Introduction
Sturge-Weber syndrome (SWS), also recognized as
cerebral trigeminal neuroangiomatosis, presents as a noteworthy
subject within the realm of rare neurocutaneous disorders,
exhibiting a prevalence of ~1 in 50,000 individuals (1). In the initial stages of this
condition, the superficial part of the primary vascular system
supplies blood to the facial skin, scalp, middle meninges and other
regions, whereas its deeper counterpart caters to the needs of the
brain. During this phase, reduced blood flow may lead to hypoxia,
triggering vascular wall degeneration, calcification and the
formation of characteristic high-density shadows detectable in
neuroimaging (1). This process can
culminate in the development of distinctive hemangiomas. Despite
being congenital, this unique cerebrovascular anomaly is not
familial, but arises sporadically due to mutations in the G protein
subunit α q (GNAQ) gene (2,3). The
clinical manifestations of SWS vary widely, ranging from vascular
birthmarks to seizures and ocular complications, prompting
researchers to delineate the syndrome into three specific types
(4,5). Of particular consideration is SWS
type III, notable for the conspicuous absence of facial
hemangiomas, rendering it exceedingly rare and diagnostically
challenging (6). Predominant
symptoms include contralateral hemiplegia, hemiatrophy, headaches,
seizures and cognitive impairment (7). Given the dearth of literature on SWS
type III, along with its unique diagnostic and therapeutic
challenges, the present study seeks to bridge several vital
knowledge gaps. The rarity of SWS type III, coupled with the
absence of facial hemangiomas typically observed in other SWS
variants, underscores the necessity for more focused research.
Current diagnostic guidelines for SWS, as outlined by Sabeti et
al (5), often remain broad and
non-specific, failing to adequately capture the distinct features
of this subtype, potentially leading to delayed or missed
diagnoses. Furthermore, current treatment approaches, largely
extrapolated from methods used in more common forms of SWS, lack
rigorous validation for their effectiveness in type III cases
requiring surgical interventions (7-9).
With these challenges in mind, the present study
aims to shed light on the intricacies of SWS type III by presenting
two pediatric cases treated at the Children's Hospital Affiliated
to Shandong University (Jinan, China), thereby laying a crucial
