Introduction
Retinal cone dystrophy (COD) is a heterogeneous
inherited retinal disease with a prevalence of 1 in 30,000-40,000
individuals (1). The disease is
characterized by decreased central vision, color vision impairment
and increased sensitivity to light, due to abnormal photoreceptor
function in early-stage retinal cone cells. As the disease
progresses, the photoreceptor function of rods deteriorates,
resulting in night blindness and loss of peripheral vision. COD can
be classified as stable or progressive. The stable form presents
with congenital or early infantile onset, which is primarily
characterized by cone cell dysfunction, whereas the progressive
type presents with later onset, typically in mid-adolescence or
later, and progressively involves rod photoreceptors (2).
Cyclic nucleotide-gated channel subunit α 3
(CNGA3), located on chromosome 2q11.2, encodes a member of
the CNG cation-channel protein family that is crucial for normal
vision (3). CNGA3 mutations
are associated with color vision disorders, including color
blindness (4), and even cone cell
dystrophy and Leber congenital amaurosis in rare cases (5). To date, 36 CNGA3 mutations
have been associated with cone cell dystrophy and 102 have been
associated with color blindness recorded in the Human Gene Mutation
Database (https://www.hgmd.cf.ac.uk/ac/index.php).
The present study reports a case of COD in a young
girl exhibiting bilateral reduced visual acuity and mild nystagmus.
Whole-exome sequencing was performed on the proband and the
unaffected parents, followed by bioinformatic filtering of rare
variants and validation via Sanger sequencing. Identified
CNGA3 mutations were cloned into expression plasmids,
transfected into 293T cells and analyzed by western blotting. The
protein stability and tissue expression patterns were assessed by
bioinformatics tools. The findings expand the CNGA3 mutation
spectrum in COD pathogenesis and suggest that specific CNGA3
structural domains may represent mutation hotspots in COD,
highlighting potential targets for developing therapies for
CNGA3-related retinal diseases.
Materials and methods
Sample collection and whole-exome
sequencing
Peripheral blood was obtained from the patient, a
9-year-old girl with COD, in September 2020 at Tianjin Eye Hospital
(Tianjin, China). During the same hospital visit, peripheral blood
samples were also collected from the parents, who had not been
diagnosed with any associated eye disease. The patient underwent
eye examinations, including fundus photography, optical coherence
tomography (OCT) and electroretinography (ERG). Follow-up was
performed once yearly.
High-throughput sequencing of the samples was
performed by Novogene Co., Ltd. DNA libraries were prepared for
sequencing using the NEBNext® Ultra™ DNA Library Prep
Kit (E7370L; New England Biolabs, Inc.). The quality of the samples
was verified by the 5400 Fragment Analyzer System (Agilent
Technologies Inc.). Exome sequencing was performed using an NovaSeq
6000 Sequencing System (Illumina, Inc.) using paired-end 150-bp
reads. The NovaSeq 6000 S4 Reagent Kit v1.5 (Illumina, Inc.) was
used for sequencing. Library concentrations were assessed using the
CFX96 Touch Real-Time PCR Detection System (Bio-Rad Laboratories
Co., Ltd.) and an Invitrogen Qubit 4 Fluorometer (Thermo Fisher
Scientific, Inc.).
Whole exome sequencing data
analysis
The data files obtained from high-throughput
sequencing were analyzed by Base Calling and converted into
Sequenced Reads, and then sequencing data were aligned with the
human reference genome (Genome Reference Consortium Human Build 37)
using BWA (version 0.7.8-r455; https://github.com/). Variant calling was performed
using SAMtools (version 1.21) (6)
and the resulting VCF files were annotated using ANNOVAR (version
2022-01-13) (7).
Pathogenic variants were screened based on the
following criteria: i) Variants with an allele frequency of
#x003C;1% in the 1000 Genomes (8)
Exome Sequencing Project 6500 (https://www.ebi.ac.uk/ena), Genome Aggregation
databases (9) and ChinaMap
(http://www.mbiobank.com/) were retained; ii) only
variants located in the coding (exonic) or in the splice site (±10
bp) regions were retained; iii) nonsynonymous mutations in highly
conserved regions (grep++ score >2) and those affecting splicing
(dbscSNV score >0.6) were retained, whereas small (#x003C;10 bp)
fragments of non-displaced indel mutations located in the repeat
region were excluded; iv) variants predicted to be deleterious
using two of the following four functional annotation algorithms
were retained: Sorting Intolerant from Tolerant (SIFT) (10), Polymorphism Phenotyping v2
(PolyPhen-2) high-diversity model (11), MutationTaster (12) and Combined Annotation Dependent
Depletion (13); and v) alignment
with autosomal recessive or compound heterozygous inheritance
patterns.
Molecular cloning and Sanger
sequencing
Genomic DNA was extracted from peripheral blood
samples using the Magnetic Blood DNA Extraction Kit (Vazyme Biotech
Co., Ltd.) and primers were designed using Primer3 (https://primer3.org/). The primer sequences are listed
in Table SI.
To analyze the mutant allele c.566_567insT:p.R189fs,
PCR was performed to amplify the target locus. The thermal cycling
protocol consisted of initial denaturation at 95˚C for 5 min,
followed by 35 cycles of denaturation at 95˚C for 30 sec, annealing
at 57˚C for 4 min and extension at 72˚C for 4 min. The resulting
PCR products were cloned into the pEASY-Blunt Zero Cloning vector
and transformed into Trans1-T1 receptor cells using the
pEASY-Blunt Zero Cloning Kit (TransGen Biotech Co., Ltd.) following
the manufacturer's instructions. Sanger sequencing was performed on
individual clones using M13 forward and reverse primers to confirm
the presence of the mutation in the patient samples.
CNGA3 plasmid construction
The pcDNA3.1 plasmid containing the CNGA3
cDNA sequence (Table SII) with a
C-terminal FLAG tag (Table SIII)
was synthesized by Beijing Tsingke Biotech Co., Ltd. The
CNGA3 sequence in the National Center for Biotechnology
Information database (accession number, NM_001298.3) was used. This
matched the sequences of the alleles from the father and mother of
the proband, confirming they carried wild-type alleles. The
CNGA3 wild-type plasmid was designated pcDNA3.1-CNGA3
(Fig. S1).
To introduce a p.S334F mutation, the Mut Express II
Fast Mutagenesis Kit V2 (Vazyme Biotech Co., Ltd.) was used. The
pcDNA3.1-CNGA3-p.S334F (c.C1001T) mutant plasmid was generated
using pcDNA3.1-CNGA3 as a template. Mutation-specific primers were
designed as follows: Forward primer (p.S334F-F):
5'-GACAGACTtCTGGGTCTACCCAAACATCTCAA-3' (plasmid positions,
1,984-2,015), which corresponds to bases 993-1,024 in the
CNGA3 coding sequence (CDS), with the lower case ‘t’
indicating the mutation site between bases 1,000 and 1,002; and
reverse primer (p.S334F-R): 5'-AGACCCAGaAGTCTGTCCCAAAACCAATGAACT-3'
(plasmid positions, 1,968-2,000), which corresponds to the reverse
complementary sequence to bases 977-1,009 in the CNGA3 CDS,
with lower case ‘a’ representing the mutation site. The amplified
product was treated with DpnI to remove the methylated
template plasmid, and ExNase II was used to cyclize the linear DNA.
Subsequently, the resulting pcDNA3.1-CNGA3-p.S334F plasmid was
transformed into E.coli DH5α competent cells (Takara Bio
Inc.), inoculated into Luria-Bertani (LB) solid medium (Thermo
Fisher Scientific Inc.) supplemented with ampicillin (Amp+)
(Beyotime Biotechnology) and subjected to 12 h of inverted culture.
Single clones were selected and inoculated into LB liquid medium
(Amp+) at 37˚C for amplification. Plasmid DNA was then extracted
from the bacteria using an EndoFree Plasmid Midi Kit (Jiangsu CoWin
Biotech Co., Ltd.), and the experimental steps were as described by
the manufacturer.
A pcDNA3.1-CNGA3-p.R189fs mutant plasmid was also
generated. The mutation-specific primers were as follows: Forward
primer (p.R189fs-F): 5'-ATTTGCAGtGGCCTGTTTCGATGAGCTGCAGT-3'
(plasmid positions, 1,550-1,580) which corresponds to bases 559-589
in the CNGA3 CDS, with lower case ‘t’ indicating the
mutation site between 566 and 567; and reverse primer (p.R189fs-R):
5'-AACAGGCCaCTGCAAATAAGCAGATACCAGTTATAGA-3' (plasmid positions,
1,530-1,565), which corresponds to the reverse complementary
sequence to bases 539-574, with lower case ‘a’ indicating the
mutation site. The plasmid construction process was analogous to
that described for pcDNA3.1-CNGA3-p.S334F. All constructed plasmids
were verified by sequencing.
Cell culture and transfection
293T cells (Guangzhou Ubigene Biosciences Co., Ltd.)
were cultured at 37˚C in a 5% CO2 atmosphere in
Dulbecco's modified Eagle's medium (Thermo Fisher Scientific Inc.),
supplemented with 10% fetal bovine serum (Thermo Fisher Scientific
Inc.) and 1% penicillin-streptomycin. When cell confluence reached
70-80%, the plasmids were mixed with polyethyleneimine (PEI) to
form a DNA-PEI complex. DNA (2 µg) was added to the cells and
incubated at 37˚C for 48 h prior to the execution of subsequent
experiments.
Western blotting
After rinsing with PBS, total protein was extracted
from the cells by treatment with RIPA buffer (Beijing Solarbio
Science & Technology Co., Ltd.; cat. no. R0020) and PMSF (cat.
no. P0100). The total protein concentration was then determined
using the BCA method with BSA as the standard. A total of 20 µg
total protein/lane was separated using SDS-PAGE (separation gel
10%, concentrating gel 5%) and transferred onto a polyvinylidene
fluoride (PVDF) membrane. The membrane was blocked with 5% skimmed
milk powder for 1 h at 37˚C to prevent nonspecific binding.
