Introduction
According to gene expression profiling, diffuse
large B-cell lymphoma (DLBCL) can be classified into three main
subtypes: i) Activated B-cell-like (ABC); ii) germinal center
B-cell-like; and iii) primary mediastinal B-cell lymphoma (1-3).
Standard chemoimmunotherapy drugs, such as rituximab,
cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP),
as well as novel drugs combined with chemotherapy (R-CHOP+X), have
resulted in a notable therapeutic effect in ~40% of patients with
DLBCL (4,5). However, the ABC-DLBCL subtype has
been found to be more resistant to the aforementioned standard
treatments (6,7). One contributing factor to this
resistance is a mutation in CD79B, which encodes a subunit of the
B-cell receptor (BCR) (8). Several
studies have demonstrated that BCR signaling remains active in
ABC-DLBCL (2,8,9).
Another possible explanation for the resistance of ABC-DLBCL is the
inability to undergo class switch recombination (CSR) due to the
deletion of the Sµ region in immunoglobulin (Ig) genes (10).
Following antigen stimulation, the BCR triggers a
continuous proliferation of B cells. As a result, a significant
number of large B cells accumulate in the germinal center (GC), but
they fail to differentiate into plasma cells and memory B cells,
ultimately leading to the development of DLBCL (2,9).
During B cell differentiation, activation-induced cytidine
deaminase (AID) is upregulated by TLR signaling through BCR
signaling. AID converts cytosine (C) to uracil (U) to generate the
U:G mismatch, and the U:G mismatch is subsequently repaired through
DNA replication, base excision repair or mismatch repair (11,12).
However, incomplete repair leads to mutations or deletions, which
are essential in inducing somatic hypermutation (SHM) in the
variable region of Ig genes and CSR in the constant region of Ig
genes (11,12). Accurate and effective SHM and CSR
could facilitate antibody affinity maturation (11,12).
ABC-DLBCL is a type of lymphoma that arises from the abnormal
process of large B cells that possess high levels of AID, but fail
to complete antibody affinity maturation (13-15).
In approximately half of patients with ABC-DLBCL, the Sµ region in
Ig genes, which is a key target of AID in SHM and CSR, is deleted
(10). Generally, it is considered
that Sµ deletion or translocation are the direct result of
AID-mediated abnormal CSR (10).
At present, the majority of studies focus on the role of AID in
DLBCL, including deamination induced mutations and chromosome
translocations (1,2). It has been widely reported that the
overexpression of AID of is associated with B-cell derived
malignancies including ABC-DLBCL (13), and AID expression contributes to
poor prognosis of patients with DLBCL treated with CHOP-based
chemotherapy (16). This indicates
a negative function of AID in the clinical treatment of DLBCL.
However, previous research has shown that AID has a positive effect
in inhibiting DLBCL progression (14). Therefore, restoring and enhancing
the ability of AID to mediate antibody affinity maturation may be a
viable option for developing effective therapy for ABC-DLBCL.
Proteasome inhibitors, such as MG132, is
well-established for their ability to inhibit the
ubiquitin-proteasome system, which regulates the levels of numerous
cancer-related molecules in cells, such as cyclins,
cyclin-dependent kinases, tumor suppressors and nuclear factor-κB
(17-20).
A previous study demonstrated the potent anti-lymphoma effect of
MG132 in DLBCL (13). The present
study aims to elucidate the mechanism of the anti-lymphoma effect
of MG132 on ABC-DLBCL, which has been shown to be more refractory
in clinical settings (1-3).
It has been reported that the proteasome pathway plays a role in
the post-transcriptional regulation of AID (21). Therefore, the present study aims to
explore the ability of MG132 to inhibit AID degradation through the
proteasome pathway, and develop the possibility of proteasome
inhibitors in the treatment for ABC-DLBCL.
Materials and methods
In vivo tumor cell engraftment and
treatment of mice
The present study utilized 10 female NOD/SCID mice
(age, 6-8 weeks; Henan Skobes Biotechnology Co., Ltd.) raised in an
independent ventilation system with a constant temperature of 22˚C
and a light/dark cycle of 12/12 h. These mice (weight, 18-22 g)
were maintained in specific pathogen-free conditions at Xinxiang
Medical University (Xinxiang, China). A mouse model of human DLBCL
was established by subcutaneously injecting 2x107
OCI-LY10 cells into the right flank of the NOD/SCID mice (14). Tumor volume was quantified using
the formula: Volume=(length x width2)/2. Once the
average tumor volume reached 80-100 mm3, treatment with
MG132 commenced. The NOD/SCID mice bearing OCI-LY10 tumors were
then allocated into a control group (n=5) or a treatment group
(n=5). Mice in the treatment group received intraperitoneal
injections of MG132 at a concentration of 50 mg/kg (200 µl), while
the control group was administered an equal volume of the solvent
(4% DMSO + 30% PEG300 + 20% propylene glycol + ddH2O)
via the same route (14). Tumor
growth was monitored every two days. All mice were sacrificed 45
days following the initiation of MG132 therapy. Euthanasia was
performed using an overdose of sodium pentobarbital (100 mg/kg),
administered intravenously to ensure rapid and painless loss of
consciousness. The tumors in the control and treatment groups were
excised and weighed. The present study was approved by The Ethics
Committee of Xinxiang Medical University (Xinxiang, China; approval
no. XYLL-2022001253), and all the animal procedures were performed
in accordance with the ‘Guide for the Care and Use of Laboratory
Animals’ published by the National Institutes of Health (22).
Constructs and cells
The pCas9-AID constructs were successfully generated
through the ligation of gRNA targeting AID into the
pL-CRISPR.EFS.PAC plasmids (cat. no. 57828; Addgene, Inc.)
(12,13). The specific target sequences are
detailed in Table SI (NCBI
accession no. NM_020661.4). These gRNAs were designed using online
software E-CRISPR (http://www.e-crisp.org/) to generate AID knock out.
Additionally, the primer sequences utilized for amplifying the AID
cDNA to produce the pWPI-AID-GFP lentivirus constructs are provided
in Table SII.
The OCI-LY10 ABC-DLBCL cell line was acquired from
the BeNa Culture Collection (Beijing Beina Chunglian Institute of
Biotechnology; cat. no. BNCC337742). The 293T cells were maintained
in the cell bank of Xinxiang Key Laboratory of Tumor Vaccine and
Immunotherapy (Xinxiang, China). The AIDKO OCI-LY10 ABC-DLBCL cell
line was generated by the AIDKO lentivirus, and the lentivirus was
obtained from the supernatant of 293T cells co-transfected by
pCas9-AID constructs (or pL-CRISPR.EFS.PAC plasmids as control) and
virus generation plasmids including ΔR9 and pVSVG. Both cell lines
were cultured in a humidified incubator at 37˚C and 5%
CO2, using either Iscove's modified Dulbecco's medium or
Dulbecco's modified Eagle's medium (Hyclone; Cytiva) as the base
medium, supplemented with 10% fetal bovine serum (MilliporeSigma),
non-essential amino acids and a 1% penicillin-streptomycin
mixture.
To detect AID ubiquitination, pWPI-AID-GFP and Ub-HA
constructs were co-transfected into 293T cells and cultured for 24,
48 and 72 h at 37˚C, respectively. Following these time points,
cells were harvested for total protein extraction and subsequent
immunoprecipitation analysis.
To assess MG132-induced cell death, cells were
incubated with MG132 (10 µM; Selleck Chemicals; cat. no. S2619) for
8 h at 37˚C. The MG132 powder was prepared using DMSO as a solvent.
As a control, an equivalent volume of DMSO was used in the
treatment of the control cells.
RNA extraction and RT-qPCR
Total RNA was extracted from OCI-LY10 ABC-DLBCL cell
pellets using TRIzol® reagent (Invitrogen; Thermo Fisher
Scientific, Inc.; cat. no. 15596026) in accordance with the
manufacturer's protocol. Subsequently, cDNA was synthesized
utilizing the PrimeScript™ RT Reagent Kit (Takara Bio, Inc.; cat.
no. RR037A) using reverse transcription for 15 min at 37˚C and
terminated for 5 sec at 85˚C. qPCR was conducted on the
QuantStudio™ 5 System (Applied Biosystems; Thermo Fisher
Scientific, Inc.). The thermocycling conditions were as follows: 15
sec at 95˚C, 30 sec at 60˚C and 30 sec at 72˚C, for a total of 40
cycles. The relative mRNA levels were determined using the
2-ΔΔCq method (23)
calculated using Microsoft Office 2016, with the expression of
β-actin taken as the internal control. The primer sequences
employed for the quantification of AID and β-actin transcription
are provided in Table SIII.
