Introduction
Inherited neuromuscular disorder (IND) comprises a
large and diverse group of conditions caused by neurosystem, muscle
tissue structural and/or functional dysfunction [involving either
myofibers or corresponding extracellular matrix (ECM)] and muscle
fiber innervation (1). After the
etiology of an inherited neuromuscular disorder named Duchenne
muscular dystrophy (DMD) was first discovered in 1987(2), the understanding of the genetic basis
of these disorders has markedly progressed, with pathogenic
variants on <500 genes detected and reported to be the factors
causing neuromuscular diseases (1). Proteins encoded by these genes with
pathogenic variants may be associated with skeletal muscles, motor
neurons or neuromuscular junctions. In addition, the various
diseases caused by the defects in these proteins may have
phenotypic overlaps. Consequently, more advanced and potent
molecular genetic approaches are needed for precise typing and a
conclusive diagnosis (3), which
would ultimately be beneficial for follow-up prognostic analysis,
genetic counseling and treatment planning. The present study
discussed 3 rare and typical cases of these neuromuscular
diseases.
Collagen VI-related dystrophies (COL6-RDs) may
impair the function of the basal lamina, which is a thin layer of
specialized connective tissue surrounding myofibers. COL6-RDs,
which mainly display the features of joint contractures combined
with muscle weakness, including diverse overlapped phenotypes,
ranging from mild Bethlem muscular dystrophy to severe Ullrich
congenital muscular dystrophy. The phenotypes within these two have
been referred to as intermediate COL6-RD (4). COL6-RD can be diagnosed based on its
typical clinical manifestations and muscle imaging findings
together with muscle immunohistochemical analysis results. It is
verified by identifying the biallelic or heterozygous pathogenic
variant(s) within the collagen type VI α3 chain 1 (COL6A1),
COL6A2 or COL6A3 genes [Mendelian Inheritance in Man
(MIM) *120220, *120240 and *120250, respectively] (5-7).
COL6-RDs mostly conform to the autosomal dominant (AD) inheritance
pattern, with ~50-75% of cases resulting from the de novo
collagen VI pathogenic variants (8,9).
However, COL6-RD cases with an autosomal recessive pattern are also
reported and mostly involve nonsense or frameshift variants
(10-12).
Therefore, it is important to elucidate the clinical and genetic
classification of COL6-RD cases for the management of this
disease.
Hereditary spastic paraplegia (HSP), another
neuromuscular disorder, is a congenital neurodegenerative disorder
characterized by dieback corticospinal tract degeneration and
axonal swelling, both of which lead to gait deficiency (13). HSP has a prevalence of 1-5 cases
per 100,000 individuals. The predominant HSP signs include lower
limb spasticity and weakness. HSP is associated with at least 80
genes, and its most common subtype is referred to as spastic
paraplegia type IV (SPG4), also known as SPAST-HSP. SPG4 is induced
by Spastin (SPAST) gene mutations (MIM *604277) (13,14),
which accounts for 20 and 40% of simplex HSPs and AD-HSPs,
respectively (15-17).
SPG4 shows the typical features of insidious progression of gait
spasticity in both lower extremities, with more than half of the
patients having weak lower extremities together with decreased
ankle vibration sense (18). The
different onset ages and phenotypic manifestation are the main
challenges encountered with SPG4 in clinical practice (19).
Dystrophinopathies represent a wide-spectrum
X-linked muscle disease group, including DMD and Becker muscular
dystrophy, along with dilated cardiomyopathy related to DMD with
increasing severity (20). Of
note, the most serious conditions include delayed motor milestones
and progressive muscle disorders, which ultimately result in
mortality around puberty (21). In
~95% of cases, mutational analysis of the DMD gene (MIM
*300377) enables a definitive diagnosis (21). Although a variety of novel and
promising therapies are on the horizon, with a few having entered
clinical trials as well, the treatment approach currently in use is
symptomatic treatment. Therefore, genetic diagnosis, screening and
counseling become particularly meaningful in this regard.
In the present study, 3 cases with the above
neuromuscular disorders were analyzed and diverse genetic and
clinical examinations were performed. The clinical phenotypes and
genetic variations in these patients are distinctive and worthy of
attention.
Materials and methods
Subjects
Approval of the present study was granted by the
Ethics Committee of Shijiazhuang Obstetrics and Gynecology Hospital
(Shijiazhuang, China; no. 2021-0068).
A total of three cases with myopathy-like symptoms
who visited our centers between January 2018 and December 2021 were
enrolled in the present study and analyzed retrospectively. A
thorough clinical evaluation with/without X-ray imaging and
magnetic resonance imaging (MRI) examination was performed.
Subsequently, peripheral blood was extracted from the probands and
their parents to conduct a genetic evaluation.
Each participant or their guardians as applicable
provided written informed consent before participating in the
present study. Every participant or their guardians consented to
genetic analysis and publication of the results. The protocols of
the present study were in line with the Declaration of Helsinki
from 1964, its associated amendments and related ethical
criteria.
DNA isolation
The genomic DNA (gDNA) was collected in the samples
with the QIAamp DNA Blood Mini-Kit (Qiagen GmbH). Thereafter, the
quality of the extracted DNA was confirmed on 1% agarose gel using
the Qbit DNA Assay Kit in a Qubit 2.0 Fluorometer (Thermo Fisher
Scientific, Inc.).
Whole-exome sequencing (WES)
WES was conducted according to previous reports
(22-24).
In brief, the exonic sequences were enriched with the use of the
Sure Select Human Exon Sequence Capture Kit (Agilent Technologies,
Inc.). Thereafter, Illumina DNA Standards and Primer Premix Kit
(Kapa Biosystems; Roche Diagnostics) were employed for quantifying
sequencing libraries, which was followed by massively parallel
sequencing using the Illumina Novaseq6000 platform (Illumina,
Inc.). After sequencing and screening low-quality reads, the
high-quality reads (quality level Q30 >89%) were compared
against the human reference genome[hg19/GRCh37] using
Burrows-Wheeler Aligner tool. The GATK software was then utilized
to identify those potential pathogenic variants (https://software.broadinstitute.org/gatk/). The
variants in the sample sequences were identified by aligning the
sequences against those in the National Center for Bioinformatics
(NCBI) database (https://www.ncbi.nlm.nih.gov/) using the ‘NCBI
Reference Sequence’ in Chromas v2.33 (Technelysium Pty Ltd.). In
line with the American College of Medical Genetics and Genomics
(ACMG) (25) guidelines and
related databases [1000 g2015aug_eas (https://www.internationalgenome.org/); ExAC_EAS
(http://exac.broadinstitute.org);gnomAD_exome_EAS
(http://gnomad.broadinstitute.org/);
Human Gene Mutation Database (HGMD) (professional version 2019.4)
(hgmd.cf.ac.uk/ac/index); ClinVar
(https://www.ncbi.nlm.nih.gov/clinvar/) with the
Enliven® Variants Annotation Interpretation system
(Berry Genomics)], detected variants were assessed for their
pathogenicity. The REVEL score was used to predict the
pathogenicity of missense variation.