FLAG-tagged CNGA3 protein was then detected using a FLAG antibody
(product no. F3165-1MG; 1:5,000; Sigma-Aldrich; Merck KGaA) and
incubated at 4˚C for 14 h. GAPDH served as a loading control and
was detected using a GAPDH antibody (cat. no. 2118S; 1:5,000; Cell
Signaling Technology) and incubated at 4˚C for 14 h. The blot was
then incubated with a horseradish peroxidase-conjugated secondary
antibody (cat. no. SE134; 1:5,000; Beijing Solarbio Science &
Technology Co., Ltd.) at room temperature for 1 h. Subsequently, a
luminescent solution was applied to the PVDF membrane. NcmECL Ultra
Luminol/Enhancer Reagent (A) was mixed with NcmECL Ultra Stabilized
Peroxide Reagent (B) in equal proportions to make a luminescent
solution (New Cell & Molecular Biotech Co., Ltd.). Images were
captured using NiceAlliance Q9 software (Uvitec Ltd.). Experiments
were repeated three times.
Bioinformatics analysis of CNGA3
protein expression
Protein distribution data were retrieved from the
Human Protein Atlas (HPA; https://www.proteinatlas.org/). CNGA3 RNA levels in
the brain were retrieved from the HPA Brain Resource (https://www.proteinatlas.org/ENSG00000144191-CNGA3/brain),
which integrates and normalizes RNAseq data from the
Genotype-Tissue Expression (GTEx) project for the brain and retina;
the GTEx project collects and analyzes human postmortem tissues,
with RNA-sequencing performed on 36 tissue types using RSEMv1.3.0
(v8) (https://deweylab.github.io/RSEM/). The GTEx retina
data are based on EyeGEx data reported by Ratnapriya et al
(14), with transcript abundance
estimated using Kallisto v0.48.0 (https://pachterlab.github.io/kallisto/) with Ensembl
version 109 as the reference genome. The RNA tissue expression
results for CNGA3 were obtained by searching the consensus dataset
in the HPA Tissue Resource for ‘CNGA3’ (https://www.proteinatlas.org/ENSG00000144191-CNGA3/tissue).
The consensus dataset in the HPA integrates transcriptomics data
from HPA and GTEx datasets for 55 tissue types through an internal
normalization pipeline. CNGA3 single-cell expression results were
obtained by searching the Single Cell Type and Single Cell Resource
sections of the HPA for ‘CNGA3’ (https://www.proteinatlas.org/ENSG00000144191-CNGA3/single+cell).
This analysis integrates single-cell information for 31 human
tissues from various databases, including the Single Cell
Expression Atlas, Human Cell Atlas, Gene Expression Omnibus, Tabula
Sapiens, Allen Brain Atlas and European Genome-phenome Archive.
Transcript per million (TPM) values were rescaled to a sum of one
million TPM following the removal of non-coding transcripts. Within
each data source, the TPM values of all samples were normalized
separately using the trimmed mean of the M values to allow
between-sample comparisons. The final normalized transcript
expression values, denoted as normalized parts per million (nTPM)
markers, were calculated for each gene in each sample, with nTPM
values #x003C;0.1 excluded from visualization in the HPA.
Prediction of protein stability after CNGA3 mutation was
performed using the DEZYME tool (https://soft.dezyme.com/query/create/pop), with ΔΔG
#x003C;0 meaning a stabilizing mutation.
Statistical analysis
Protein expression results in the in vitro
experiment are presented as the mean ± SD. The difference between
wild-type and CNGA3:p.S334F groups was analyzed using an unpaired
Student's t-test. P#x003C;0.05 was considered to indicate a
statistically significant difference. In order to assess whether
the distribution of CNGA3 mutation sites in the structural
and non-structural domains was statistically significant, the
distributional characteristics of the mutation sites were
systematically analyzed in this study using a one-sample hypothesis
test. The null hypothesis (H0) of the test posited that there was
no statistically significant difference in the distribution of
mutation sites between the structural and non-structural domains.
By contrast, the alternative hypothesis (H1) predicted that the
proportion of mutation sites in the structural domains would be
significantly higher than that in the non-structural domains. The
significance level of the test was set at α=0.05. The statistical
analysis was conducted using GraphPad Prism (version 10.4) and R
software (version 4.1.2).
Results
Clinical data of the patient
The patient was a 9-year-old girl with COD whose
parents exhibited no signs of any associated eye disease. The child
was first diagnosed with COD in September 2020. She had a visual
acuity of 20/100 in both eyes and mild nystagmus. Fundus
photography revealed a loss of foveal reflex in both eyes (Fig. 1A and B). OCT revealed low ganglion cell
thickness and a retinal nerve fiber layer thinner than normal
(Fig. 1C-F) (15). ERG demonstrated decreased
dark-adapted b-wave and a-wave amplitudes and approximately normal
oscillatory potential amplitudes in both eyes (Fig. 2). The patient was prescribed
glasses for full correction of hyperopia and astigmatism. However,
her visual acuity improved by no more than one line during
follow-up. Upon re-examination on December 5, 2024, her visual
acuity was unchanged at 20/100 in each eye. Her electroretinogram
exhibited a slight improvement, although it remained markedly
abnormal (Fig. S2).
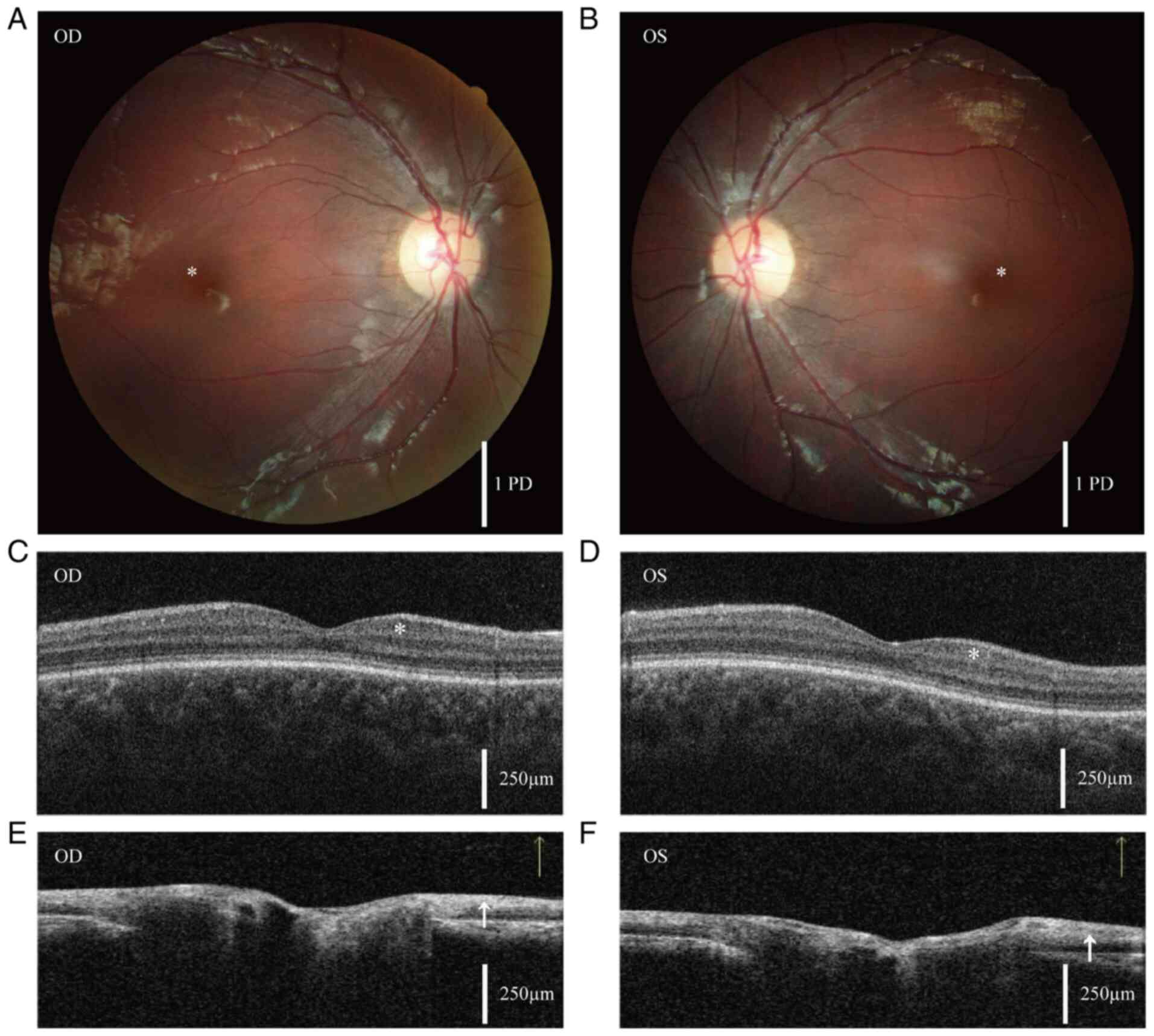 | Figure 1Fundus photography and OCT. (A and B)
Fundus photographs of the right and left eyes of the patient,
respectively. The asterisk indicates disappearance of the foveal
reflex. Scale bar, 1 optic disc PD. Since PD varies among
individuals, it serves as a standard for distance and range in
fundus examinations. (C and D) Measurements of the GCC in the
macula of the right and left eyes, respectively. The asterisk
indicates that OCT examination revealed a thinner than normal
ganglion cell layer, with an average GCC thickness of 84 µm in the
right eye and 85 µm in the left eye. (E and F) Measurements of the
optic nerve head in the right and left eyes, respectively, reveal
that the retinal nerve fiber layer, indicated by the arrows, is
also thinner than normal. The average retinal nerve fiber layer
thickness is 99 µm in the right eye and 95 µm in the left eye.