Flow cytometry and antibodies
To assess the apoptosis rate of OCI-LY10 ABC-DLBCL
cells induced by MG132, the treated cells were harvested and twice
washed with 1X PBS at 4˚C. The cells were then incubated with
anti-PI and anti-Annexin V (BD Biosciences; cat. no. 556547) for 15
min at room temperature. Following incubation, the cells were
resuspended in flow cytometry buffer and subjected to analysis via
flow cytometry. All data were acquired and processed using a
CytoFLEX Flow Cytometer (with BECKMAN CytoExpert version 2.4)
(Beckman Coulter, Inc.).
Immunoblot analysis
For protein extraction, the OCI-LY10 cell pellet was
lysed in RIPA buffer (cat. no. P0013B; Beyotime Institute of
Biotechnology) for 30 min. The RIPA preparation included 50 mM Tris
(pH 7.4), 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate and
0.1% SDS. The lysates were subsequently centrifuged at 13,000 x g
for 20 min at 4˚C, and the protein-containing supernatant was
collected. The quality and concentration of the total protein were
determined by BCA protein assay Kit (Beyotime Institute of
Biotechnology). Protein samples (20-100 µg per lane) were then
loaded onto 10% (w/v) Tris-HCl SDS-PAGE gels for electrophoresis,
transferred to a PVDF membrane (MilliporeSigma), and subjected to
blotting. The membrane was blocked with 5% (w/v) skimmed milk, then
probed with an anti-AID antibody (1:1,000; cat. no. 4959; CST
Biological Reagents Co., Ltd.) and anti-GAPDH (1:5,000; cat. no.
ab9485; Abcam) served as the loading control. The primary antibody
incubation was performed at 4˚C overnight. The signal was detected
using a goat anti-rabbit secondary antibody conjugated with
horseradish peroxidase (1:1,000; cat. no. 31460; Thermo Fisher
Scientific, Inc.). The secondary anti-rabbit antibody was incubated
with the membrane for 1 h at room temperature. The band signals
were captured using the FUSION FX7 system (Vilber Lourmat).
Immunoprecipitation
For protein extraction, the OCI-LY10 cell pellet was
lysed in RIPA buffer (Beyotime Institute of Biotechnology) for 30
min. The lysates were subsequently centrifuged at 13,000 x g for 20
min at 4˚C, and the protein-containing supernatant was collected.
The quality and concentration of the total protein were determined
by BCA protein assay Kit (Beyotime Institute of Biotechnology).
Following pre-clearance of the chromatin using Dynabeads Protein G
beads (2X; 25 µl for each IP reaction; cat. no. 10003D; Invitrogen;
Thermo Fisher Scientific, Inc.), a portion of the aliquot was set
aside as the input sample. Subsequently, proteins from
5x106 cells were incubated with 5 µg of a specific
antibody or normal goat IgG (Santa Cruz Biotechnology, Inc.; cat.
no. sc2346) overnight at 4˚C. Anti-GFP (Abcam; cat. no. ab13970)
immune complexes were then captured by incubating with Dynabeads
Protein G beads (2X; 25 µl for each IP reaction) (2X; Invitrogen;
Thermo Fisher Scientific, Inc.; cat. no. 10003D) for 3 h. The beads
were washed at 4˚C using RIPA buffer containing varying
concentrations of NaCl. The proteins from the pull down were
denatured at 100˚C and loaded onto SDS-PAGE gels for subsequent
immunoblotting.
Detection of CSR
The AID-/- mice were a gift from
Professor Ji of Xi'an Jiaotong University (Xi'an, China) (14,15),
and were bred in the Laboratory Animal Centre of Xinxiang Medical
University (Xinxiang, China). The WT and AID-/- C57 mice
were confirmed to carry the homozygous disruption of the AID gene
according to the generation of AID-/- mice reported by
Honjo T (24). C57 and
AID-/- mice were administered a tail vein injection of
MG132 (10 µg/kg/day) for 24 h (25). Splenic resting B cells were
purified by negative selection using anti-CD43-conjugated
microbeads (Miltenyi Biotec GmbH) (26). A total of ~3-5x107
splenic resting B cells were obtained from one mouse spleen.
Purified B cells were cultured at 5x105 cells/ml in RPMI
medium supplemented with 15% fetal bovine serum (MilliporeSigma),
non-essential amino acids, penicillin-streptomycin (1%) and
β-mercaptoethanol (50 µM). Cells were cultured in a humidified
incubator at 37˚C and 5% CO2. Cells were cultured in the
presence of LPS and IL-4 for 5 days: i) LPS (50 µg/ml); and ii) LPS
(3 µg/ml) plus IL-4 (50 ng/ml), to induce class switching to IgG1,
IgG3 and IgE. MG132 (5 µm) treatment was sustained throughout the
stimulation process. The antibody class switch was detected using
RT-qPCR. The relative mRNA levels were determined using the
2-ΔΔCq method (23)
calculated using Microsoft Office 2016, with the expression of
β-actin taken as the internal control. The primers for RT-qPCR
performed as aforementioned are listed in Table SIV (NCBI accession no. MF430613.1,
V00818.1 and KU613484.1).
Bioinformatics
The expression levels of AID in clinical specimens
were accomplished through interrogating the online databases GENT2
(http://gent2.appex.kr/gent2/) by
searching tissue, blood disease and gene symbols. The significance
cut-off level was defined as P<0.05 and |fold-change|>2.
Statistical analysis
Data analysis were performed using GraphPad Prism
8.0 software (Dotmatics). Unpaired t-tests were conducted when
comparing datasets of two groups. One-way ANOVA and Dunnett's
multiple comparisons test were used to perform the comparison of
multiple groups. Data shown are representative of 3 independent
experiments and represented as the mean ± SD. P<0.05 was
considered to indicate a statistically significant difference.
Results
Upregulation of AID in DLBCL and the
anti-lymphoma effect of MG132 on ABC-DLBCL
The association between the expression of AID and
several lymphoma subtypes including Hodgkin lymphoma (HL) and
non-Hodgkin lymphoma (NHL) was examined using the GENT2 database,
which showed that AID expression was significantly higher in the
NHL subtype compared with the HL subtype (Fig. 1A). The association between the
expression of AID and the subtypes of NHL including follicular
lymphoma, mantle cell lymphoma, DLBCL and chronic lymphocytic
leukemia were evaluated using the GENT2 database. AID showed a
significantly higher expression in DLBCL in comparison with the
other NHL subtypes (Fig. 1B).
These data indicated that the upregulation of AID is associated
with DLBCL progression.
