molecular basis of major depressive disorder: Towards a unified
model for understanding clinical depression. Mol Biol Rep.
47:753–770. 2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Lindqvist D, Dhabhar FS, James SJ, Hough
CM, Jain FA, Bersani FS, Reus VI, Verhoeven JE, Epel ES, Mahan L,
et al: Oxidative stress, inflammation and treatment response in
major depression. Psychoneuroendocrinology. 76:197–205.
2017.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Chang CC, Jou SH, Lin TT, Lai TJ and Liu
CS: Mitochondria DNA change and oxidative damage in clinically
stable patients with major depressive disorder. PLoS One.
10(e0125855)2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Czarny P, Wigner P, Galecki P and
Sliwinski T: The interplay between inflammation, oxidative stress,
DNA damage, DNA repair and mitochondrial dysfunction in depression.
Prog Neuropsychopharmacol Biol Psychiatry. 80:309–321.
2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Sharma S and Akundi RS: Mitochondria: A
connecting link in the major depressive disorder jigsaw. Curr
Neuropharmacol. 17:550–562. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Maher P, van Leyen K, Dey PN, Honrath B,
Dolga A and Methner A: The role of Ca2+ in cell death caused by
oxidative glutamate toxicity and ferroptosis. Cell Calcium.
70:47–55. 2018.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Xing Y, Zhang M, Li WB, Dong F and Zhang
F: Mechanisms involved in the neuroprotection of electroacupuncture
therapy for ischemic stroke. Front Neurosci. 12(929)2018.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yang S, Hu B, Wang Z, Zhang C, Jiao H, Mao
Z, Wei L, Jia J and Zhao J: Cannabinoid CB1 receptor agonist ACEA
alleviates brain ischemia/reperfusion injury via CB1-Drp1 pathway.
Cell Death Discov. 6(102)2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Jia J, Zhang L, Shi X, Wu M, Zhou X, Liu X
and Huo T: SOD2 mediates amifostine-induced protection against
glutamate in PC12 cells. Oxid Med Cell Longev.
2016(4202437)2016.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhang Q, Hong S, Cao J, Zhou Y, Xu X, Ai M
and Kuang L: Hippocampal subfield volumes in major depressive
disorder adolescents with a history of suicide attempt. Biomed Res
Int. 2021(5524846)2021.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Cipriani A, Furukawa TA, Salanti G,
Chaimani A, Atkinson LZ, Ogawa Y, Leucht S, Ruhe HG, Turner EH,
Higgins JPT, et al: Comparative efficacy and acceptability of 21
antidepressant drugs for the acute treatment of adults with major
depressive disorder: A systematic review and network meta-analysis.
Focus (Am Psychiatr Publ). 16:420–429. 2018.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Xu W, Li H, Wang L, Zhang J, Liu C, Wan X,
Liu X, Hu Y, Fang Q, Xiao Y, et al: Endocannabinoid signaling
regulates the reinforcing and psychostimulant effects of ketamine
in mice. Nat Commun. 11(5962)2020.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Chen DJ, Gao M, Gao FF, Su QX and Wu J:
Brain cannabinoid receptor 2: Expression, function and modulation.
Acta Pharmacol Sin. 38:312–316. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Vaváková M, Ďuračková Z and Trebatická J:
Markers of oxidative stress and neuroprogression in depression
disorder. Oxid Med Cell Longev. 2015(898393)2015.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Giménez-Palomo A, Dodd S, Anmella G,
Carvalho AF, Scaini G, Quevedo J, Pacchiarotti I, Vieta E and Berk
M: The role of mitochondria in mood disorders: From physiology to
pathophysiology and to treatment. Front Psychiatry.
12(546801)2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Lener MS, Niciu MJ, Ballard ED, Park M,
Park LT, Nugent AC and Zarate CA Jr: Glutamate and
gamma-aminobutyric acid systems in the pathophysiology of major
depression and antidepressant response to ketamine. Biol
Psychiatry. 81:886–897. 2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Zelko IN, Mariani TJ and Folz RJ:
Superoxide dismutase multigene family: A comparison of the CuZn-SOD
(SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures,
evolution, and expression. Free Radic Biol Med. 33:337–349.
2002.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Ma L, Jia J, Niu W, Jiang T, Zhai Q, Yang
L, Bai F, Wang Q and Xiong L: Mitochondrial CB1 receptor is
involved in ACEA-induced protective effects on neurons and
mitochondrial functions. Sci Rep. 5(12440)2015.PubMed/NCBI View Article : Google Scholar
|