Scale bar in the OCT images, 250 µm. OCT, optical coherence
tomography; OD, oculus dexter/right eye; OS, oculus
sinister/left eye; PD, papilla diameter; GCC, ganglion cell
complex. |
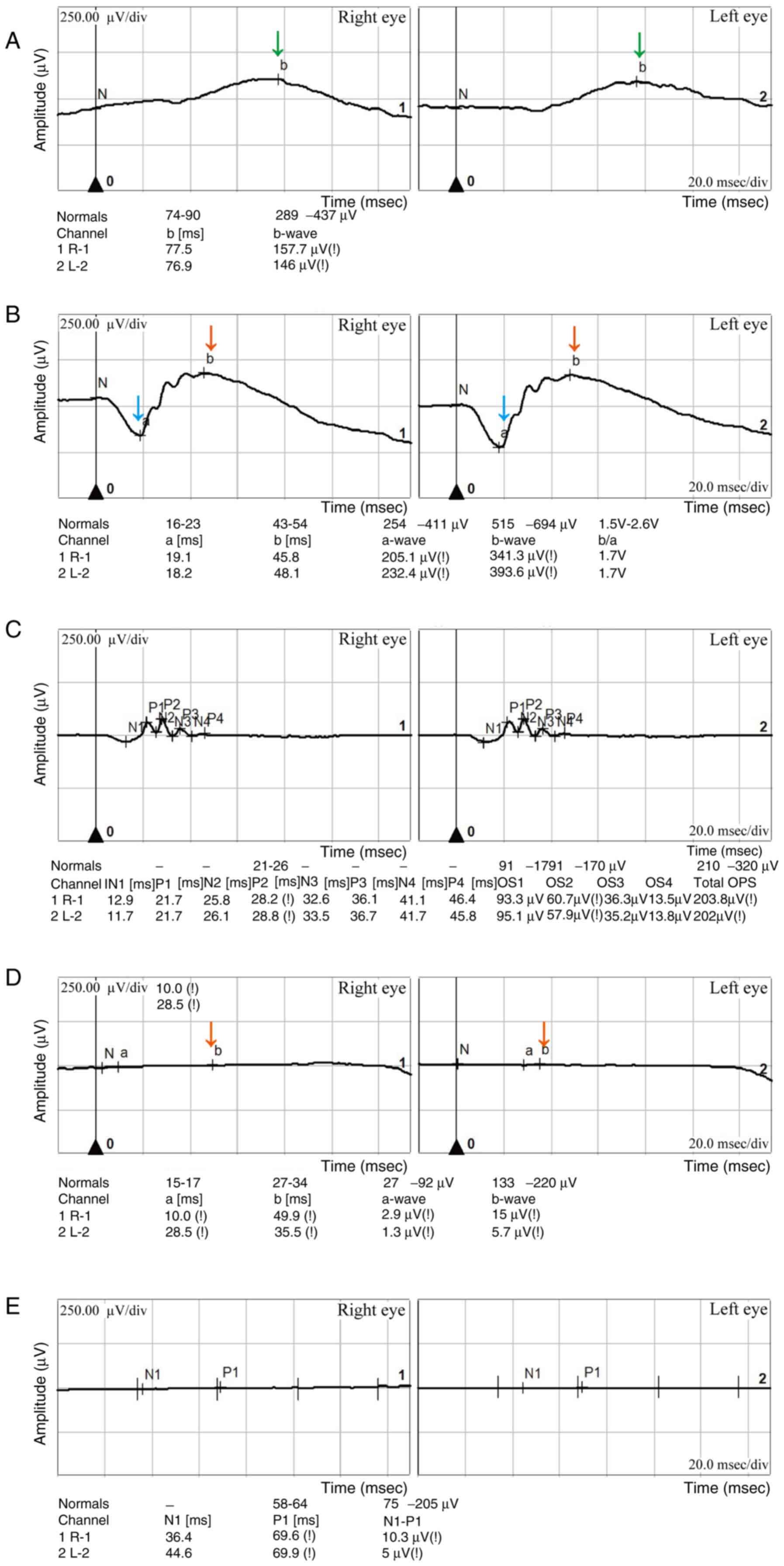 | Figure 2Retinal electroretinogram results.
(A) Dark-adapted 0.01 ERG showing b-wave amplitudes of 157.7 and
146 µV in the right and left eye, respectively. The dark-adapted
b-wave amplitudes (indicated by the green arrows) are diminished in
both eyes compared with the normal range (289-437 µV). This
dysfunction may involve rod photoreceptors or be selective to
post-phototransduction processes or rod bipolar cells within the
retina. (B) Dark-adapted 3.0 ERG showing a-wave amplitudes of 205.1
and 232.4 µV in the right and left eye, respectively. This
indicates a reduction in the a-wave amplitude (indicated by the
blue arrows), with both eyes measuring below the normal range
(254-411 µV). The b-wave amplitudes (indicated by the red arrows)
are 341.3 and 393.6 µV in the right and left eye, respectively,
both below the normal range (515-694 µV). The dark-adapted 3.0 ERG
reflects both the rod and cone cell activity, with the rod cell
system being the predominant contributor in the normal retina.
Therefore, these findings indicate that the cellular photoreceptors
in the patient are dysfunctional. (C) Oscillatory potential ERG
indicates an approximately normal oscillatory potentials response,
reflecting the absence of long-synapse cell signaling, consistent
with normal expectations. (D) Light-adapted 3.0 ERG demonstrates a
pronounced reduction in b-wave amplitude (indicated by the red
arrow). The amplitudes of the right and left eye are 15 and 5.7 µV,
respectively, markedly below the normal range (133-220 µV). These
results suggest the possibility of cone cell photoreceptor
dysfunction. (E) Analysis by 30-Hz flicker ERG demonstrates an
N1-P1 wave amplitude of 10.3 and 5 µV in the right and left eye,
respectively, below the typical range (75-205 µV) in both eyes.
This indicates the potential presence of anomalies in cone cells
and their posterior retinal structures. The clinical manifestations
of decreased visual acuity, night blindness and impaired color
vision in the patient may be attributed to dysfunction of the cone
and rod photoreceptors and their associated retinal structures in
both eyes. ERG, electroretinography. |
CNGA3 mutations contributing to
COD
Whole-exome sequencing of the proband and her
parents (Fig. 3A) was performed
(16). Genetic analysis revealed
compound heterozygous CNGA3 mutations in the proband,
including a known missense single-nucleotide variant
(CNGA3:c.C1001T) and a previously unreported frameshift
mutation (CNGA3:c.566_567insT) (Table I). The mother was a carrier of the
c.C1001T mutation, which results in substitution of serine with
phenylalanine at codon 334 (p.S334F). The novel c.566_567insT
frameshift mutation was inherited from her father, who was a
carrier. Fig. S3 presents the
nucleic acid sequence following the
CNGA3:c.566_567insT:p.R189fs mutation, highlighting the
insertion of base T between positions 566 and 567 in the cDNA
sequence. This mutation leads to a TGA premature stop codon at
position 194, thereby reducing the number of amino acids in the
reading frame from 694 to 193, resulting in a shorter, less stable
protein. These mutations were validated using Sanger sequencing,
which confirmed the CNGA3:c.C1001T:p.S334F mutation in the
patient and her mother (Fig. 3B)
and the CNGA3:c.566_567insT:p.R189fs mutation in the patient
and her father (Fig. 3C). Clonal
sequencing of CNGA3:c.566_567insT:p.R189fs yielded results
consistent with this mutation (Fig.
3D). Bioinformatics analysis reveals that CNGA3 is highly
expressed in the brain (Fig.
S4A), retina (UniProtKB, Q16281; Fig. S4B), neuronal protrusions, axons
and cone photoreceptor cells (Fig.
S4C).
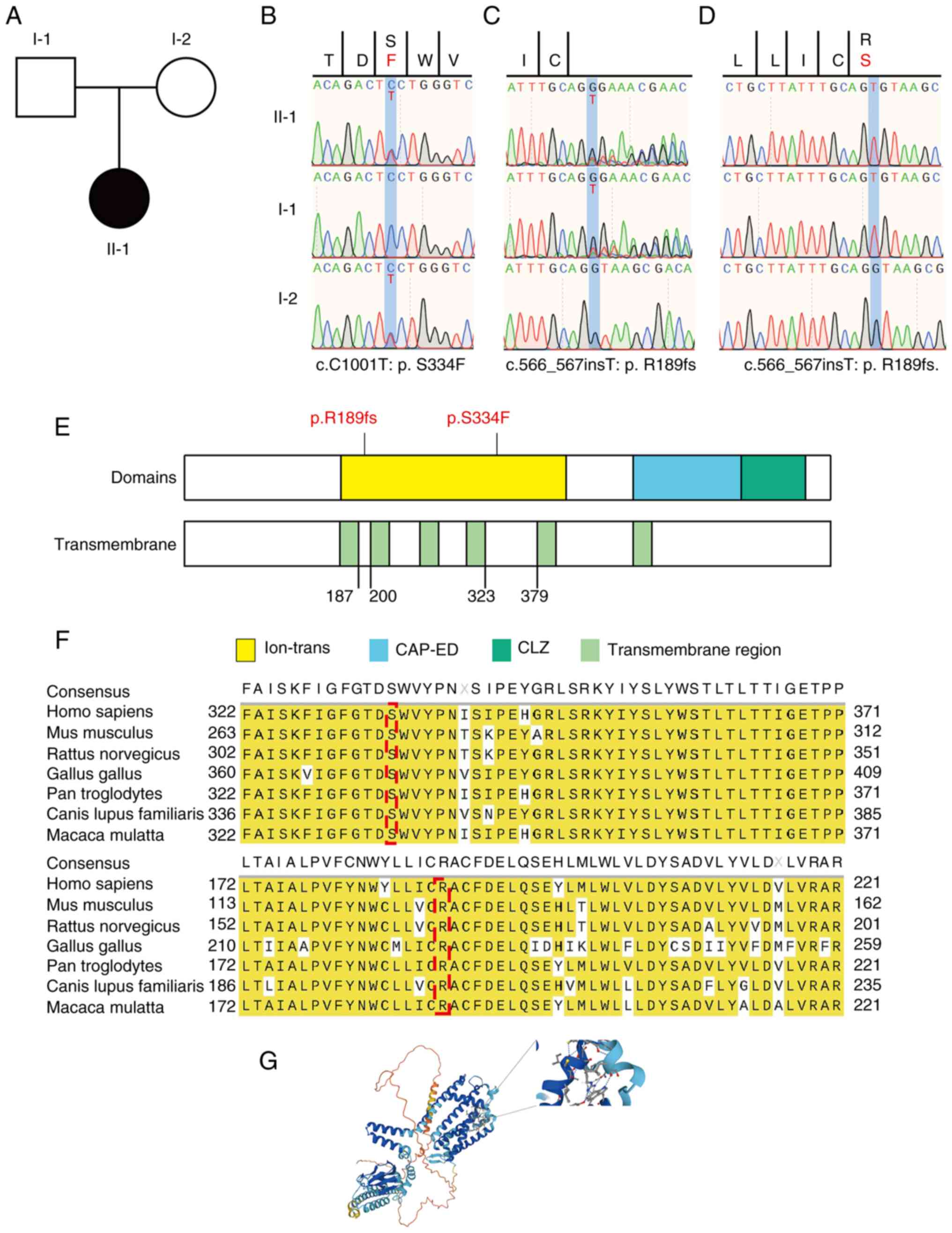 | Figure 3CNGA3 mutations associated
with retinal cell dystrophy. (A) Family relationship diagram of the
patient, a 9-year-old girl with evidence of retinal dystrophy.