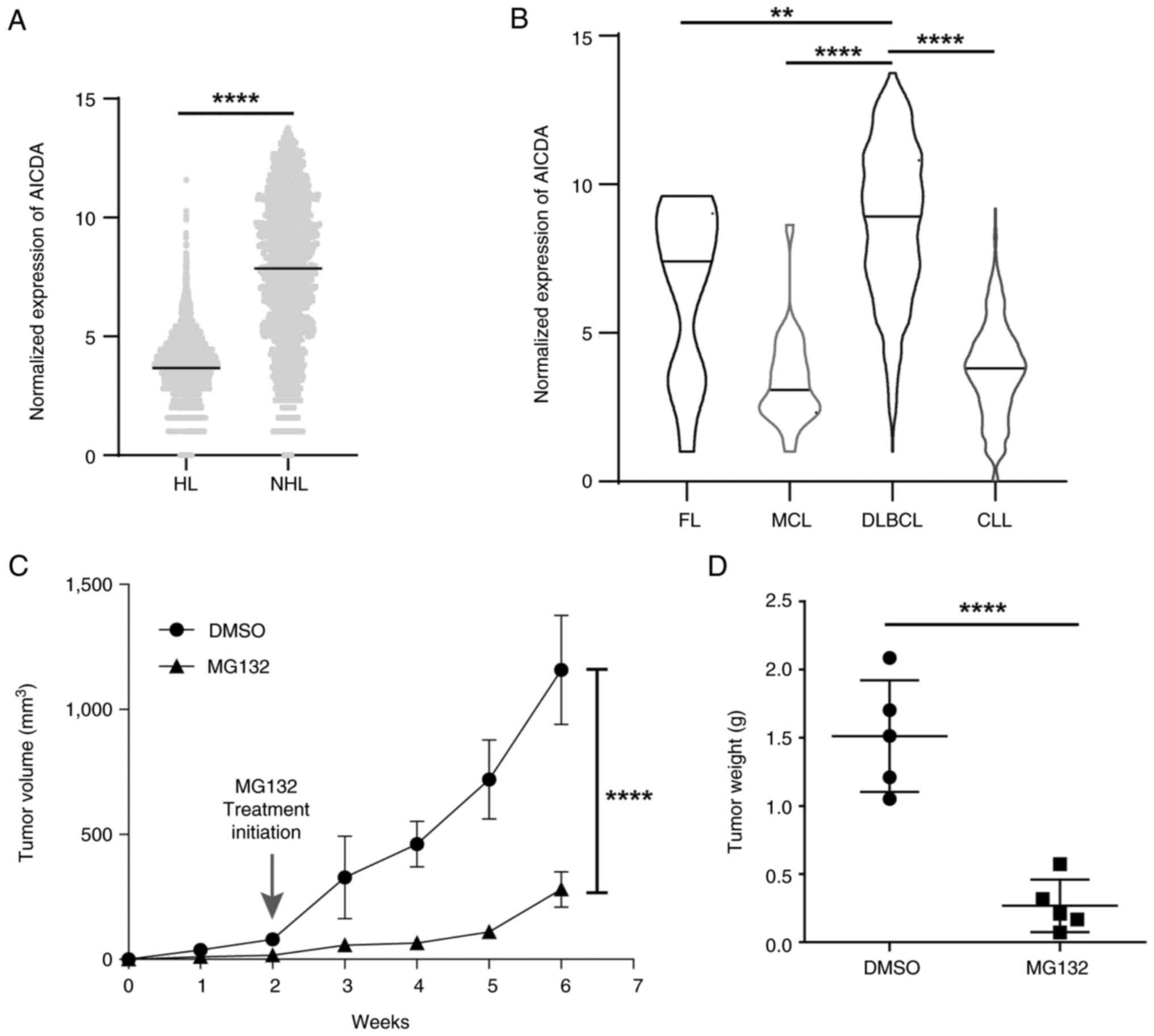 | Figure 1MG132 manifests anti-lymphoma effects
on ABC-DLBCL. (A) AID mRNA expression levels in HL and NHL, and (B)
the subtypes of NHL (FL, MCL, DLBCL and CLL) were analyzed using an
online database (http://gent2.appex.kr/gent2/). (C) Tumor volume of
OCI-LY10 tumor bearing mice after MG132 treatment (n=5). All groups
compared with the DLBCL group by pairwise comparison. (D) Averaged
weights of tumors collected from with or without MG132 treatment
groups on day 45 (n=5). Data are represented as the mean ± SD.
**P<0.01 and ****P<0.0001. HL, Hodgkin
lymphoma; NHL, non-Hodgkin lymphoma; ABC-DLBCL, activated
B-cell-like diffuse large B-cell lymphoma; AID, activation-induced
cytidine deaminase; FL, follicular lymphoma; MCL, mantle cell
lymphoma; CLL, chronic lymphocytic leukemia. |
To investigate therapeutic strategies to treat the
refractory ABC-DLBCL subtype, MG132 was introduced on the basis of
previous studies (14,15). ABC-DLBCL cell xenograft
tumor-bearing mice were established to explore the MG132 treatment
effect in vivo. Tumor growth was monitored for 45 days,
while MG132 treatment was initiated on day 14, when tumor volume
had reached 80-100 mm3. The results showed that tumor
volume was decreased in the MG132 treated tumor-bearing mice in
comparison with the mice in the DMSO-treated group (Fig. 1C; Fig. S1), and the tumor weight showed the
same trend (Fig. 1D). These
results indicate the inhibitory effect of MG132 on tumor growth in
ABC-DLBCL xenograft tumors. Collectively, these results imply an
association between MG132 treatment and AID in ABC-DLBCL.
AID suppresses ABC-DLBCL
progression
The post-transcription regulation of AID is achieved
through the proteasome pathway (21). Considering the significant effect
of AID in DLBCL (Fig. 1) and the
inhibition of MG132 to ubiquitination through the proteasome
pathway (19-21),
AID was taken as a key factor to link the mechanism of MG132
induced anti-lymphoma to ABC-DLBCL. The OCI-LY10 ABC-DLBCL cells
were transduced using CRISPR/Cas9 with three gRNAs for AID to
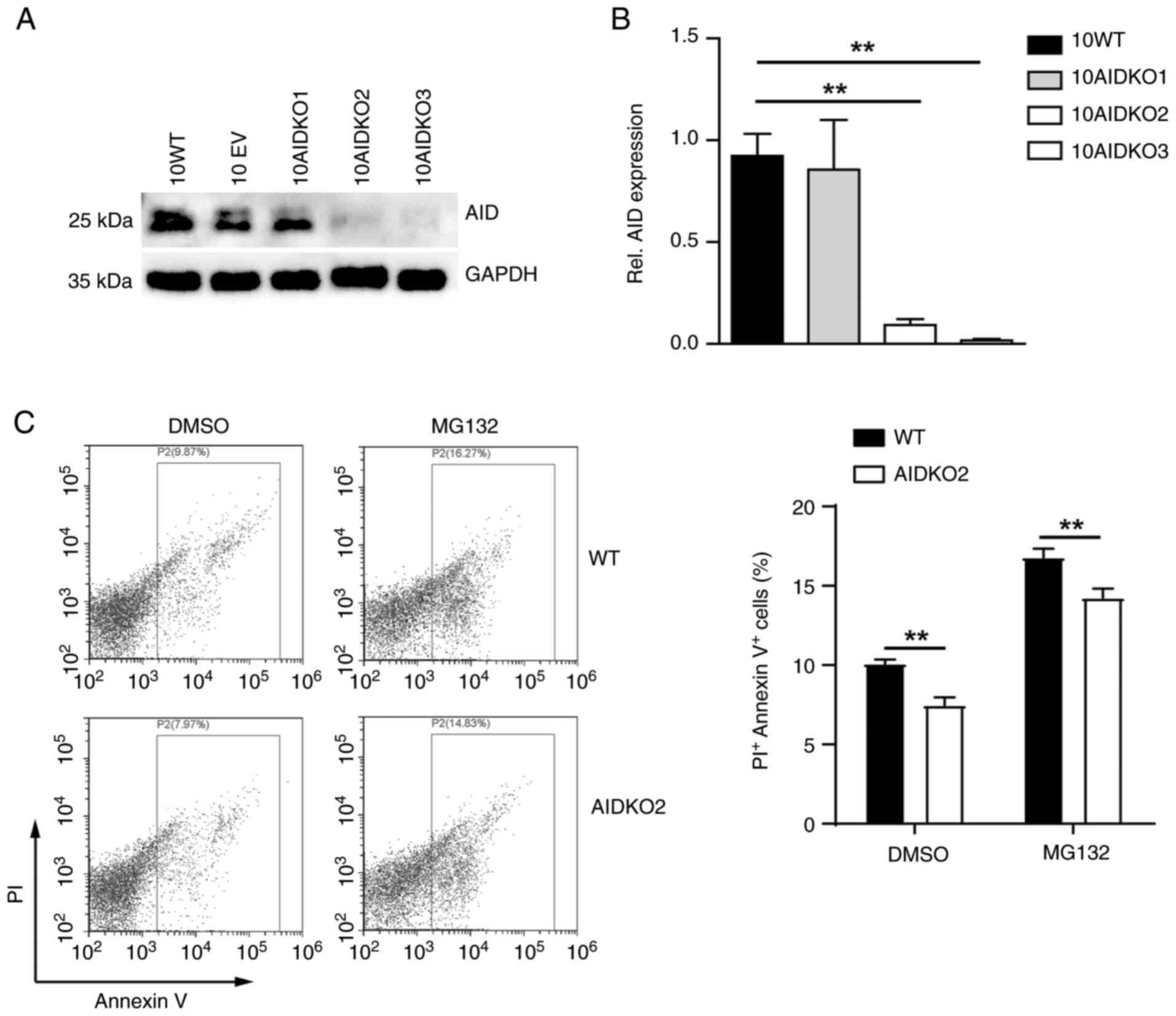
generate the AID knockout OCI-LY10 (10AIDKO) cell lines. The levels
of mRNA and protein for AID were significantly depleted in 10AIDKO
cells compared with their wild-type (WT) counterparts and the
negative control (EV; Fig. 2A and
B). Specifically, two OCI-LY10
cell lines (10AIDKO2 and 10AIDKO3) with efficient AID deficiency
were identified.
To determine whether AID loss had an impact on
cellular function, the apoptosis rate of 10AIDKO2 cells was
examined using Annexin V and PI staining in presence of DMSO. The
AIDKO2 cells presented significantly less Annexin
V+PI+ populations compared with the WT cells
(Fig. 2C), indicating reduced
apoptosis rate of ABC-DLBCL cells after AID deficiency. By
contrast, these results showed an anti-lymphoma effect of AID to
ABC-DLBCL cells.