Variant verification
Verification through Sanger sequencing was carried
out using the 3500DX Genetic Analyzer (Applied Biosystems; Thermo
Fisher Scientific, Inc.) to validate suspected diagnostic variants.
By adopting MEGA7 (http://www.megasoftware.net), the evolutionary
conservatism of amino acids was analyzed under the influence of
certain missense variants using default parameters.
To determine the gDNA copy number of the proband and
the proband's mother at the variant site in the COL6A3 gene
in Case 1, a set of copy number assays based on quantitative
fluorescence PCR (qfPCR) were designed and conducted. Total RNA was
extracted from tissue using the RNA kit II (cat. no. R6934; Omega
Bio-Tek, Inc.) according to the manufacturer's instructions. The
concentration and quality of total RNA were checked using a
NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Inc.).
The first-strand cDNA was synthesized using MonScript™
RTIII Super Mix with dsDNase (two-Step) and oligonucleotide (dT)
primers. RT-qPCR analysis of mRNA using the following primers was
performed on the ABI 7500 FAST system using TB Green®
Premix Ex Taq™ GC (Takara Biotechnology Co., Ltd.). The
primers used in this study were as follows: COL6A3-exon19
forward, 5'-CCTGTCCGCCTTATTCCCTC-3' and reverse,
3'-AAGGTCACACCTGCTGCAAT-5' (amplicon size, 167 bp) and β-globin
forward, 5'-ACACAACTGTGTTCACTAGC-3' and reverse,
3'-CAACTTCATCCACGTTCACC-5' (amplicon size, 110 bp). The PCR
conditions were as follows: Initial denaturation at 95˚C for 30
sec, followed by 40 cycles of 10 sec at 95˚C and 30 sec at the
annealing temperature of 60˚C. Relative gene expression levels were
calculated using the 2-∆∆Cq method (26). The outcome data of the qfPCR were
reported as the mean ± standard deviation.
Structural analysis
Wild-type (WT) and spastin: p.T389K mutant models of
Spast protein segments were modeled using the Alphafold program
(https://alphafold.ebi.ac.uk/entry/E5KRP5). Molecular
dynamics (MD) predictions were obtained and analyzed using GROMACS
(version 2020.6) (27). MD
simulations (60 nsec each) were conducted for the SPAST-WT and
SPAST-T389K models. In addition, hydrogen atoms and C- and
N-terminal patches were added to the models by applying the
CHARMM36 force field (28). Later mutant or WT protein
structures were enclosed within the water-containing cubic box,
with a distance of ≥1.0 nm away from box edges. Neutralization was
completed using Cl- and Na+ ions. MD
simulations were performed for a 60-nsec period at 300 K following
energy equilibration and minimization. Furthermore, the following
GROMACS distribution programs were utilized for generating the MD
trajectories: gmxrms, gmxrmsf, gmx gyrate, gmx sasa and gmx hbond.
The root-mean-square fluctuation, the radius of gyration,
root-mean-square deviation, solvent accessible surface area and
hydrogen bond (H-bond) number values were generated.
Statistical analysis
Each experiment was carried out three times. Only
representative data are presented. Statistical analysis was
completed with Microsoft Excel 2016 (Microsoft Corporation) and
GraphPad Prism v.6 (GraphPad Software; Dotmatics). ANOVA
(parametric) test was applied in determining significance of
difference, followed by Tukey's post hoc multiple comparisons test
in the event of a significant result. P<0.05 was considered to
indicate statistical significance.
Results
Clinical manifestations and family