groundwork for future multidisciplinary research and the evolution
of patient care protocols.
Case report
Case 1. A 4-year-old boy was treated at the
Children's Hospital Affiliated to Shandong University in February
2020 for recurrent right-sided hemiplegia and epileptic seizures,
with these symptoms first occurring ~1 year prior to the hospital
visit. Initially presenting with impaired mobility in the right
limb, which took 2-3 h to recover, the patient suffered
progressively worsening episodes, lasting 4-5 h daily. The
exacerbation led to continuous hemiplegic states interspersed with
seizures characterized by twitching of the right mouth corner, eye
and fingers, along with drooling, which lasted minutes to
hours.
The patient's comprehensive medical history revealed
no significant personal, familial or prior health issues. A
physical examination showed consciousness but reduced mental
responsiveness, a diminished right nasolabial fold, a leftward
mouth angle shift and normal eyelid function. Neurological
assessment, utilizing the Medical Research Council (MRC) scale for
muscle strength, revealed left limb muscle strength at grade V
(normal function), with the distal and proximal muscle strengths of
the right limb at grades III and II, respectively (10). According to the MRC scale, grade V
represents normal muscle strength, grade III indicates active
movement against gravity but not against resistance and grade II
signifies active movement with gravity eliminated. Enhanced
hyper-reflexia on the right side, compared with normal reflexes,
and the presence of the Babinski sign were noted.
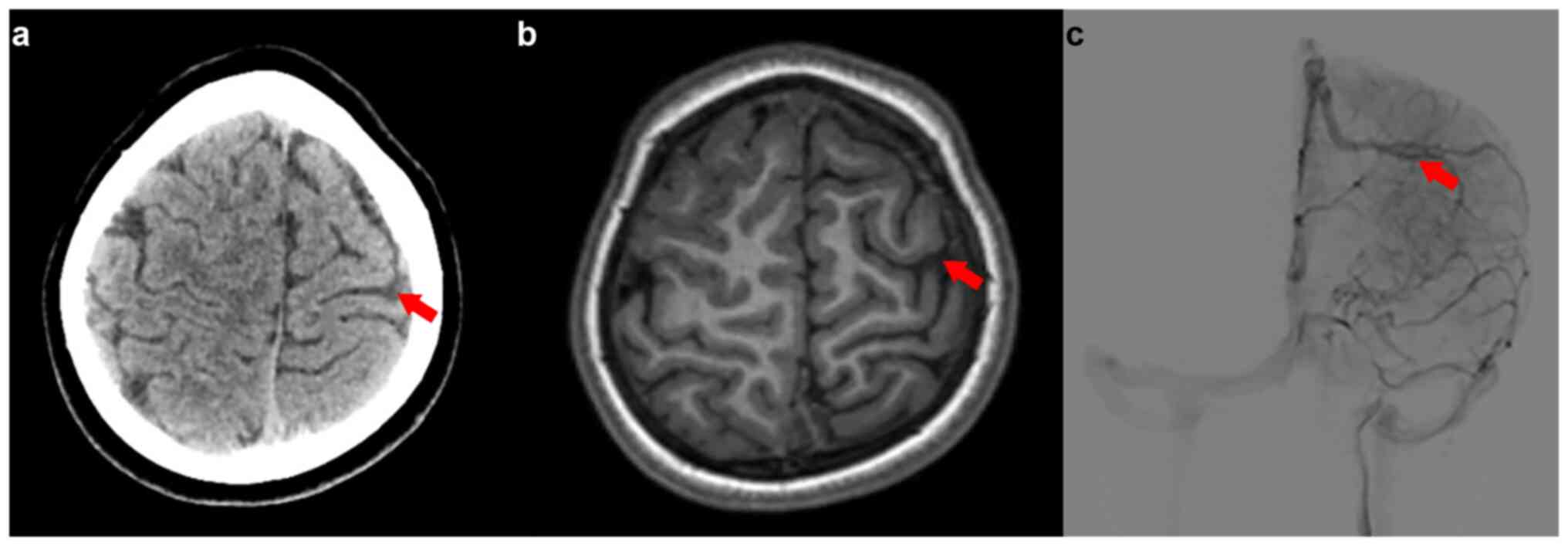
Radiological evaluation using magnetic resonance
imaging (MRI) revealed a marginally reduced left frontal-parietal
lobe compared with the right parietal lobe, abnormal local
myelination and a size disparity in the A1 segment of the left
anterior cerebral artery. Notably, a venous malformation in the
left frontal region and numerous small, winding superficial veins
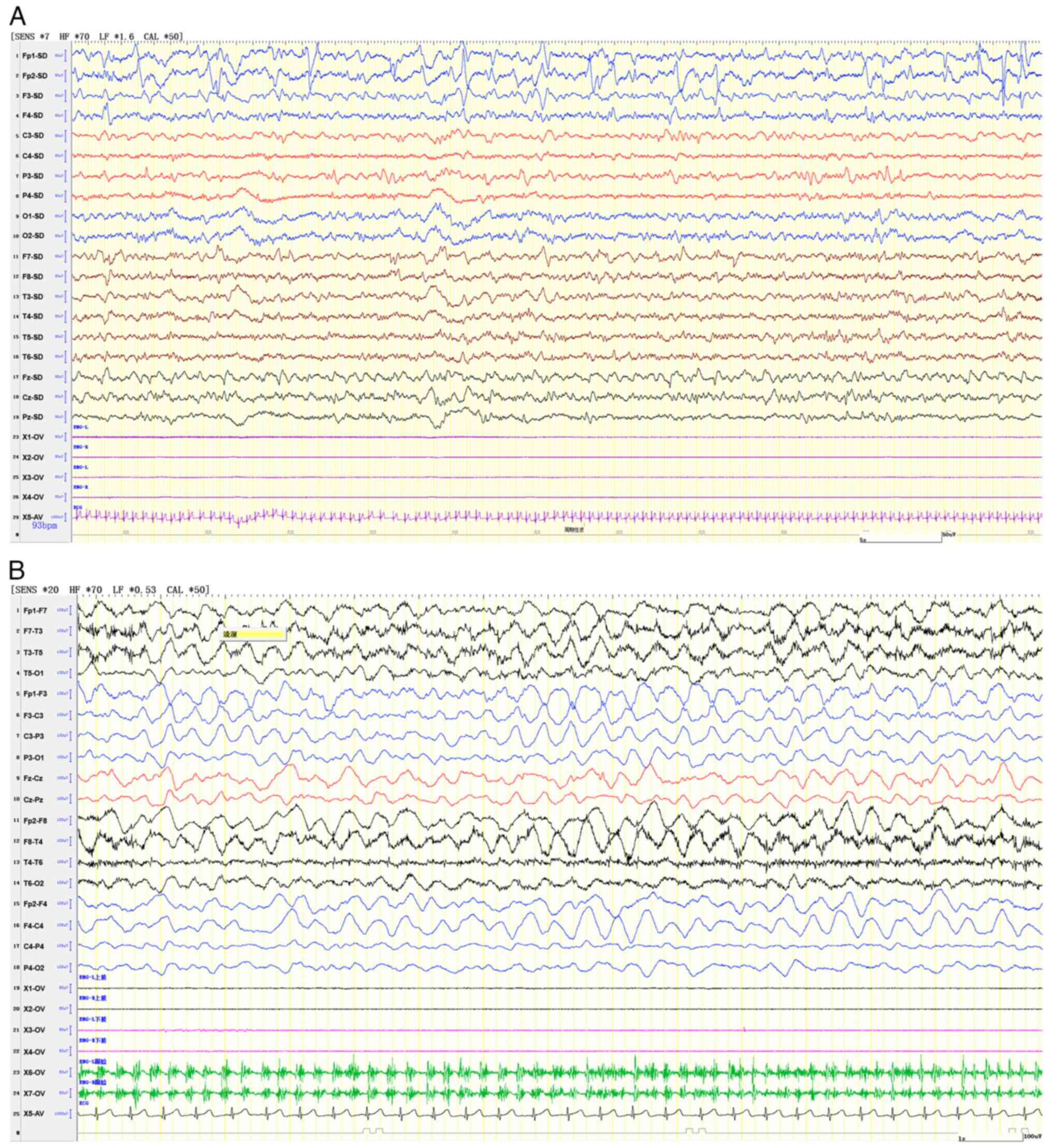
in the left subarachnoid space were observed (Fig. 1). An electroencephalogram (EEG)
indicated slow background activity with left-right asymmetry and
slow-wave discharges, sometimes classified as periodic lateralized
epileptiform discharges, from the anterior and temporal head
regions, pinpointing a focal epileptic episode on the right side
(Fig. 2).