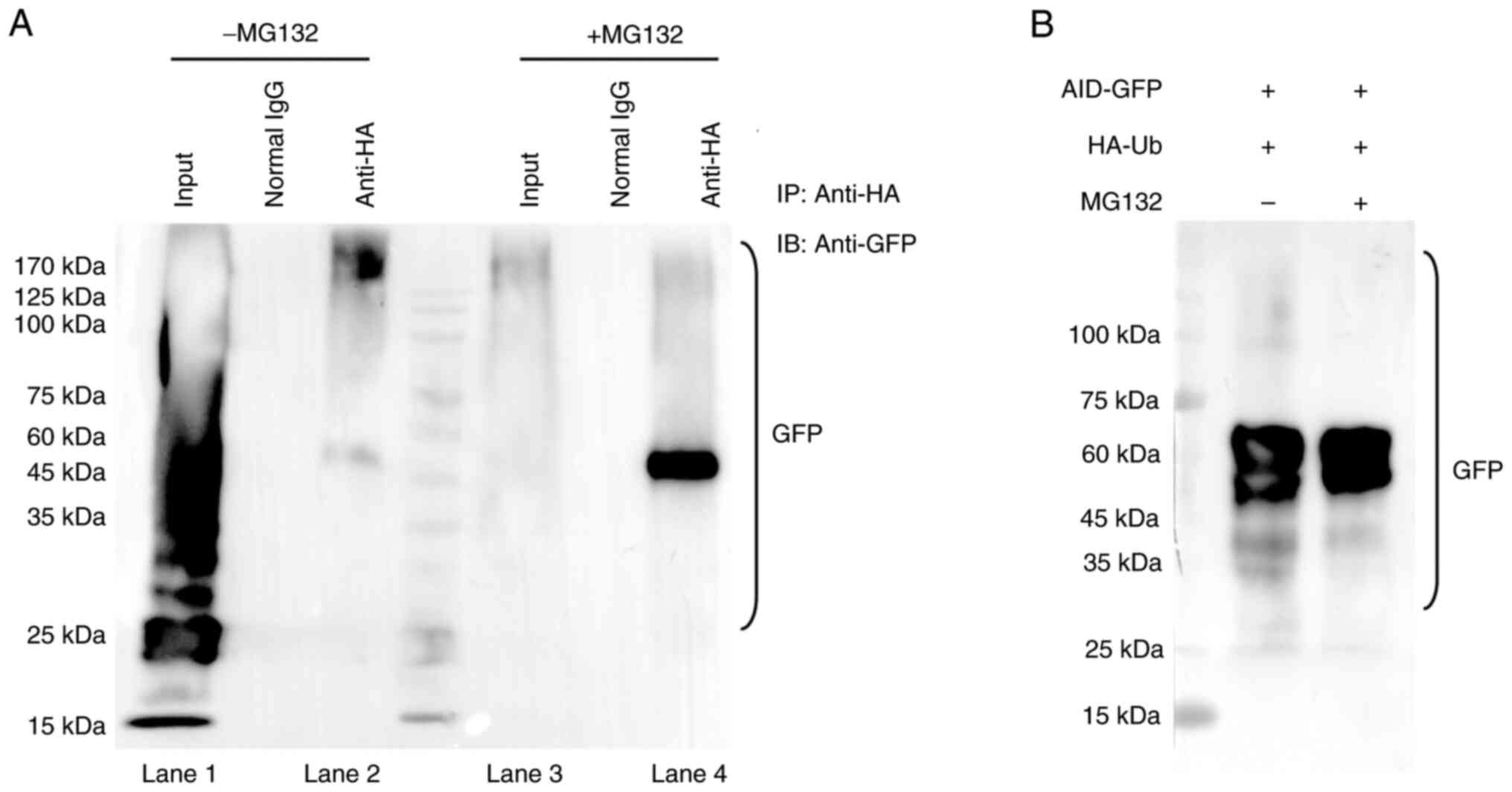
MG132 induces AID accumulation by
inhibiting ubiquitination
To assess the effect of MG132 on AID degradation
through proteasomes, the anti-lymphoma effect of MG132 on ABC-DLBCL
was confirmed through inhibiting AID degradation. The
ubiquitination of AID after MG132 treatment was detected, with the
anti-GFP purified proteins indicating the ubiquitination of AID
with MG132 treatment. The results showed that AID underwent
degradation without MG132 administration (Fig. S2; Fig. 3A, lanes 1 and 2; Fig. 3B), while MG132 administration
resulted in almost a complete disappearance of ubiquitination and a
single AID band in the immunoblots (Fig. 3A, lane 3 and lane 4; Fig. 3B), suggesting that MG132 resulted
in the inhibition of proteasome degradation of AID. Therefore, the
results indicate that MG132 induces AID accumulation by inhibiting
ubiquitination.
MG132 modulates AID upregulation and
elevates CSR
To further explore the mechanism of MG132 treating
ABC-DLBCL, the inhibitory effects of MG132 on AID and the important
role of AID in CSR was assessed. Therefore, class switch in WT and
AID-/- mice treated with or without MG132 in vivo
was detected (Fig. 4A). AID
accumulation was observed after MG132 treatment (WT + MG132 group;
Fig. 4B), suggesting that MG132
induced AID accumulation, which may be associated with the
anti-lymphoma effect of MG132 on ABC-DLBCL.
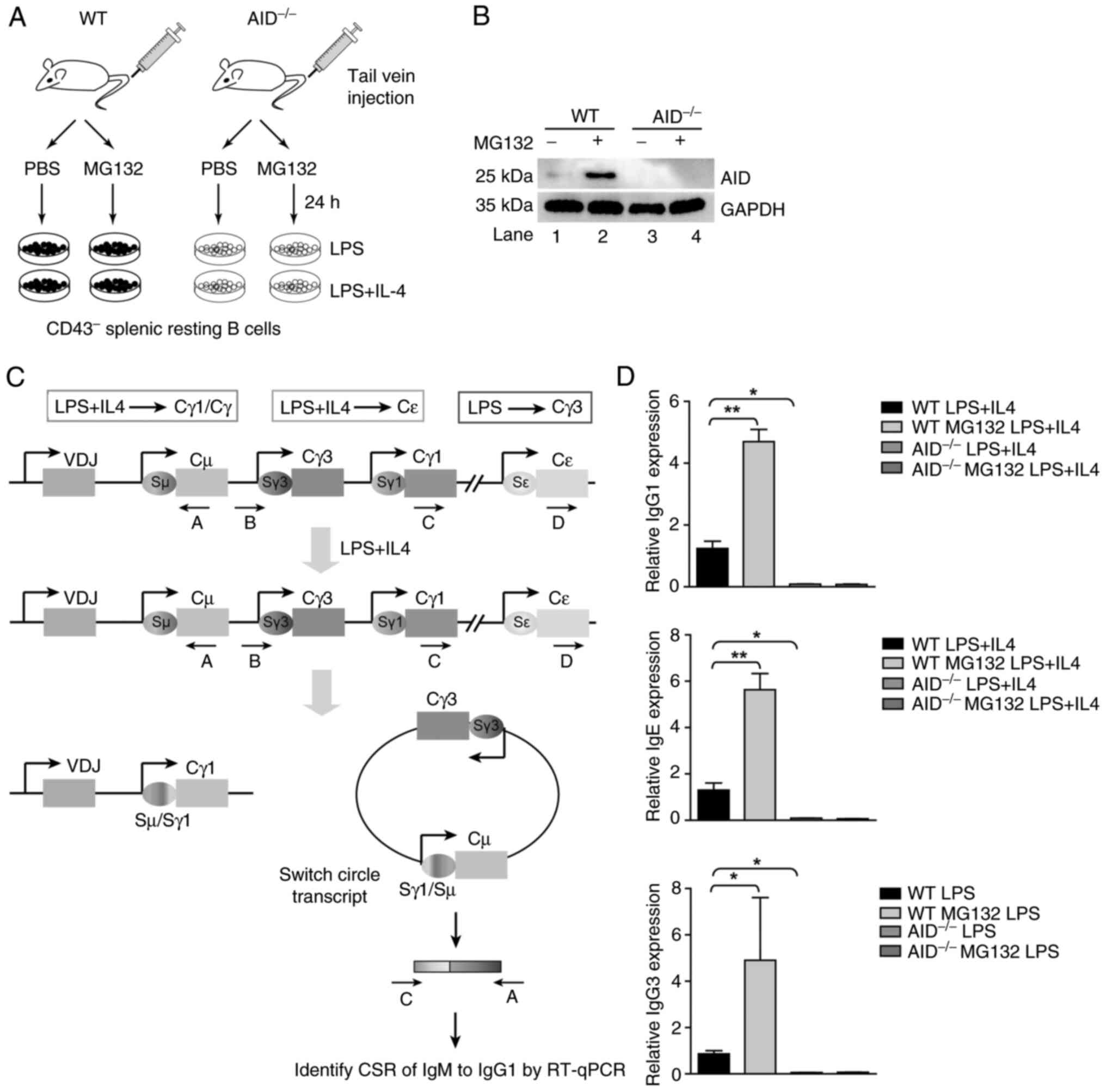 | Figure 4MG132 inhibited AID ubiquitination.
