Introduction
Peroxisome proliferator-activated receptor α (PPARα)
is a critical regulator of cardiac lipid metabolism and has a
significant effect on various functions, such as glucose and lipid
homeostasis, cardiac metabolism substrate conversion, and
inflammation and autoimmune disease development (1). PPARα can also reduce the release of
pro-inflammatory factors, inhibit the production of chemokines, and
promote T cell differentiation (2). Fenofibrate lowers cholesterol and
triglyceride levels and is a widely used as a PPARα agonist in
clinical practice. It has multiple effects on the heart, including
prevention of myocardial inflammation, attenuation of
isoproterenol-induced acute myocardial ischemic injury, and
inhibition of macrophage and T lymphocyte infiltration into the
left ventricle (3). Experimental
autoimmune myocarditis (EAM) is an autoimmune disease induced by
CD4(+) T cells. Histopathology has shown that CD4(+) T cells
infiltrate the myocardium in the acute phase, leading to severe
myocardial damage and subsequent cardiac fibrosis (4-6).
Fenofibrate treatment can alleviate EAM (7,8). It
has been previously shown that PPARα plays a crucial role in T
helper 17 (Th17) cell differentiation, and fenofibrate can
alleviate EAM (9).
Methyl-β-cyclodextrin, a specific cholesterol-depleting agent, also
ameliorates EAM by suppressing myocarditis-induced apoptosis
(10).
IκBζ is the most recently identified member of the
IκB family and interacts with the NF-κBp50 subunit to positively
regulate the expression of numerous inflammatory factors (11,12).
IκBζ plays a central role in inflammatory diseases and various
autoimmune diseases, making it a key Th17-associated factor
(13,14). IκBζ mediates the inflammation
response to TNF-α and IL-17, and its inhibition mediates toll-like
receptor transcriptional responses (15,16).
However, the role of IκBζ in EAM and the underlying mechanisms
remain unclear.
In the present study, the authors investigated
whether fenofibrate could regulate the IκBζ signaling pathway in
rat EAM and explore its possible mechanisms. The results showed
that fenofibrate treatment ameliorated EAM by preventing myocardial
inflammation and fibrosis. It was also found that IκBζ is a key
factor in EAM, and the PPARα/IκBζ signaling pathway is involved in
the pathogenesis of EAM. This suggested that IκBζ may be a new
molecular target of fenofibrate for treating autoimmune
myocarditis.
Materials and methods
Animals
Male Lewis rat aged 6-8 weeks (180-200 g) and PPARα
(-/-) mice (129S4/SvJae) aged 8-10 weeks (20-25 g) were acquired
from Beijing Vital River Lab Animal Technology Co., Ltd. and
Jackson ImmunoResearch Laboratories, Inc., respectively. Male
C57BL/6J mice as PPARα (+/+) wild-type (WT) mice were purchased
from Shanghai SLAC Laboratory Animal Company, Ltd. Animal care and
experiments were conducted in accordance with the procedures
approved by the Animal Care and Use Committee of Xiamen University
(approval no. XMULAC 20220111; Xiamen, China). Animals were
provided free access to standard rodent chow and water and were
housed in a SPF conditions at 23±2˚C and a relative humidity of
60±5%. Throughout the studies, all animals were treated in
accordance with the guidelines for animal experiments of our
institution.
Rat EAM model
EAM model was established as previously described
(5,6). Throughout the studies, all the
experiment operations were performed under the anesthesia
environment to animal welfare consideration. The duration of the
experiment was 21 days. On day 0 the rats were anesthetized with 2%
isoflurane inhalation and immunized once by subcutaneous injection
with a 0.2-ml emulsion containing 1 mg of cardiac myosin and an
equal volume of complete Freud's adjuvant in both footpads; the
morbidity and survival rate were 100% in rats immunized by this
method (4). A total of 24 rats
were divided into three groups (n=8 for each group). The control
group rats received only complete Freund's adjuvant for
immunization. The immunized EAM rats underwent daily oral gavage
administration with fenofibrate or a solvent alone from day 14 to
day 21 for 7 consecutive days. All the rats' health and behavior
were monitored and body weight were measured every 3 days. It was
previously reported that rat EAM cardiac inflammation occurred in
the acute phase and peaked on day 14 to day 21, characterized by
severe heart failure (5,6). On day 21 all the experimental rats
were anesthetized with 2% isoflurane inhalation and euthanized
through cervical dislocation, the area of the chest were sterilized
with 75% alcohol, and an aseptic surgical knife was subsequently
used to fully expose the heart. A total of 5 ml blood from the
inferior vena cava were drawn out. After dissecting the heart,
heart tissues were carefully harvested, washed, and weighted. Body
weight (BW) and heart weight (HW) were measured to calculate the
ratio of HW/BW.
Histopathological examination
Heart tissues were fixed in 10% formalin at room
temperature for 3 days and 4% paraformaldehyde in PBS and embedded
in paraffin wax. Sections were cut at 5-µm thickness for
hematoxylin & eosin (H&E) staining and scored
macroscopically as follows: i) 0, no inflammation; ii) 1, presence
of a small discolored focus; iii) 2, presence of multiple small
discolored foci; iv) 3, diffuse discolored areas not exceeding a
total of 1/3 of the cardiac surface; and v) 4, diffuse discolored
areas totaling >1/3 of the cardiac surface (5,9,10).
Inflammatory cell infiltration was examined under a light
microscope, the ratio of the area of inflammatory cell infiltration
in each field to the area of the whole field was calculated, and
its mean value was used for microscopic scoring as follows: i) 0,
no inflammation; ii) 1, <25% of the heart section involved; iii)
2, 25-50%; iv) 3, 50-75%; and v) 4, >75% (5).
Cardiac function assessment
Echocardiography was performed on day 21 for all the
experimental rats using a 14-MHz probe (Vivid 7; GE Healthcare).
Wall thickness and left ventricular (LV) dimensions (including LV
internal dimensions in systole, LV internal dimensions in diastole,
and LV posterior wall of diastole), interventricular septal
thickness at diastolic (IVS), and heart rate were measured. LV
ejection fraction (LVEF) and LV fractional shortening (FS) were
assessed as previously described (9,10).
CD4(+) T cell isolation
CD4(+) T cells were isolated and purified from rats
with EAM and PPARα(-/-) mice spleens using microbeads [CD4(+) T
cell isolation kit; MiltenyiBiotec, Inc.] as previously described
(9). CD4(+) T cells from rats with
EAM were induced Th17 cell differentiation and incubated with
fenofibrate 20 µM (Abcam) and PPARα antagonist MK886 20 µM (Abcam)
for 24 h for reverse transcription-quantitative PCR (RT-qPCR)
analyses. CD4(+) T cells from PPARα(-/-) mice were activated using
1 µg/ml anti-CD3 (BD Biosciences) bound to plates and 2 µg/ml
anti-CD28 (BD Biosciences) in solution for 3, 6, 12 and 24 h.
CD4(+) T cells from PPARα(+/+) mice were added and incubated with
three PPARα agonists, fenofibrate 100 µmol/l (Abcam), Wy14643 50
µmol/l (Abcam) or GW7646 1 µmol/l (Abcam) for 24 h. Cells were
collected for western blotting and RT-qPCR analyses.
Western blot analysis
Cardiac ventricles from the EAM rats and cells were
homogenized in a lysis buffer composed of 8 M urea, 1 mM
dithiothreitol, 1 mM ethylenediaminetetraacetic acid (EDTA), and 50
mM Tris-HCl at pH 8.0. Following precipitation with
trichloro-acetate and sodium deoxycholate, the protein samples were
quantified using Lowry's method. SDS-PAGE (12.5%) was used to
isolate 10 µg of proteins, which were subsequently transferred to a
PVDF membrane, the membrane was blocked with 5% non-fat milk or 5%
BSA (Beijing Solarbio Science & Technology Co., Ltd.) for 1 h
at room temperature and incubated overnight at 4˚C with antibodies
against anti-vimentin (1:1,000; Thermo Fisher Scientific, Inc.),
anti-collagen I (1:1,000; Abcam), anti-RAR-related orphan receptor
gamma (RORγt; 1:1,000; BD Biosciences), anti-IκBζ (1:1,000; Cell
Signaling Technology, Inc.), anti-pNF-κBp65 (1:1,000; Thermo Fisher
Scientific, Inc.), anti-β-actin (1:2,000; Thermo Fisher Scientific,
Inc.), anti-GAPDH (1:2,000; Thermo Fisher Scientific, Inc.)
followed by incubation with goat anti-mouse IgG antibody (1:2,000;
Cell Signaling Technology, Inc.) or goat anti-rabbit IgG antibody
(1:2,000; Cell Signaling Technology, Inc.) at room temperature for
2 h. Membranes were eventually visualized with the ChemiDoc Touch
Imaging System (Bio-Rad Laboratories, Inc.). Quantification of the
resulting bands was achieved using densitometry software ImageJ
(version 1.5.4; National Institutes of Health).
