Osteoporosis has become a significant health concern
worldwide as a number of countries develop aging populations
(1-3).
Previous studies have focused on the relationship between
cardiovascular diseases (CVDs) and osteoporosis, and have reported
that hyperlipidemia forms the common pathophysiological basis of
the two disorders (4-7).
Under hyperlipidemia, excessive lipids in the serum are delivered
to the bone marrow, changing the microenvironment and dysregulating
bone cell function. Osteoblastic cells are the major functional
cells during osteogenic differentiation and bone formation, and
their dysfunction results in insufficient bone formation and bone
loss (8). At the cellular level,
excessive lipids accumulate in osteoblastic cells and further
impair their function as reported by Kim et al (9). This previous study investigated the
biodistribution of lipids by administering radiolabeled fatty acid
tracers (3H-bromopalmitate and 14C-oleate)
through gavage to C57BL/6 mice, and found significant uptake of
both tracers in the femur, tibia and calvaria, which indicated that
except for the heart and liver, bone was the main organ of lipid
uptake. A similar study was conducted by injecting
125I-tyramine cellobiose chylomicron remnants (CR) into
C57BL/6 mice, with radioactivity measured to assess internalized CR
particles 20 min after injection. The results showed that CR uptake
by bone was ~17% of uptake by the liver, and was higher than uptake
by the lung, muscle, heart and kidney. Further study indicated that
CR uptake by apolipoprotein E (ApoE)-deficient osteoblasts was ~50%
that of wild-type osteoblasts (10). The aforementioned results
illustrate that osteoblasts possess a high lipoprotein uptake
capacity and that endogenous ApoE is necessary for efficient lipid
internalization for osteoblasts. However, how lipids taken up by
osteoblastic cells affect cell function is not entirely clear.
Autophagy serves an essential role in the
homeostasis of osteoblastic cells and impaired autophagy leads to
decreased bone mass (11-13).
Autophagy of lipids, termed lipophagy, is an important lipid
catabolism event that degrades triglycerides (TGs) and cholesterol
(CH) in lipid droplets (LDs) via the autophagy-lysosome system
(14,15). During lipophagy, the LD coat
proteins are identified and selectively removed, inducing the
release of free fatty acids (FFAs) (16,17).
The generated FFAs improve the rate of mitochondrial β-oxidation,
to produce ATP to meet the need for nutrients in cells (18). Therefore, lipophagy is required for
mediating lipid content, preventing the formation of potentially
toxic lipids and maintaining cellular energy homeostasis (19). Impaired lipophagy leads to
excessive tissue lipid accumulation (20,21).
A previous study reported high-fat environments suppress osteoblast
mineralization and activate lipophagy (22). However, the role of lipophagy in
regulating bone metabolism and the mechanism by which lipophagy
affects osteoblastic cell function remain largely unknown.
In the present review, current knowledge of the role
of autophagy/lipophagy in osteoblastic cell dysfunction is
summarized. This is expected to provide insight into the mechanism
of hyperlipidemia-induced osteoporosis, providing a theoretical
foundation and potential therapeutic targets for the disorder.
Lipids are stored in LDs, which are primarily found
in the cytoplasm; however, in certain cell types such as Huh7
cells, LDs are also located in the nucleus (23). These LDs form, expand, shrink and
dissolve in response to changes in the cell energy status. In the
case of energy demand, FAs escaping from LDs are substrates for
β-oxidation and ultimately generate ATP to meet the energy
requirements for cell survival. On a whole organism level, the
degradation of LDs in white adipose tissue is important to supply
fuel during nutrient insufficiency (24). It has previously been reported that
the accumulation of excess LDs in cells may be the cause of
osteoporosis (24). Pirih et
al (25) reported that a
high-fat diet (HFD) significantly decreased the cortical bone
volume fraction (BV/TV), and, reduced femoral bone strength and
stiffness while increasing cortical porosity. It was also observed
that the serum levels of parathyroid hormone, calcium, phosphorus
and TNF-α were markedly increased, whereas procollagen type I
N-terminal pro-peptide, a serum marker of bone formation, was
decreased in a Ldlr-/- mouse model but not in the
wild-type mice, which indicated that hyperlipidemia impaired bone
regeneration and mechanical strength, and induced secondary
hyperparathyroidism. Further study showed that the adverse effects
of hyperlipidemia on bone tissue were regulated by oxidized lipids
and could be blunted after administration of D-4F, an ApoA-I
mimetic peptide. Almeida et al (26) also reported that HFD-fed
Ldlr-/- mice had significantly different collagen
orientations and decreased volumetric tissue mineral density
assessed using micro-computed tomography, which suggested that
hyperlipidemia affected bone microstructure and density. Similarly,
C57BL/6 mice fed a high-CH diet (40% of calories from fat, 1.25% of
calories from CH) exhibited an osteoporotic bone phenotype,
including trabeculae loss, and thinning of the trabeculae and
cortex (27). Female mice fed a
Western diet (1.1 mg CH/g) or high-CH diet demonstrated a notable
decrease in bone mass, and reduced bone mineral content and bone
mineral density (BMD) in the femur compared with mice fed a
meat-supplemented diet (28,29).
As expected, a high-CH diet also led to decreased bone formation
and reduced BMD in rats (30).
Previous studies reported that the loss of lysosomal acid lipase
(LAL), the only known essential CH ester (CE) hydrolysis enzyme,
increased CE and TG accumulation in numerous cells and tissues
(31,32). A further study reported that global
Lal-/- mice exhibited lower cortical bone thickness and
strength along with fewer osteoblasts, which resulted in altered
lipid metabolism and was connected with the Wnt, Notch and bone
morphogenetic protein (BMP) signaling pathways, which indicated
that hyperlipidemia had adverse effects on bone metabolism and
induced osteoporosis via another mechanism (33). However, although the mechanisms in
other tissues, such as the liver and heart, have been extensively
illustrated, the mechanisms underlying the impact of hyperlipidemia
on bone metabolism are inadequately understood (18,34,35).
Hyperlipidemia leads to abnormal accumulation of
lipids in bone tissue compartments, which has a wide range of
effects on bone cell function through varied mechanisms, causing
bone loss (36-38).