(A) C57 mice and AID-/- mice were administered MG132
through tail intravenous injection. The mice in four treatment
groups: i) WT + PBS; ii) WT + MG132; iii) AID-/- + PBS;
and iv) AID-/- + MG132 were sacrificed 24 h after
injection. The CD43- B cells were further divided into
three groups and were stimulated by LPS (50 µg/ml), LPS (3 µg/ml)
plus IL-4 (500 U/ml) for 5 days. (B) The AID and GAPDH of four
treatment groups was detected by immunoblotting. (C) A scheme
depicting the primers used to detect the CSR of IgM to IgG1, IgG3
and IgE assessed by RT-qPCR. CSR occurs between a Sµ switch region
and a S region located upstream of another C region, such as Sγ1,
which will result in the switching from IgM to IgG1. The
combinations of cytokines and LPS that lead to the switching of Ig
isotypes are indicated on the top. Ovals indicate switch regions,
arrows mark the promoters and rectangles are constant region exons
(Cµ, Cγ3, Cγ1 and Cε). Class-switching can result in the formation
of a switch circle, from which a hybrid transcript is expressed.
These transcripts can be reverse-transcribed for cDNA, and using
specific primers (such as arrows marked with C and A for detecting
the switching to Cγ1 isotype, with B and A for detecting the
switching to Cγ3 isotype or D and A for detecting the switching to
Cε), quantitate switch circle formation by qPCR. (D) The class
switch of IgM to IgG1, IgG3 and IgE in the resting B cells was
identified by RT-qPCR. All groups were compared with the WT LPS +
IL4 group by pairwise comparison. Data shown are representative of
three independent experiments. Data are represented as the mean ±
SD. *P<0.05 and **P<0.01. WT, wild
type; AID, activation-induced cytidine deaminase; CSR, class switch
recombination; VDJ, variable diversity joining. |
After confirmation of MG132 inhibition of AID
ubiquitination, the AID-induced class switch in the presence or
absence of MG132 was also assessed in vivo. By stimulating
with LPS or/and LPS plus IL-4, the purified spleen resting B cells
underwent class switch to IgG1, IgG3 and IgE (Fig. 4C). Through stimulation of LPS (3
µg/ml) plus IL-4 (50 ng/ml), a notable elevated class switch to
IgG1 was detected in mice with complete AID expression, while no
class switch to IgG1 was observed in spleen resting B cells from
AID-/- mice treated with MG132 (Fig. 4D). Similar trends were also
observed in the detection of class switch to IgG3 and IgE with
stimulation by LPS (50 µg/ml) only and LPS (3 µg/ml) plus IL-4 (50
ng/ml) for 5 days (Fig. 4D). As
hypothesized, notable class switching to IgE and IgG3 was detected
by RT-qPCR in spleen resting B cells of WT mice, while
AID-/- mice had no class switching to IgE and IgG3 due
to AID deficiency (Fig. 4D).
Collectively, MG132 inhibits AID ubiquitination to elevate AID
levels, which thus induces apparent CSR.
MG132-mediated AID upregulation
rescues abnormal CSR in ABC-DLBCL cells
To evaluate the impact of MG132 on ABC-DLBCL cells,
ABC-DLBCL OCI-LY10 cells were treated with MG132 (5 µM) in the
presence or absence of AID. The ubiquitination of MG132 treatment
at different times (24, 48 and 72 h) was detected by immunoblotting
(Fig. 5A). The results showed that
MG132-treated WT OCI-LY10 cells, specifically in the 24 h treatment
group, showed increased AID accumulation compared with the
untreated counterparts (Fig. 5A,
lanes 2-4 compared with lane 1). In addition, as time progressed,
AID accumulation reduced, which could be attributed to the declined
drug efficiency along with increased time (Fig. 5A, lane 2-4).
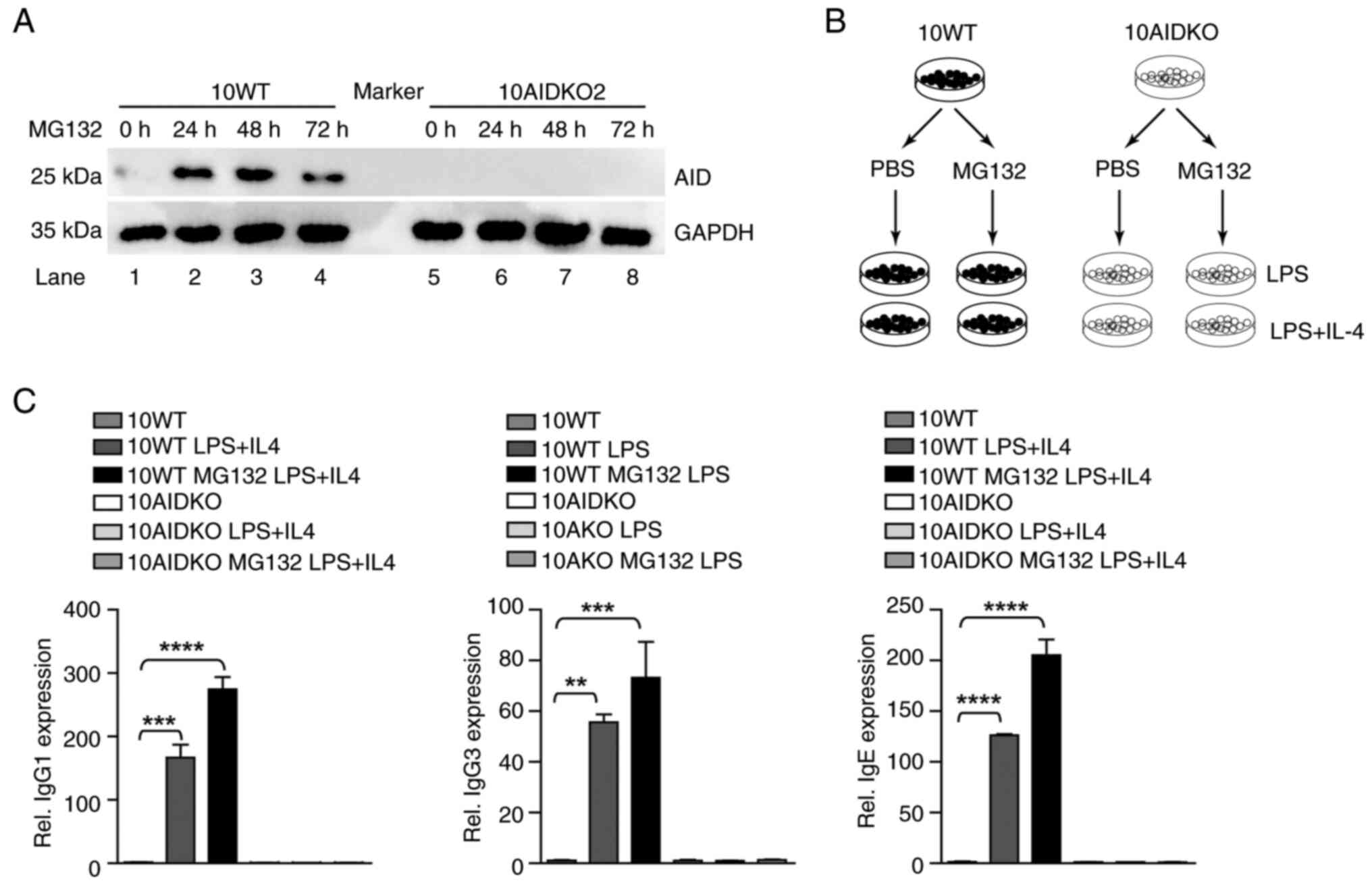 | Figure 5MG132 induced-AID upregulation
restores abnormal CSR in ABC-DLBCL cells. (A) 10WT and 10AIDKO2
cells were treated by MG132 (5 µM) for 24, 48 and 72 h,
respectively. The ubiquitin, AID and GAPDH expression of different
treatment groups was detected by immunoblotting. (B) Scheme
depicting LPS and IL-4 stimulation. 10WT and 10AIDKO2 cells were
stimulated by LPS (50 µg/ml) or LPS (3 µg/ml) plus IL-4 (50 ng/ml)
for 3 days, respectively. (C) The class switch of IgM to IgG1, IgG3
and IgE in WT and AIDKO was detected by RT-qPCR. All groups were
compared with the WT LPS + IL4 group by pairwise comparison. Data
shown are representative of three independent experiments. Data are
represented as the mean ± SD. **P<0.01,
***P<0.001 and ****P<0.001. WT, wild
type; AIDKO, AID knockout OCI-LY10; AID, activation-induced
cytidine deaminase; CSR, class switch recombination; Rel.,
relative. |
In order to further detect MG132 induced CSR in
ABC-DLBCL, 10WT and 10AIDKO cells were stimulated by LPS or/and LPS
plus IL-4 (Fig. 5B). A significant
increase in class switching to IgG1, IgG3 and IgE was detected in
WT ABC-DLBCL cells stimulated by LPS (3 µg/ml) alone or LPS plus
IL-4 (50 ng/ml), but not in AIDKO cells (Fig. 5C). Thus, the data indicate that
MG132 inhibits AID ubiquitination to elevate AID levels, thus
inducing apparent CSR in ABC-DLBCL cells.