RT-qPCR analysis
Total RNA was extracted from the rat hearts, spleen,
and isolated CD4(+) T cells using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) in accordance with the
manufacturer's protocol. Random primers and reverse transcriptase
were employed to synthesize cDNA from 2 µg of the total RNA using
the PrimeScript RT reagent Kit (Takara Bio, Inc.) according to the
manufacturer's protocol. The reaction temperature and time were set
according to the same protocol. qPCR was performed using SuperReal
PreMix Plus (SYBR Green; Tiangen Biotech Co., Ltd.) on an ABI 7500
Fast Real-Time PCR Detection system (Applied Biosystems; Thermo
Fisher Scientific, Inc.). Gene amplification was performed using
specific primer pairs (Table SI).
The reaction program was determined as 3 min at 95˚C, followed by
40 cycles of denaturation at 95˚C for 30 sec, and annealing at 60˚C
for 60 sec, and extension at 75˚C for 60 sec. Comparative analysis
of qPCR results by utilizing the 2-ΔΔCq method (17) was conducted to assess the relative
levels of these molecules. This analysis was carried out after
normalizing the results to GAPDH or β-actin expression.
Cytokine ELISA
The serum protein levels of IL-17 (eBioscience;
Thermo Fisher Scientific, Inc.), IFN-γ (R&D Systems), and IL-4
(R&D Systems) in the control, EAM, and fenofibrate-treated EAM
rats were measured using rat ELISA kit (Table SII) at day 21.
Immunohistochemistry staining
(IHC)
Deparaffinized cardiac tissue sections (5 µm) were
heated in EDTA buffer, treated with 3% H2O2
in methanol for 10 min and blocked with 5% normal serum at room
temperature for 30 min. These sections were incubated with the
primary antibodies anti-RORγt (1:100; Abcam), anti-vimentin (1:100;
Thermo Fisher Scientific, Inc.), and anti-IκBζ (1:100; Cell
Signaling Technology, Inc.) at 4˚C overnight, followed by secondary
antibodies rabbit anti-mouse IgG-HRP (1:5,000; Abcam) or goat
anti-rabbit IgG-HRP (1:5,000; Abcam) (Table SII) for 1 h at room temperature.
The sections were then visualized with DAB chromogen,
counterstained with hematoxylin for 10 sec at room temperature,
mounted with an antifade mounting medium (Applygen Technologies,
Inc.) and analyzed using an Olympus fluorescent Microscope (IX51;
Olympus Corporation) for the immunohistochemical examinations of
vimentin, IκBζ, and RORγt.
Statistical analysis
The results are presented as the mean values with
standard deviations. Statistical analysis was conducted using
one-way ANOVA, followed by Bonferroni's test for multiple
comparisons. Statistical significance was established at P<0.05.
All statistical analyses were performed using GraphPad Prism 7.0
software (GraphPad; Dotmatics).
Results
Fenofibrate treatment ameliorates rat
EAM
Preliminary studies about the long-term effects of
fenofibrate on EAM were conducted using different dosages from day
0 to day 21. The results showed that both 100 mg/kg and 200 mg/kg
of fenofibrate ameliorated EAM while the dosage of 200 mg/kg had
improved treatment effect compared with 100 mg/kg (Fig. S1). Therefore, fenofibrate was
administered at a dosage of 200 mg/kg to evaluate its long-term
effects in a previous study conducted by the authors (9). In the present study, fenofibrate (200
mg/kg) was used to evaluate its short-term effect. H&E staining
of transverse cardiac ventricle sections revealed severe
myocarditis in the rats with EAM, characterized by extensive
inflammatory cells infiltration. Fenofibrate treatment
significantly reduced inflammatory cells infiltration and mitigated
EAM-induced myocardial inflammation (Fig. 1A). EAM in rats caused macroscopic
and microscopic alterations with marked inflammatory cells
infiltration and necrosis, and fenofibrate treatment ameliorated
EAM as evidenced by a decrease in HW/BW, myocardial damage scores
and inflammatory cell infiltrate scores (Fig. 1B-D). According to echocardiographic
parameters, the rats with EAM showed enlarged LV, thick IVS and
decreased LVEF leading to heart failure, Meanwhile, fenofibrate
treatment improved the cardiac function as revealed by a reduction
in LV end diameter at systole and diastole and an increase in LVEF
and FS (Table I).
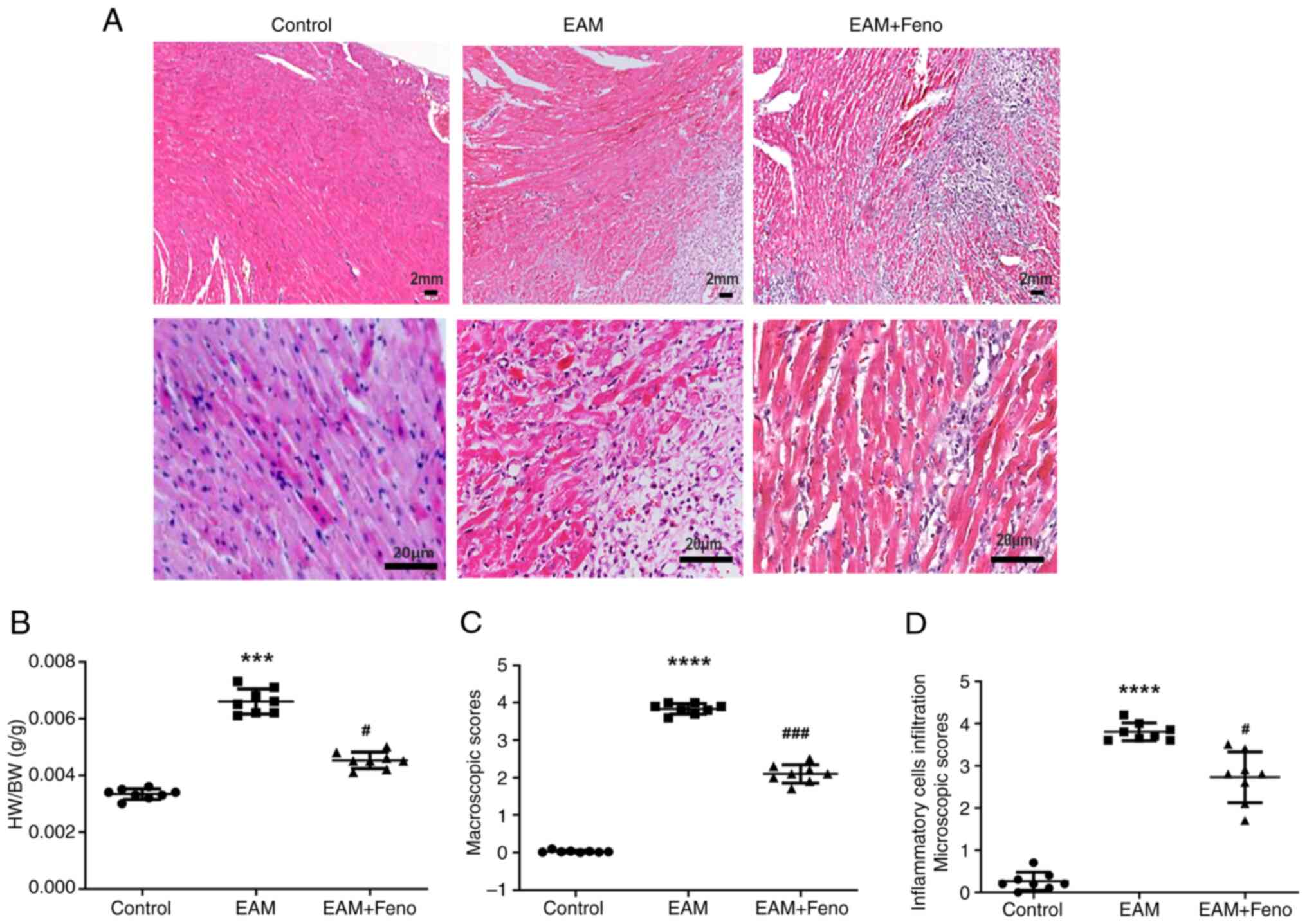 | Figure 1Feno alleviates rat EAM. (A)
Representative hematoxylin & eosin staining of ventricular
sections from control, EAM and feno-treated rats with EAM (upper
images, 10x10; scale bar, 2 mm and lower images, 10x40; scale bar,
20 µm). (B) Ratio of heart HW/BW. (C) EAM macroscopic scores. (D)
Microscopic scores (inflammatory cell infiltration) (n=8 for each
group). EAM vs. control, ***P<0.001 and
****P<0.0001. EAM + Feno vs. EAM,
#P<0.05 and ###P<0.001. EAM,
experimental autoimmune myocarditis; EAM + Feno, EAM and
fenofibrate; HW/BW, heart weight to body weight. |
 | Table IEchocardiographic parameters. |
Table I
Echocardiographic parameters.