Hyperlipidemia causes osteogenic cell dysfunction through certain
pathways, which eventually induce bone loss and osteoporosis.
Marrow stromal cells (MSCs) isolated from C57BL/6 mice fed a HFD
were reported to have failed to undergo osteogenic differentiation
in vitro. In addition, the osteogenic differentiation of the
murine MSCs, M2-10B4, was inhibited after treatment with minimally
oxidized LDL (MM-LDL), along with decreased alkaline phosphatase
(ALP) activity, decreased levels of collagen I (Col I) and
suppressed mineralization, which were all related to the MAPK
pathway (39). In addition,
MM-LDL, but not native LDL, promoted the adipogenic differentiation
of M2-10B4 and 3T3-L1 preadipocytes by activating PPARα (39). These observations indicate that LDL
oxidation products induce osteoporotic loss of bone by directing
osteoprogenitor cells to undergo adipogenic rather than osteogenic
differentiation. Moreover, it has been reported that CH inhibited
osteoblastic differentiation by downregulating osteogenic
differentiation marker genes, such as Bmp2, Cbfa1, Alp and Col I,
and inhibiting matrix calcium deposition, which is regulated by
intracellular reactive oxygen species (ROS) (40,41).
CH serves a dual role in mediating osteoblastic cell function
because it is not only the structural component of the cell
membrane but also possesses the ability to regulate cellular
function. A previous study reported that exogenous CH inhibited
osteoblast differentiation, whereas endogenous CH at physiological
levels was essential for bone marrow stem cell (BMSC) osteogenesis
(42). Zhang et al
(43) reported that CH retarded
BMSC senescence in a dose-dependent manner when cells were treated
with H2O2 for 30 h, which was associated with
enhanced autophagy regulated by the ROS/p53/p21Cip1/Waf1 signaling
pathway. These observations indicate that the effect of CH on
osteoblastic cell function is more complex than that of either
‘bad’ or ‘good’ (42).
However, accumulated lipids indirectly influence
osteogenic cell function by altering the microenvironment of the
bone marrow (44,45). One of the major functions of bone
marrow is to provide mature blood cells to the circulation, where
they are involved in blood clotting and innate immunity. As well as
the close association of hyperlipidemia with CVD, CH content is
closely associated with the bone marrow microenvironment and
influences hematopoiesis (46).
LDL, which accumulates in the subendothelial matrices of arteries,
undergoes oxidation and produces modified forms, such as MM-LDL and
oxidized-LDL. Modified LDL further induces potent inflammatory
responses, such as the induction of chemotactic factors in
endothelial cells, recruitment of monocytes to the arterial wall
and adhesion of monocytes to endothelial cells (47). In chronic inflammatory conditions,
such as certain rheumatological diseases, systemic bone loss has
been observed in both patients and experimental models (48-51).
Chronic inflammatory diseases weaken the function of osteogenic
cells in maintaining the balance of bone remodeling, inducing the
occurrence of osteoporosis. Redlich et al (51) reported that osteogenic cells were
present at local erosion sites in rheumatoid arthritis, but their
number and activity were too low to counteract osteoclast action
owing to the role of proinflammatory cytokines. TNF, for example,
inhibits osteoblast differentiation through the p55 TNF receptor by
inhibiting Runx2, which is regulated in part by inducing Runx2
ubiquitylation (52-54).
Other pro-inflammatory cytokines, such as IL-1 and IL-6, lead to
osteogenic cell dysfunction and inhibit osteoblastogenesis
(55,56). Moreover, a number of cytokines
negatively affect osteoblast function by activating the NF-κB
signaling pathway, thought to be via inhibition of the JUN
N-terminal kinase 1 and thus decreasing the transcription factor
AP1(57). In addition, the
negative effects of proinflammatory cytokines on osteogenic cells
can be further regulated byDickkopf-1, an inhibitor of the Wnt
signaling pathway and induced by Tnf32. Sclerostin, another
inhibitor of the Wnt signaling pathway, can bind to and antagonize
BMPs to suppress osteogenesis (58-60).
The aforementioned studies indicate that an inflammatory
environment impairs osteogenic cell function, which strongly
suggests that the inflammatory response induced by hyperlipidemia
in the bone marrow leads to osteogenic cell dysfunction and bone
loss. However, the mechanism by which hyperlipidemia impairs
osteogenic cell function has not been clearly elucidated.
Previous studies have indicated that abnormal
autophagy can lead to imbalances in bone metabolism and serve a
critical role in bone metabolism disorders (73,74).
Piemontese et al (75)
reported that the autophagy level in primary osteoblasts from
Atg7-/- mice was inhibited, which caused the
accumulation of endoplasmic reticulum stress, and resulted in low
bone mass and a greater number of fractures compared with wild-type
mice. Further study indicated that the effect of Atg7 deficiency on
bone tissue might be associated with a decreased number of
osteoblasts. Another study reported that 17 β-estradiol could
induce autophagy to protect osteoblast function in women with
postmenopausal osteoporosis through the G protein-coupled receptor
30 (GPR30) and extracellular-regulated protein kinases 1/2
signaling pathway (76). However,
this protective effect could be abolished by G15, a selective GPR30
antagonist (77). In a type 2
diabetes mouse model, the acceleration of autophagy in osteoblasts
protected their ability to survive and differentiate by increasing
ROS and protein oxidation induced by the high glucose environment
(78). Autophagy was also reported
to regulate MSC function to control the development of
postmenopausal osteoporosis, which was mediated by the mTOR
signaling pathway (79). Moreover,
numerous studies have reported that autophagy is significantly
enhanced during osteoblast differentiation and mineralization, and
its inhibition rapidly causes dysfunction of osteoblasts in
vitro (80-83).
The aforementioned studies demonstrate that autophagy serves a key
role in osteoporosis by regulating osteoblastic cell function and
differentiation.