Neither the father (I-1) or mother (I-2e) of the child is afflicted
with the disease, and the girl (II-1) was the first to manifest
this condition. The pattern of inheritance may be autosomal
recessive. (B) The CNGA3:c.C1001T:p.S334F mutation was
identified in the precursor and her mother and (C) the
CNGA3:c.566_567insT:p.R189fs mutation was detected in the
patient and her father by the whole-exome sequencing and data
analysis of peripheral blood. (D) Clonal sequencing of
CNGA3:c.566_567insT:p.R189fs yielded results consistent with
those in (C). These data indicate that the patient inherited
distinct compound heterozygous mutations from both parents. (E)
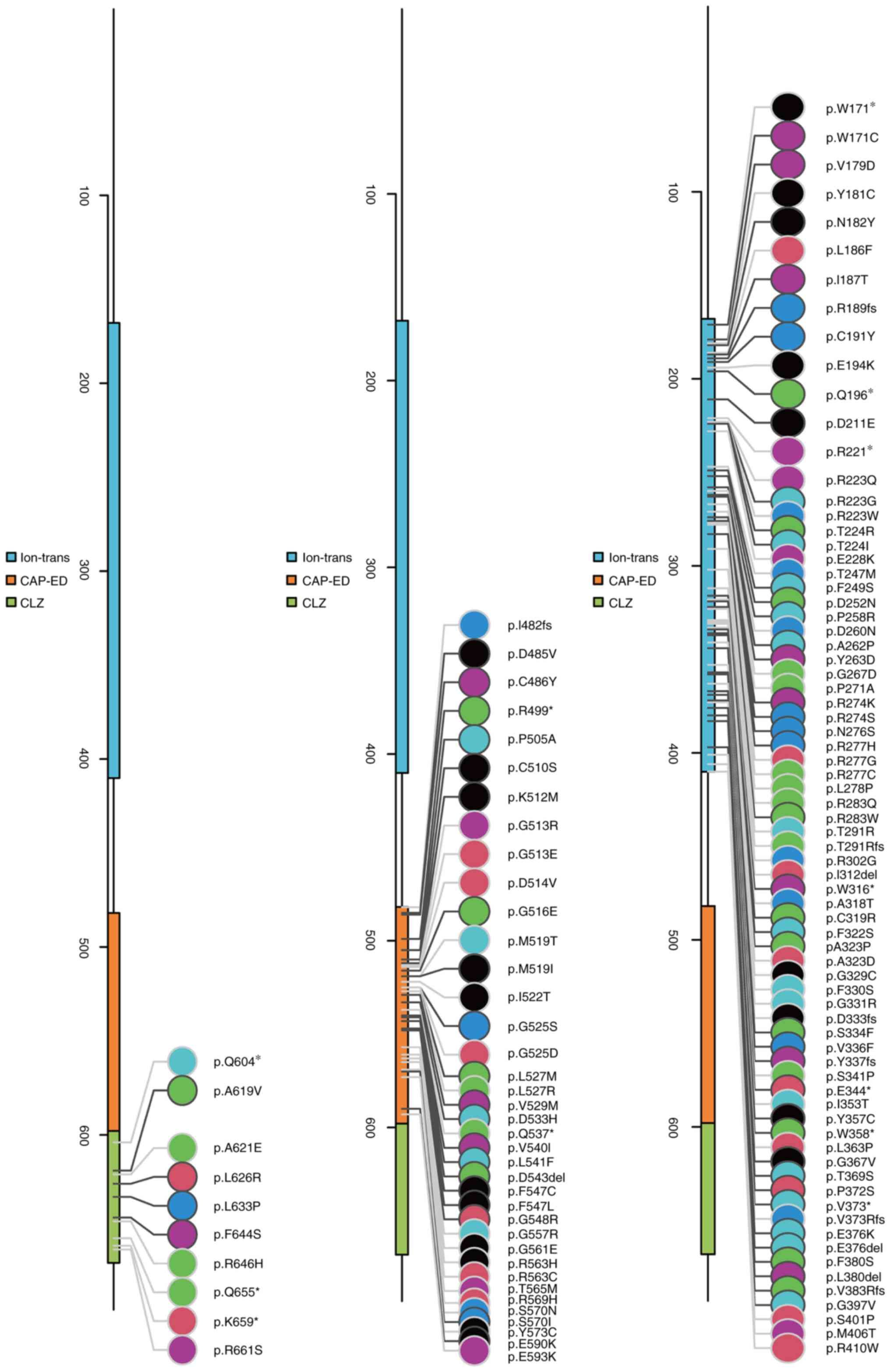
Schematic of the CNGA3 structural domains, namely ion-trans, CAP-ED
and CLZ. The p.S334F and p.R189fs mutations both occur in the
ion-trans structural domain, indicating that they may affect the
transmembrane transport of ions. (F) Conservativeness analysis of
the two mutant loci, p.R189fs and p.S334F, reveals that the
mutations are located in regions that are highly conserved across
species. This suggests that they are likely to impact protein
function. (G) Schematic showing the location of the
CNGA3:p.S334F mutant amino acids in the molecular structure.
CNGA3, cyclic nucleotide-gated channel subunit α 3; ion-trans, ion
transport; CAP-ED, cysteine-rich CAP domain-extended domain; CLZ,
cyclic nucleotide-gated ligand-binding zinc finger-like. |
 | Table IDetailed information of the variants
CNGA3(p. R189fs) and CNGA3(p.S334F). |
Table I
Detailed information of the variants
CNGA3(p. R189fs) and CNGA3(p.S334F).
| Carriers | Chr | Pos (GRCh37) | Ref. | Alt | AD | ChinaMap | ESP | GnomAD | 1000G | SIFT | Polyphen-2 | CADD |
|---|
| Ⅰ-1 and Ⅱ-1 | 2 | 99006237 | G | GT | Ⅰ-1: 8,8 and Ⅱ-1:
7,11 | - | - | - | - | - | - | 5.571691 |
| Ⅰ-2 and Ⅱ-1 | 2 | 99012634 | C | T | Ⅰ-2: 32,24 and Ⅱ-1
:38,50 | - | - | - | - | 0.003, D | 0.173, B | - |
Effect of CNGA3 mutations on protein
structure and function
The p.S334F and p.R189fs mutations are located on
the ion-transport (ion-trans) structural domain of CNGA3 protein.
Notably, p.R189fs results in a truncated protein product that only
retains the ion-trans domain and lacks the cysteine-rich CAP
domain-extended domain (CAP-ED) and cyclic nucleotide-gated
ligand-binding zinc finger-like (CLZ) domain (Fig. 3E). The conservativeness of the two
mutant loci, p. R189fs and p.S334F, was evaluated, and the
mutations were found to be located in regions that are highly
conserved across species (Fig.
3F). Mutations in highly conserved protein regions may impact
their functionality (17). The
p.S334F mutation affects a conserved residue in the ion-trans
domain and is predicted to be harmful based on SIFT and PolyPhen
analyses (SIFT 0.003, damaging; Polyphen2_HVAR 0.863, possibly
damaging; PolyPhen2_HDIV 0.965, probably damaging). The protein
structure and stability were then predicted to gain insights into
the impact of the mutation on the protein. The p.S334F mutation was
predicted to decrease protein stability, with a ΔΔG value of 0.74
kcal/mol. A schematic showing the location of the CNGA3
mutation in the molecular structure is presented in Fig. 3G. Of the total 151 CNGA3
mutations reported in humans, 40 are associated with COD (Table II). Further hypothesis testing of
the relationship between the mutations and structural domains of
CNGA3 proteins yielded P#x003C;0.0011 (α=0.05), indicating the
enrichment of mutations in structural domains (Fig. 4).
| Table IICNGA3 mutations associated
with COD and associated clinical information. |
Table II
CNGA3 mutations associated
with COD and associated clinical information.