| Parameters | Control | EAM | EAM + Feno |
|---|
| LVEDs (mm) | 3.055±0.22 |
6.291±0.884a |
3.904±0.604c |
| LVEDd (mm) | 5.816±0.186 |
8.591±0.768a |
5.753±0.931c |
| LVPW (mm) | 1.347±0.18 | 1.732±0.475 | 1.513±0.185 |
| IVS (mm) | 1.208±0.137 |
2.520±0.285a | 1.399±0.168 |
| LVEF (%) | 77.87±3.823 |
52.014±6.184b |
54.319±8.652c |
| LVFS (%) | 47.327±3.972 |
27.999±3.807b |
30.297±5.603c |
| Heart rate
(time/min) | 294±13 | 458±24b | 342±28d |
Fenofibrate inhibits the expression of
Th17-related inflammatory cytokines
The experimental rats were euthanized after 21 days.
Hearts, splenocytes and CD4(+) T cells were isolated. RT-qPCR
quantified IL-6, TGF-β, IL-23 and RORγt expression levels,
revealing that fenofibrate treatment suppressed the expression of
these inflammatory cytokines in the hearts, splenocytes and
CD4+ T cells of the rats with EAM (Fig. 2A-C). Fenofibrate treatment also
significantly reduced the expression of Th17-related factors TNF-α,
IL-1β, IFNγ, IL-4 and IL-17 (Fig.
3A-E). ELISA confirmed that fenofibrate treatment suppressed
the serum protein levels of IFNγ, IL-4 and IL-17 in the rats with
EAM (Fig. 3F).
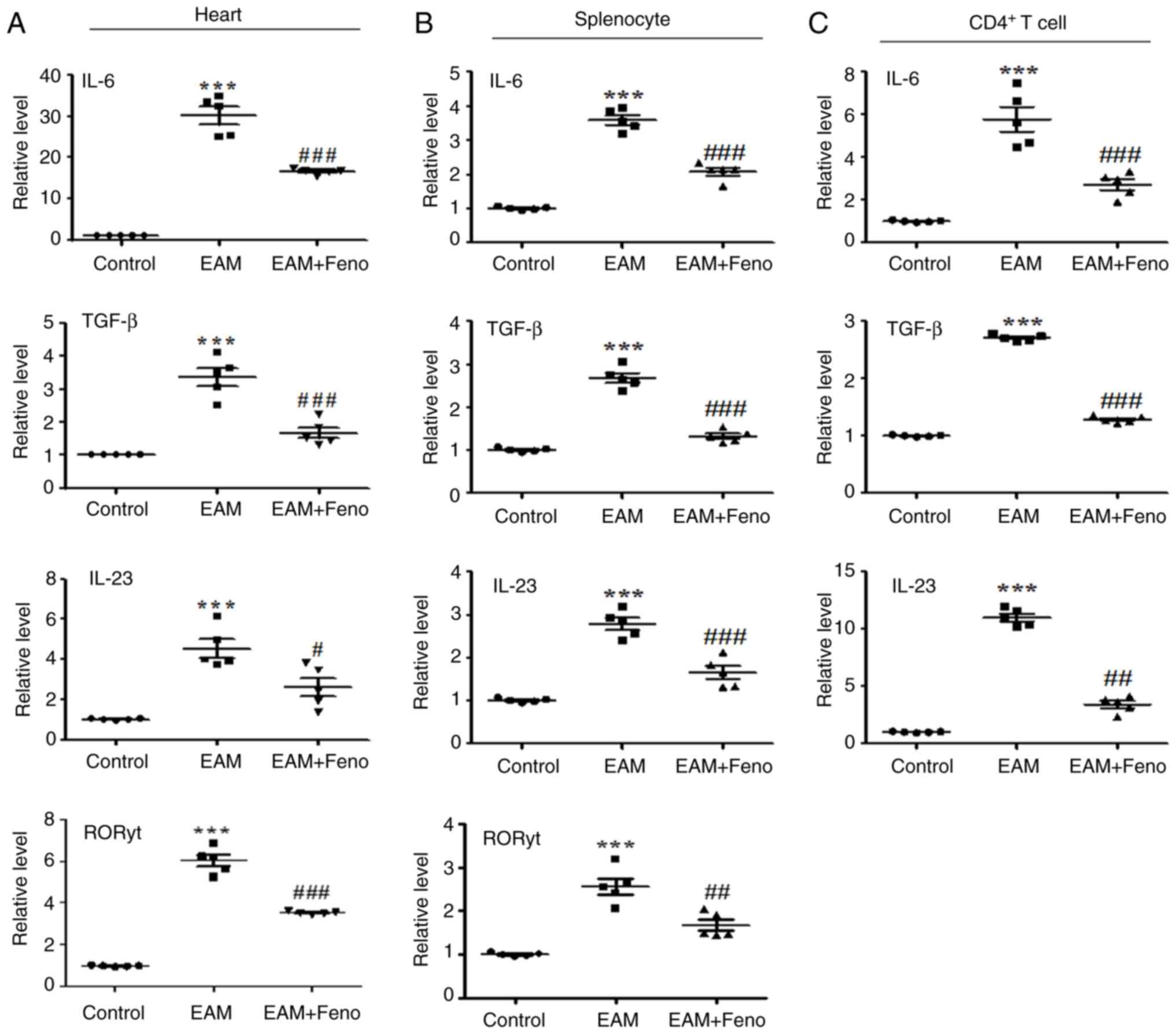 | Figure 2Feno inhibits IL-6, TGF-β, IL-23 and
RORγt expression in the heart, splenocyte and CD4(+) T cells. (A)
Heart, (B) splenocytes and (C) CD4(+) T cells were purified from
the spleens of rats. Reverse transcription-quantitative PCR was
employed to measure the relative transcript levels, with GAPDH
serving as the internal reference for normalization (n=5 for each
group). EAM vs. control, ***P<0.001. EAM + Feno vs.