Autophagy can be activated in osteoblastic cells by
LDs in a high-fat environment (84). Putative links between autophagy and
LDs were identified following the observation that mutations in
LAL, a lipase responsible for lysosomal LD degradation, caused LD
accumulation in certain organs (17). However, the contribution of
lipophagy to LD accumulation is unknown. Singh et al
(15) clearly demonstrated that,
in hepatocytes, autophagy was termed the ‘lipophagy’ when it was
linked to LD degradation, which suggested new avenues of study of
the role of the regulation of lipid metabolism in cellular
physiology and pathophysiology. Lipophagy is a type of selective
autophagy that can occur via both macro- and micro-based
mechanisms. Macrolipophagy refers to the classical
autophagosome-mediated manner in which LD budding occurs and LDs
are sequestered for subsequent delivery to autolysosomes.
Microlipohagy refers to the transient and direct interactions
between LDs and lysosomes as a means of degrading LD-derived lipids
(17).
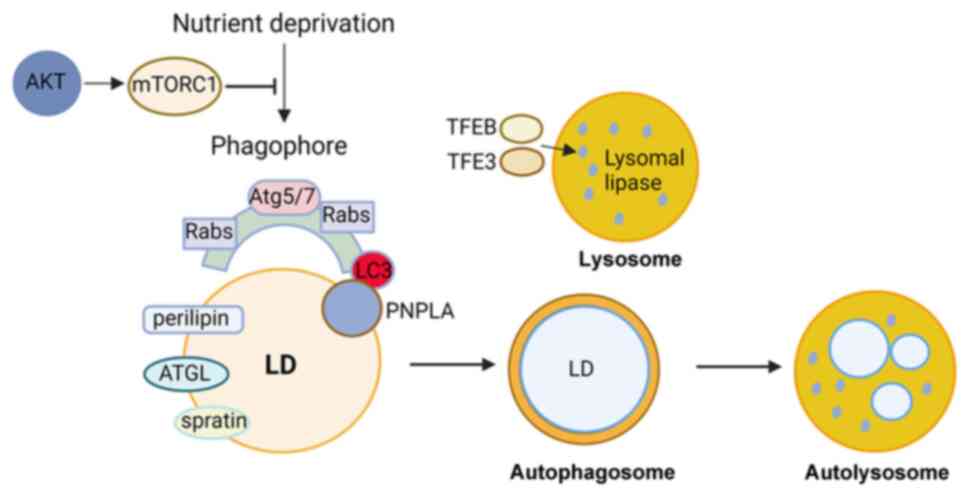
Lipophagy is regulated by certain proteins and
pathways. ATGs that mediate membrane fusion and the subsequent
degradation processes have been identified in recent studies.
Lipophagy is reported to begin with the identification of cargo by
the autophagosomal membrane through interaction with LC3 (85,86).
LC3 promotes the movement of cytoplasmic ATGL, another important
protein during lipophagy, to LDs by interacting with ATGL to induce
lipophagy. In the liver, ATGL accelerates lipophagy to mediate
catabolism of hepatic LDs via SIRT1 activity (87). Small regulatory Rab GTPase (Rab)
molecular switch families are indispensable in lipophagy (88). Rab7 serves an essential role in the
regulation of autolysosome-mediated lipid degradation in adipocytes
(89). Moreover, Rab7 can be
activated to enhance the recruitment of lysosomes and
multi-vesicular bodies to the surface of LDs during lipophagy under
nutrient deprivation conditions (89). The deficiency of Rab7 leads to
morphological alterations of multi-vesicular bodies, lysosomes and
autophagosomes, resulting in decreased lipophagy in hepatocellular
lysosomes (90). Rab10 forms a
complex with EH domain-binding protein 1 and EH domain-containing 2
to promote LC3-positive autophagic membrane migration to the LD
surface. Deletion of Rab10 causes lipophagy dysfunction and LD
accumulation (91). Lipases, such
as patatin-like phospholipase domain-containing enzyme (PNPLA)5
have been reported to contribute to lipophagy and autophagic
proteolysis (92). These lipases
serve key roles in the initial stage of lipophagy by recruiting
triglycerides and sterol esters, resulting in the formation of
autophagosomes (93,94). PNPLA8 regulates SREBP-2 to drive
lipophagy by interacting with LC3 in the hepatocytes of HFD-fed
mice (95). In energy-deprived
conditions, PNPLA3 mediates the formation of autophagosomes during
the lipophagy process in human hepatocytes, (96). Moreover, perilipin, which exists on
the surface of LDs, is removed before degradation by lipophagy,
which is mediated by chaperones through AMP-activated protein
kinase (AMPK) (97). Under
nutrient deprivation conditions, lipophagy is regulated by
falconoid X receptor, cAMP response element-binding protein, mTOR
or AMPK (98-101).
To meet the energy requirements of cells, lipophagy is activated,
leading to the breakdown of triglycerides in LDs under fasting
conditions. During this process, LDs are targeted by
autophagosomes, captured and broken down by LAL (15,102,103). Lysosomal lipase expression is
mediated by the lysosomal biogenesis transcription factor EB in
mouse hepatocytes and Caenorhabditis elegans (104). Furthermore, fork head homeobox
transcription factor1 is associated with lysosomal lipase and
induces lipophagy in adipocytes under dietary restriction (105). A recent study reported that
spartin, as a receptor localized to the LD surface, interacts with
the core autophagy machinery, and that spartin is required for the
delivery of LDs to lysosomes and spartin-deficiency in neurons
leads to LD accumulation in cultured human neurons or the murine
brain (106). Numerous functions
of lipophagy have been reported in different cellular processes
ranging from transdifferentiation to resistance to apoptosis
(Fig. 1). However, the role of
lipophagy in bone cells remains unclear.