The patient's treatment included immunoglobulin (7.5
g daily, intravenously), dexamethasone (4 mg daily, intravenously),
mannitol (60 mg every 12 h, intravenous push) and topiramate
(initially 12.5 mg daily, adjusted to 25 mg twice daily),
transitioning to oral topiramate post-discharge (25 mg twice daily,
continued until the last follow-up visit). This regimen effectively
controlled the seizures and normalized muscle strength, allowing
for a stable discharge. Following discharge, the patient underwent
a systematic follow-up regimen at 1, 3 and 6 months, and then once
a year subsequently. Over a 2-year period, this rigorous monitoring
confirmed the absence of any recurrence of hemiplegia or epileptic
episodes. These findings indicate a stable condition and a
favorable prognosis for the patient.
Case 2
In July 2021, a 2.5-year-old male patient was
admitted to the Children's Hospital Affiliated to Shandong
University with recurrent seizures characterized by leftward eye
deviation, the mouth skewing to the left and involuntary shaking of
the left side of the body. These brief episodes, lasting 3-5 sec,
recurred daily for 3-4 days. The seizures varied in presentation,
with some leading to unconsciousness, eye rolling, limb stiffness
and temporary left limb dysfunction post-seizure. Initial
improvement was noted with levetiracetam treatment (200 mg every 12
h, orally), but seizure recurrence ensued upon medication
discontinuation, presenting as rhythmic convulsions affecting the
left upper limb.
Upon presentation at the hospital, the patient was
preliminarily diagnosed with epilepsy. Despite oxcarbazepine (210
mg every 12 h, orally) and perampanel treatment (3 mg nightly,
orally), the seizure patterns changed, involving abrupt shakes,
falls and limb tremors. No significant personal, familial or prior
health issues were reported. A physical assessment revealed a
conscious yet minimally responsive patient with bilateral muscle
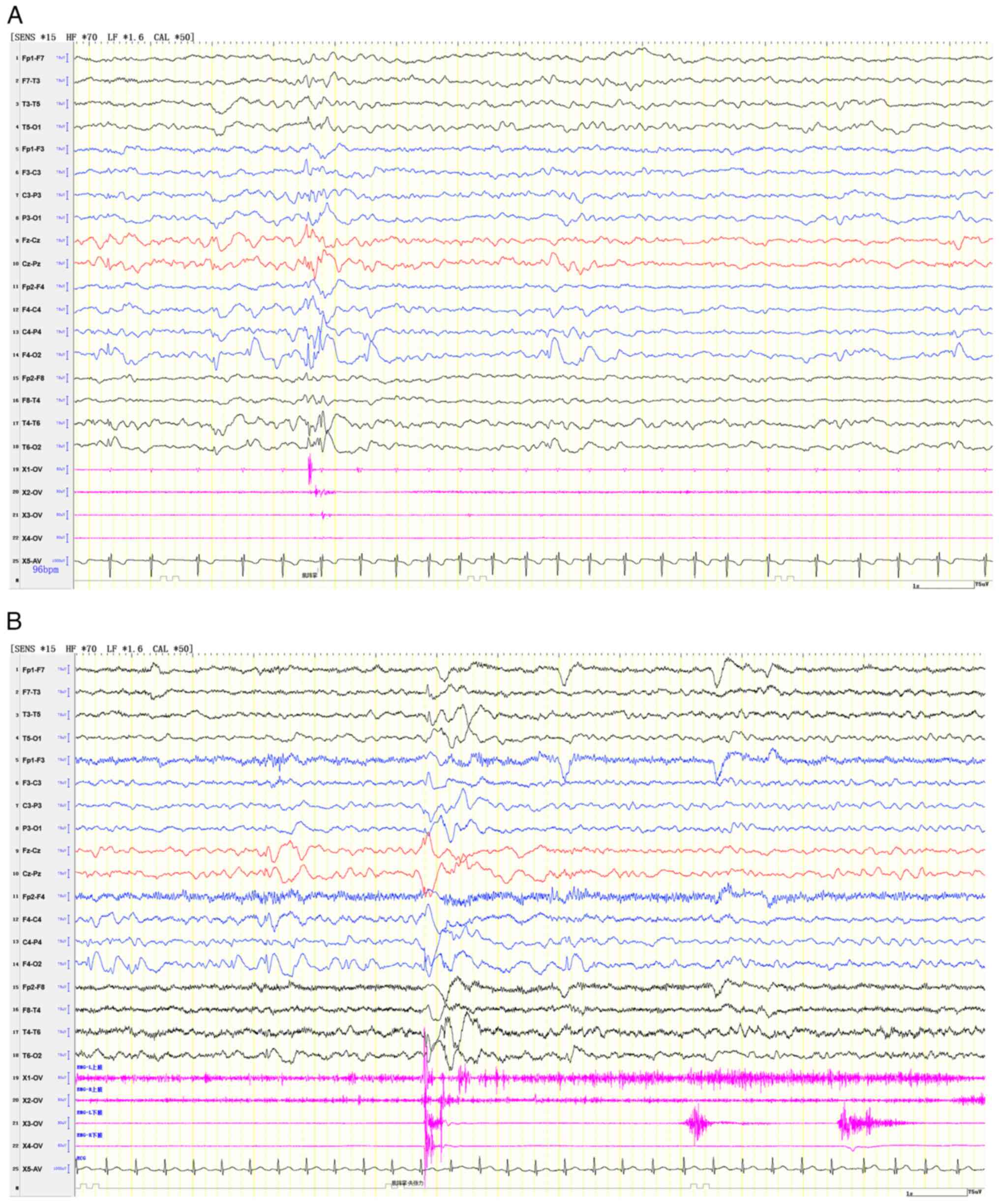
strength grade V. An EEG showed generalized or right focal
myoclonic-atonic seizures (Fig.