EAM, #P<0.05; ##P<0.01 and
###P<0.001. EAM, experimental autoimmune myocarditis;
EAM + Feno, EAM and fenofibrate; IL, interleukin; TGF-β,
transforming growth factor beta; RORγt, RAR-related orphan receptor
gamma t. |
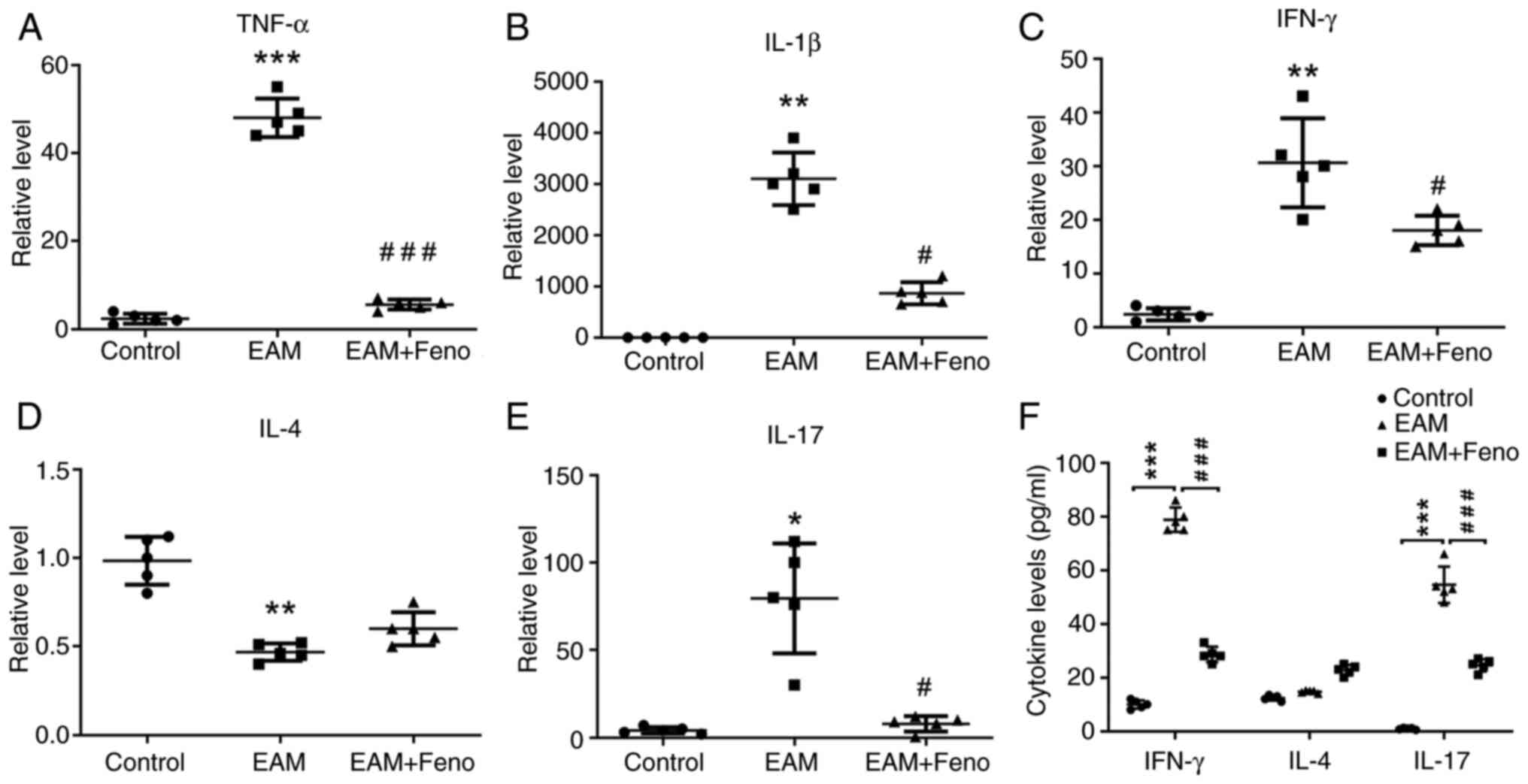 | Figure 3Feno inhibits the expression of
Th17-related factors in the heart of rats with EAM. (A-E) TNF-α,
IL-1β, IFN-γ, IL-4 and IL-17. Rat hearts were isolated and examined
by reverse transcription-quantitative PCR analysis, with GAPDH
serving as the internal reference for normalization (n=5 for each
group). (F) Serum levels of IFNγ, IL-4 and IL-17 on day 21 of EAM
rats were tested by ELISA (n=5 for each group). EAM vs. control,
*P<0.05; **P<0.01 and
***P<0.001. EAM + Feno vs. EAM, #P<0.05
and ###P<0.001. EAM, experimental autoimmune
myocarditis; EAM + Feno, EAM and fenofibrate; Th17, T helper 17;
TNF-α, tumor necrosis factor alpha; IL, interleukin; IFN-γ,
interferon gamma. |
Fenofibrate inhibits the expression of
fibrosis-associated factors
PPARα can suppress TGF-β-induced myocardial fibrosis
(18). Therefore, the expression
of fibrosis-associated factors TGF-β, tissue inhibitors of
metalloproteinase (TIMP), fibronectin (FN) and galectin-3 (GAL3),
and fibrosis markers vimentin and collagen type I (collagen I) were
assessed in the hearts of rats with EAM. Fenofibrate treatment
significantly reduced their expression (Fig. 4A-D). IHC and western blot analysis
revealed that fenofibrate treatment significantly inhibited the
expression of vimentin and collagen I (Fig. 4E and F).
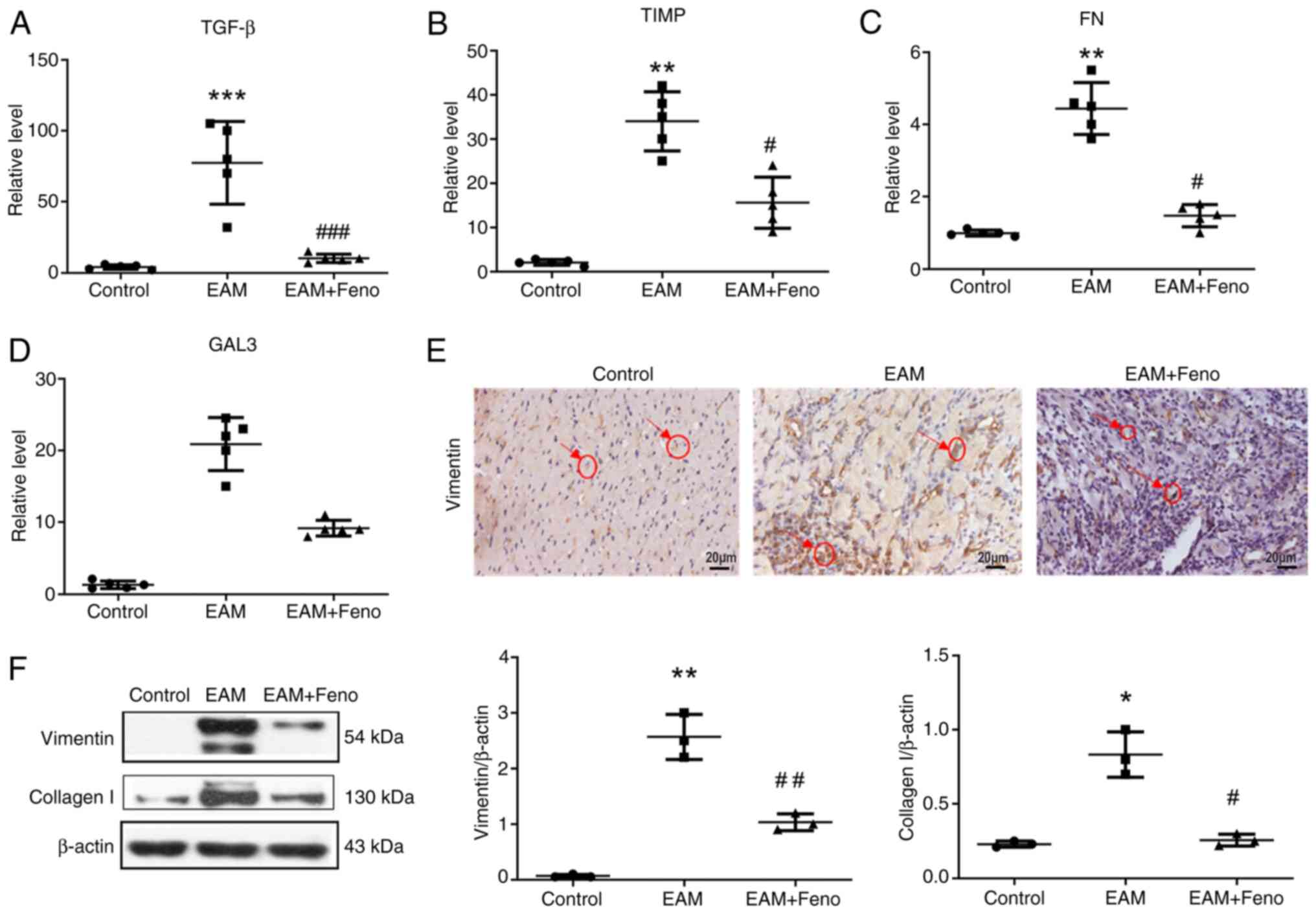 | Figure 4Feno inhibits the expression of
fibrosis-associated factors in the heart of rats with EAM. mRNA
levels of (A) TGF-β, (B) TIMPs, (C) FN and (D) GAL3 were determined
using reverse transcription-quantitative PCR analysis with GAPDH
serving as the internal reference for normalization (n=5 for each
group). (E) Vimentin, representative immunohistochemistry image of
ventricular sections. Scale bar, 20 µm. (F) Vimentin and collagen I
protein expression in the heart examined by western blot analysis.