Previous studies have reported that lipophagy is one
of the pivotal mechanisms by which patients experience lipotoxic
effects on cells in bone tissue, inducing osteoblastic cell
dysfunction (107,108). Autophagy/lipophagy in
osteoblastic cells has previously been verified by
co-administration of rapamycin (RAP), an autophagy promoter, and
3-MA, an autophagy inhibitor, with different concentrations of
high-lipid medium. In moderately-high lipid conditions, RAP
promoted the co-localization of LDs and autophagy-associated
proteins, which contributed to a reduction in lipid deposition in
osteoblasts and relieved the adverse effects of high-lipid
conditions on the proliferation and osteogenic differentiation of
osteoblasts, accompanied by increased activity of ALP,
mineralization of nodules and high expression of
osteogenic-associated proteins. Treatment with 3-MA decreased
lipophagy, weakened lipolysis and produced inverted oil-red
granules, and further suppressed osteoblast proliferation and
osteogenesis. However, at high-lipid concentrations, RAP inhibited
both osteoblast proliferation and osteogenic differentiation, and
3-MA improved the inhibitory effect of high-lipid conditions on
proliferation and osteogenesis by suppressing autophagy/lipophagy,
although the change in lipid deposition was reported to not be
significant (22). Subsequent
in vivo studies reported that autophagy/lipophagy was
activated in the bone tissue defect of a mouse model of
hyperlipidemia and in osteoblasts cultured in high-fat medium.
Specifically, a defect created on the femurs of mice fed a HFD was
assessed after 2 weeks of healing; the results showed that BV/TV
and BMD were decreased, and the number of new bones was
significantly reduced in the hyperlipidemia mouse model compared
with in wild-type mice. Although bone healing was promoted in
hyperlipidemic mice when RAP was used to enhance
autophagy/lipophagy, new bone was further reduced in hyperlipidemic
mice treated with 3-MA to inhibit autophagy/lipophagy (22). This study illustrates that local
promotion of autophagy/lipophagy reduces osteogenesis in
hyperlipidemia and provides a potential therapeutic strategy for
patients with poor bone metabolism induced by acquired
hyperlipidemia. A similar study reported that CH retarded
senescence in BMSCs in a dose-dependent manner by altering
autophagy and regulating LC3 expression (43). Furthermore, palmitate (PA) induced
normal human osteoblast apoptosis, which led to decreased
osteoblastogenesis and bone mineralization, whereas 3-MA-induced
inhibition of autophagy reduced apoptosis. These studies indicated
that lipophagy could be activated by lipids, such as CH and PA, in
osteoblastic cells and regulate cell function (109). These contradictory findings
indicate the need for more comprehensive and systematic studies on
the influence of lipophagy on the function of osteoblastic cells
and bone metabolism. Elucidating the role of lipophagy in bone
metabolism under hyperlipidemia conditions and the mechanism of
hyperlipidemia-induced dysfunction of osteoblastic cells is
challenging because of the complexity of cross-talk between
multiple organs.
Hyperlipidemia affects the function of osteoblastic
cells, the major functional cell of bone formation, by inducing an
inflammatory response in the bone cavity and osteoblastic cell
dysfunction by internalizing lipids. However, how lipids affect
osteoblastic cell function remains unknown. The present review
described the research progress on hyperlipidemia-induced
osteoporosis and discussed the possible mechanisms. Lipophagy in
osteoblastic cells can be activated by excessive lipid levels under
hyperlipidemia conditions to regulate lipid metabolism, and mediate
osteoblastic cell differentiation and bone formation, which would
be a novel mechanism for hyperlipidemia-induced bone disorders in
the future, although Pirih et al (25) preliminarily illustrated the role of
lipophagy in the disease. However, there are numerous issues that
need to be addressed during future investigation of the role of
lipophagy in regulating osteoblastic cell function. Firstly, the
types of lipids which affect osteoblastic cell function need to be
elucidated. Secondly, how osteoblastic cells respond to
hyperlipidemia conditions, and which molecules or pathways mediate
osteoblastic cell differentiation under hyperlipidemia conditions
need to be identified. Lastly, the potential for the use of
lipophagy as a therapeutic target for bone metabolism disorder
needs to be further assessed. Clarification of these points would
provide insight into the mechanism of hyperlipidemia-induced
osteoporosis, and provide references for the prevention and
treatment of osteoporosis for patients with hyperlipidemia.
Not applicable.
Funding: This work was supported by the Natural Science
Foundation of China (grant no. 82204543), the Shaanxi Province
Natural Science Foundation (grant nos. 2022JQ-822 and
2023-JC-QN-2486), the Natural Science Research Project of Shaanxi
Provincial Education Department (grant no. 22JS032), the Project of
Youth Innovation Team of Shaanxi Universities (grant no. 202056)
and the Xi'an Medical University Scientific Research Fund (grant
nos. 2021DOC11 and 2021DOC13).
Not applicable.
YH and PS wrote the manuscript. YL and QY wrote the
outline of the manuscript. YH, XC and RL collected and prepared the
related references. YH and AX drafted the manuscript. YH and PS
drew the figure. YH, XC and RL made substantial contributions to
data interpretation and analysis. Data authentication is not
applicable. All authors contributed to the article, and read and
approved the final version of the manuscript.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Hu LF, Yin C, Zhao F, Ali A, Ma J and Qian
A: Mesenchymal stem cells: Cell fate decision to osteoblast or
adipocyte and application in osteoporosis treatment. Int J Mol Sci.
19(360)2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Johnell O and Kanis JA: An estimate of the
worldwide prevalence and disability associated with osteoporotic
fractures. Osteoporos Int. 17:1726–1733. 2006.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Van Staa TP, Dennison EM, Leufkens HG and
Cooper C: Epidemiology of fractures in England and Wales. Bone.
29:517–522. 2001.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Hu X, Ma S, Yang C, Wang W and Chen L:
Relationship between senile osteoporosis and cardiovascular and
cerebrovascular diseases. Exp Ther Med. 17:4417–4420.
2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Tanko LB, Bagger YZ, Nielsen SB and
Christiansen C: Does serum cholesterol contribute to vertebral bone
loss in postmenopausal women? Bone. 32:8–14. 2003.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Trimpou P, Oden A, Simonsson T, Wilhelmsen
L and Landin-Wilhelmsen K: High serum total cholesterol is a
long-term cause of osteoporotic fracture. Osteoporos Int.
22:1615–1620. 2011.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Polyzos SA, Anastasilakis AD, Efstathiadou
ZA, Yavropoulou MP and Makras P: Postmenopausal osteoporosis
coexisting with other metabolic diseases: Treatment considerations.