Discussion
At present, the standard treatment strategy for
DLBCL in clinical practice is R+CHOP therapy, which involves the
combination of rituximab with cyclophosphamide, doxorubicin,
vincristine and prednisone (1-3).
This approach has led to significant improvements in the survival
of patients with DLBCL, with a success rate of ~40% (1-5).
However, the standard first-line R-CHOP is used to treat all
subtypes of DLBCL instead of individual therapy targeting the
subtypes of DLBCL (1-3).
The majority of patients eventually suffer from refractory DLBCL,
particularly those with the ABC-DLBCL subtype, following relapse in
R-CHOP or R-CHOP-like chemotherapy (6,7). To
address this issue, various strategies have been developed to
improve the outcome of ABC-DLBCL, including the addition of new
targets such as CD20 and CD47 to R-CHOP therapy (termed R-CHOP+X)
(27,28), inhibitors targeting genetic
aberrations involving oncogenic signaling pathways such as BCR,
NOTCH, NF-κB and PI3K/AKT, and strategies targeting epigenetic
modification factors (2,9,29,30).
However, the efficacy of these therapeutic approaches in clinical
practice remains unsatisfactory. In the present study, MG132
demonstrated a significant cell killing and tumor inhibition
effect, indicating the feasibility of using proteasome inhibitors
to treat ABC-DLBCL. However, MG132 is a peptide-like compound with
poor protein kinase activity and is not the ideal proteasome
inhibitor for in vivo use (18-20).
The present study utilized the proteasome inhibitor MG132 as a tool
to inhibit the ubiquitination of AID through the proteasome
pathway, resulting in the accumulation of AID. These findings shed
light on the anti-lymphoma effect of proteasome inhibitors on
ABC-DLBCL. Notably, bortezomib, another proteasome inhibitor, is
currently being investigated in 10 clinical trials listed on
ClinicalTrials.gov (http://www.clinicaltrials.gov) (31-33),
but the mechanisms of bortezomib in treating DLBCL have not been
investigated. The present study elucidates the mechanism of MG132
treating DLBCL by inhibiting AID degradation. However, as a
comprehensive proteasome instead of a 20S proteasome (such as
bortezomib), the use of MG132 in clinical treatment may cause
systematic targeting but not specific targeting (17-20).
Therefore, the clinical translation of MG132 requires more research
into the routes of drug administration and the dosage. The results
of the present study demonstrate the mechanism of the proteasome
inhibitor-mediated anti-lymphoma effect on ABC-DLBCL, showing that
proteasome inhibitors elevate AID levels to rescue abnormal CSR in
ABC-DLBCL. These findings enhance the potential clinical utility of
proteasome inhibitors in treating refractory ABC-DLBCL in the
future.
The BCR plays a crucial role in the activation and
maturation of B cells. During the development of GC, BCR transduces
the antigen stimulation signal, leading to an increase in BCL6
expression and the proliferation of B cells to form the dark zone.
AID mediates CSR and SHM in the dark zone (34). As B cells differentiate into plasma
cells and memory B cells in the bright zone of GC, the cells with
high-affinity to antigens proliferate and leave the GC structure,
while those with low-affinity to antigens undergo apoptosis
(Fig. 6A) (34,35).
Abnormalities in BCR are associated with an increasing number of
B-cell malignancies (2,8,9).
Mutations in signaling transducing components of the BCR pathway,
such as CD79B and CARD11, have been identified in ABC-DLBCL
(2,8,9).
These mutations result in sustained and chronic active BCR
signaling, leading to continuous activation of the NF-κB pathway
(34). Sustained BCR signaling
prevents large B cells from differentiating into plasma cells and
memory B cells efficiently (36).
Furthermore, ABC-DLBCL is deficient in CSR due to Sµ region
deletions (10). ABC-DLBCL is
characterized by the continuous proliferation and accumulation of
large B cells, which prevents the cells from undergoing CSR
(9). Therefore, increasing the
level of AID could be a potential strategy to drive the
differentiation of these accumulated large B cells. In the present
study, it was demonstrated that using the proteasome inhibitor
MG132 to inhibit AID degradation through the proteasome pathway can
effectively up-regulate AID protein levels. This, in turn, promotes
AID-induced CSR and facilitates the differentiation of GC B cells
into mature B cells (Fig. 6B). The
effect of MG132 on AID accumulation in ABC-DLBCL was investigated
by detecting CSR to IgG1, IgG3 and IgE. The results demonstrate
that MG132 mediates AID accumulation and apparent CSR using WT and
AID-/- mice. It is hypothesized that AID may not induce
CSR in vitro. However, notable CSR was observed in the
detection of CSR in ABC-DLBCL cells with the stimulation of
cytokines (LPS and/or IL4), indicating MG132 induced AID
accumulation to mediate the restoration of abnormal CSR in
ABC-DLBCL. This indicates that cytokines such as LPS and IL-4 may
simulate the in vivo environment for CSR onset. It has been
reported that half of patients with ABC-DLBCL have internal
deletions in Sµ (10). Although
apparent CSR was observed after LPS and/or LPS + IL4 stimulation,
the effect of excessive AID on the Sµ region was not assessed in
the present study. This may be due to various processes such as DNA
repair, cell division and clone selection (37). Nonetheless, the restoration of CSR
is a promising outcome induced by the elevated level of AID in
ABC-DLBCL.