β-actin serving as the internal reference (n=3). EAM vs. control,
*P<0.05, **P<0.01 and
***P<0.001. EAM + Feno vs. EAM,
#P<0.05, ##P<0.01 and
###P<0.001. EAM, experimental autoimmune myocarditis;
EAM + Feno, EAM and fenofibrate; TGF-β, transforming growth factor
beta; TIMPs, tissue inhibitors of metalloproteinase; FN,
fibronectin; GAL3, galectin 3. |
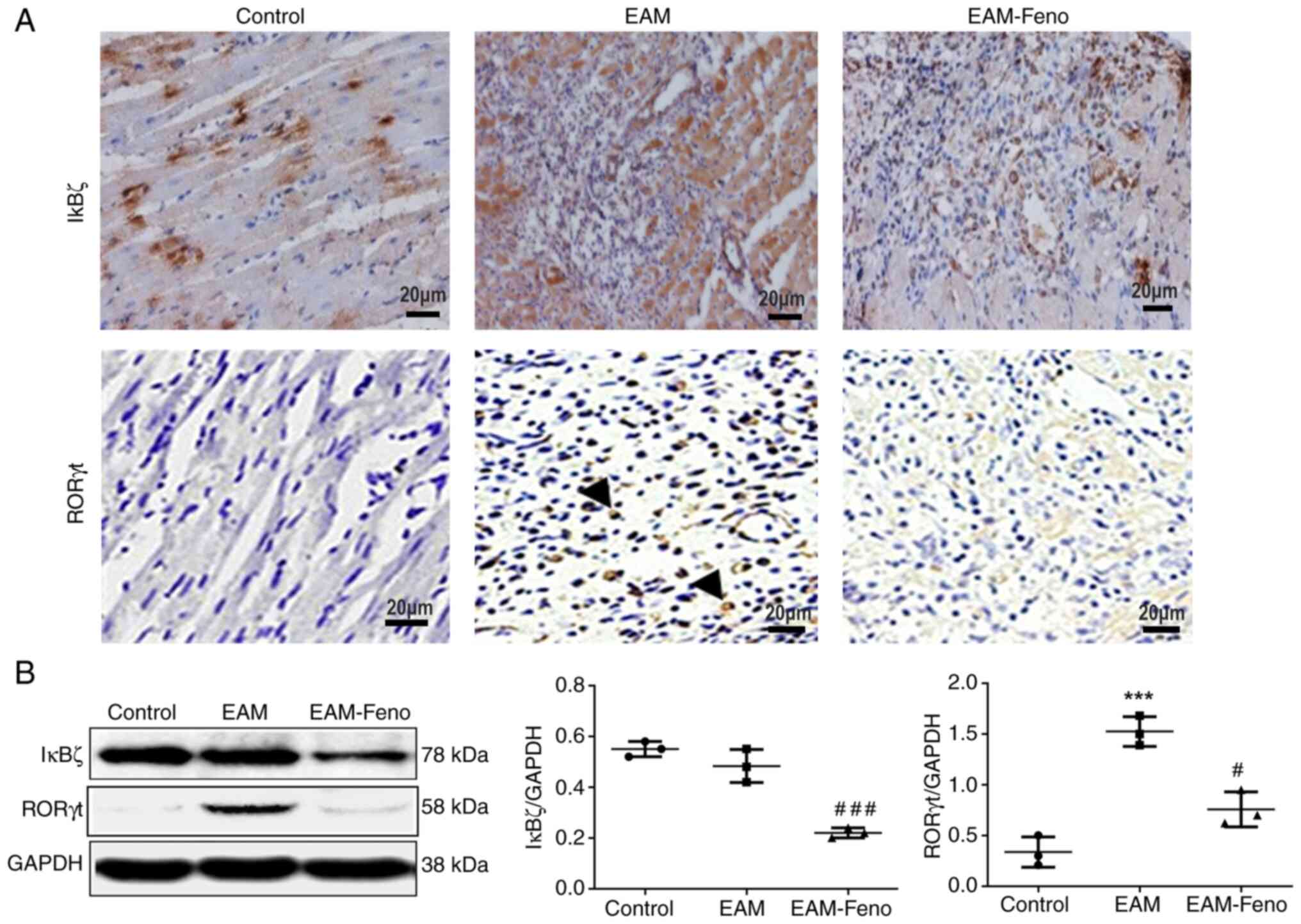
Fenofibrate inhibits IκBζ expression
in the heart of rats with EAM
IκBζ is critical in inflammatory and autoimmune
diseases, and RORγt is essential for Th17 differentiation in EAM
(19). IHC staining of transverse
cardiac sections revealed the extensive infiltration of
IκBζ-positive and RORγt-positive cells in the myocardium at EAM
lesions, which were significantly reduced by fenofibrate treatment
(Fig. 5A). Western blot analysis
confirmed that fenofibrate suppressed IκBζ and RORγt expression
(Fig. 5B).
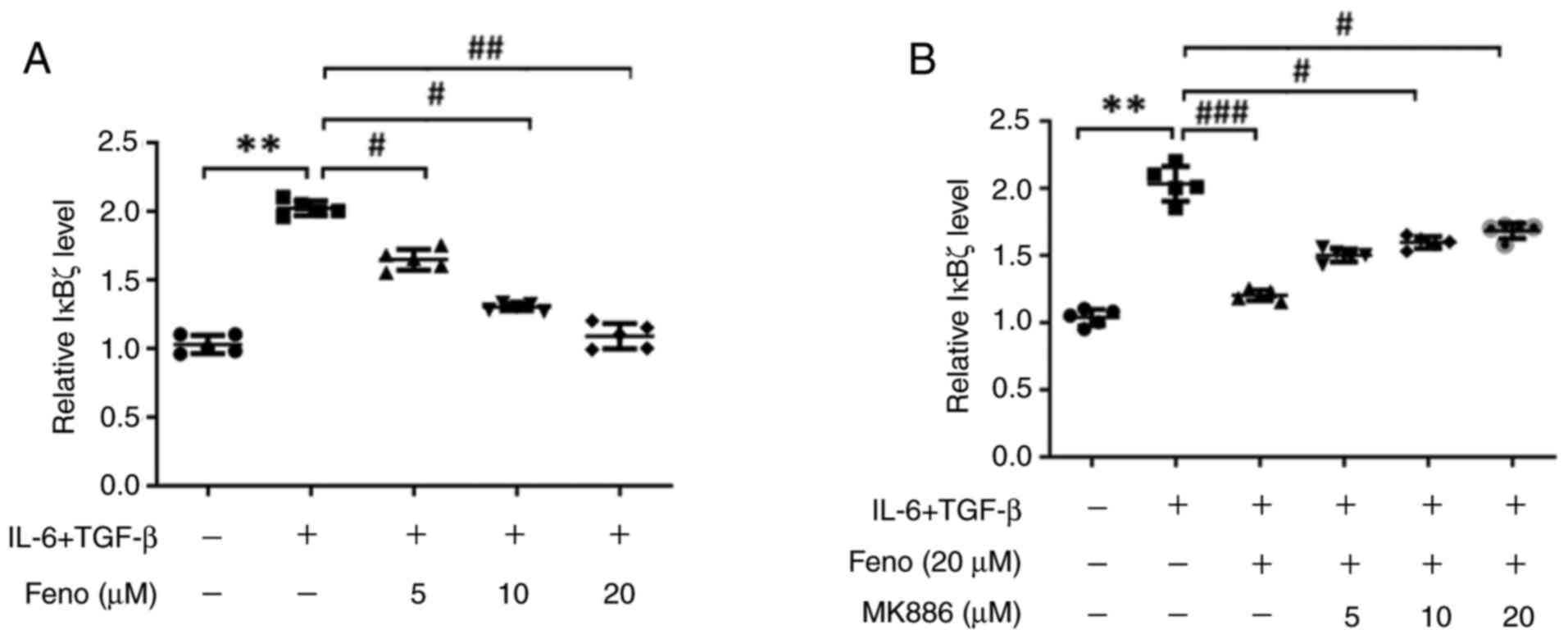
Fenofibrate inhibits IκBζ expression
in the CD4(+) T cells from rats with EAM
The CD4(+) T cells were purified from the spleen of
rats with EAM and induced Th17 cell differentiation using
recombinant (r) IL-6 and rTGF-β. RT-qPCR analysis demonstrated that
fenofibrate, at concentrations ranging from 0 to 20 µM,
dose-dependently decreased IκBζ (Fig.
6A). MK886, which acts as a PPARα antagonist, displayed a
dose-dependent reversal of these effects (Fig. 6B). These findings indicated that
IκBζ is involved in Th17 differentiation during EAM
development.