Maturitas. 147:19–25. 2021.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yin C, Tian Y, Hu L, Yu Y, Wu Z, Zhang Y,
Wang X, Miao Z and Qian A: MACF1 alleviates aging-related
osteoporosis via HES1. J Cell Mol Med. 25:6242–6257.
2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kim SP, Li Z, Zoch ML, Frey JL, Bowman CE,
Kushwaha P, Ryan KA, Goh BC, Scafidi S, Pickett JE, et al: Fatty
acid oxidation by the osteoblast is required for normal bone
acquisition in a sex- and diet-dependent manner. JCI Insight.
2(e92704)2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Niemeier A, Niedzielska D, Secer R,
Schilling A, Merkel M, Enrich C, Rensen PCN and Heeren J: Uptake of
postprandial lipoproteins into bone in vivo: Impact on osteoblast
function. Bone. 43:230–237. 2008.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:1845–1846.
2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Pierrefite-Carle V, Santucci-Darmanin S,
Breuil V, Camuzard O and Carle GF: Autophagy in bone: Self-eating
to stay in balance. Ageing Res Rev. 24:206–217. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Wang J, Zhang Y, Cao J, Wang Y, Anwar N,
Zhang Z, Zhang D, Ma Y, Xiao Y, Xiao L and Wang X: The role of
autophagy in bone metabolism and clinical significance. Autophagy.
19:2409–2427. 2023.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Shin DW: Lipophagy: Molecular mechanisms
and implications in metabolic disorders. Mol Cells. 43:686–693.
2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Singh R, Kaushik S, Wang Y, Xiang Y, Novak
I, Komatsu M, Tanaka K, Cuervo AM and Czaja MJ: Autophagy regulates
lipid metabolism. Nature. 458:1131–1135. 2009.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Li W, He P, Huang Y, Li YF, Lu J, Li M,
Kurihara H, Luo Z, Meng T, Onishi M, et al: Selective autophagy of
intracellular organelles: Recent research advances. Theranostics.
11:222–256. 2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Schulze RJ, Sathyanarayan A and Mashek DG:
Breaking fat: The regulation and mechanisms of lipophagy. Biochim
Biophys Acta Mol Cell Biol Lipids. 1862:1178–1187. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Miao J, Zang X, Cui X and Zhang J:
Autophagy, hyperlipidemia, and atherosclerosis. Adv Exp Med Biol.
1207:237–264. 2020.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Barros JAS, Siqueira JAB, Cavalcanti JHF,
Araújo WL and Avin-Wittenberg T: Multifaceted roles of plant
autophagy in lipid and energy metabolism. Trends Plant Sci.
25:1141–1153. 2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Maan M, Peters JM, Dutta M and Patterson
AD: Lipid metabolism and lipophagy in cancer. Biochem Biophys Res
Commun. 504:582–589. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Liu Q, Wang YM and Gu HF: Lipophagy in
atherosclerosis. Clin Chim Acta. 511:208–214. 2020.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ji C, Zhang Z, Xu X, Song D and Zhang D:
Hyperlipidemia impacts osteogenesis via lipophagy. Bone.
167(116643)2023.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Ohsaki Y, Kawai T, Yoshikawa Y, Cheng J,
Jokitalo E and Fujimoto T: PML isoform II plays a critical role in
nuclear lipid droplet formation. J Cell Biol. 212:29–38.
2016.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Greenberg AS, Coleman RA, Kraemer FB,
McManaman JL, Obin MS, Puri V, Yan QW, Miyoshi H and Mashek DG: The
role of lipid droplets in metabolic disease in rodents and humans.
J Clin Invest. 121:2102–2110. 2011.PubMed/NCBI View
Article : Google Scholar
|
|
25
|
Pirih F, Lu J, Ye F, Bezouglaia O, Atti E,
Ascenzi MG, Tetradis S, Demer L, Aghaloo T and Tintut Y: Adverse
effects of hyperlipidemia on bone regeneration and strength. J Bone
Miner Res. 27:309–318. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
26
|
Almeida M, Ambrogini E, Han L, Manolagas
SC and Jilka RL: Increased lipid oxidation causes oxidative stress,
increased peroxisome proliferator-activated receptor-gamma
expression, and diminished pro-osteogenic Wnt signaling in the
skeleton. J Biol Chem. 284:27438–2748. 2009.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Pelton K, Krieder J, Joiner D, Freeman MR,
Goldstein SA and Solomon KR: Hypercholesterolemia promotes an
osteoporotic phenotype. Am J Pathol. 181:928–936. 2012.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Demigne C, Bloch-Faure M, Picard N, Sabboh
H, Besson C, Rémésy C, Geoffroy V, Gaston AT, Nicoletti A, Hagège
A, et al: Mice chronically fed a westernized experimental diet as a
model of obesity, metabolic syndrome and osteoporosis. Eur J Nutr.
45:298–306. 2006.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Parhami F, Tintut Y, Beamer WG, Gharavi N,
Goodman W and Demer LL: Atherogenic high-fat diet reduces bone
mineralization in mice. J Bone Miner Res. 16:182–188.
2001.PubMed/NCBI View Article : Google Scholar
|
|
30
|
You L, Sheng ZY, Tang CL, Chen L, Pan L
and Chen JY: High cholesterol diet increases osteoporosis risk via
inhibiting bone formation in rats. Acta Pharmacol Sin.
32:1498–1504. 2011.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Li F and Zhang H: Lysosomal acid lipase in
lipid metabolism and beyond. Arterioscler Thromb Vasc Biol.
39:850–856. 2019.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Du H, Heur M, Duanmu M, Grabowski GA, Hui
DY, Witte DP and Mishra J: Lysosomal acid lipase-deficient mice:
Depletion of white and brown fat, severe hepatosplenomegaly, and
shortened life span. J Lipid Res. 42:489–500. 2001.PubMed/NCBI
|
|
33
|
Helderman RC, Whitney DG, Duta-Mare M,
Akhmetshina A, Vujic N, Jayapalan S, Nyman JS, Misra BB, Rosen CJ,
Czech MP, et al: Loss of function of lysosomal acid lipase (LAL)
profoundly impacts osteoblastogenesis and increases fracture risk
in humans. Bone. 148(115946)2021.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Nakamichi R, Hayashi K and Itoh H: Effects
of high glucose and lipotoxicity on diabetic podocytes. Nutrients.