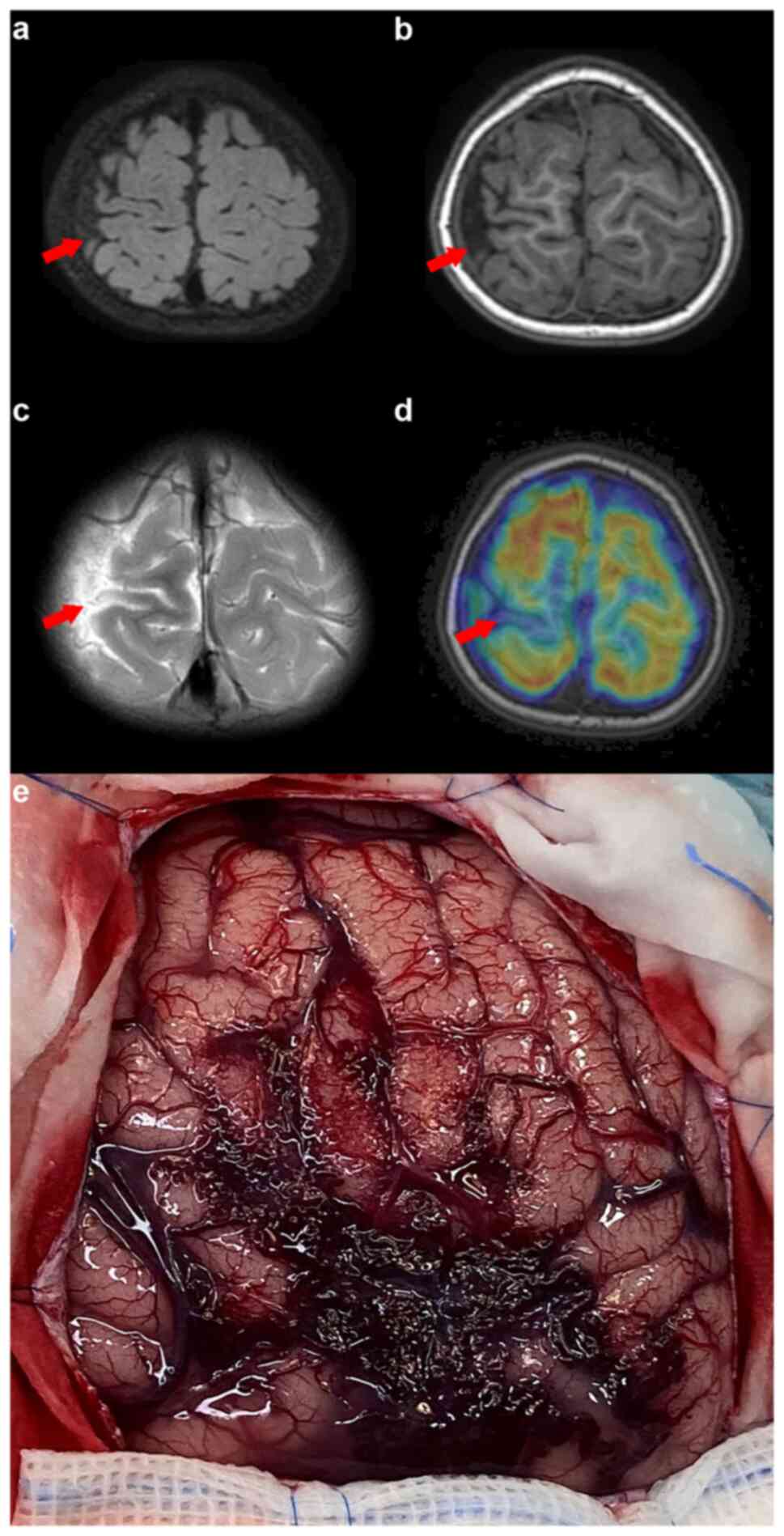
3). MRI, MR angiography and MR venography indicated a smaller
right parietal gyrus portion and localized cortical dysplasia, with
multiple enhanced vascular shadows in the right parietal space
suggesting vascular malformation (Fig.
4). A preoperative evaluation, including positron emission
tomography (PET)/CT and skull MR fusion, localized an epileptogenic
focus in the right central parietal area, indicative of focal
cortical dysplasia (FCD). A craniotomy performed to resect the
epileptic focus revealed a vascular mass flexion in the right
parietal region. The clinical and surgical findings led to a
diagnosis of SWS type III (Fig.
4). Post-surgery, the patient was enrolled in a detailed
follow-up program, including visits at ~1, 3, 6 and 12 months, to
closely monitor recovery and seizure activity. Over the 1-year
follow-up period, the patient experienced a stable recovery and
remained seizure-free. This follow-up regimen facilitated the early
detection and management of any potential complications,
contributing to the patient's positive prognosis. The continued
absence of seizures post-surgery underscores the effectiveness of
the surgical treatment and supports a favorable outlook for the
patient's continued neurological health.