history. Report of case 1
A male, born to non-consanguineous parents in
October 2014, had gait abnormalities appearing since learning to
walk. In May 2016, the patient was admitted to the Prenatal
Diagnosis Center of Shijiazhuang Obstetrics and Gynecology Hospital
for examination. The X-ray revealed ‘left hip dislocation, right
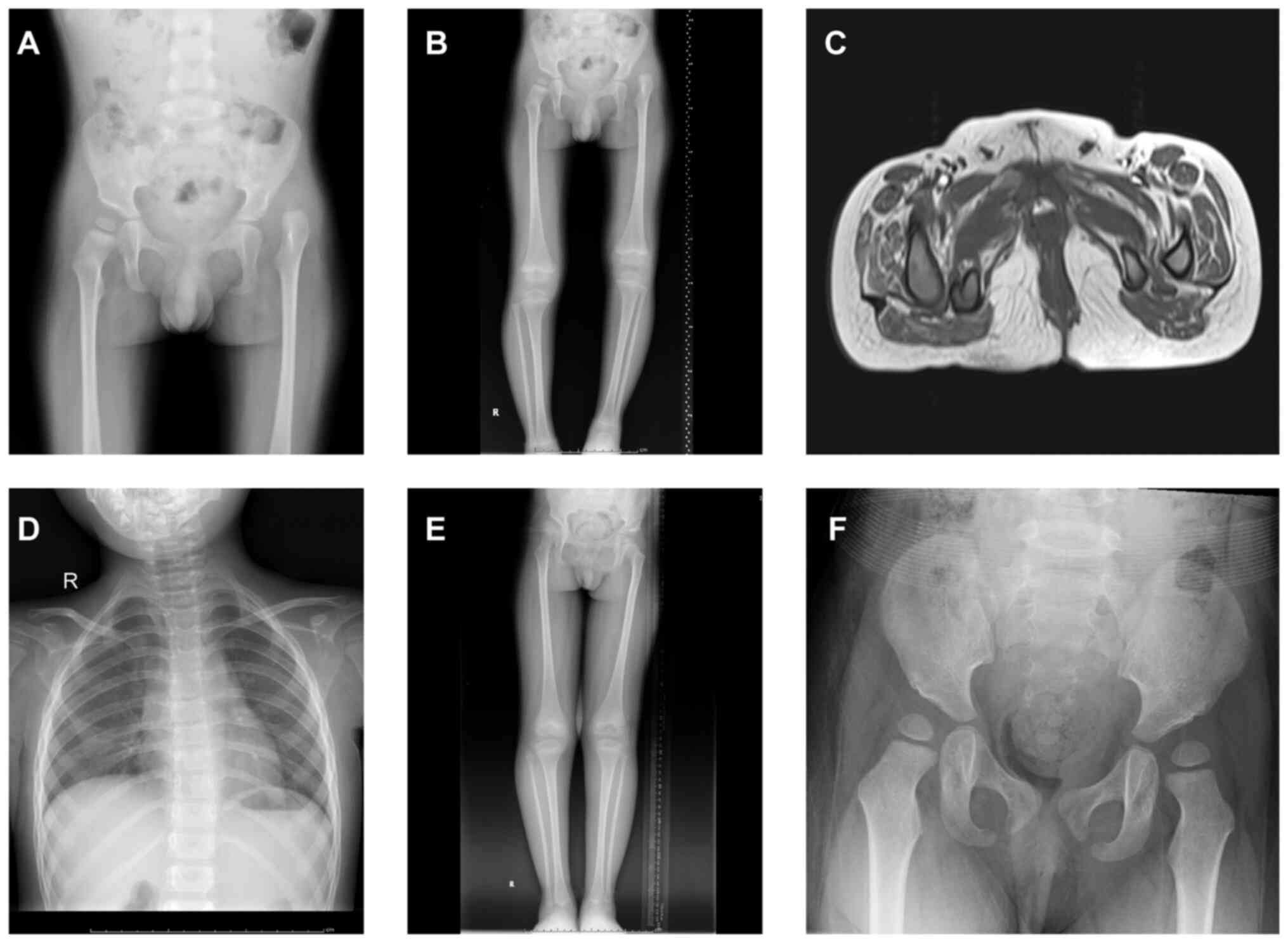
hip subluxation and dysplasia of the right acetabulum’ (Fig. 1A). Physical and X-ray examination
revealed slight muscle weakness and unequal length of the lower
limbs (Fig. 1B). MRI of the root
of the thigh revealed abnormal signals in the center and normal
margins (Fig. 1C). With growth,
mild scoliosis appeared in the year 2019 (Fig. 1D). In May 2017, incision reduction
of left hip dislocation, salter osteotomy of the pelvis and
internal fixation of the proximal femur were performed, and the
postoperative healing was satisfactory (Fig. 1E). Follow-up until the age of 8
years revealed that the patient had not developed any respiratory
or dermatological symptoms.
Report of case 2. A male, born in September
2017, had been unable to walk since birth. The physical examination
conducted in the Prenatal Diagnosis Center of Shijiazhuang
Obstetrics and Gynecology Hospital in September 2019 revealed
significantly increased muscle tension, poor activity and joint
contracture in both lower limbs. X-ray examination demonstrated
dysplasia of the hip joints (Fig.
1F). The patient's parents reported cognitive impairment
evidenced by intellectual disability, dysfunction of the bladder
sphincter evidenced by urinary urgency and urinary incontinence
symptoms in the patient at 2 years old. Continuous follow-up of
this case is necessary to see if its recent clinical phenotype is
consistent with the clinical diagnosis of SPG4.
Report of case 3. A male, born in August
2017, presented to the Prenatal Diagnosis Center of Shijiazhuang
Obstetrics and Gynecology Hospital due to muscle weakness and gait
instability. The laboratory evaluation conducted in February 2019
(multiple times) indicated that the patient's serum creatine
phosphokinase (CK) concentration was significantly elevated (9-13
times the normal level). A recent follow-up confirmed that the
patient frequently experienced an unsteady gait and falls, had a
typical presentation of proximal muscle weakness and a high serum
CK concentration, although the patient had not yet developed any
evident indication of calf hypertrophy. The patient was clinically
considered as a case of dystrophinopathy. Calf muscle
pseudohypertrophy is the typical clinical feature of
dystrophinopathy. Attention should be paid to calf muscle
pseudohypertrophy, and dilated cardiomyopathy that may develop in
the proband during development (clinical indication images not
presented, as per the wish of the affected family).
Genetic and relative structural
findings. Case 1
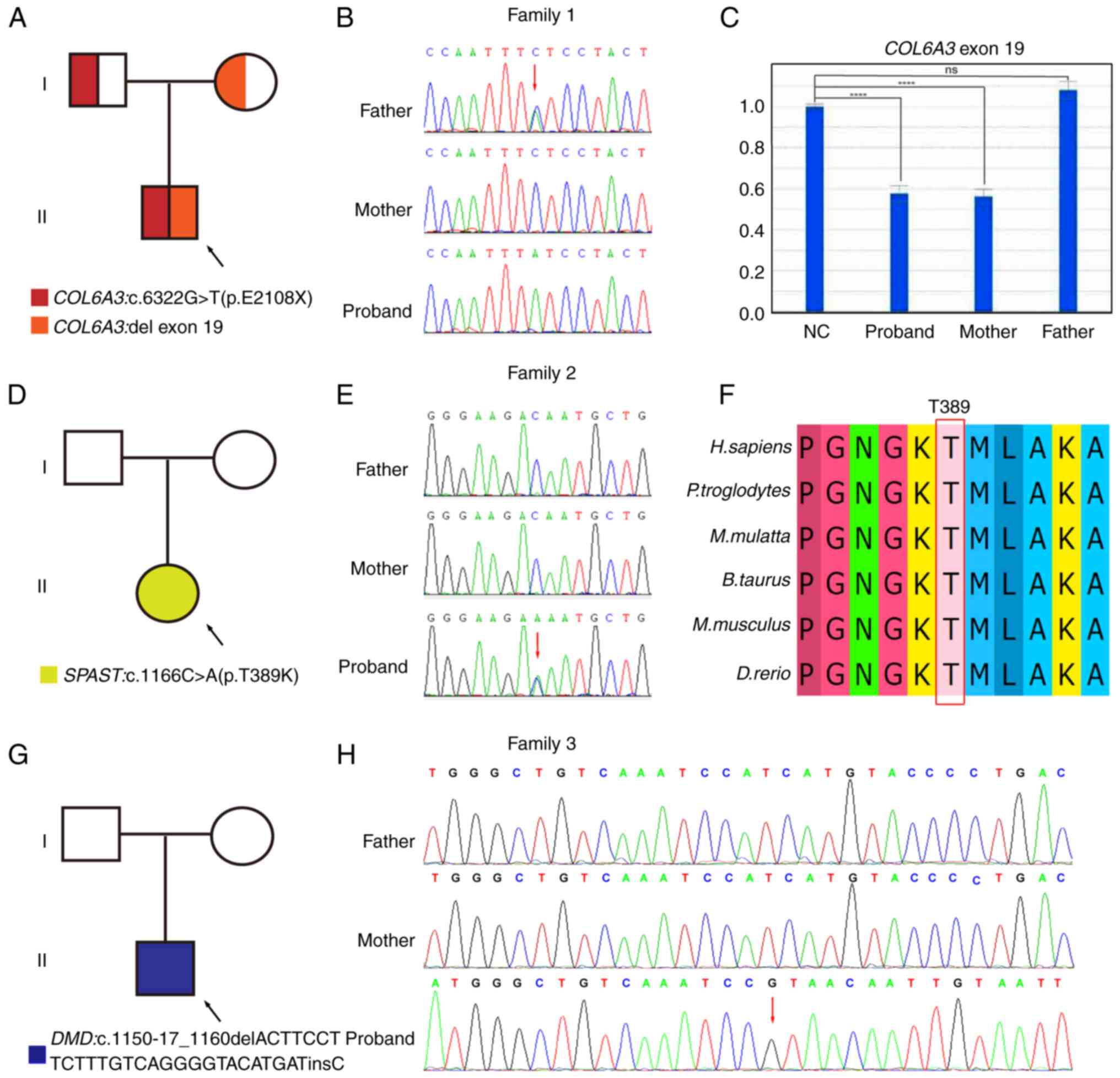
Fig. 2A displays a
pedigree chart along with the carrier status for each variant in
this family. The WES detected a homozygous nonsense variation of
the COL6A3 gene-(NM_004369; exon19) c.6322G>T (p.E1208*).
According to further Sanger sequencing-based familial verification,
the father was heterozygous, while the mother was a WT at this site
(Fig. 2B). To provide a clear
determination of this special case, a qfPCR-based verification of
the COL6A3 gDNA copy number analysis of the flanking region