13(241)2021.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Zhang B, Li X, Liu G, Zhang C, Zhang X,
Shen Q, Sun G and Sun X: Peroxiredomin-4 ameliorates
lipotoxicity-induced oxidative stress and apoptosis in diabetic
cardiomyopathy. Biomed Pharmacother. 141(111780)2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Singh L, Tyagi S, Myers D and Duque G:
Good, bad, or ugly: The biological roles of bone marrow fat. Curr
Osteoporos Rep. 16:130–137. 2018.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Veldhuis-Vlug AG and Rosen CJ: Clinical
implications of bone marrow adiposity. J Intern Med. 283:121–139.
2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Al Saedi A, Bermeo S, Plotkin L, Myers DE
and Duque G: Mechanisms of palmitate-induced lipotoxicity in
osteocytes. Bone. 127:353–359. 2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Parhami F, Jackson SM, Tintut Y, Le V,
Balucan JP, Territo M and Demer LL: Atherogenic diet and minimally
oxidized low density lipoprotein inhibit osteogenic and promote
adipogenic differentiation of marrow stromal cells. J Bone Miner
Res. 14:2067–2078. 1999.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Yin W, Li Z and Zhang W: Modulation of
bone and marrow niche by cholesterol. Nutrients.
11(1394)2019.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Arai M, Shibata Y, Pugdee K, Abiko Y and
Ogata Y: Effects of reactive oxygen species (ROS) on antioxidant
system and osteoblastic differentiation in MC3T3-E1 cells. IUBMB
Life. 59:27–33. 2007.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Li K, Xiu C, Zhou Q, Ni L, Du J, Gong T,
Li M, Saijilafu Yang H and Chen J: A dual role of cholesterol in
osteogenic differentiation of bone marrow stromal cells. J Cell
Physiol. 234:2058–2066. 2019.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Zhang M, Du Y, Lu R, Shu Y, Zhao W, Li Z,
Zhang Y, Liu R, Yang T, Luo S, et al: Cholesterol retards
senescence in bone marrow mesenchymal stem cells by modulating
autophagy and ROS/p53/p21(Cip1/Waf1) pathway. Oxid Med Cell Longev.
2016(7524308)2016.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Parhami F: Possible role of oxidized
lipids in osteoporosis: Could hyperlipidemia be a risk factor?
Prostaglandins Leukot Essent Fatty Acids. 68:373–378.
2003.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Lim HY, Rutkowski JM, Helft J, Reddy ST,
Swartz MA, Randolph GJ and Angeli V: Hypercholesterolemic mice
exhibit lymphatic vessel dysfunction and degeneration. Am J Pathol.
175:1328–1337. 2009.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Soehnlein O and Swirski FK:
Hypercholesterolemia links hematopoiesis with atherosclerosis.
Trends Endocrinol Metab. 24:129–136. 2013.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Gleissner CA, Leitinger N and Ley K:
Effects of native and modified low-density lipoproteins on monocyte
recruitment in atherosclerosis. Hypertension. 50:276–283.
2007.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Gough AK, Lilley J, Eyre S, Holder RL and
Emery P: Generalised bone loss in patients with early rheumatoid
arthritis. Lancet. 344:23–27. 1994.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Romas E: Bone loss in inflammatory
arthritis: Mechanisms and therapeutic approaches with
bisphosphonates. Best Pract Res Clin Rheumatol. 19:1065–1079.
2005.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Roldan JF, Del Rincon I and Escalante A:
Loss of cortical bone from the metacarpal diaphysis in patients
with rheumatoid arthritis: Independent effects of systemic
inflammation and glucocorticoids. J Rheumatol. 33:508–516.
2006.PubMed/NCBI
|
|
51
|
Redlich K, Gortz B, Hayer S, Zwerina J,
Doerr N, Kostenuik P, Bergmeister H, Kollias G, Steiner G, Smolen
JS and Schett G: Repair of local bone erosions and reversal of
systemic bone loss upon therapy with anti-tumor necrosis factor in
combination with osteoprotegerin or parathyroid hormone in tumor
necrosis factor-mediated arthritis. Am J Pathol. 164:543–555.
2004.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Gilbert L, He X, Farmer P, Rubin J, Drissi
H, van Wijnen AJ, Lian JB, Stein GS and Nanes MS: Expression of the
osteoblast differentiation factor RUNX2 (Cbfa1/AML3/Pebp2alpha A)
is inhibited by tumor necrosis factor-alpha. J Biol Chem.
277:2695–2701. 2002.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Kaneki H, Guo R, Chen D, Yao Z, Schwarz
EM, Zhang YE, Boyce BF and Xing L: Tumor necrosis factor promotes
Runx2 degradation through up-regulation of Smurf1 and Smurf2 in
osteoblasts. J Biol Chem. 281:4326–4333. 2006.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Takano M, Otsuka F, Matsumoto Y, Inagaki
K, Takeda M, Nakamura E, Tsukamoto N, Miyoshi T, Sada KE and Makino
H: Peroxisome proliferator-activated receptor activity is involved
in the osteoblastic differentiation regulated by bone morphogenetic
proteins and tumor necrosis factor-alpha. Mol Cell Endocrinol.
348:224–232. 2012.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Ding J, Ghali O, Lencel P, Broux O,
Chauveau C, Devedjian JC, Hardouin P and Magne D: TNF-alpha and
IL-1beta inhibit RUNX2 and collagen expression but increase
alkaline phosphatase activity and mineralization in human
mesenchymal stem cells. Life Sci. 84:499–504. 2009.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Hughes FJ and Howells GL: Interleukin-6
inhibits bone formation in vitro. Bone Miner. 21:21–28.
1993.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Krum SA, Chang J, Miranda-Carboni G and
Wang CY: Novel functions for NFκB: Inhibition of bone formation.