| Mutation | | Best visual
acuity | Fundus |
Electroretinogram | |
|---|
| DNA | Amino acid | State | Sex | Age at examination,
years | Right | Left | Right | Left | Rods | Cones | First author, year
(Refs.) |
|---|
| c.62C>G | p.S21* | Heterozygous | M | 2.0 | - | - | PFR | PFR | Moderately
reduced | Extinguished | Li et al,
2014(32) |
| c.67C>T | p.R23* | Heterozygous | F | 0.4 | PO | PO | TDP, PM | TDP, PM | Mildly reduced | Extinguished | Li et al,
2014a (32); Johnson et al, 2004(67); Ellingford et al,
2016(68) |
| c.284C>T | p.P95L | Heterozygous | - | - | - | - | - | - | - | - | Thiadens et
al, 2010(31) |
| c.396-11C>G | - | Heterozygous | F | 4.0 | - | - | Normal | Normal | Moderately
reduced | Severely
reduced | Li et al,
2014(32) |
| | | Homozygous | M | 0.6 | 0.20 | 0.20 | ARA, TDP | ARA, TDP | Moderately
reduced | Severely
reduced | Li et al,
2014(32) |
| | | Heterozygous | M | 5.0 | 0.40 | 0.40 | TDP, PM | TDP, PM | Moderately
reduced | Extinguished | Li et al,
2014(32) |
| c.512G>A | p.W171* | Homozygous | F | 1.2 | - | - | Normal | Normal | Mildly reduced | Mildly reduced | Li et al,
2014(32) |
| c.513G>T | p.W171C | Heterozygous | F | 1.5 | - | - | PM | PM | Mildly reduced | Extinguished | Li et al,
2014(32) |
| c.566_567insT | p.R189fs | Heterozygous | F | 9.0 | 0.20 | 0.20 | PM | PM | Moderately
reduced | Severely
reduced | Present study |
| c.661C>T | p.R221* | Heterozygous | F | 2.0 | - | - | - | - | Moderately
reduced | Severely
reduced | Li et al,
2014a (32); Johnson et al, 2004(67) |
| | | Heterozygous | M | 4.0 | PO | PO | ARA | ARA | Moderately
reduced | Severely
reduced | |
| c.667C>T | p.R223W | Homozygous | F | 2.0 | - | - | - | - | Mildly reduced | Severely
reduced | Li et al,
2014(32); Wissinger et al,
2001(28) |
| c.671C>T | p.T224I | Heterozygous | M | 3.0 | - | - | TDP | TDP | Moderately
reduced | Severely
reduced | Li et al,
2014(32) |
| | | Heterozygous | M | 2.3 | - | - | NFR | NFR | Moderately
reduced | Moderately
reduced | Li et al,
2014(32) |
| c.674-2A>C | - | Heterozygous | M | 2.3 | - | - | NFR | NFR | Moderately
reduced | Moderately
reduced | Li et al,
2014(32) |
| c.682G>A | p.E228K | Heterozygous | M | 47.0 | - | - | - | MA | - | - | Thiadens et
al, 2010a(31);
Reuter et al, 2008(69) |
| c.773C>G | p.P258R | Heterozygous | F | 4.0 | - | - | Normal | Normal | Moderately
reduced | Extinguished | Li et al,
2014(32) |
| c.778G>A | p.D260N | Heterozygous | M | 2.0 | - | - | PFR | PFR | Moderately
reduced | Extinguished | Li et al,
2014a (32); Wissinger et al,
2001(28) |
| c.829C>T | p.R277C | Heterozygous | F | 15.0 | 0.50 | 0.50 | - | - | - | - | Wissinger et
al, 2001(28) |
| | | Heterozygous | F | 2.0 | - | - | - | - | Moderately
reduced | Severely
reduced | Li et al,
2014(32) |
| c.830G>A | p.R277H | Heterozygous | M | 1.5 | - | - | TDP, ARA | TDP, ARA | Mildly reduced | Extinguished | Li et al,
2014a (32); Wissinger et al,
2001(28) |
| c.847C>T | p.R283W | Heterozygous | F | 15.0 | 0.50 | 0.50 | - | - | - | - | Wissinger et
al, 2001a(28);
Kohl et al, 1998(37) |
| | | Heterozygous | M | 3.0 | - | - | TDP | TDP | Moderately
reduced | Severely
reduced | Li et al,
2014a (32); Kohl et al, 1998(37) |
| | | Heterozygous | F | 1.5 | - | - | PM | PM | Mildly reduced | Extinguished | |
| c.872_873del | p.T291Rfs*77 | Heterozygous | F | 8.0 | - | - | MA | MA | - | - | Li et al,
2014(32) |
| | | Heterozygous | F | 4.5 | 0.10 | 0.20 | TDP | TDP | Mildly reduced | Severely
reduced | Li et al,
2014(32) |
| c.955T>C | p.C319R | Homozygous | F | 10.0 | CF (1 M) | CF (1 M) | MA | MA | Moderately
reduced | Severely
reduced | Shaikh et
al, 2015(5) |
| c.967G>C | p.A323P | Heterozygous | F | - | - | - | - | - | - | - | Carss et al,
2017(70) |
| c.989T>C | p.F330S | Heterozygous | M | 0.3 | PL | PL | - | - | Mildly reduced | Severely
reduced | Li et al,
2014(32) |
| | | Heterozygous | F | 6.0 | 0.08 | 0.08 | NFR, TDP | TDP | Mildly reduced | Severely
reduced | Li et al,
2014(32) |
| c.1001C>T | p.S334F | Heterozygous | M | 2.5 | 0.40 | 0.40 | ARA | ARA | Moderately
reduced | Extinguished | Li et al,
2014(32) |
| c.1074G>A | p.W358* | Heterozygous | M | 28.0 | 0.10 | 0.10 | ARA, MA | ARA, MA | Moderately
reduced | Extinguished | Li et al,
2014(32) |
| | | Heterozygous | M | 0.5 | PL | PL | ARA, NFR | ARA, NFR | Mildly reduced | Severely
reduced | Li et al,
2014(32) |
| | | Heterozygous | F | 4.0 | - | - | Normal | Normal | Moderately
reduced | Severely
reduced | Li et al,
2014(32) |
| c.1116dup | p.V373Rfs*4 | Heterozygous | F | 4.0 | - | - | Normal | Normal | Moderately
reduced | Extinguished | Li et al,
2014(32) |
| c.1306C>T | p.R436W | Heterozygous | F | 1.0 | PO | PO | Normal | Normal | Mildly reduced | Extinguished | Li et al,
2014a (32); Wissinger et al,
2001(28) |
| | | Homozygous | F | 2.0 | PO | PO | PM | PM | Moderately
reduced | Extinguished | |
| c. 1315C>T | p.R439W | Heterozygous | M | 0.3 | PL | PL | - | - | Mildly reduced | Severely
reduced | Li et al,
2014a (32); Reuter et al, 2008(69) |
| | | Heterozygous | M | 2.5 | 0.40 | 0.40 | ARA | ARA | Moderately
reduced | Extinguished | |
| c.1495C>T | p.R499* | Heterozygous | F | 1.0 | PO | PO | Normal | Normal | Mildly reduced | Extinguished | Li et al,
2014(32) |
| | | Heterozygous | M | 5.0 | 0.40 | 0.40 | TDP, PM | TDP, PM | Moderately
reduced | Extinguished | Li et al,
2014(32) |
| c.1513C>G | p.P505A | Heterozygous | - | - | - | - | - | - | - | - | Huang et al,
2016(29) |
| c.1537G>C | p.G513R | Heterozygous | - | - | - | - | - | - | - | - | Huang et al,
2016(29) |
| c.1556T>C | p.M519T | Heterozygous | - | - | - | - | - | - | - | - | Huang et al,
2016(29) |
| c.1585G>A | p.V529M | Homozygous | F | 1.1 | - | - | Normal | Normal | Moderately
reduced | Severely
reduced | Li et al,
2014a (32); Kohl et al, 1998(37) |
| | | Homozygous | M | 5.0 | - | - | Normal | Normal | Mildly reduced | Severely
reduced | |
| | | Homozygous | F | 0.6 | PO | PO | - | - | Mildly reduced | Severely
reduced | |
| | | Heterozygous | M | 28.0 | 0.10 | 0.10 | ARA, MA | ARA, MA | Moderately
reduced | Extinguished | |
| | | Heterozygous | M | 4.0 | PO | PO | ARA | ARA | Moderately
reduced | Severely
reduced | |
| | | Heterozygous | F | 6.0 | 0.08 | 0.08 | NFR, TDP | TDP | Mildly reduced | Severely
reduced | |
| | | Heterozygous | M | 17.0 | 0.10 | 0.10 | PM | PM | Mildly reduced | Moderately
reduced | |
| | | Heterozygous | M | 1.5 | - | - | TDP, ARA | TDP, ARA | Mildly reduced | Extinguished | |
| | | Heterozygous | F | 4.5 | 0.10 | 0.20 | TDP | TDP | Mildly reduced | Severely
reduced | |
| c.1597G>C | p.D533H | Homozygous | F | 3.0 | - | - | ARA | ARA | Mildly reduced | Extinguished | Li et al,
2014(32) |
| c.1618G>A | p.V540I | Heterozygous | - | - | - | - | - | - | - | - | Thiadens et
al, 2010(31) |
| c.1641C>A | p.F547L | Heterozygous | F | 32.0 | #x003C;0.50 | #x003C;0.50 | - | - | - | - | Wissinger et
al, 2001(28); Kohl et
al, 1998(37) |
| c.1688G>A | p.R563H | Heterozygous | F | 32.0 | #x003C;0.50 | #x003C;0.50 | - | - | - | - | Wissinger et
al, 2001(28); Ellingford
et al, 2016(68) |
| c.1709G>A | p.S570N | Heterozygous | M | 17.0 | 0.10 | 0.10 | PM | PM | Mildly reduced | Moderately
reduced | Li et al,
2014(32) |
| c.1768G>A | p.E590K | Heterozygous | M | 2.0 | - | - | PFR | PFR | Moderately
reduced | Extinguished | Li et al,
2014(32); Nishiguchi et
al, 2005(38) |
| c.1856C>T | p.A619V | Heterozygous | - | - | - | - | - | - | - | - | Thiadens et
al, 2010(31) |
| c.1877T>G | p.L626R | Heterozygous | - | - | - | - | - | - | - | - | Huang et al,
2016(29) |
| c.1975A>T | p.K659* | Heterozygous | F | 8.0 | - | - | MA | MA | - | - | Li et al,
2014(32) |
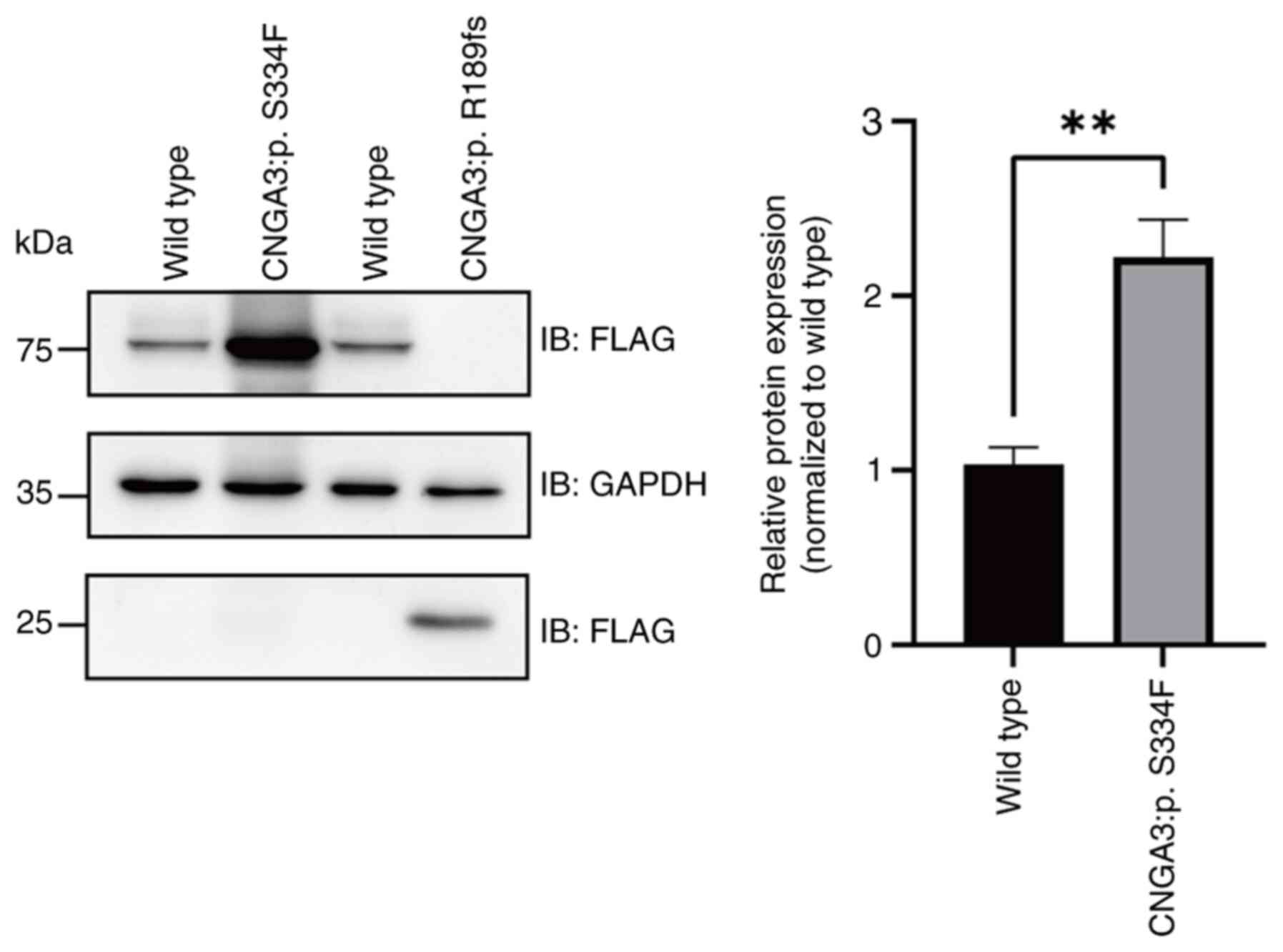
In vitro analysis of CNGA3
mutations
To further investigate the effects of these
mutations on protein expression, wild-type and mutant CNGA3
proteins were expressed in 293T cells and analyzed by western
blotting (Fig. S5). The
FLAG-tagged wild-type CNGA3 protein appeared as two distinct bands,
specifically, a main band at 81.7 kDa and an additional band of
higher molecular weight (~100 kDa), likely representing a
post-translationally modified protein. The S334F mutation resulted
in significantly increased CNGA3 protein expression compared with
that of wild-type CNGA3, but notably lacked the ~100 kDa
band (Fig. 5). Given that S334 is
a potential phosphorylation site, we hypothesize that the S334F
mutation might prevent phosphorylation at this position. By
contrast, the c.566_567insT:p.R189fs mutation produced a truncated
protein with a molecular weight of 16.0 kDa, consistent with the
predicted premature termination of protein synthesis.