Discussion
SWS has consistently captured the attention of
neurologists and clinicians due to its rarity and diverse clinical
presentations (8,9,11-13).
The syndrome is categorized into three types: Type I, encompassing
skin and neurological symptoms with or without glaucoma; type II,
characterized by skin involvement without neurological symptoms but
potential for glaucoma; and type III, marked by isolated
neurological involvement, typically without skin abnormalities or
glaucoma (14). Type III presents
unique diagnostic challenges, primarily due to the absence of
facial hemangiomas, complicating early and specific identification
(15). This necessitates a deeper
exploration of alternative diagnostic indicators and advanced
imaging techniques. The literature suggests that a diagnosis of SWS
is considered when hallmark symptoms, such as unilateral facial
erythema (port-wine stain) associated with the distribution of the
first trigeminal nerve, progressive seizures and cerebral cortex
atrophy, become clinically evident (16). The challenge in detecting SWS type
III lies not only in its rarity and the subtlety of its clinical
presentations but also in the potential for misdiagnosis or delayed
intervention, which can profoundly impact patient outcomes.
Consequently, dedicated research into SWS type III is imperative to
unravel the complexities of its pathophysiology, improve diagnostic
accuracy and explore targeted therapeutic strategies. Enhanced
understanding and innovation in this area are crucial for advancing
patient care and outcomes in this underrecognized and understudied
subtype of Sturge-Weber Syndrome.
The diagnosis of SWS type III is generally confirmed
through radiological examinations (17,18).
Emerging imaging techniques, such as high-resolution MRI and PET
scans, are increasingly crucial in the early detection of SWS type
III. These modalities can reveal subtle cerebral changes and
vascular abnormalities that are not apparent on standard imaging,
facilitating earlier diagnostic intervention (19). Key neuroradiological markers of SWS
include calcification of the retrocerebral gyrus, localized
cerebral atrophy and enlargement of the choroidal plexus on the
meningioma-affected side. CT scans tend to reveal more details,
whereas brain MRI often shows pia mater enhancement after
gadolinium injection, and MR angiography detects abnormal venous
flow into the deep venous system. Functional neuroimaging is
instrumental in identifying areas of reduced metabolism and
perfusion around vascular malformations (7,20).
EEG can also assist in diagnosing SWS, as
subclinical seizure activity in SWS may manifest as asymmetries in
rhythm and voltage, which could be more prevalent than overt
epileptiform abnormalities. Numerous research studies have
indicated the effectiveness of quantitative EEG power analysis in
both screening for and monitoring brain involvement in SWS
(19,21). Employing EEGs before the appearance
of symptoms to screen for brain involvement in SWS can facilitate
an earlier diagnosis (22). These
diagnostic tools collectively contribute to a comprehensive
understanding and identification of SWS.
Although the GNAQ mutation is the most
frequently occurring mutation, identifying it does not contribute
to determining the involvement of the brain or eyes and
necessitates a facial skin biopsy (23,24).
As a result, genetic testing is not routinely conducted for most
patients. Nevertheless, recent advancements in genetic research
have begun to shed light on the molecular underpinnings of SWS
(19). Identifying specific
biomarkers linked to SWS, particularly type III, could
revolutionize the diagnostic process. Ongoing research into these
genetic markers may soon enable more targeted and efficient
diagnoses. Among these, mutations similar to GNAQ, such as GNA11,
offer new insights, albeit primarily associated with conditions
such as uveal melanoma, suggesting a broader genetic overlap that
could inform SWS research (25).
The PIK3CA mutations, known for their role in vascular overgrowth
syndromes, present another intriguing avenue, highlighting shared
pathways that might be relevant to SWS despite not being directly
linked. Moreover, the focus on angiogenesis, the process of new
blood vessel formation, brings to light potential biomarkers such
as vascular endothelial growth factor (VEGF) and angiopoietins.
These molecules, critical in vascular development, could correlate
with SWS severity or progression (8,26).
The majority of children diagnosed with SWS
experience early brain injury and various neurological disorders,
with seizures being the most common neurological symptom, affecting
~75% of these patients (9).
Studies have suggested that the cerebral capillary-venous
malformations characteristic of SWS interfere with normal
hemodynamic responses during seizures, perpetuating ischemia and
potentially leading to more seizures (27-29).
This cycle is associated with cognitive decline and progressive
neurological deterioration.
Currently, no specific targeted therapy is available
for SWS (19). The primary
treatment for seizures in patients with SWS remains the
antiepileptic medication. The choice of antiepileptic drugs (AEDs)
is guided by a combination of factors, such as the age of onset,
the extent of brain involvement and the presence of a family
history of seizures. These elements are significant predictors of
treatment response and the need for multiple AEDs. For instance,
one study showed that patients with bilateral brain involvement,
early onset of seizures and a family history of seizures often
received a greater number of anticonvulsants due to their more
severe clinical courses (8).