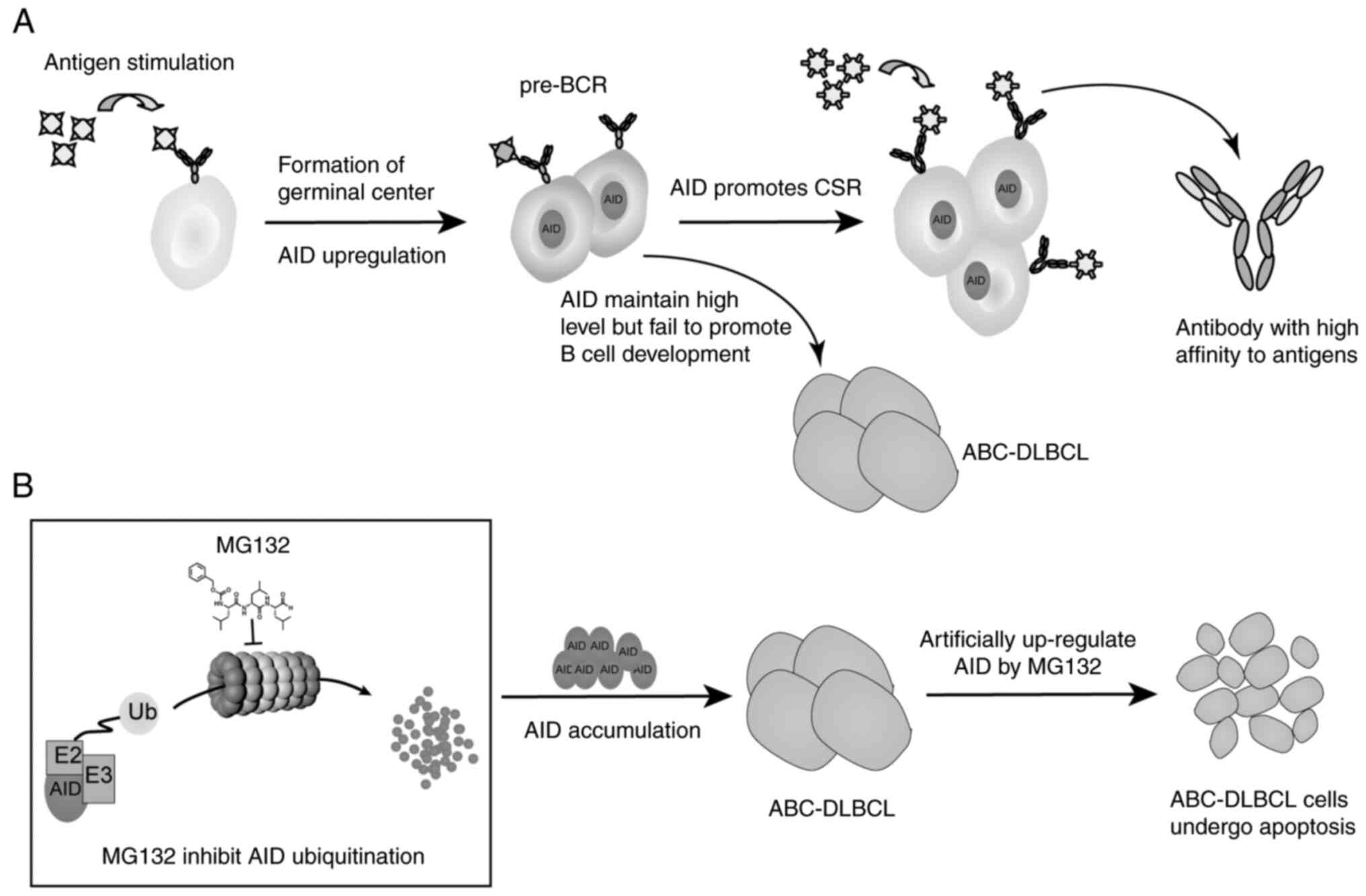 | Figure 6Proteasome inhibitor MG132 regulates
AID-induced CSR by inhibiting AID ubiquitination in ABC-DLBCL. (A)
After stimulating by antigens, BCR transduces signaling, which
induces B cells to proliferate and form germinal centers. AID
promotes SHM and CSR, which are essential to antibody affinity
maturation. ABC-DLBCL is derived from the high level of AID but
fails to promote CSR. (B) By treating ABC-DLBCL with the proteasome
inhibitor MG132, AID accumulates in ABC-DLBCL, the process of CSR
is restored and ABC-DLBCL cells undergo apoptosis. AID,
activation-induced cytidine deaminase; BCR, B-cell receptor; CSR,
class switch recombination; Ub, ubiquitin; ABC-DLBCL, activated
B-cell-like diffuse large B-cell lymphoma; SHM, somatic
hypermutation. |
AID is an enzyme that induces SHM and CSR of Ig
genes (11-13).
However, the off-target effects of AID on non-Ig genes or the
dysfunction of repairing AID-mediated double-strand breaks can
cause point mutations or chromosome translocations (38). The off-target effects of AID is
reported to be a leading cause of carcinogenesis, indicating that
high levels of AID play a negative role in promoting the occurrence
and progression of cancers (39).
However, the present study indicates a positive role of excessive
AID in treating ABC-DLBCL, providing a novel insight into
re-evaluating the function of AID in cancer. Previous research has
attempted to reveal the possible mechanism of excessive AID,
showing that AID acts as a transcriptional factor to regulate
complex gene networks (12,13).
This provides a way to identify novel functions of AID in addition
to its traditional role in carcinogenesis. In the present study,
the positive role of AID in anti-lymphoma was verified, which was
an alternative use of its traditional function, whereby excessive
AID restores the dysfunction of CSR in ABC-DLBCL. However, a
contradiction emerged between the beneficial role ascribed to AID
in ABC-DLBCL and the previously reported harmful role of AID in
DLBCL (13,16), as well as the detrimental impact of
AID inferred from TCGA data. These previous studies and the TCGA
data discussed the negative role of AID in DLBCL mainly through the
results of the off-targets of AID (40), but omitting its mechanism in
ABC-DLBCL with abnormal processing of antibody affinity maturation.
The findings of the anti-lymphoma effects of AID in the present
study may imply that the elevated AID expression in DLBCL could be
a compensatory mechanism to counteract detrimental factors.
Nonetheless, the robust expression of AID was insufficient to halt
the progression of ABC-DLBCL. The research introduces a novel
approach involving the artificial enhancement of AID levels through
proteasome inhibition, aiming to counteract negative influences and
exert an anti-tumor effect. Therefore, understanding the dual role
of AID in carcinogenesis, elucidating the mechanism of AID in
specific cancers and exploring the alternative function of AID
(except for its mutational function) could lead to the development
of future cancer treatments.
In conclusion, the findings of the present study
have aided the understanding of the mechanisms underlying how
proteasome inhibitors can effectively treat refractory ABC-DLBCL.
The crucial role of MG132 in inducing AID accumulation through
inhibition of AID degradation via the proteosome pathway was
identified, and it was demonstrated that the elevation of AID
levels rescues abnormal CSR. These results suggest the potential
therapeutic value of AID in treating ABC-DLBCL, and the promising
application of proteasome inhibitors in clinical therapy strategies
for AID-associated DLBCL. However, there are limitations that
warrant acknowledgment. Firstly, the findings on the mechanisms
underlying the effect of MG132 on ABC-DLBCL was discussed from the
aspect of AID-mediated antibody affinity maturation in ABC-DLBCL,
and it would be beneficial to further investigate the systematic
regulation mechanisms of MG132 in treating ABC-DLBCL. Secondly, for
MG132 clinical translation, the potential side-effect and patient
variability requires further investigation.
Supplementary Material
Subcutaneous tumors taken from
OCI-LY10 ABC-DLBCL cell tumor bearing mice treated with DMSO or
MG132. ABC-DLBCL, activated B-cell-like diffuse large B-cell
lymphoma.
GFP fluorescence of 293T cells
co-transfected by pWPI-AID-GFP and Ub-HA observed by fluorescence
microscope. AID, activation-induced cytidine deaminase; Ub,
ubiquitin; HA, hemagglutinin.
Sequences of the destroyed alleles in
the AID gene.
Sequences of primers for amplifying
AID cDNA.
Sequences of primers for quantitative
AID transcription.
Reverse transcription-quantitative PCR
primers for detecting antibody class switch.
Acknowledgements
The authors would like to thank Professor Yanhong Ji
of Xi'an Jiaotong University (Xi'an, China) for providing the
AID-/- mice used in the present study.
Funding
Funding: This research was funded by The Key Scientific Research
Foundation of the Higher Education Institutions of Henan Province
(grant no. 22A320041), Natural Science Foundation of Henan province
(grant no. 232300421189) and The Undergraduate Innovation and
Entrepreneurship Training Program of Henan province (grant no.
202310472044).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
JJ conceptualized the study; JJ and ZL performed the
study methodology; ZL searched the online software; CX performed
the formal analysis; ZL and ZW carried out the experiments; ZL, CX,
ZW, ZLiu obtained the resources; CX and ZL curated the data and
wrote the original draft; ZL reviewed and edited the manuscript,
and supervised the study; JJ visualized the study, performed
project administration and acquired the funding. All authors have
read and approved the final version of the manuscript. ZL, CX, ZW,
ZL and JJ confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
The present study was approved by The Ethics
Committee of Xinxiang Medical University, China (Xinxiang, China;
approval no. XYLL-2022001253). All the animal procedures were
performed in accordance with The ‘Guide for the Care and Use of
Laboratory Animals’ published by the National Institutes of Health
(22).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Li S, Young KH and Medeiros LJ: Diffuse
large B-cell lym-phoma. Pathology. 50:74–87. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Schmitz R, Wright GW, Huang DW, Johnson
CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer
AL, et al: Genetics and pathogenesis of diffuse large B-cell
lymphoma. N Engl J Med. 378:1396–1407. 2018.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Crombie J: Classifying DLBCL subtypes for
optimal treatment. Oncology (Williston Park).
33(686504)2019.PubMed/NCBI
|
|
4
|
Takahara T, Nakamura S, Tsuzuki T and
Satou A: The immunology of DLBCL. Cancers (Basel).
15(835)2023.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Alaggio R, Amador C, Anagnostopoulos I,
Attygalle AD, Araujo IBO, Berti E, Bhagat G, Borges AM, Boyer D,
Calaminici M, et al: The 5th edition of the world health
organization classification of haematolymphoid tumours: Lymphoid
neoplasms. Leukemia. 36:1720–1748. 2022.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Coiffier B and Sarkozy C: Diffuse large
B-cell lymphoma: R-CHOP failure-what to do? Hematology Am Soc
Hematol Educ Program. 2016:366–378. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Nowakowski GS, Chiappella A, Witzig TE,
Spina M, Gascoyne RD, Zhang L, Flament J, Repici J and Vitolo U:
ROBUST: Lenalidomide-R-CHOP versus placebo-R-CHOP in previously
untreated ABC-type diffuse large B-cell lymphoma. Future Oncol.