of this site was performed as described above. The result indicated
that both the mother and the proband were heterozygous carriers of
a one-copy loss at this region, while the father was WT (Fig. 2C). Accordingly, this proband was
ultimately recognized as being affected by a compound heterozygous
variation in COL6A3 comprising one allelic point mutation
and one allelic copy loss, which is being newly reported in the
present study.
Case 2. Fig.
2D displays a pedigree diagram along with the carrier status
for each variant. WES identified the new missense variant of the
SPAST gene (NM_014946; exon8) c.1166C>A (p.T389K). Sanger
sequencing indicated that this variant was de novo, i.e.,
both of the parents of the proband were WT (Fig. 2E). The MEGA7 result demonstrated
that the T389 residue of spastin protein showed a high conservation
degree across species (Fig. 2F).
To investigate the intramolecular impact resulting from this
missense variant, structural analysis and MD simulations were
performed. The structural result demonstrated that the T389K
variant affected H-bonding within the amino acids inside a protein.
Particularly, for the WT, T389 formed H-bonds along with the K393
as well as D441 residues. However, in the T389K mutant, K389 formed
an H-bond with K393 (Fig. 3A-D;
Fig. 3B and D depict the local amplifications of
Fig. 3A and C, respectively). The MD results revealed
that the K389 residue in the mutant model generated more H-bonds
with other amino acids within that protein in comparison with WT
residue T389 (Fig. 3E). In
addition, the total number of H-bonds inside the modeled protein
segment during 60 nsec was greater in the WT than in the T389K
mutant (Fig. 3F). Furthermore, the
T389K variant resulted in a change in the secondary structure
around the 390th residue (Fig. 3G and H). Specifically, in the WT, the secondary
structure at this position alternated between α-helix and TURN,
with the domination of TURN. However, in the mutant, the secondary
structure alternated between α-helix and TURN, with the domination
of α-helix. Finally, the T389K variant resulted in greater changes
in the protein structure (Fig.
3I), increased amino acid flexibility in the protein (Fig. 3J), decreased protein compactness
(Fig. 3K) and increased surface
area accessible to the protein solvent (Fig. 3L).
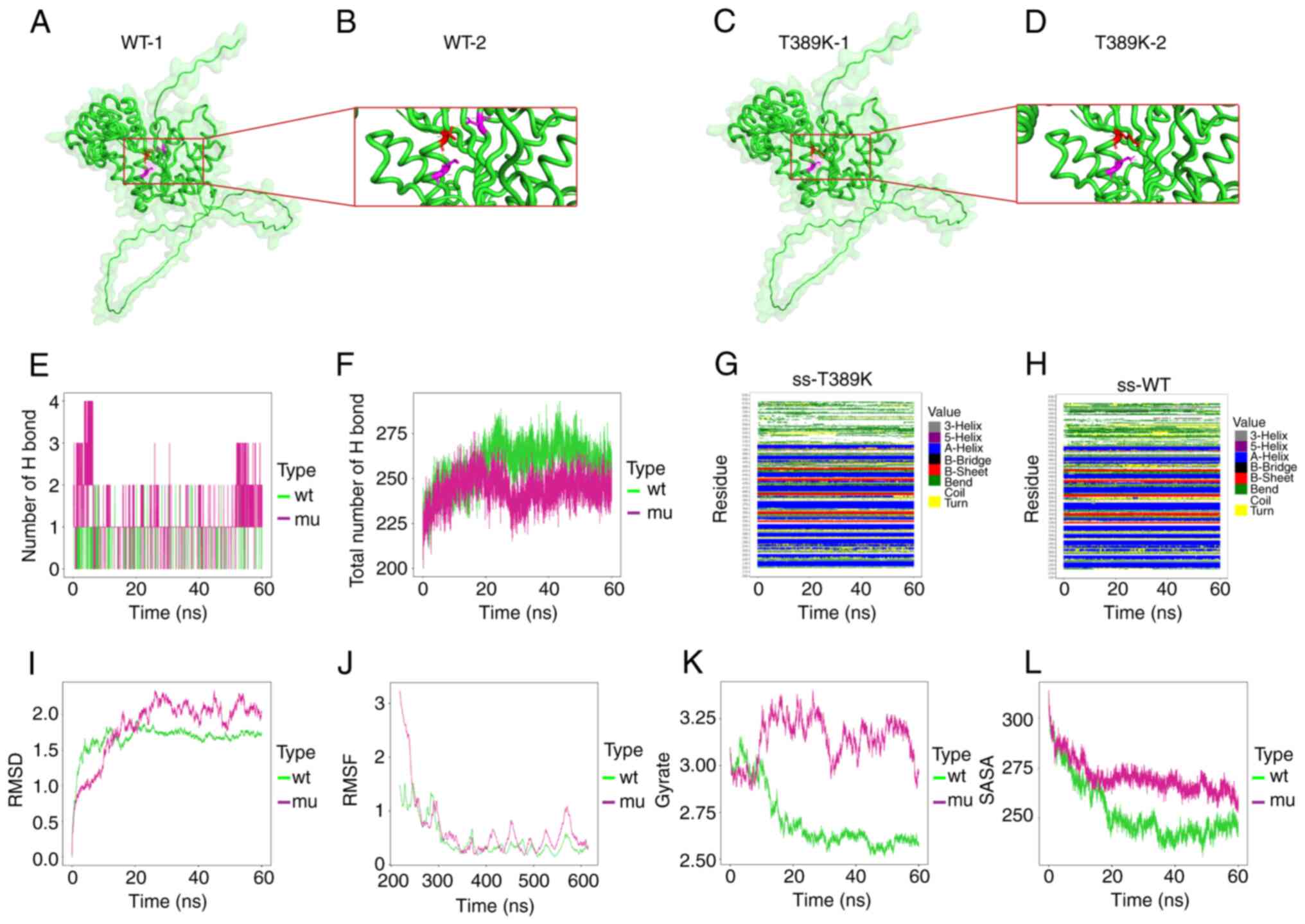 | Figure 3Results of the structure and MD
analyses on SPAST: c.1166C>A(p.T389K) variation. (A) The
WT structure of the SPAST protein and (B) enlarged image of the
segment containing the T389 residue. (C) The mutant structure of
SPAST protein and (D) enlarged segment containing the variant K389
residue. (E) The H-bond number generated in target amino acid (T389
or K389) and the other residues. (F) The total number of hydrogen
bonds in the WT and mutant models, respectively. (G and H)
Comparison of the local secondary structure data between (G) the
T389K mutant and (H) WT. (I) RMSD: Parameter indicating
heterogeneity in the two structures. (J) RMSF: Similar to RMSD,
although rather than representing the heterogeneity in the position
with time for the entire structure, this measure calculates the
flexibility of a single residue or the extent to which one specific
residue migrates (fluctuates) in the simulation process. (K)
Gyrate: Measure of the structural displacement of a protein atom
with its shared mass center during simulation, which offers