Multicenter research data showed that among the various AEDs,
levetiracetam, oxcarbazepine and low-dose aspirin were predominant
choices. Levetiracetam was used in 48% of the cases, reflecting its
favorable profile in terms of efficacy and side effects (28). Oxcarbazepine, chosen for 40% of the
patients, was another key medication due to its effectiveness in
controlling complex partial seizures and generalized tonic-clonic
seizures, which are common complications in SWS (28). Notably, low-dose aspirin has
emerged as a significant adjunctive therapy in SWS (28), with its role extending beyond
seizure control to potentially reducing stroke-like episodes and
improving neurological outcomes. This dual benefit is particularly
relevant in SWS, where thrombosis and venous stasis are underlying
pathologies (7). However, the risk
of bleeding and the development of complications such as subdural
hematoma, albeit rare, necessitate careful monitoring. The decision
for surgical intervention, particularly hemispherectomy, is
carefully considered in cases with medically refractory seizures
and unilateral brain involvement, acknowledging the risks and
potential outcomes, especially in bilaterally affected patients
(30,31). Furthermore, the recent addition of
cannabidiol (CBD) as a treatment option represents an important
development in the management of refractory seizures in SWS
(32). The neuroprotective
properties of CBD and its effectiveness in reducing seizure
frequency make it a viable option for treatment-resistant cases.
However, its use requires careful monitoring for potential side
effects, such as behavioral changes and increased seizure
activity.
It is imperative to consider a comprehensive
approach that encompasses both medical and multidisciplinary care
for pediatric patients with SWS type III in long-term management.
The role of advanced neuroimaging techniques, including
gadolinium-enhanced MRI and transcranial Doppler, plays a crucial
part in both the diagnosis and ongoing monitoring of the disease
(5,33,34).
The application of these techniques in identifying early signs of
brain involvement, coupled with their utility in assessing disease
progression, underscores their value in long-term management.
Regular EEG monitoring is also fundamental in detecting subclinical
seizure activity, rhythm and voltage asymmetry, which are
indicative of neurological involvement in SWS (21). Furthermore, the use of quantitative
EEG enhances the ability to predict the risk of brain involvement
and guides the timing of diagnostic MRI, particularly in infants
with high-risk facial capillary malformations (22). The potential of emerging
biomarkers, such as urine levels of MMP-2, MMP-9 and VEGF, offers
promising avenues for non-invasive monitoring of disease
progression and tailoring of treatment approaches (35). This aligns with the overarching
goal of early intervention and optimized management strategies to
mitigate the neurological impact of the syndrome. In addition to
medical treatment, the integration of occupational and physical
therapy is crucial for addressing the physical and cognitive
impairments associated with SWS (36). This multidisciplinary approach not
only aids in improving the quality of life for these children, but
also supports their developmental needs (7). Thus, the long-term management of SWS
should encompass regular neuropsychological evaluations to monitor
cognitive function and adjust therapies as needed. Lastly, the
importance of a coordinated multidisciplinary care team, comprising
pediatric neurologists, neurosurgeons, radiologists and therapists,
cannot be overstated (7,8,37).
This team-based approach is essential for providing comprehensive
care tailored to the individual needs of each patient, thereby
optimizing the long-term prognosis and overall well-being of
children affected by SWS type III.
In the cases presented in the current study, the
first patient experienced recurrent seizures and partial
hemiplegia. Although the exact cause was initially unclear, a
definitive diagnosis was established following a digital
subtraction angiography examination. This child showed a decrease
in seizure frequency and maintained standard cognitive abilities,
responding well to antiepileptic drugs. By contrast, for the second
child, who was unresponsive to oral medications and suffered
recurrent seizures, SWS could not be confirmed through EEG and MRI
alone, but was ultimately confirmed through later surgical
intervention, from which the child benefited. Therefore, both
medical and surgical interventions can be effective in treating
SWS. However, the choice between these approaches should be based
on the severity of epilepsy and any associated neurocognitive
decline. The cases presented here highlight the complexity and
variability of clinical manifestations in SWS type III. Although
both patients experienced recurrent seizures, the specific
characteristics, duration and accompanying clinical symptoms varied
significantly, underscoring the need for a comprehensive and
tailored diagnostic approach. The initial presentations, treatment
responses and outcomes in these pediatric cases offer crucial
insights into the broad spectrum of SWS. Notably, while seizures
were a common symptom, the pathophysiological mechanisms underlying
these seizures differed, as indicated by the distinct findings on
imaging and EEG evaluations. The discovery of a venous malformation
in the first patient and a vascular mass flexion in the second
patient further emphasize the cerebrovascular anomalies typically
associated with SWS. Furthermore, the successful management of
these cases, resulting in a seizure-free state post-treatment,
highlights the critical importance of early diagnosis, precise
treatment and consistent follow-up in managing this rare and
complex condition. The current clinical practice faces limitations
in the timely and accurate diagnosis of SWS type III, often due to
the subtlety of its symptoms and the lack of awareness among
clinicians. Investigating the progression of the disease and its
response to various interventions can offer valuable insights,
aiding in the formulation of more effective management strategies.
Future research should aim at developing standardized diagnostic
protocols for SWS, particularly type III.
In conclusion, SWS should be suspected in children
presenting with epileptic seizures, hemiplegia and intellectual
disabilities. The diagnosis involves assessing clinical symptoms,
EEG results and imaging. The condition can be misdiagnosed as FCD
due to similar symptoms. Often, a definitive diagnosis of SWS
requires comprehensive preoperative tests and may only be confirmed
during surgery. More research is needed due to the rarity of SWS
and its complex pathology.
Acknowledgements
Not applicable.
Funding
Funding: This study was supported by Jinan Municipal Health
Commission (grant no. 2023-2-146).
Availability of data and materials
The data generated in the present study are not
publicly available due to ethical and privacy restrictions but may
be requested from the corresponding author.