12:1553–1563. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Frick M, Bettstetter M, Bertz S,
Schwarz-Furlan S, Hartmann A, Richter T, Lenze D, Hummel M,
Dreyling M, Lenz G and Gaumann A: Mutational frequencies of CD79B
and MYD88 vary greatly between primary testicular DLBCL and
gastrointestinal DLBCL. Leuk Lymphoma. 59:1260–1263.
2018.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Davis RE, Ngo VN, Lenz G, Tolar P, Young
RM, Romesser PB, Kohlhammer H, Lamy L, Zhao H, Yang Y, et al:
Chronic active B-cell-receptor signalling in diffuse large B-cell
lymphoma. Nature. 463:88–92. 2010.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Lenz G, Nagel I, Siebert R, Roschke AV,
Sanger W, Wright GW, Dave SS, Tan B, Zhao H, Rosenwald A, et al:
Aberrant immunoglobulin class switch recombination and switch
translocations in activated B cell-like diffuse large B cell
lymphoma. J Exp Med. 204:633–643. 2007.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kumar R, DiMenna LJ, Chaudhuri J and Evans
T: Biological function of activation-induced cytidine deaminase
(AID). Biomed J. 37:269–283. 2014.PubMed/NCBI View Article : Google Scholar
|
|
12
|
An L, Chen C, Luo R, Zhao Y and Hang H:
Activation-induced cytidine deaminase aided in vitro antibody
evolution. Methods Mol Biol. 1707:1–14. 2018.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Teater M, Dominguez PM, Redmond D, Chen Z,
Ennishi D, Scott DW, Cimmino L, Ghione P, Chaudhuri J, Gascoyne RD,
et al: AID drives epigenetic heterogeneity and accelerates germinal
center-derived lymphomagenesis. Nat Commun. 9(222)2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Jiao J, Lv Z, Zhang P, Wang Y, Yuan M, Yu
X, Otieno Odhiambo W, Zheng M, Zhang H, Ma Y and Ji Y: AID assists
DNMT1 to attenuate BCL6 expression through DNA methylation in
diffuse large B-cell lymphoma cell lines. Neoplasia. 22:142–153.
2020.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Jiao J, Jin Y, Zheng M, Zhang H, Yuan M,
Lv Z, Odhiambo W, Yu X, Zhang P, Li C, et al: AID and TET2
co-operation modulates FANCA expression by active demethylation in
diffuse large B cell lymphoma. Clin Exp Immunol. 195:190–201.
2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kawamura K, Wada A, Wang JY, Li Q, Ishii
A, Tsujimura H, Takagi T, Itami M, Tada Y, Tatsumi K, et al:
Expression of activation-induced cytidine deaminase is associated
with a poor prognosis of diffuse large B cell lymphoma patients
treated with CHOP-based chemotherapy. J Cancer Res Clin Oncol.
142:27–36. 2016.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Varshavsky A: The ubiquitin system,
autophagy, and regulated protein degradation. Annu Rev Biochem.
86:123–128. 2017.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Thibaudeau TA and Smith DM: A practical
review of proteasome pharmacology. Pharmacol Rev. 71:170–197.
2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Guo N and Peng Z: MG132, a proteasome
inhibitor, induces apoptosis in tumor cells. Asia Pac J Clin Oncol.
9:6–11. 2013.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Schenkein D: Proteasome inhibitors in the
treatment of B-cell malignancies. Clin Lymphoma. 3:49–55.
2002.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Aoufouchi S, Faili A, Zober C, D'Orlando
O, Weller S, Weill JC and Reynaud CA: Proteasomal degradation
restricts the nuclear lifespan of AID. J Exp Med. 205:1357–1368.
2008.PubMed/NCBI View Article : Google Scholar
|
|
22
|
National Research Council: Committee for
the Update of the Guide for the Care and Use of Laboratory Animals.
Guide for the Care and Use of Laboratory Animals. 8th edition.
National Academies Press, Washington, DC, 2011.
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Muramatsu M, Kinoshita K, Fagarasan S,
Yamada S, Shinkai Y and Honjo T: Class switch recombination and
hypermutation require activation-induced cytidine deaminase (AID),
a potential RNA editing enzyme. Cell. 102:553–563. 2000.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Caron AZ, Haroun S, Leblanc E, Trensz F,
Guindi C, Amrani A and Grenier G: The proteasome inhibitor MG132
reduces immobilization-induced skeletal muscle atrophy in mice. BMC
Musculoskelet Disord. 12(185)2011.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Rupniewska ZM, Roliński J and
Bojarska-Junak A: Universal CD43 molecule. Postepy Hig Med Dosw.
54:619–638. 2000.PubMed/NCBI(In Polish).
|
|
27
|
Szydłowski M, Garbicz F, Jabłońska E,
Górniak P, Komar D, Pyrzyńska B, Bojarczuk K, Prochorec-Sobieszek
M, Szumera-Ciećkiewicz A, Rymkiewicz G, et al: Inhibition of PIM
kinases in DLBCL targets MYC transcriptional program and augments
the efficacy of anti-CD20 antibodies. Cancer Res. 81:6029–6043.
2021.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Advani R, Flinn I, Popplewell L, Forero A,
Bartlett NL, Ghosh N, Kline J, Roschewski M, LaCasce A, Collins GP,
et al: CD47 blockade by Hu5F9-G4 and rituximab in non-Hodgkin's
lymphoma. N Engl J Med. 379:1711–1721. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Schmitt A, Xu W, Bucher P, Grimm M,
Konantz M, Horn H, Zapukhlyak M, Berning P, Brändle M, Jarboui MA,
et al: Dimethyl fumarate induces ferroptosis and impairs
NF-κB/STAT3 signaling in DLBCL. Blood. 138:871–884. 2021.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Xu W, Berning P and Lenz G: Targeting
B-cell receptor and PI3K signaling in diffuse large B-cell
lymphoma. Blood. 138:1110–1119. 2021.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ruan J, Martin P, Furman RR, Lee SM,
Cheung K, Vose JM, Lacasce A, Morrison J, Elstrom R, Ely S, et al:
Bortezomib plus CHOP-rituximab for previously untreated diffuse
large B-cell lymphoma and mantle cell lymphoma. J Clin Oncol.
29:690–697. 2011.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Davies AJ, Barrans S, Stanton L, Caddy J,
Wilding S, Saunders G, Mamot C, Novak U, McMillan A, Fields P, et
al: Differential efficacy from the addition of bortezomib to R-CHOP
in diffuse large B-Cell lymphoma according to the molecular
subgroup in the REMoDL-B study with a 5-year follow-up. J Clin
Oncol. 41:2718–2723. 2023.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Lin Z, Chen X, Li Z, Zhou Y, Fang Z, Luo
Y, Zhao J and Xu B: The role of bortezomib in newly diagnosed
diffuse large B cell lymphoma: A meta-analysis. Ann Hematol.
97:2137–2144. 2018.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Victora GD and Nussenzweig MC: Germinal
centers. Annu Rev Immunol. 40:413–442. 2022.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Bao K, Zhang J, Scherl A, Ziai J,
Hadadianpour A, Xu D, Dela Cruz C, Liu J, Liang Y, Tam L, et al:
Activation-induced cytidine deaminase impacts the primary antibody
repertoire in naive mice. J Immunol. 208:2632–2642. 2022.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhou J, Zuo M, Li L, Li F, Ke P, Zhou Y,
Xu Y, Gao X, Guan Y, Xia X, et al: PIM1 and CD79B
mutation status impacts the outcome of primary diffuse large B-Cell
lymphoma of the CNS. Front Oncol. 12(824632)2022.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Yu K: AID function in somatic
hypermutation and class switch recombination. Acta Biochim Biophys
Sin (Shanghai). 54:759–766. 2022.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Çakan E and Gunaydin G: Activation induced
cytidine deaminase: An old friend with new faces. Front Immunol.
13(965312)2022.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Rios LAS, Cloete B and Mowla S:
Activation-induced cytidine deaminase: In sickness and in health. J
Cancer Res Clin Oncol. 146:2721–2730. 2020.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Jiao J, Lv Z, Wang Y, Fan L and Yang A:
The off-target effects of AID in carcinogenesis. Front Immunol.
14(1221528)2023.PubMed/NCBI View Article : Google Scholar
|