Nat Rev Rheumatol. 6:607–611. 2010.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Li X, Zhang Y, Kang H, Liu W, Liu P, Zhang
J, Harris SE and Wu D: Sclerostin binds to LRP5/6 and antagonizes
canonical Wnt signaling. J Biol Chem. 280:19883–19887.
2005.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Winkler DG, Sutherland MK, Geoghegan JC,
Yu C, Hayes T, Skonier JE, Shpektor D, Jonas M, Kovacevich BR,
Staehling-Hampton K, et al: Osteocyte control of bone formation via
sclerostin, a novel BMP antagonist. EMBO J. 22:6267–6276.
2003.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Mason JJ and Williams BO: SOST and DKK:
Antagonists of LRP family signaling as targets for treating bone
disease. J Osteoporos. 2010(460120)2010.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Tanida I: Autophagosome formation and
molecular mechanism of autophagy. Antioxid Redox Signal.
14:2201–2214. 2011.PubMed/NCBI View Article : Google Scholar
|
|
62
|
He C and Klionsky DJ: Regulation
mechanisms and signaling pathways of autophagy. Annu Rev Genet.
43:67–93. 2009.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Parzych KR and Klionsky DJ: An overview of
autophagy: Morphology, mechanism, and regulation. Antioxid Redox
Signal. 20:460–473. 2014.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Guan JL, Simon AK, Prescott M, Menendez
JA, Liu F, Wang F, Wang C, Wolvetang E, Vazquez-Martin A and Zhang
J: Autophagy in stem cells. Autophagy. 9:830–849. 2013.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Mizushima N and Levine B: Autophagy in
mammalian development and differentiation. Nat Cell Biol.
12:823–830. 2010.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Rubinsztein DC, Marino G and Kroemer G:
Autophagy and aging. Cell. 146:682–695. 2011.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Rubinsztein DC: The roles of intracellular
protein-degradation pathways in neurodegeneration. Nature.
443:780–786. 2006.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Ciechanover A: Intracellular protein
degradation: From a vague idea thru the lysosome and the
ubiquitin-proteasome system and onto human diseases and drug
targeting. Cell Death Differ. 12:1178–1190. 2005.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Mizushima N and Klionsky DJ: Protein
turnover via autophagy: Implications for metabolism. Annu Rev Nutr.
27:19–40. 2007.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Wei H, Wei S, Gan B, Peng X, Zou W and
Guan JL: Suppression of autophagy by FIP200 deletion inhibits
mammary tumorigenesis. Genes Dev. 25:1510–1527. 2011.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Kimmelman AC: The dynamic nature of
autophagy in cancer. Genes Dev. 25:1999–2010. 2011.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Wang Z, Deng Z, Gan J, Zhou G, Shi T, Wang
Z, Huang Z, Qian H, Bao N, Guo T, et al: TiAl (6)V(4) particles
promote osteoclast formation via autophagy-mediated downregulation
of interferon-beta in osteocytes. Acta Biomater. 48:489–498.
2017.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Wang Z, Liu N, Liu K, Zhou G, Gan J, Wang
Z, Shi T, He W, Wang L, Guo T, et al: Autophagy mediated CoCrMo
particle-induced peri-implant osteolysis by promoting osteoblast
apoptosis. Autophagy. 11:2358–2369. 2015.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Piemontese M, Onal M, Xiong J, Han L,
Thostenson JD, Almeida M and O'Brien CA: Low bone mass and changes
in the osteocyte network in mice lacking autophagy in the
osteoblast lineage. Sci Rep. 6(24262)2016.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Kanda N and Watanabe S: 17-beta-estradiol
inhibits oxidative stress induced apoptosis in keratinocytes by
promoting Bcl-2 expression. J Invest Dermatol. 121:1500–1509.
2003.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Sun X, Yang X, Zhao Y, Li Y and Guo L:
Effects of 17β-estradiol on mitophagy in the murine MC3T3-E1
osteoblast cell line is mediated via g protein-coupled estrogen
receptor and the ERK1/2 signaling pathway. Med Sci Monit.
24:903–911. 2018.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Bartolome A, Lopez-Herradon A,
Portal-Nunez S, García-Aguilar A, Esbrit P, Benito M and Guillén C:
Autophagy impairment aggravates the inhibitory effects of high
glucose on osteoblast viability and function. Biochem J.
455:329–337. 2013.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Qi M, Zhang L, Ma Y, Shuai Y, Li L, Luo K,
Liu W and Jin Y: Autophagy maintains the function of bone marrow
mesenchymal stem cells to prevent estrogen deficiency-induced
osteoporosis. Theranostics. 7:4498–4516. 2017.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Nollet M, Santucci-Darmanin S, Breuil V,
Al-Sahlanee R, Cros C, Topi M, Momier D, Samson M, Pagnotta S,
Cailleteau L, et al: Autophagy in osteoblasts is involved in
mineralization and bone homeostasis. Autophagy. 10:1965–1977.
2014.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Li H, Li D, Ma Z, Qian Z, Kang X, Jin X,
Li F, Wang X, Chen Q, Sun H and Wu S: Defective autophagy in
osteoblasts induces endoplasmic reticulum stress and causes
remarkable bone loss. Autophagy. 14:1726–1741. 2018.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Li W, Zhang S, Liu J, Liu Y and Liang Q:
Vitamin K2 stimulates MC3T3-E1 osteoblast differentiation and
mineralization through autophagy induction. Mol Med Rep.
19:3676–3684. 2019.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Vidoni C, Ferraresi A, Secomandi E,
Vallino L, Gardin C, Zavan B, Mortellaro C and Isidoro C: Autophagy
drives osteogenic differentiation of human gingival mesenchymal
stem cells. Cell Commun Signal. 17(98)2019.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Al Saedi A, Myers DE, Stupka N and Duque
G: 1,25(OH)2D3 ameliorates palmitate-induced
lipotoxicity in human primary osteoblasts leading to improved
viability and function. Bone. 141(115672)2020.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Singh R and Cuervo AM: Lipophagy:
Connecting autophagy and lipid metabolism. Int J Cell Biol.