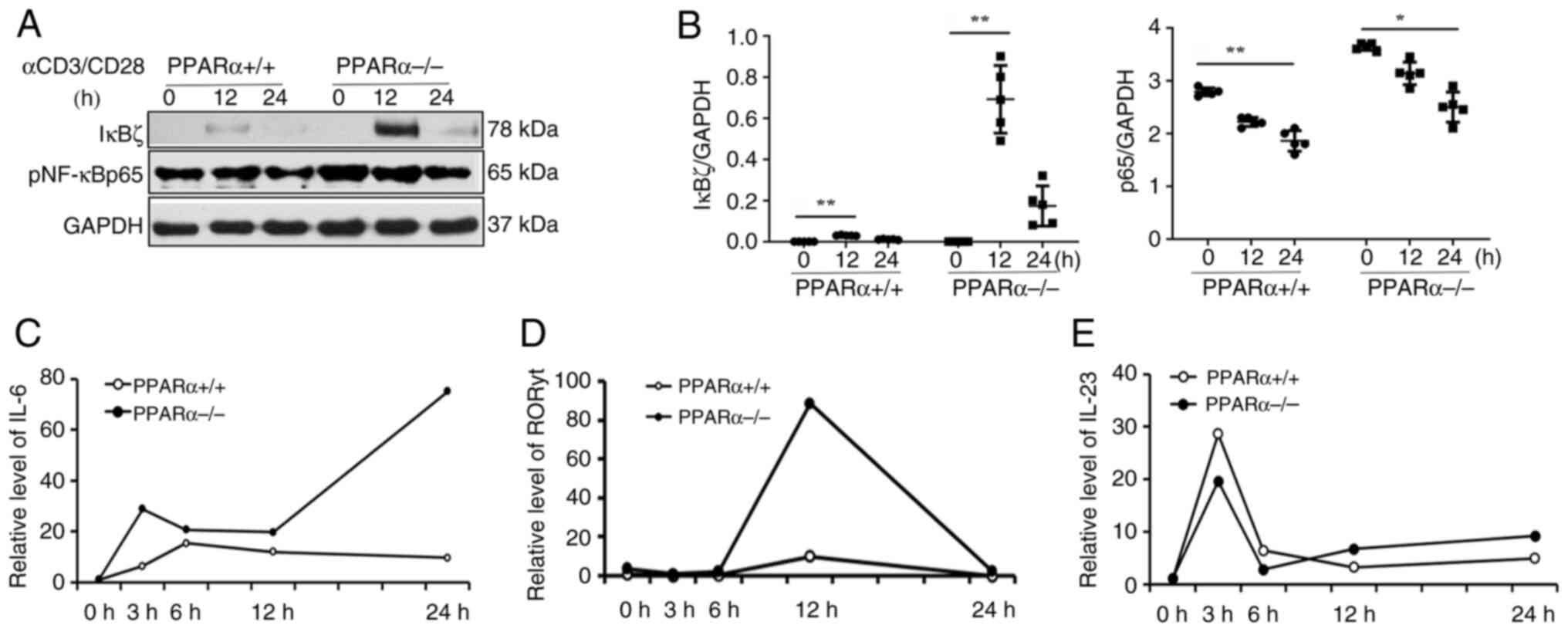
PPARα deficiency upregulates IκBζ and
IL-6 expression
Spleen-derived CD4(+) T cells from PPARα(-/-) mice
were exposed to anti-CD3 (1 µg/ml) and anti-CD28 (2 µg/ml)
monoclonal antibodies and incubated for periods of 3, 6, 12 and 24
h. Western blot analysis demonstrated that IκBζ and pNF-κBp65
levels were significantly increased in the CD4(+) T cells of
PPARα(-/-) mice compared with that in PPARα(+/+) mice (Fig. 7A and B). IL-6 is regulated by IκB-ζ and NF-κB
activation. IκBζ controls Th17 differentiation by RORγt and IL-23
activation (20). It was observed
that the levels of IL-6 and RORγt were higher at all tested time
points in the CD4(+) T cells of PPARα (-/-) mice. However, the
IL-23 expression did not change significantly (Fig. 7C-E).
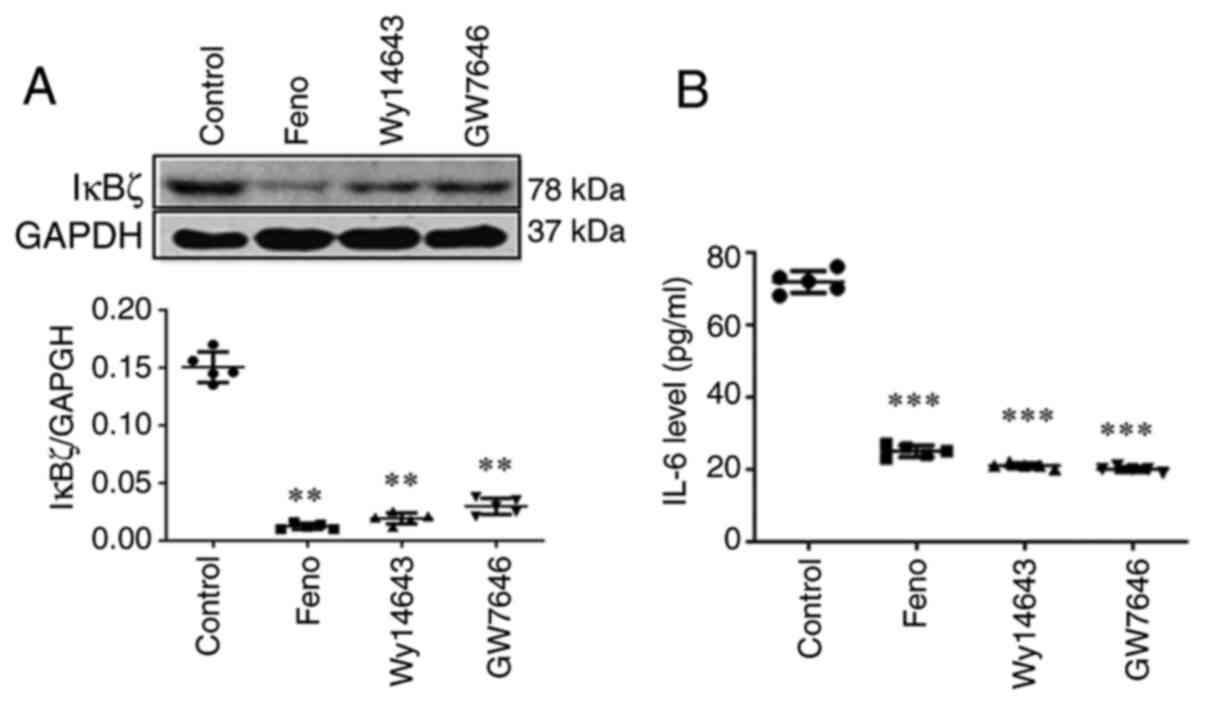
Activation of PPARα inhibits IκBζ and
IL-6 expression
CD4(+) T cells from PPARα(+/+) mice were added and
incubated with three PPARα agonists (fenofibrate 100 µmol/l,
Wy14643 50 µmol/l, GW7646 1 µmol/l) to study whether PPARα affects
IL-6 expression. Western blot analysis revealed that these PPARα
agonists suppressed IκBζ expression (Fig. 8A). ELISA results also indicated
that these PPARα agonists significantly reduced IL-6 secretion
(Fig. 8B). All these findings
indicated that IL-6 mediates Th17 differentiation via the
PPARα/IκBζ pathway.
Discussion
Myocarditis is an inflammatory cardiomyopathy that
can lead to acute heart failure and dilated cardiomyopathy and
currently has no specific treatment. EAM in rats is similar to
human giant cell myocarditis, and recurrent forms can lead to
dilated cardiomyopathy (4-6).
In a previous study conducted by the authors, it was found that the
peak of EAM cardiac inflammation occurred on days 14 to 21 and was
characterized by infiltration of the CD4(+) T cells from the spleen
into the myocardium. It was also revealed that Th17 cells play an
important role in the development of EAM and that fenofibrate can
improve EAM by inhibiting Th17 differentiation (9).
In the present study, it was demonstrated that
short-term administration of fenofibrate alleviated EAM. The
expression levels of typical Th17-related factors including IL-6,
TGF-β and IL-23 in the heart and splenic CD4(+) T cells from rats
with EAM were significantly increased. The proinflammatory
cytokines TNF-α, IL-1β, IFNγ and IL-17 were also significantly
upregulated. The levels of these factors were reduced significantly
by fenofibrate treatment. These findings suggested that fenofibrate
attenuates cardiac inflammation and exerts anti-inflammatory
effects by suppressing Th17-related inflammatory cytokines
secretion. Thus, this provides a more comprehensive understanding
of fenofibrate's therapeutic potential on autoimmune myocarditis.
This result is consistent with the effect of fenofibrate on other
inflammatory diseases (21,22).
In the pathological process of EAM, the infiltration
of inflammatory cells into myocardium leads to myocardial fibrosis.