integrated data regarding protein tightness with time. (L) SASA:
Parameter indicating the surface exposed within a protein
structure, which can be accessed by the solvent molecules. WT,
wild-type; mu, mutant; ns, nanoseconds; SPAST, Spastin; RMSD, root
mean square deviation; RMSF, root mean square fluctuation; SASA,
solvent accessible surface area. |
Case 3. The pedigree diagram and the carrying
status of each variant are presented in Fig. 2G. WES identified the following
insertion and deletion (InDel) variation in the DMD gene:
(NM_004006; exon11)
c.1150-17_1160delACTTCCTTCTTTGTCAGGGGTACATGATinsC. Accordingly, it
was preliminarily concluded that the mutation would affect the
normal splicing of this gene. Familial validation demonstrated that
this variation was de novo (Fig. 2H).
Table I lists the
main clinical and genetic indications of the 3 cases described
above.
 | Table IMain clinical and genetic indications
of the 3 cases. |
Table I
Main clinical and genetic indications
of the 3 cases.
| Case no. | Sex | Age, years | Major clinical
features | Gene | Genomic
variation | Peptide
alteration | Frequency in three
databasesa | Prediction by
REVELb |
HGMD/ClinVarc rating | Pathogenicity
ratingd
(evidence) |
|---|
| 1 | M | 4 | Hip dislocation;
hip subluxation; acetabulum dysplasia; scoliosis | COL6A3 | c.6322G> T | p.E2108 X | 0; 0; 0 | / | / | LP (PVS1+PM2) |
| | | | | COL6A3 | Exonic one-copy
loss | Uncertain | 0; 0; 0 | / | / | LP (PVS1+
PM2+PM3) |
| 2 | F | 2 | Increased muscle
tension; poor activity; joint contracture; hip dysplasia | SPAST | c.1166C>A | p. T389K | 0; 0; 0 | D | / | LP (PS2+
PM2+PP3) |
| 3 | M | 2 | Delay of motor
development; calf hypertrophy | DMD |
c.1150-17_1160delACT TCCTTCTTTGTCAG
GGGTACATGATinsC | Splicing error | 0; 0; 0 | / | / | P (PVS+
PS2+PM2) |
Discussion
IND attracts much attention in the research field
due to its wide variability along with severe clinical impact
(29). Although hundreds of
disease-causing genes have been identified, only a small number of
effective treatments have been developed so far, and more genetic
research is necessary, particularly in the area of
genotype-phenotype association (30). Whether the knowledge of new
mutations could offer potential treatment options requires further
investigation. Reporting additional cases associated with these
mutations may help identify genotype-phenotype correlations and
lead to clinical trials in the future. Although gene therapy has
been developed for certain rare genetic diseases, there are still
several challenges in gene therapy. The first challenge in treating
hereditary heterogeneous diseases is to identify the pathogenic
genes. New mutations identified in this study are potential targets
for future gene therapy. In the present study, 3 cases, including
Bethlem muscular dystrophy, AD-HSP and dystrophinopathy, were
analyzed clinically and genetically, and their specific features
were described. All variants identified in the three cases were
divided into pathogenic (P) and likely pathogenic (LP) variants.
According to the ACMG criteria (25), the COL6A3:c.6322G>T (p.E1208*)
variant was categorized as LP, with the evidence of PVS1+PM2, the
SPAST: c.1166C>A (p.T389K) variant was classified as LP, with
the evidence of PS2+PM2+PP3, and the DMD gene:
c.1150-17_1160delACTTCCTTCTTTGTCAGGGGTACATGATinsC variant was
categorized as P, with the evidence of PVS1+PS2+PM2.