2012(282041)2012.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Wang CW: Lipid droplets, lipophagy, and
beyond. Biochim Biophys Acta. 1861:793–805. 2016.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Sathyanarayan A, Mashek MT and Mashek DG:
ATGL promotes autophagy/lipophagy via SIRT1 to control hepatic
lipid droplet catabolism. Cell Rep. 19:1–9. 2017.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Kiss RS and Nilsson T: Rab proteins
implicated in lipid storage and mobilization. J Biomed Res.
28:169–177. 2014.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Lizaso A, Tan KT and Lee YH: β-adrenergic
receptor-stimulated lipolysis requires the RAB7-mediated
autolysosomal lipid degradation. Autophagy. 9:1228–1243.
2013.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Schroeder B, Schulze RJ, Weller SG,
Sletten AC, Casey CA and McNiven MA: The small GTPase Rab7 as a
central regulator of hepatocellular lipophagy. Hepatology.
61:1896–1907. 2015.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Li Z, Schulze RJ, Weller SG, Krueger EW,
Schott MB, Zhang X, Casey CA, Liu J, Stöckli J, James DE and
McNiven MA: A novel Rab10-EHBP1-EHD2 complex essential for the
autophagic engulfment of lipid droplets. Sci Adv.
2(e1601470)2016.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Dupont N, Chauhan S, Arko-Mensah J,
Castillo EF, Masedunskas A, Weigert R, Robenek H, Proikas-Cezanne T
and Deretic V: Neutral lipid stores and lipase PNPLA5 contribute to
autophagosome biogenesis. Curr Biol. 24:609–620. 2014.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Shpilka T, Welter E, Borovsky N, Amar N,
Mari M, Reggiori F and Elazar Z: Lipid droplets and their component
triglycerides and steryl esters regulate autophagosome biogenesis.
EMBO J. 34:2117–2131. 2015.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Ward C, Martinez-Lopez N, Otten EG,
Carroll B, Maetzel D, Singh R, Sarkar S and Korolchuk VI:
Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim
Biophys Acta. 1861:269–284. 2016.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Kim KY, Jang HJ, Yang YR, Park KI, Seo J,
Shin IW, Jeon TI, Ahn SC, Suh PG, Osborne TF and Seo YK:
Corrigendum: SREBP-2/PNPLA8 axis improves non-alcoholic fatty liver
disease through activation of autophagy. Sci Rep.
6(37794)2016.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Negoita F, Blomdahl J, Wasserstrom S,
Winberg ME, Osmark P, Larsson S, Stenkula KG, Ekstedt M, Kechagias
S, Holm C and Jones HA: PNPLA3 variant M148 causes resistance to
starvation-mediated lipid droplet autophagy in human hepatocytes. J
Cell Biochem. 120:343–356. 2019.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Kaushik S and Cuervo AM: Degradation of
lipid droplet-associated proteins by chaperone-mediated autophagy
facilitates lipolysis. Nat Cell Biol. 17:759–770. 2015.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Li Y, Yang P, Zhao L, Chen Y, Zhang X,
Zeng S, Wei L, Varghese Z, Moorhead JF, Chen Y and Ruan XZ: CD36
plays a negative role in the regulation of lipophagy in hepatocytes
through an AMPK-dependent pathway. J Lipid Res. 60:844–855.
2019.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Seok S, Fu T, Choi SE, Li Y, Zhu R, Kumar
S, Sun X, Yoon G, Kang Y, Zhong W, et al: Transcriptional
regulation of autophagy by an FXR-CREB axis. Nature. 516:108–111.
2014.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Zhang H, Yan S, Khambu B, Ma F, Li Y, Chen
X, Martina JA, Puertollano R, Li Y, Chalasani N and Yin XM: Dynamic
MTORC1-TFEB feedback signaling regulates hepatic autophagy,
steatosis and liver injury in long-term nutrient oversupply.
Autophagy. 14:1779–1795. 2018.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Zhang Z, Yao Z, Chen Y, Qian L, Jiang S,
Zhou J, Shao J, Chen A, Zhang F and Zheng S: Lipophagy and liver
disease: New perspectives to better understanding and therapy.
Biomed Pharmacother. 97:339–348. 2018.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Grumet L, Eichmann TO, Taschler U, Zierler
KA, Leopold C, Moustafa T, Radovic B, Romauch M, Yan C, Du H, et
al: Lysosomal acid lipase hydrolyzes retinyl ester and affects
retinoid turnover. J Biol Chem. 291:17977–17987. 2016.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Zechner R, Madeo F and Kratky D: Cytosolic
lipolysis and lipophagy: Two sides of the same coin. Nat Rev Mol
Cell Biol. 18:671–684. 2017.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Settembre C, De Cegli R, Mansueto G, Saha
PK, Vetrini F, Visvikis O, Huynh T, Carissimo A, Palmer D, Klisch
TJ, et al: TFEB controls cellular lipid metabolism through a
starvation-induced autoregulatory loop. Nat Cell Biol. 15:647–558.
2013.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Barbato DL, Tatulli G, Aquilano K and
Ciriolo MR: FoxO1 controls lysosomal acid lipase in adipocytes:
Implication of lipophagy during nutrient restriction and metformin
treatment. Cell Death Dis. 4(e861)2013.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Chung J, Park J, Lai ZW, Lambert TJ,
Richards RC, Zhang J, Walther TC and Farese RV Jr: The Troyer
syndrome protein spartin mediates selective autophagy of lipid
droplets. Nat Cell Biol. 25:1101–1110. 2023.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Zhang X, Evans TD, Jeong SJ and Razani B:
Classical and alternative roles for autophagy in lipid metabolism.
Curr Opin Lipidol. 29:203–211. 2018.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Nguyen TB and Olzmann JA: Lipid droplets
and lipotoxicity during autophagy. Autophagy. 13:2002–2003.
2017.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Gunaratnam K, Vidal C, Boadle R, Thekkedam
C and Duque G: Mechanisms of palmitate-induced cell death in human
osteoblasts. Biol Open. 2:1382–1389. 2013.PubMed/NCBI View Article : Google Scholar
|