Discussion
The present study reports a case of COD with novel
compound heterozygous CNGA3 mutations. Although the
CNGA3:p.S334F mutation was predicted to reduce CNGA3 protein
stability, western blotting results revealed that CNGA3 protein
expression was abnormally increased following this mutation. It is
hypothesized that this may be attributed to the mutation preventing
protein phosphorylation. In addition, no additional protein band
was evident above the CNGA3:p.S334F protein, whereas a band was
clearly visible above the wild-type protein. Serine is a common
phosphorylation site (18,19). In a study of spastin
phosphorylation, Zhang et al (20) compared wild-type spastin with a
mutant in which 2 potential phosphorylation sites were mutated to
alanine. They found that the quantity of the wild-type protein was
markedly diminished compared with that of the protein with mutated
phosphorylation sites, indicating that protein degradation may
occur as a result of protein phosphorylation. Kaushik and Cuervo
(21) found that the treatment of
cells with oleate-conjugated albumin increased total perilipin 2
(PLIN2) content 1.5-fold, with a more pronounced (5-fold) increase
in the phosphorylated form. The phosphorylation of PLIN2 was
undetectable when chaperone-mediated autophagy was blocked by
lysosome-associated membrane protein 2A knockdown, while the amount
of PLIN2 protein was concurrently increased. This suggests that
phosphorylation may have played a role in promoting PLIN2
degradation or preventing its accumulation. These findings for
other proteins support the hypothesis that the CNGA3:p.S334F
mutation prevents protein phosphorylation, which would otherwise
lead to protein degradation. Although the inherent structural
instability of the mutation may also contribute to protein
degradation, this effect appears to have been less pronounced than
the impact of phosphorylation changes on the protein. Consequently,
the expression of the CNGA3 protein following mutation was
significantly higher than that of the wild-type protein, likely due
to a lack of degradation. However, it must be acknowledged that the
lack of detection of protein phosphorylation levels in the western
blot experiments is a limitation of the present study. The novel
CNGA3:p.R189fs mutation creates a premature stop codon that
leads to the deletion of the key CAP-ED and CLZ structural domains,
which is predicted to disrupt the normal tetrameric structure of
the CNGA3 protein. This structural disruption would be expected to
impair the normal function of the ion channel in the retina,
leading to symptoms such as decreased visual acuity and other
visual disturbances. To the best of our knowledge, the present
study is the first to identify the CNGA3:p.R189fs mutation
in COD, and the only in vitro study of CNGA3
mutations in COD.
The main features of COD include decreased visual
acuity, impaired color recognition, and increased sensitivity to
light, typically manifested during the first or second decade of
life (22). Mutations in genes
such as CNGA3, calcium voltage-gated channel subunit α2δ4,
CNGB3, phosphodiesterase 6C (PDE6C), PDE6H and
ATP-binding cassette subfamily A member 4 have been implicated in
the pathogenesis of COD (23-27).
The association between CNGA3 and COD was first reported by
Wissinger et al (28) in a
study of 258 individuals with hereditary cone photoreceptor disease
in 2001. Since then, several studies have identified additional
CNGA3 mutations in patients with COD (29-31).
COD caused by CNGA3 mutations follows an autosomal recessive
inheritance pattern (32,33), and mutations of CNG channels often
alter their plasma membrane localization and gating properties
(34,35).
CNGA3 mutations are associated with
hereditary cone photoreceptor disorders, particularly color
blindness (OMIM: 600053) (36).
Due to the absence of functional cone photoreceptors in the retina,
the disease is characterized by complete color blindness, low
vision, photophobia and nystagmus (37,38).
The CNG3 channel comprises CNGA3 and CNGB3, which form a
heterotetrametric structure with two α and two β subunits (3). Both CNGA3 and CNGB3 are implicated in
cone photoreceptor disorders (39,40),
and mice with CNGA3 and CNGB3 knockout exhibit
reduced electroretinographic responses, decreased phototransduction
expression and significantly increased expression of endoplasmic
reticulum stress marker proteins (41). These findings suggest that
mitochondrial damage may contribute to endoplasmic reticulum
stress-mediated retinal cone cell death. Impaired calcium
homeostasis and the mislocalization of retinal proteins may also
accelerate rod cell death, while cGMP accumulation can lead to
retinal cone cell stress and damage (42,43).
cGMP activates CNGA3, which triggers the
G-protein-coupled cascade and leading to the opening of cation
channels and depolarization of cone photoreceptors, which is
essential for normal vision (44-46).
CNGA3 also plays an important role in the light-evoked electrical
responses in red-, green- and blue-sensitive cones (47). The mutations in CNGA3
identified in the present case, namely p.S334F and p.R189fs, will
disrupt the transporter function of ion channels, preventing the
normal depolarization of cone photoreceptor cells and thereby
impair the ability to discriminate colors. In the present study,
the patient exhibited reduced visual acuity and color vision
deficiency. Considering that the patient was at an early stage of
OCD, fundus photography did not reveal any distinctive fundus
lesions and showed only a loss of foveal reflex. However, as the
disease progresses, fundus imaging may be expected to reveal the
presence of macular lesions or retinal pigment epithelial lesions
in a bull's-eye configuration (48). Furthermore, OCT may detect the
absence of the interdigitation zone early in the disease, followed
by progressive destruction of the ellipsoid zone (49-51).
In addition, ERG examination of the single-flash response may
reveal delayed a- and b-waves and reduced light-adapted a- and
b-wave amplitudes (52).
Definitive treatments to stop COD progression or
severe vision loss are unavailable, with only symptomatic
management through refractive correction and tinted glasses being
available (53). Notably, several
complementary treatments are currently being investigated, and gene
therapy could emerge as a key treatment modality for hereditary
retinal diseases such as COD in the future. Gene editing with
CRISPR/Cas9 is currently the primary therapeutic modality in
clinical trials, aiming to introduce specific nucleotide
alterations into the target genome to restore normal gene
expression (54-56).
In addition, gene replacement therapy has been shown to enable the
sustained expression of normal genes in mice with retinal disease
and promote functional improvement, with treatments approved by the
US Food and Drug Administration already being available (57-59).
In addition, silencing the gene associated with a dominant retinal
degeneration mouse model, namely guanylate cyclase activating
protein 1, has been found to significantly improve photoreceptor
survival, delay disease onset and enhance visual function (60-62).
Other adjuvant therapies are also being investigated. These include
brain-derived neurotrophic factor, pigment epithelium-derived
neurotrophic factor, basic fibroblast growth factor, ciliary
neurotrophic factor and rod cone viability factor, which are being
explored for their potential to slow down retinal degeneration in
COD (63-66).
In conclusion, the present study identified a
compound heterozygous mutation in CNGA3, with a
c.C1001T:p.S334F variant and a novel frameshift mutation
c.566_567insT:p.R189fs, in a patient with COD, and demonstrated
their potential impact on protein stability. The findings not only
expand the spectrum of disease-causing mutations in CNGA3
but also provide crucial insights into its role in COD
pathogenesis. Integrating a literature analysis allowed the results
to further reveal that specific CNGA3 structural domains may
represent mutation hotspots in COD. These findings highlight
potential targets for developing therapies for CNGA3-related
retinal diseases.
Supplementary Material
pcDNA3.1-CNGA3 plasmid mapping. The
cloning vector was pcDNA3.1(+), which contains ori, human CMV
promoter and enhancer, SV40 promoter, AmpR, Neo/KanR, multiple
restriction endonuclease sites, bGH poly A signal, and SV40 poly A
signal components. Insertion of the CNGA3 coding sequence was
achieved using HindIII and BamHI cloning sites, with
the addition of a 3xFLAG tag after the Kozak sequence. The
pcDNA3.1-CNGA3 plasmid contained 7,576 bases. p.S334F-F and
p.S334F-R correspond to positions 1,984-2,015 and 1,968-2,000 in
the plasmid, and include T and A as mutated bases, respectively.
p.R189fs-F and p.R189fs-R correspond to positions 1,550-1,580 and
1,530-1,565 of the plasmid, and include T and A in as mutated
bases, respectively. CNGA3, cyclic nucleotide-gated channel subunit
α 3; ori, origin of replication; CMV, cytomegalovirus; AmpR,
ampicillin resistance gene; Neo/KanR, neomycin/kanamycin resistance
gene; bGH poly A, bovine growth hormone polyadenylation; SV40 poly
A, SV40 polyadenylation; p.S334F, serine at position 334 of the
protein is replaced by phenylalanine; p.R189fs, frameshift mutation
where arginine is inserted at position 189; F, forward; R,
reverse.