Authors' contributions
HWZ and JGS were instrumental in the study
conception and design. YPW, WDH and GFG were responsible for
drafting of the manuscript, as well as in the acquisition, analysis
and interpretation of data. GFG and JGS contributed significantly
through the performance of a novel surgical technique. HZ, YL and
ZFG provided critical revision of the manuscript for important
intellectual content. ZFG was responsible for choosing critical
medical imaging, HZ tailored the treatment plan and YL outlined the
study design. YPW played a crucial role in analyzing patient data
and revising the manuscript critically, ensuring the integrity of
the work. HWZ and JGS confirm the authenticity of all the raw data.
All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
Jinan Children's Hospital (approval no. SDFE-IRB/P-2023038; Jinan,
China). Written informed consent to participate in the present
study was provided by the participants' legal guardian/next of
kin.
Patient consent for publication
Written informed consent was obtained from the
patients' parents for the publication of any potentially
identifiable images or data included in this article.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zeka N, Zeka B, Gerguri A, Bejiqi R,
Retkoceri R, Maloku A and Zogaj L: Sturge-Weber syndrome and
variability of clinical presentation. Med J Malaysia. 78:145–148.
2023.PubMed/NCBI
|
|
2
|
Formisano M, di Pippo MC, Scuderi L and
Abdolrahimzadeh S: Current concepts on diffuse choroidal hemangioma
in Sturge Weber syndrome. Ophthalmic Genet. 42:375–382.
2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wetzel-Strong SE, Galeffi F, Benavides C,
Patrucco M, Bullock JL, Gallione CJ, Lee HK and Marchuk DA:
Developmental expression of the Sturge-Weber syndrome-associated
genetic mutation in Gnaq: A formal test of Happle's paradominant
inheritance hypothesis. Genetics. 224(iyad077)2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Ejike O, Odume C, Ekwochi U, Ndu I and
Imanyikwa U: A rare type of congenital Sturge-Weber Syndrome:
Presenting with history of perinatal asphyxia. Clin Case Rep.
4:725–727. 2016.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Sabeti S, Ball KL, Bhattacharya SK,
Bitrian E, Blieden LS, Brandt JD, Burkhart C, Chugani HT, Falchek
SJ, Jain BG, et al: Consensus statement for the management and
treatment of sturge-weber syndrome: Neurology, neuroimaging, and
ophthalmology recommendations. Pediatr Neurol. 121:59–66.
2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Manohari S, Banyal P, Saini AG and Vyas S:
Sturge-Weber syndrome type III: An important stroke mimic. BMJ Case
Rep. 16(e248742)2023.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Yeom S and Comi AM: Updates on
sturge-weber syndrome. Stroke. 53:3769–3779. 2022.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Sanchez-Espino LF, Ivars M, Antonanzas J
and Baselga E: Sturge-Weber Syndrome: A review of pathophysiology,
genetics, clinical features, and current management approache. Appl
Clin Genet. 16:63–81. 2023.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Bianchi F, Auricchio AM, Battaglia DI,
Chieffo DRP and Massimi L: Sturge-Weber syndrome: An update on the
relevant issues for neurosurgeons. Childs Nerv Syst. 36:2553–2570.
2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Vanhoutte EK, Faber CG, van Nes SI, Jacobs
BC, van Doorn PA, van Koningsveld R, Cornblath DR, van der Kooi AJ,
Cats EA, van den Berg LH, et al: Modifying the Medical Research
Council grading system through Rasch analyses. Brain. 135(Pt
5):1639–1649. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Raval DM, Rathod VM, Patel AB, Sharma B
and Lukhi PD: Sturge-Weber Syndrome: A rare case report. Cureus.
14(e28786)2022.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Poliner A, Fernandez Faith E, Blieden L,
Kelly KM and Metry D: Port-wine Birthmarks: Update on diagnosis,
risk assessment for sturge-weber syndrome, and management. Pediatr
Rev. 43:507–516. 2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Sabeti S, Ball KL, Burkhart C, Eichenfield
L, Fernandez Faith E, Frieden IJ, Geronemus R, Gupta D, Krakowski
AC, Levy ML, et al: Consensus statement for the management and
treatment of port-wine birthmarks in sturge-weber syndrome. JAMA
Dermatol. 157:98–104. 2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Roach ES: Neurocutaneous syndromes.
Pediatr Clin North Am. 39:591–620. 1992.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Mozaffari K, Krishnakumar A, Chen JS, Goel
K, Wang A, Shlobin NA, Weil AG and Fallah A: Seizure outcomes in
children with Sturge-Weber syndrome undergoing epilepsy surgery: An
individual participant data meta-analysis. Seizure. 107:43–51.
2023.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Singh AK and Keenaghan M: Sturge-Weber
Syndrome. In: StatPearls [Internet]. StatPearls Publishing,
Treasure Island, FL, 2024.
|
|
17
|
Mukherjee D, Kundu R and Niyogi PC:
Sturge-Weber syndrome type III. Indian J Pediatr. 82:97–98.
2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Mishra B, Singh RK, Garg A, Vibha D,
Bhargavi R and Tripathi M: Tram track sign in sturge-weber syndrome
type 3. Ann Indian Acad Neurol. 24(592)2021.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Garcia Caride S, Fernandez-Vigo JI and
Valverde-Megias A: Update on the diagnosis and treatment of
choroidal hemangioma. Arch Soc Esp Oftalmol (Engl Ed). 98:281–291.