In Case 1, the initial manifestation of the proband
was hip dislocation. The physical examination did not reveal any
significant hypotonia or joint contracture. Furthermore, the MRI
results were only marginally suggestive. The patient was clinically
considered as a case of Bethlem muscular dystrophy. The main
clinical features of this disease include joint contractures and
respiratory dysfunction caused by muscle weakness.
The follow-up until the age of 8 years demonstrated
that the patient had not developed any respiratory or skin
involvement thus far. Attention should be paid to muscle weakness,
respiratory and dermatological symptoms that may develop in the
proband during development. Genetic evaluation with WES initially
identified a homozygous COL6A3 stop-gain variation
designated as c.6322G>T (p.E1208*), which was a rare occurrence
(10,31). A noteworthy observation in this
case, which was consistent with a previous report, was that the
pathogenic variants responsible for recessive COL6-RD tended to be
nonsense or frameshift variants (12). Interestingly, the proband's father
carried the heterozygous variant, while the mother was of the WT,
which may allow for the hypothesis that this variation in this
patient may have been due to a de novo mutation in the
oocyte, a uniparental isodisomy, maternal mosaicism (32), or, more likely, maternal copy
number loss (4,33,34).
A simple qfPCR assay revealed that both the patient and the mother
carried one-copy-loss of the COL6A3: exon 19 segment.
Bethlem muscular dystrophy is caused by mutation in the
COL6A3 gene, and it was inferred that this patient should be
clinically classified as a case of mild Bethlem muscular dystrophy.
However, due to methodological limitations, the specific breakpoint
causing the deletion remained unknown, although it did not
influence the subsequent processing. Further verification as to
whether the symptoms of the patient in Case 1 are caused by the
mutation at this site requires further investigation and the lack
of this verification is a limitation of the present study.
Genetically, the family continues to have a 25% probability of
conceiving another child suffering from this condition. Therefore,
evidence-based counseling and fertility guidance must be provided
to the parents. In addition, attention should be paid to subsequent
symptoms that may develop in the proband during development.
SPAST-HSP (SPG4) covers as many as 40% of all AD-HSP
cases (17,35). For the patient in Case 2, clinical
indications were consistent with previous reports (13,14,36,37).
At the level of large cohort clinical studies, neuroimaging and
structural biology, researchers have recently tried to explain some
of the hallmarks of SPG4, such as the ‘double peaks’ of the onset
age and substantial phenotypic variability (18,38,39).
Parodi et al (18)
indicated that missense variations and truncated mutations of the
SPAST gene affected protein function at varying degrees,
which may be responsible for the younger age of onset in the former
and the older age of onset in the latter. Due to interest in the
missense variant SPAST: c.1166C>A (p.T389K) detected in
Case 2 of the present study, structural analysis was performed. The
SPAST-encoded Spastin represents the microtubule-severing
enzyme combining with and generating internal breaks within the
microtubule lattice and is, therefore, being increasingly
considered a key modulator of the microtubule cytoskeleton in
neuron biology (40).
Structurally, microtubule severing is performed mainly in the
region between the 228 to 616 residues, and the region between 382
and 389 residues constitutes the ATP binding site, which is exactly
where the p.T389K is located (41). According to the results of the
structural and MD analyses, the T389K variant most likely
significantly affected both local and global H-bond formation,
thereby leading to altered protein stability, while also disrupting
the desired secondary structure of the protein, disrupting the
binding of the protein to ATP. However, this hypothesis requires to
be validated through solid evidence from in vitro functional
experiments.
Dystrophinopathy is probably the most well-studied
neuromuscular disease to date, with a prevalence of ~1/3,500 in
male newborns and one of the few diseases to reach the gene therapy
stage (20). The DMD gene
is located on chromosome Xp21 and is the only recognized pathogenic
gene related to this disease. This gene is known for its large
span, diverse mutation types and high proportion of de novo
variants (42). The association
between various DMD variants and the specific subtypes of
dystrophinopathy, which is referred to as the genotype-phenotype
association, has been widely discussed in the literature (43). In Case 3 of the present study, a
de novo deletion-insertion variant was detected in the
proband, which spanned the splice site and involved both exonic and
intronic regions. This variant most likely affects the splicing
process, although its ultimate effect on protein formation remains
to be elucidated and validated. Immunohistochemical analysis of
dystrophin in patient tissue and in vitro functional study
by cell transfection should be performed to understand the role of
the variant in the entire process of disease development. The
current evidence suggests that this variant is de novo, and
gonadal mosaicism in the proband's mother cannot be ruled out.
Therefore, it is prudent to recommend targeted prenatal diagnosis
for any subsequent pregnancies in this family. The same suggested
measure applies to Case 2. The multiplex ligation-dependent probe
amplification (MLPA) method was not used and is a limitation of the
present study. MLPA helps to detect large fragment
deletions/duplications (multiple consecutive exon or intron
deletions/duplications). However, MLPA may lead to a missed
diagnosis of single-exon deletion, micro-deletion,
micro-duplication and point variations. In the present study, WES
identified an InDel variation in exon 11 of the DMD gene.
MLPA may have led to a missed diagnosis of this microinsertion and
deletion variant.
In conclusion, the present study meticulously
indexed the clinical manifestations in three patients, along with
the related family members. WES detected the diagnostic variants of
the COL6A3, SPAST and DMD genes in these three
patients. In total, four variations were found and verified. Among
all variants, to the best of our knowledge, three were identified
for the first time, namely COL6A3 (NM_004369; exon19):
c.6322G>T (p.E2108X) in addition to one-copy loss of
COL6A3, exon19; SPAST (NM_014946; exon8): c.1166C>A
(p.T389K); and DMD (NM_004006; exon11):
c.1150-17_1160delACTTCCTTCTTTGTCAGGGGTACATGATinsC. The results of
the present study contributed to genetically diagnosing IND in
three cases. These findings extend the spectrum of mutations and
the understanding of the genotype-phenotypic associations in these
diseases.
Acknowledgements
Not applicable.
Funding
Funding: The present study was funded by the Medical Scientific
Research Project of the Health Commission of Hebei Province (grant
no. 20231661), the Medical Scientific Research Project of the
Health Commission of Hebei Province (grant no. 20231649) and the
Science and Technology Research and Development Program of Langfang
(grant no. 2022013136).