Although cardiac fibrosis is beneficial for enhancing the
structural stability of the heart, it can also lead to heart
structure remodeling and impaired cardiac function. Therefore,
improving cardiac fibrosis may help avoid further deterioration
caused by EAM. The expression levels of types I, III and IV
collagen, FN, matrix metalloproteinases, and TIMP are increased
during the progression of myocardial fibrosis (23). PPARα activation can inhibit the
TGF-β-induced cardiac fibrosis pathway. Fenofibrate can also reduce
myocardial inflammation and collagen deposition by modulating the
PPARα pathway and therefore, fenofibrate can relieve cardiac
fibrosis and reverse cardiac dysfunction (24,25).
In the present study, the expression levels of fibrosis-related
factors TIMP, FN and GAL3 as well as fibrosis markers vimentin and
collagen I, were detected by western blotting and RT-qPCR.
Consistent with previous studies, the results also showed that
these fibrosis-related factors were significantly upregulated in
the hearts of EAM rats, and that fenofibrate treatment inhibited
the levels of these factors and improved cardiac fibrosis.
IκBζ is a key Th17-related factor that plays an
important role in the development of autoimmune diseases such as
psoriasis (26,27). In mice with IkBz-deficiency, the
development of psoriasis induced by IL-17, IL-23 and imiquimod was
significantly inhibited (28,29).
IL-17 and its family members, produced by CD4(+) T cells and
various innate immune cells, are implicated in the pathogenesis of
EAM (30); however, its role in
EAM remains to be investigated. The results of the present study
indicated that fenofibrate significantly inhibited the expression
of IκBζ in the heart of rats with EAM. IL-6 plays an important role
in EAM initiation through RORγt-mediated Th17 differentiation. The
present study also revealed that fenofibrate treatment
significantly inhibited the upregulation of RORγt expression in the
hearts of rats with EAM. These findings suggested that IκBζ is a
molecular target involved in Th17 differentiation in autoimmune
myocarditis. By stimulating CD4(+) T cells isolated from the spleen
of EAM rats to induce Th17 cell differentiation, it was found that
PPARα agonist fenofibrate upregulated IκBζ expression in a
dose-dependent manner. This effect was reversed by PPARα antagonist
MK886 in a dose-dependent manner. These results suggested that
PPARα promotes Th17 cell differentiation through the IκBζ signaling
pathway.
To further explore the potential mechanism of
fenofibrate in treating EAM, CD4(+) T cells isolated from the
spleen of PPARα-/- mice were activated and it was revealed that the
mRNA and protein levels of IκBζ were upregulated in activated
PPARα(-/-) mice CD4(+) T cells compared with those in PPARα(+/+)
mice. IκBζ interacts with the NF-κBp50 subunit to positively
regulate the expression of pro-inflammatory cytokines such as IL-6,
IL-12 and CCL2(31). The
activation of NF-κB and stimulation of Toll-like receptor ligands
and IL-1β are required to induce IκBζ expression (32). Previously, several studies have
reported that IκBζ deficiency in LPS-induced macrophages prevents
the production of the key pro-inflammatory cytokine IL-6(33). In the absence of IκBζ, T cells
exhibit serious defects in the development of Th17 cells (34). IκBζ is induced by IL-17R and
collaboratively regulates IL-17 expression with RORγt. IL-6, which
is a key factor in inducing Th17 cell differentiation, activates
STAT3 and increases the expression of RORγt (35). Th17 cell differentiation and IL-6
secretion can be inhibited by PPARα agonists (36).
In the present study, the correlation between PPARα
and IκBζ was confirmed. In vivo, it was shown that PPARα
activation inhibited IκBζ expression on EAM. In vitro, it
was revealed that PPARα deficiency upregulated IκBζ and IL-6
expression in the CD4(+) T cells from PPARα(-/-) mice. By
conducting Chromatin immunoprecipitation (ChIP) assays, Muromoto
et al (34,37) found that two different IκB-ζ
promoter regions and STAT3 constitutively binds to the genomic
promoter region of IκB-ζ TSS1. Luciferase reporter assays of the
IκB-ζ promoter activity, revealed that catalytic activity of TYK2
and its substrate transcription factor STAT3, is required for IκB-ζ
promoter activity. The limitation of the present study is the lack
of direct molecular evidence of PPARα binding or transcriptional
modulation with IκB-ζ. In a future study, the authors will provide
further molecular evidences and focus on specific mechanisms
regarding how PPARα and fenofibrate interacts with IκBζ and whether
PPARα directly interacts with or binds the promoter regions of IκBζ
gene. Luciferase reporter assays will be performed to evaluate the
effects of fenofibrate on the PPARα/IκBζ pathway activation, and
the promoter activity of IκBζ and mRNA stability will be examined
using ChIP assay. Further mechanistic studies will be
continued.
In summary, it was demonstrated that IκBζ
contributes to the pathogenesis of autoimmune myocarditis, and
fenofibrate treatment ameliorates EAM by preventing myocardial
inflammation and fibrosis possibly through the PPARα/IκBζ signaling
pathway. Thus, IκBζ may be a new molecular target for fenofibrate
treatment in autoimmune myocarditis.
Supplementary Material
Long-term effects of different dosages
of fenofibrate on rat EAM. (A) Representative whole heart images
(scale bar, 2 mm). (B) Representative hematoxylin and eosin
staining of ventricular sections images (scale bar, 20 μm).
(C) Ratio of heart HW/BW. (D) The relative transcript levels of
heart failure marker ANP were examined by reverse
transcription-quantitative PCR analysis, with GAPDH serving as the
internal reference for normalization. Echocardiograph parameters:
(E) LVEDd (mm); (F) EF (%). The immunized EAM rats underwent daily
oral gavage administration with fenofibrate (100 and 200 mg/kg) or
a solvent from day 0 to day 21. All the experiment rats were
euthanized on day 21. N=5 for each group. **P<0.01
and ***P<0.001 vs. control. #P<0.05 and
##P<0.01 vs. EAM. ANP, Atrial natriuretic peptide;
LVEDd, left ventricular end-diastolic internal diameter; EF, left
ventricular ejection fraction; EAM, experimental autoimmune
myocarditis; HW/BW, heart weight/body weight.
Rat primers for reverse
transcription-quantitative PCR.
Catalogue number of antibodies and
kits used in the present study.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Natural Science
Foundation of Fujian Province of China (grant no. 2021J01014), the
Scientific Research Foundation for Advanced Talents, Xiang'an
Hospital of Xiamen University, Fujian, China (grant no.
PM201809170018).
Availability of data and materials
The data generated in this study may be requested
from the corresponding author.
Authors' contributions
HC and ZQ designed the study, wrote and revised the
manuscript. YWa and YWu conducted the experiments to collected
data. SLS analyzed the data. HC and ZQ confirm the authenticity of
all the raw data. All authors read and approved the final version
of the manuscript.
Ethics approval and consent to
participate
Animal care and experiments were conducted in
accordance with the procedures approved by the Ethics Committee of
Animal Care and Use of Xiamen University (approval no. XMULAC
20220111; Xiamen, China). All animals were treated in accordance
with the principles of The Declaration of Helsinki and welfare
considerations were taken to minimize the number of animals used
and their suffering.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Manoharan I, Suryawanshi A, Hong Y,
Ranganathan P, Shanmugam A, Ahmad S, Swafford D, Manicassamy B,
Ramesh G, Koni PA, et al: Homeostatic PPARα signaling limits
inflammatory responses to commensal microbiota in the intestine. J
Immunol. 196:4739–4749. 2016.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Riaz F, Wei P and Pan F: PPARs at the
crossroads of T cell differentiation and type 1 diabetes. Front
Immunol. 14(1292238)2023.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Balakumar P, Rohilla A and Mahadevan N:
Pleiotropic actions of fenofibrate on the heart. Pharmacol Res.
63:8–12. 2011.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Kodama M, Matsumoto Y, Fujiwara M, Masani
F, Izumi T and Shibata A: A novel experimental model of giant cell
myocarditis induced in rats by immunization with cardiac myosin
fraction. Clin Immunol Immunopathol. 57:250–262. 1990.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Zhong C, Wu Y, Chang H, Liu C, Zhou L, Zou
J and Qi Z: Effect of PKC inhibitor on experimental autoimmune
myocarditis in Lewis rats. Oncotarget. 8:54187–54198.
2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Zhong C, Chang H, Wu Y, Zhou L, Wang Y,
Wang M, Wu P, Qi Z and Zou J: Up-regulated Cx43 phosphorylation at
Ser368 prolongs QRS duration in myocarditis. J Cell Mol Med.