2023.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Cousyn L, Leclercq D, Ta MC, Gilbert F, Di
Meglio L, Marois C, Haddad A, Mathon B, Eyries M and Navarro V:
Late-Onset status epilepticus associated with isolated
leptomeningeal angioma and sturge-weber syndrome-related GNA11
pathogenic variation. Neurology. 101:1021–1022. 2023.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Ewen JB, Kossoff EH, Crone NE, Lin DD,
Lakshmanan BM, Ferenc LM and Comi AM: Use of quantitative EEG in
infants with port-wine birthmark to assess for Sturge-Weber brain
involvement. Clin Neurophysiol. 120:1433–1440. 2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Gill RE, Tang B, Smegal L, Adamek JH,
McAuliffe D, Lakshmanan BM, Srivastava S, Quain AM, Sebold AJ, Lin
DDM, et al: Quantitative EEG improves prediction of Sturge-Weber
syndrome in infants with port-wine birthmark. Clin Neurophysiol.
132:2440–2446. 2021.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Waelchli R, Aylett SE, Robinson K, Chong
WK, Martinez AE and Kinsler VA: New vascular classification of
port-wine stains: Improving prediction of Sturge-Weber risk. Br J
Dermatol. 171:861–867. 2014.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Shirley MD, Tang H, Gallione CJ, Baugher
JD, Frelin LP, Cohen B, North PE, Marchuk DA, Comi AM and Pevsner
J: Sturge-Weber syndrome and port-wine stains caused by somatic
mutation in GNAQ. N Engl J Med. 368:1971–1979. 2013.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Thorpe J, Frelin LP, McCann M, Pardo CA,
Cohen BA, Comi AM and Pevsner J: Identification of a mosaic
activating mutation in GNA11 in atypical sturge-weber syndrome. J
Invest Dermatol. 141:685–688. 2021.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Nguyen V, Hochman M, Mihm MC Jr, Nelson JS
and Tan W: The pathogenesis of port wine stain and sturge weber
syndrome: Complex interactions between genetic alterations and
aberrant MAPK and PI3K Activation. Int J Mol Sci.
20(2243)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Comi AM, Fischer R and Kossoff EH:
Encephalofacial angiomatosis sparing the occipital lobe and without
facial nevus: On the spectrum of Sturge-Weber syndrome variants? J
Child Neurol. 18:35–38. 2003.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Smegal LF, Sebold AJ, Hammill AM, Juhász
C, Lo WD, Miles DK, Wilfong AA, Levin AV, Fisher B, Ball KL, et al:
Multicenter research data of epilepsy management in patients with
sturge-weber syndrome. Pediatr Neurol. 119:3–10. 2021.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Martinez-Lopez A, Salvador-Rodriguez L,
Montero-Vilchez T, Molina-Leyva A, Tercedor-Sanchez J and
Arias-Santiago S: Vascular malformations syndromes: An update. Curr
Opin Pediatr. 31:747–753. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wang S, Pan J, Zhao M, Wang X, Zhang C, Li
T, Wang M, Wang J, Zhou J, Liu C, et al: Characteristics, surgical
outcomes, and influential factors of epilepsy in Sturge-Weber
syndrome. Brain. 145:3431–3443. 2022.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ainuz BY, Wolfe EM and Wolfe SA: Surgical
management of facial port-wine stain in sturge weber syndrome.
Cureus. 13(e12637)2021.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Kaplan EH, Offermann EA, Sievers JW and
Comi AM: Cannabidiol treatment for refractory seizures in
sturge-weber syndrome. Pediatr Neurol. 71:18–23 e2. 2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Jimenez-Legido M, Martinez-de-Azagra-Garde
A, Bernardino-Cuesta B, Solís-Muñiz I, Soto-Insuga V,
Cantarín-Extremera V, García-Salido A, Duat-Rodríguez A,
García-Peñas JJ and Ruíz-Falcó-Rojas ML: Utility of the
transcranial doppler in the evaluation and follow-up of children
with Sturge-Weber Syndrome. Eur J Paediatr Neurol. 27:60–66.
2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Offermann EA, Sreenivasan A, DeJong MR,
Lin DDM, McCulloch CE, Chung MG and Comi AM: National Institute of
Health Sponsor; Rare Disease Clinical Research Consortium (RDCRN);
Brain and Vascular Malformation Consortium (BVMC); National
Sturge-Weber Syndrome Workgroup. Reliability and clinical
correlation of transcranial doppler ultrasound in sturge-weber
syndrome. Pediatr Neurol. 74:15–23 e5. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Sreenivasan AK, Bachur CD, Lanier KE,
Curatolo AS, Connors SM, Moses MA and Comi AM: Urine vascular
biomarkers in Sturge-Weber syndrome. Vasc Med. 18:122–128.
2013.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Harmon KA, Day AM, Hammill AM, Pinto AL,
McCulloch CE and Comi AM: National Institutes of Health Rare
Disease Clinical Research Consortium (RDCRN) Brain and Vascular
Malformation Consortium (BVMC) SWS Investigator Group. Quality of
life in children with sturge-weber syndrome. Pediatr Neurol.
101:26–32. 2019.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Comi AM: Presentation, diagnosis,
pathophysiology, and treatment of the neurological features of
Sturge-Weber syndrome. Neurologist. 17:179–184. 2011.PubMed/NCBI View Article : Google Scholar
|