Availability of data and materials
The sequencing data generated in the present study
may be found in the Figshare repository at the following URLs:
[COL6A3 (https://figshare.com/articles/dataset/COL6A3_NM_004369_exon19_c_6322G_T_p_E1208_/21548523);
SPAST (https://figshare.com/articles/dataset/SPAST_NM_014946_exon8_c_1166C_A_p_T389K_/21548727);
and DMD (https://figshare.com/articles/dataset/WES_identified_an_InDel_insertion_and_deletion_variation_in_the_DMD_gene/21548733)].
The other data generated in the present study are included in the
figures and/or tables of this article.
Authors' contributions
YZL and JZ made substantial contributions to
conception and design. WQC and YFY were involved in drafting the
manuscript and performed the statistical analysis. KNH, DLS and QBH
performed the experiments and agreed to be accountable for all
aspects of the work in ensuring that questions related to the
accuracy were investigated and resolved. SWW, YML and CYH were
involved in revising the manuscript critically for important
intellectual content and analyzed the data. YZL gave final approval
of the version to be published. All authors have read and approved
the final version of the manuscript. YZL and JZ confirm the
authenticity of all the raw data.
Ethics approval and consent to
participate
The current study was approved by the Ethics
Committee in Shijiazhuang Obstetrics and Gynecology Hospital
(Shijiazhuang, China; approval no. 2021-0068). Each subject or the
corresponding guardian provided informed consent before
participating in this work. Every participant or their guardians
consented to genetic analysis and publication of the results. Every
procedure was conducted in accordance with the Declaration of
Helsinki from 1964, related amendments and associated ethical
guidelines.
Patient consent for publication
Each subject or corresponding guardian provided
informed consent for publication of patient information, genetic
data and images in this manuscript. Clinical indication images are
not presented for case 3, as per the wish of the affected
family.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Dowling JJ, Weihl CC and Spencer MJ:
Molecular and cellular basis of genetically inherited skeletal
muscle disorders. Nat Rev Mol Cell Biol. 22:713–732.
2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Koenig M, Hoffman EP, Bertelson CJ, Monaco
AP, Feener C and Kunkel LM: Complete cloning of the Duchenne
muscular dystrophy (DMD) cDNA and preliminary genomic organization
of the DMD gene in normal and affected individuals. Cell.
50:509–517. 1987.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Butterfield RJ: Congenital muscular
dystrophy and congenital myopathy. Continuum (Minneap Minn).
25:1640–1661. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Foley AR, Mohassel P, Donkervoort S,
Bolduc V and Bönnemann CG: Collagen VI-related dystrophies. In:
GeneReviews® [Internet]. Adam MP, Feldman J, Mirzaa GM,
Pagon RA, Wallace SE, Bean LH, Gripp KW and Amemiya A (eds).
University of Washington, Seattle, WA, 1993.
|
|
5
|
Pepea G, Bertini E, Bonaldo P, Bushby K,
Giusti B, de Visser M, Guicheney P, Lattanzi G, Merlini L, Muntoni
F, et al: Bethlem myopathy (BETHLEM) and Ullrich scleroatonic
muscular dystrophy: 100th ENMC international workshop, 23-24
November 2001, Naarden, The Netherlands. Neuromuscul Disord.
12:984–993. 2002.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Briñas L, Richard P, Quijano-Roy S,
Gartioux C, Ledeuil C, Lacène E, Makri S, Ferreiro A, Maugenre S,
Topaloglu H, et al: Early onset collagen VI myopathies: Genetic and
clinical correlations. Ann Neurol. 68:511–520. 2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Bönnemann CG: The collagen VI-related
myopathies: Muscle meets its matrix. Nat Rev Neurol. 7:379–390.
2011.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Allamand V, Merlini L and Bushby K:
Consortium for Collagen VI-Related Myopathies. 166th ENMC
international workshop on collagen type VI-related myopathies,
22-24 May 2009, Naarden, The Netherlands. Neuromuscul Disord.
20:346–354. 2010.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Allamand V, Briñas L, Richard P, Stojkovic
T, Quijano-Roy S and Bonne G: ColVI myopathies: Where do we stand,
where do we go? Skelet Muscle. 1(30)2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Foley AR, Hu Y, Zou Y, Columbus A,
Shoffner J, Dunn DM, Weiss RB and Bönnemann CG: Autosomal recessive
inheritance of classic Bethlem myopathy. Neuromuscul Disord.
19:813–817. 2009.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zamurs LK, Idoate MA, Hanssen E,
Gomez-Ibañez A, Pastor P and Lamandé SR: Aberrant mitochondria in a
Bethlem myopathy patient with a homozygous amino acid substitution
that destabilizes the collagen VI α2(VI) chain. J Biol Chem.
290:4272–4281. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Camacho Vanegas O, Bertini E, Zhang RZ,
Petrini S, Minosse C, Sabatelli P, Giusti B, Chu ML and Pepe G:
Ullrich scleroatonic muscular dystrophy is caused by recessive
mutations in collagen type VI. Proc Natl Acad Sci USA.
98:7516–7521. 2001.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Hedera P: Hereditary spastic paraplegia
overview. In: GeneReviews® [Internet]. Adam MP, Feldman
J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW and Amemiya A
(eds). University of Washington, Seattle, WA, 1993.
|
|
14
|
Parodi L, Rydning SL, Tallaksen C and Durr
A: Spastic paraplegia 4. In: GeneReviews® [Internet].
Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp
KW and Amemiya A (eds). University of Washington, Seattle, WA,
1993.
|
|
15
|
Erichsen AK, Koht J, Stray-Pedersen A,
Abdelnoor M and Tallaksen CM: Prevalence of hereditary ataxia and
spastic paraplegia in southeast Norway: A population-based study.
Brain. 132:1577–1588. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Fei QZ, Tang WG, Rong TY, Tang HD, Liu JR,
Guo ZL, Fu Y, Xiao Q, Wang XJ, He SB, et al: Two novel mutations in
the Spastin gene of Chinese patients with hereditary spastic
paraplegia. Eur J Neurol. 18:1194–1196. 2011.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Ruano L, Melo C, Silva MC and Coutinho P:
The global epidemiology of hereditary ataxia and spastic
paraplegia: A systematic review of prevalence studies.