22:3537–3547. 2018.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Cheng H, Xi Y, Chi X, Wu Y and Liu G:
Fenofibrate treatment of rats with experimental autoimmune
myocarditis by alleviating Treg/Th17 disorder. Cent Eur J Immunol.
41:64–70. 2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Maruyama S, Kato K, Kodama M, Hirono S,
Fuse K, Nakagawa O, Nakazawa M, Miida T, Yamamoto T, Watanabe K and
Aizawa Y: Fenofibrate, a peroxisome proliferator-activated receptor
alpha activator, suppresses experimental autoimmune myocarditis by
stimulating the interleukin-10 pathway in rats. J Atheroscler
Thromb. 9:87–92. 2002.PubMed/NCBI View
Article : Google Scholar
|
|
9
|
Chang H, Zhao F, Xie X, Liao Y, Song Y,
Liu C, Wu Y, Wang Y, Liu D, Wang Y, et al: PPARα suppresses Th17
cell differentiation through IL-6/STAT3/RORγt pathway in
experimental autoimmune myocarditis. Exp Cell Res. 375:22–30.
2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Chang H, Wang Y, Wu Y, Ma P, Song Y, Liu
C, Ye Y, Qi JH and Qi Z: Cardiac apoptosis caused by elevated
cholesterol level in experimental autoimmune myocarditis. Exp Cell
Res. 395(112169)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Feng Y, Chen Z, Xu Y, Han Y, Jia X, Wang
Z, Zhang N and Lv W: The central inflammatory regulator IκBζ:
Induction, regulation and physiological functions. Front Immunol.
14(1188253)2023.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Hövelmeyer N, Schmidt-Supprian M and
Ohnmacht C: NF-κB in control of regulatory T cell development,
identity, and function. J Mol Med (Berl). 100:985–995.
2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Kakiuchi N, Yoshida K, Uchino M, Kihara T,
Akaki K, Inoue Y, Kawada K, Nagayama S, Yokoyama A, Yamamoto S, et
al: Frequent mutations that converge on the NFKBIZ pathway in
ulcerative colitis. Nature. 577:260–265. 2020.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Choi MC, MaruYama T, Chun CH and Park Y:
Alleviation of murine osteoarthritis by cartilage-specific deletion
of IκBζ. Arthritis Rheumatol. 70:1440–1449. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Bambouskova M, Gorvel L, Lampropoulou V,
Sergushichev A, Loginicheva E, Johnson K, Korenfeld D, Mathyer ME,
Kim H, Huang LH, et al: Electrophilic properties of itaconate and
derivatives regulate the IκBζ-ATF3 inflammatory axis. Nature.
556:501–504. 2018.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Slowikowski K, Nguyen HN, Noss EH, Simmons
DP, Mizoguchi F, Watts GFM, Gurish MF, Brenner MB and Raychaudhuri
S: CUX1 and IκBζ (NFKBIZ) mediate the synergistic inflammatory
response to TNF and IL-17A in stromal fibroblasts. Proc Natl Acad
Sci USA. 117:5532–5541. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Rao X, Huang X, Zhou Z and Lin X: An
improvement of the 2ˆ(-delta delta CT) method for quantitative
real-time polymerase chain reaction data analysis. Biostat
Bioinforma Biomath. 3:71–85. 2013.PubMed/NCBI
|
|
18
|
Bansal T, Chatterjee E, Singh J, Ray A,
Kundu B, Thankamani V, Sengupta S and Sarkar S: Arjunolic acid, a
peroxisome proliferator-activated receptor α agonist, regresses
cardiac fibrosis by inhibiting non-canonical TGF-β signaling. J
Biol Chem. 292:16440–16462. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Yamazaki S: The nuclear NF-κB regulator
IκBζ: Updates on its molecular functions and pathophysiological
roles. Cells. 13(1467)2024.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Hawkes JE, Yan BY, Chan TC and Krueger JG:
Discovery of the IL-23/IL-17 signaling pathway and the treatment of
psoriasis. J Immunol. 201:1605–1613. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Park A and Heo TH: IL-17A-targeting
fenofibrate attenuates inflammation in psoriasis by inducing
autophagy. Life Sci. 326(121755)2023.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Elaidy SM, Essawy SS, Hussain MA,
El-Kherbetawy MK and Hamed ER: Modulation of the IL-23/IL-17 axis
by fenofibrate ameliorates the ovalbumin/lipopolysaccharide-induced
airway inflammation and bronchial asthma in rats. Naunyn
Schmiedebergs Arch Pharmacol. 391:309–321. 2018.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Cao JW, Duan SY, Zhang HX, Chen Y and Guo
M: Zinc deficiency promoted fibrosis via ROS and TIMP/MMPs in the
myocardium of mice. Biol Trace Elem Res. 196:145–152.
2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Zhang Y, Ji H, Qiao O, Li Z, Pecoraro L,
Zhang X, Han X, Wang W, Zhang X, Man S, et al: Nanoparticle
conjugation of ginsenoside Rb3 inhibits myocardial fibrosis by
regulating PPARα pathway. Biomed Pharmacother.
139(111630)2021.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Qiu Z, Zhao Y, Tao T, Guo W, Liu R, Huang
J and Xu G: Activation of PPARα ameliorates cardiac fibrosis in
Dsg2-deficient arrhythmogenic cardiomyopathy. Cells.
11(3184)2022.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Bertelsen T, Ljungberg C, Litman T,
Huppertz C, Hennze R, Rønholt K, Iversen L and Johansen C: IκBζ is
a key player in the antipsoriatic effects of secukinumab. J Allergy
Clin Immunol. 145:379–90. 2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Gautam P, Maenner S, Cailotto F, Reboul P,
Labialle S, Jouzeau JY, Bourgaud F and Moulin D: Emerging role of
IκBζ in inflammation: Emphasis on psoriasis. Clin Transl Med.
12(e1032)2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Bertelsen T, Iversen L and Johansen C: The
human IL-17A/F heterodimer regulates psoriasis-associated genes
through IκBζ. Exp Dermatol. 27:1048–1052. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Mok BR, Kim AR, Baek SH, Ahn JH, Seok SH,
Shin JU and Kim DH: PFN1 prevents psoriasis pathogenesis through
IκBζ regulation. J Invest Dermatol. 142:2455–2463.e9.
2022.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Mills KHG: IL-17 and IL-17-producing cells
in protection versus pathology. Nat Rev Immunol. 23:38–54.
2023.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Ohto-Ozaki H, Hayakawa M, Kamoshita N,
Maruyama T, Tominaga SI and Ohmori T: Induction of IκBζ augments
cytokine and chemokine production by IL-33 in mast cells. J
Immunol. 204:2033–2042. 2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Yamamoto M, Yamazaki S, Uematsu S, Sato S,
Hemmi H, Hoshino K, Kaisho T, Kuwata H, Takeuchi O, Takeshige K, et
al: Regulation of Toll/IL-1-receptor-mediated gene expression by
the inducible nuclear protein IkappaBzeta. Nature. 430:218–222.
2004.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Lorscheid S, Müller A, Löffler J, Resch C,
Bucher P, Kurschus FC, Waisman A, Schäkel K, Hailfinger S,
Schulze-Osthoff K and Kramer D: Keratinocyte-derived IκBζ drives
psoriasis and associated systemic inflammation. JCI Insight.
4(e130835)2019.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Muromoto R, Sato A, Komori Y, Nariya K,
Kitai Y, Kashiwakura JI and Matsuda T: Regulation of NFKBIZ gene
promoter activity by STAT3, C/EBPβ, and STAT1. Biochem Biophys Res
Commun. 613:61–66. 2022.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Wang J, Liu T, Chen X, Jin Q, Chen Y,
Zhang L, Han Z, Chen D, Li Y, Lv Q and Xie M: Bazedoxifene
regulates Th17 immune response to ameliorate experimental
autoimmune myocarditis via inhibition of STAT3 activation. Front
Pharmacol. 11(613160)2021.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zhou Z, Sun W, Liang Y, Gao Y, Kong W,
Guan Y, Feng J and Wang X: Fenofibrate inhibited the
differentiation of T helper 17 cells in vitro. PPAR Res.
2012(145654)2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Muromoto R, Tawa K, Ohgakiuchi Y, Sato A,
Saino Y, Hirashima K, Minoguchi H, Kitai Y, Kashiwakura JI, Shimoda
K, et al: IκB-ζ expression requires both TYK2/STAT3 activity and
IL-17-regulated mRNA stabilization. Immunohorizons. 3:172–185.
2019.PubMed/NCBI View Article : Google Scholar
|