Retinal electroretinogram results
during the follow-up visit. (A) Dark-adapted 0.01 ERG showing
b-wave amplitudes (indicated by the green arrows) of 289.1 and
299.8 μV in the right and left eye, respectively. (B)
Dark-adapted 3.0 ERG with a-wave amplitudes (indicated by the blue
arrows) of 268.6 and 256.8 μV, and b-wave amplitudes
(indicated by the red arrows) of 464.4 and 451.2 μV in the
right and left eye, respectively. The b-wave amplitudes are below
the normal range in both eyes (515-694 μV). (C) Oscillatory
potential ERG indicates an approximately normal oscillatory
potentials response. (D) Light-adapted 3. 0 ERG demonstrates a
pronounced reduction in the b-wave amplitude (indicated by the red
arrow), with amplitudes of the right and left eye being 14.8 and
13.3 μV, respectively, which are markedly below the normal
range (133-220 μV). (E) Analysis by 30-Hz flicker ERG
reveals an N1-P1 wave amplitude of 8.7 and 8.4 μV in the
right and left eye, respectively, below the typical range (75-205
μV) for both eyes. This suggests the presence of anomalies
in cone cells and their posterior retinal structures. ERG,
electroretinography.
CNGA3:c.566_567insT mutation
leads to amino acid changes. Nucleic acid sequence following
CNGA3:c.566_567insT:p. R189fs mutation, with the shortened
amino acid sequence (yellow arrow), and the insertion of base T
between positions 566 and 567 in the cDNA sequence (marked in
blue). This insertion results in the premature stop codon TGA at
position 194 (marked in red) and a reduction in the number of amino
acids encoded by the reading frame from 694 to 193, ultimately
leading to a shorter and less stable protein. The
CNGA3:c.566_567insT:p.R189fs mutation is located within the
ion-trans domain of the CNGA3 protein, as shown in Fig. 3F. CNGA3, cyclic nucleotide-gated
channel subunit α 3.
Sites of CNGA3 expression. (A)
CNGA3 expression in the brain. The brain image on the left
highlights the regions of CNGA3 expression in the brain, with
darker colors indicating higher levels. The data are based on the
bulk RNA sequencing of micro-dissected brain regions and nuclei.
Protein expression data are grouped into 13 major brain structures,
with values representing the maximum expression in any region
within each structure. The brain image on the right shows the
color-coding used in the bar graph, which presents the expression
of CNGA3 across brain regions. (B) Specific CNGA3 expression in
body tissues. Elevated expression is observed in the brain,
intestine, pituitary gland and retina. The bar graph shows
normalized expression by tissue type, with different tissue groups
shown in different colors. This RNA expression overview shows RNA
data from two different sources: Internally generated Human Protein
Atlas and Genotype-Tissue Expression project RNA-sequencing data,
and a consensus dataset based on a combination of these two data
sources. (C) Summary of normalized CNGA3 RNA expression across all
single-cell types. The highest expression may be observed in cone
cells. Color-coding represents cell-type groups, each consisting of
different cell types with functional features in common. CNGA3,
cyclic nucleotide-gated channel subunit α 3; nTPM, normalized parts
per million.
Original uncropped western blotting
image of CNGA3 wild-type and mutant proteins in cells transfected
with pcDNA3.1-CNGA3, pcDNA3.1-CNGA3-p.S334F and
pcDNA3.1-CNGA3-p.R189fs plasmids. CNGA3, cyclic nucleotide-gated
channel subunit α 3: IB, immunoblot.
Primers for Sanger sequencing.
CDS sequence of the protein encoded by
CNGA3.
Restriction enzyme recognition site,
3xFLAG tag and Kozak sequences of the CNGA3 wild-type plasmid.
Acknowledgements
The authors thank Dr Feng Dong of the Department of
Cell Biology, School of Basic Medical Sciences, Tianjin Medical
University (Tianjin, China) for technical support with plasmid
construction.
Funding
Funding: This study was supported by the Project of Tianjin 131
Innovative Talent Team (grant no. 201936), Science and Technology
Fund for Health of Tianjin (grant no. TJWJ2023ZD008), Science Fund
for Distinguished Young Scholars of Tianjin (grant no.
17JCJQJC46000), Science and Technology Planning Project of Tianjin
(grant no. 21JCYBJC00780), Jinmen Medical Talent Project of
Tianjin, and Tianjin Key Medical Discipline (Specialty)
Construction Project (grant no. TJYXZDXK-016A).
Availability of data and materials
The CNGA3:p. S334F mutation identified in the
present study may be found in dbSNP (https://www.ncbi.nlm.nih.gov/snp/) under accession
number rs1692907593. The datasets analyzed in the current study are
not publicly available because the family did not consent to the
release of their full sequencing information due to privacy
concerns; however, they may be requested from corresponding author
XS.
Authors' contributions
RS and YW performed data screening, data analysis
and statistical analysis, and wrote the manuscript. SC performed
Sanger sequencing. WZ and MP participated in data analysis. RS and
YW constructed plasmids. RS, YL, and DJ performed cell
transfection, protein extraction and western blotting experiments.
JL and XS conceived and designed the study, reviewed the
manuscript, and confirm the authenticity of all the raw data. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Tianjin Eye Hospital (Tianjin, China; approval no.
202015) and was performed in accordance with Declaration of
Helsinki guidelines. Written informed consent for participation in
the study was obtained from the participants and the legal guardian
of the child.
Patient consent for publication
The participants and the legal guardian of the child
provided written consent for publication.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Tsang SH and Sharma T: Progressive cone
dystrophy and cone-rod dystrophy (XL, AD, and AR). Adv Exp Med
Biol. 1085:53–60. 2018.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Gill JS, Georgiou M, Kalitzeos A, Moore AT
and Michaelides M: Progressive cone and cone-rod dystrophies:
Clinical features, molecular genetics and prospects for therapy. Br
J Ophthalmol. 103:711–720. 2019.PubMed/NCBI View Article : Google Scholar : (Epub ahead of
print).
|
|
3
|
Zheng X, Hu Z, Li H and Yang J: Structure
of the human cone photoreceptor cyclic nucleotide-gated channel.
Nat Struct Mol Biol. 29:40–46. 2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sun W and Zhang Q: Diseases associated
with mutations in CNGA3: Genotype-phenotype correlation and
diagnostic guideline. Prog Mol Biol Transl Sci. 161:1–27.
2019.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Shaikh RS, Reuter P, Sisk RA, Kausar T,
Shahzad M, Maqsood MI, Yousif A, Ali M, Riazuddin S, Wissinger B
and Ahmed ZM: Homozygous missense variant in the human CNGA3
channel causes cone-rod dystrophy. Eur J Hum Genet. 23:473–480.
2015.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Li H, Handsaker B, Wysoker A, Fennell T,
Ruan J, Homer N, Marth G, Abecasis G and Durbin R: 1000 Genome
Project Data Processing Subgroup. The sequence alignment/map format
and SAMtools. Bioinformatics. 25:2078–2079. 2009.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Wang K, Li M and Hakonarson H: ANNOVAR:
Functional annotation of genetic variants from high-throughput
sequencing data. Nucleic Acids Res. 38(e164)2010.PubMed/NCBI View Article : Google Scholar
|
|
8
|
1000 Genomes Project Consortium. Auton A,
Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL,
McCarthy S, McVean GA and Abecasis GR: A global reference for human
genetic variation. Nature. 526:68–74. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Karczewski KJ, Francioli LC, Tiao G,
Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A,
Birnbaum DP, et al: The mutational constraint spectrum quantified
from variation in 141,456 humans. Nature. 581:434–443.
2020.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Sim NL, Kumar P, Hu J, Henikoff S,
Schneider G and Ng PC: SIFT web server: Predicting effects of amino
acid substitutions on proteins. Nucleic Acids Res. 40 (Web Server
Issue):W452–W457. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Steinhaus R, Proft S, Schuelke M, Cooper
DN, Schwarz JM and Seelow D: MutationTaster2021. Nucleic Acids Re.
49 (W1):W446–W451. 2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Rentzsch P, Witten D, Cooper GM, Shendure
J and Kircher M: CADD: Predicting the deleteriousness of variants
throughout the human genome. Nucleic Acids Res. 47 (D1):D886–D894.
2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Ratnapriya R, Sosina OA, Starostik MR,
Kwicklis M, Kapphahn RJ, Fritsche LG, Walton A, Arvanitis M, Gieser
L, Pietraszkiewicz A, et al: Retinal transcriptome and eQTL
analyses identify genes associated with age-related macular
degeneration. Nat Genet. 51:606–610. 2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Yao Y, Fu J, Li L, Chen W, Meng Z, Su H
and Dai W: Retinal and circumpapillary nerve fiber layer thickness
and associated factors in children. Eye (Lond). 35:2802–2811.
2021.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hansen MC, Haferlach T and Nyvold CG: A
decade with whole exome sequencing in haematology. Br J Haematol.
188:367–382. 2020.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kaupp UB and Seifert R: Cyclic
nucleotide-gated ion channels. Physiol Rev. 82:769–824.
2002.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Hornbeck PV, Zhang B, Murray B, Kornhauser
JM, Latham V and Skrzypek E: PhosphoSitePlus, 2014: Mutations, PTMs
and recalibrations. Nucleic Acids Res. 43(Database
Issue):D512–D520. 2015.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hardman G, Perkins S, Brownridge PJ,
Clarke CJ, Byrne DP, Campbell AE, Kalyuzhnyy A, Myall A, Eyers PA,
Jones AR and Eyers CE: Strong anion exchange-mediated
phosphoproteomics reveals extensive human non-canonical
phosphorylation. EMBO J. 38(e100847)2019.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhang Y, He X, Zou J, Yang J, Ma A and Tan
M: Phosphorylation mutation impairs the promoting effect of spastin
on neurite outgrowth without affecting its microtubule severing
ability. Eur J Histochem. 67(3594)2023.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kaushik S and Cuervo AM: AMPK-dependent
phosphorylation of lipid droplet protein PLIN2 triggers its
degradation by CMA. Autophagy. 12:432–438. 2016.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Michaelides M, Hunt DM and Moore AT: The
cone dysfunction syndromes. Br J Ophthalmol. 88:291–297.