Neuroepidemiology. 42:174–183. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Parodi L, Fenu S, Barbier M, Banneau G,
Duyckaerts C, Tezenas du Montcel S, Monin ML, Ait Said S, Guegan J,
Tallaksen CME, et al: Spastic paraplegia due to SPAST mutations is
modified by the underlying mutation and sex. Brain. 141:3331–3342.
2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Méreaux JL, Banneau G, Papin M, Coarelli
G, Valter R, Raymond L, Kol B, Ariste O, Parodi L, Tissier L, et
al: Clinical and genetic spectra of 1550 index patients with
hereditary spastic paraplegia. Brain. 145:1029–1037.
2022.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Darras BT, Urion DK and Ghosh PS:
Dystrophinopathies. 2000 Sep 5 (Updated 2022 Jan 20). In: Adam MP,
Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJ, Gripp KW and
Amemiya A (eds). GeneReviews® [Internet]. Seattle (WA):
University of Washington, Seattle, 1993.
|
|
21
|
Flanigan KM: Duchenne and Becker muscular
dystrophies. Neurol Clin. 32:671–688, viii. 2014.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zhang J, Hu H, Mu W, Yu M, Chen W, Mi D,
Yang K and Guo Q: Case report: Exome sequencing identified a novel
compound heterozygous variation in PLOD2 causing bruck syndrome
type 2. Front Genet. 12(619948)2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Huang YX, Gao CY, Zheng CY, Chen X, Yan
YS, Sun YQ, Dong XY, Yang K and Zhang DL: Investigation of a novel
LRP6 variant causing autosomal-dominant tooth agenesis. Front
Genet. 12(688241)2021.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yang F, Yang RJ, Li Q, Zhang J, Meng YX,
Liu XJ and Yao YF: Whole-exome sequencing facilitates the
differential diagnosis of Ehlers-Danlos syndrome (EDS). Mol Genet
Genomic Med. 10(e1885)2022.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Rakhshani H, Dehghanian E and Rahati A:
Enhanced GROMACS: Toward a better numerical simulation framework. J
Mol Model. 25(355)2019.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Soteras Gutiérrez I, Lin FY,
Vanommeslaeghe K, Lemkul JA, Armacost KA, Brooks CL III and
MacKerell AD Jr: Parametrization of halogen bonds in the CHARMM
general force field: Improved treatment of ligand-protein
interactions. Bioorg Med Chem. 24:4812–4825. 2016.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Pelin K and Wallgren-Pettersson C: Update
on the genetics of congenital myopathies. Semin Pediatr Neurol.
29:12–22. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Scoto M, Finkel R, Mercuri E and Muntoni
F: Genetic therapies for inherited neuromuscular disorders. Lancet
Child Adolesc Health. 2:600–609. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Gualandi F, Urciuolo A, Martoni E,
Sabatelli P, Squarzoni S, Bovolenta M, Messina S, Mercuri E,
Franchella A, Ferlini A, et al: Autosomal recessive Bethlem
myopathy. Neurology. 73:1883–1891. 2009.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Armaroli A, Trabanelli C, Scotton C,
Venturoli A, Selvatici R, Brisca G, Merlini L, Bruno C, Ferlini A
and Gualandi F: Paternal germline mosaicism in collagen VI related
myopathies. Eur J Paediatr Neurol. 19:533–536. 2015.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Chen GL and Li DZ: Germline mosaicism in a
collagen VI-related myopathy family: A cause of autosomal recessive
inheritance. Congenit Anom (Kyoto). 61:197–198. 2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Inoue M, Saito Y, Yonekawa T, Ogawa M,
Iida A, Nishino I and Noguchi S: Causative variant profile of
collagen VI-related dystrophy in Japan. Orphanet J Rare Dis.
16(284)2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Varghaei P, Estiar MA, Ashtiani S, Veyron
S, Mufti K, Leveille E, Yu E, Spiegelman D, Rioux MF, Yoon G, et
al: Genetic, structural and clinical analysis of spastic paraplegia
4. Parkinsonism Relat Disord. 98:62–69. 2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Orlacchio A, Kawarai T, Totaro A, Errico
A, St George-Hyslop PH, Rugarli EI and Bernardi G: Hereditary
spastic paraplegia: Clinical genetic study of 15 families. Arch
Neurol. 61:849–855. 2004.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Giordani GM, Diniz F, Fussiger H,
Gonzalez-Salazar C, Donis KC, Freua F, Ortega RPM, de Freitas JL,
Barsottini OGP, Rosemberg S, et al: Clinical and molecular
characterization of a large cohort of childhood onset hereditary
spastic paraplegias. Sci Rep. 11(22248)2021.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Piermarini E, Akarsu S, Connors T,
Kneussel M, Lane MA, Morfini G, Karabay A, Baas PW and Qiang L:
Modeling gain-of-function and loss-of-function components of
SPAST-based hereditary spastic paraplegia using transgenic mice.
Hum Mol Genet. 31:1844–1859. 2022.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Karle KN, Schüle R, Klebe S, Otto S,
Frischholz C, Liepelt-Scarfone I and Schöls L: Electrophysiological
characterisation of motor and sensory tracts in patients with
hereditary spastic paraplegia (HSP). Orphanet J Rare Dis.
8(158)2013.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Costa AC and Sousa MM: The role of spastin
in axon biology. Front Cell Dev Biol. 10(934522)2022.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Liu Q, Zhang G, Ji Z and Lin H: Molecular
and cellular mechanisms of spastin in neural development and
disease (Review). Int J Mol Med. 48(218)2021.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Aartsma-Rus A, Van Deutekom JC, Fokkema
IF, Van Ommen GJ and Den Dunnen JT: Entries in the Leiden Duchenne
muscular dystrophy mutation database: An overview of mutation types
and paradoxical cases that confirm the reading-frame rule. Muscle
Nerve. 34:135–144. 2006.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Monaco AP, Bertelson CJ, Liechti-Gallati
S, Moser H and Kunkel LM: An explanation for the phenotypic
differences between patients bearing partial deletions of the DMD
locus. Genomics. 2:90–95. 1988.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Ioannidis NM, Rothstein JH, Pejaver V,
Middha S, McDonnell SK, Baheti S, Musolf A, Li Q, Holzinger E,
Karyadi D, et al: REVEL: An ensemble method for predicting the
pathogenicity of rare missense variants. Am J Hum Genet.
99:877–885. 2016.PubMed/NCBI View Article : Google Scholar
|