2004.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Michaelides M, Hardcastle AJ, Hunt DM and
Moore AT: Progressive cone and cone-rod dystrophies: Phenotypes and
underlying molecular genetic basis. Surv Ophthalmol. 51:232–258.
2006.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Michaelides M, Aligianis IA, Ainsworth JR,
Good P, Mollon JD, Maher ER, Moore AT and Hunt DM: Progressive cone
dystrophy associated with mutation in CNGB3. Invest Ophthalmol Vis
Sci. 45:1975–1982. 2004.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Wycisk KA, Zeitz C, Feil S, Wittmer M,
Forster U, Neidhardt J, Wissinger B, Zrenner E, Wilke R, Kohl S and
Berger W: Mutation in the auxiliary calcium-channel subunit
CACNA2D4 causes autosomal recessive cone dystrophy. Am J Hum Genet.
79:973–977. 2006.PubMed/NCBI View
Article : Google Scholar
|
|
26
|
Fujinami K, Zernant J, Chana RK, Wright
GA, Tsunoda K, Ozawa Y, Tsubota K, Robson AG, Holder GE, Allikmets
R, et al: Clinical and molecular characteristics of childhood-onset
Stargardt disease. Ophthalmology. 122:326–334. 2015.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Xu K, Xie Y, Sun T, Zhang X, Chen C and Li
Y: Genetic and clinical findings in a Chinese cohort with Leber
congenital amaurosis and early onset severe retinal dystrophy. Br J
Ophthalmol. 104:932–937. 2020.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wissinger B, Gamer D, Jägle H, Giorda R,
Marx T, Mayer S, Tippmann S, Broghammer M, Jurklies B, Rosenberg T,
et al: CNGA3 mutations in hereditary cone photoreceptor disorders.
Am J Hum Genet. 69:722–737. 2001.PubMed/NCBI View
Article : Google Scholar
|
|
29
|
Huang L, Xiao X, Li S, Jia X, Wang P, Sun
W, Xu Y, Xin W, Guo X and Zhang Q: Molecular genetics of cone-rod
dystrophy in Chinese patients: New data from 61 probands and
mutation overview of 163 probands. Exp Eye Res. 146:252–258.
2016.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Kuniyoshi K, Muraki-Oda S, Ueyama H,
Toyoda F, Sakuramoto H, Ogita H, Irifune M, Yamamoto S, Nakao A,
Tsunoda K, et al: Novel mutations in the gene for α-subunit of
retinal cone cyclic nucleotide-gated channels in a Japanese patient
with congenital achromatopsia. Jpn J Ophthalmol. 60:187–197.
2016.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Thiadens AAHJ, Roosing S, Collin RWJ, van
Moll-Ramirez N, van Lith-Verhoeven JJC, van Schooneveld MJ, den
Hollander AI, van den Born LI, Hoyng CB, Cremers FPM and Klaver
CCW: Comprehensive analysis of the achromatopsia genes CNGA3 and
CNGB3 in progressive cone dystrophy. Ophthalmology. 117:825–830.e1.
2010.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Li S, Huang L, Xiao X, Jia X, Guo X and
Zhang Q: Identification of CNGA3 mutations in 46 families: Common
cause of achromatopsia and cone-rod dystrophies in Chinese
patients. JAMA Ophthalmol. 132:1076–1083. 2014.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Saqib MA, Nikopoulos K, Ullah E, Sher Khan
F, Iqbal J, Bibi R, Jarral A, Sajid S, Nishiguchi KM, Venturini G,
et al: Homozygosity mapping reveals novel and known mutations in
Pakistani families with inherited retinal dystrophies. Sci Rep.
5(9965)2015.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Liu C and Varnum MD: Functional
consequences of progressive cone dystrophy-associated mutations in
the human cone photoreceptor cyclic nucleotide-gated channel CNGA3
subunit. Am J Physiol Cell Physiol. 289:C187–C198. 2005.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Muraki-Oda S, Toyoda F, Okada A, Tanabe S,
Yamade S, Ueyama H, Matsuura H and Ohji M: Functional analysis of
rod monochromacy-associated missense mutations in the CNGA3 subunit
of the cone photoreceptor cGMP-gated channel. Biochem Biophys Res
Commun. 362:88–93. 2007.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Zelinger L, Cideciyan AV, Kohl S, Schwartz
SB, Rosenmann A, Eli D, Sumaroka A, Roman AJ, Luo X, Brown C, et
al: Genetics and disease expression in the CNGA3 form of
achromatopsia: Steps on the path to gene therapy. Ophthalmology.
122:997–1007. 2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kohl S, Marx T, Giddings I, Jägle H,
Jacobson SG, Apfelstedt-Sylla E, Zrenner E, Sharpe LT and Wissinger
B: Total colourblindness is caused by mutations in the gene
encoding the alpha-subunit of the cone photoreceptor cGMP-gated
cation channel. Nat Genet. 19:257–259. 1998.PubMed/NCBI View
Article : Google Scholar
|
|
38
|
Nishiguchi KM, Sandberg MA, Gorji N,
Berson EL and Dryja TP: Cone cGMP-gated channel mutations and
clinical findings in patients with achromatopsia, macular
degeneration, and other hereditary cone diseases. Hum Mutat.
25:248–258. 2005.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Koeppen K, Reuter P, Ladewig T, Kohl S,
Baumann B, Jacobson SG, Plomp AS, Hamel CP, Janecke AR and
Wissinger B: Dissecting the pathogenic mechanisms of mutations in
the pore region of the human cone photoreceptor cyclic
nucleotide-gated channel. Hum Mutat. 31:830–839. 2010.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Azam M, Collin RW, Shah ST, Shah AA, Khan
MI, Hussain A, Sadeque A, Strom TM, Thiadens AA, Roosing S, et al:
Novel CNGA3 and CNGB3 mutations in two Pakistani families with
achromatopsia. Mol Vis. 16:774–781. 2010.PubMed/NCBI
|
|
41
|
Thapa A, Morris L, Xu J, Ma H, Michalakis
S, Biel M and Ding XQ: Endoplasmic reticulum stress-associated cone
photoreceptor degeneration in cyclic nucleotide-gated channel
deficiency. J Biol Chem. 287:18018–18029. 2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Paquet-Durand F, Beck S, Michalakis S,
Goldmann T, Huber G, Mühlfriedel R, Trifunović D, Fischer MD, Fahl
E, Duetsch G, et al: A key role for cyclic nucleotide gated (CNG)
channels in cGMP-related retinitis pigmentosa. Hum Mol Genet.
20:941–947. 2011.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Xu J, Morris L, Thapa A, Ma H, Michalakis
S, Biel M, Baehr W, Peshenko IV, Dizhoor AM and Ding XQ: cGMP
accumulation causes photoreceptor degeneration in CNG channel
deficiency: Evidence of cGMP cytotoxicity independently of enhanced
CNG channel function. J Neurosci. 33:14939–14948. 2013.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Yau KW: Cyclic nucleotide-gated channels:
An expanding new family of ion channels. Proc Natl Acad Sci USA.
91:3481–3483. 1994.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Nakamura T and Gold GH: A cyclic
nucleotide-gated conductance in olfactory receptor cilia. Nature.
325:442–444. 1987.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Tanaka JC, Eccleston JF and Furman RE:
Photoreceptor channel activation by nucleotide derivatives.
Biochemistry. 28:2776–2784. 1989.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Sundin OH, Yang JM, Li Y, Zhu D, Hurd JN,
Mitchell TN, Silva ED and Maumenee IH: Genetic basis of total
colourblindness among the Pingelapese islanders. Nat Genet.
25:289–293. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
48
|
Georgiou M, Fujinami K and Michaelides M:
Retinal imaging in inherited retinal diseases. Ann Eye Sci.
5(25)2020.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Lima LH, Sallum JMF and Spaide RF: Outer
retina analysis by optical coherence tomography in cone-rod
dystrophy patients. Retina. 33:1877–1880. 2013.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Cho SC, Woo SJ, Park KH and Hwang JM:
Morphologic characteristics of the outer retina in cone dystrophy
on spectral-domain optical coherence tomography. Korean J
Ophthalmol. 27:19–27. 2013.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Inui E, Oishi A, Oishi M, Ogino K,
Makiyama Y, Gotoh N, Kurimoto M and Yoshimura N: Tomographic
comparison of cone-rod and rod-cone retinal dystrophies. Graefes
Arch Clin Exp Ophthalmol. 252:1065–1069. 2014.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Hamel CP: Cone rod dystrophies. Orphanet J
Rare Dis. 2(7)2007.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Sahel JA, Marazova K and Audo I: Clinical
characteristics and current therapies for inherited retinal
degenerations. Cold Spring Harb Perspect Med.
5(a017111)2014.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Prado DA, Acosta-Acero M and Maldonado RS:
Gene therapy beyond luxturna: A new horizon of the treatment for
inherited retinal disease. Curr Opin Ophthalmol. 31:147–154.
2020.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Maeder ML, Stefanidakis M, Wilson CJ,
Baral R, Barrera LA, Bounoutas GS, Bumcrot D, Chao H, Ciulla DM,
DaSilva JA, et al: Development of a gene-editing approach to
restore vision loss in Leber congenital amaurosis type 10. Nat Med.
25:229–233. 2019.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Farrar GJ, Millington-Ward S, Chadderton
N, Humphries P and Kenna PF: Gene-based therapies for dominantly
inherited retinopathies. Gene Ther. 19:137–144. 2012.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Han Z, Conley SM, Makkia RS, Cooper MJ and
Naash MI: DNA nanoparticle-mediated ABCA4 delivery rescues
Stargardt dystrophy in mice. J Clin Invest. 122:3221–3226.
2012.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Sarra GM, Stephens C, de Alwis M,
Bainbridge JW, Smith AJ, Thrasher AJ and Ali RR: Gene replacement
therapy in the retinal degeneration slow (rds) mouse: The effect on
retinal degeneration following partial transduction of the retina.
Hum Mol Genet. 10:2353–2361. 2001.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Russell S, Bennett J, Wellman JA, Chung
DC, Yu ZF, Tillman A, Wittes J, Pappas J, Elci O, McCague S, et al:
Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in
patients with RPE65-mediated inherited retinal dystrophy: A
randomised, controlled, open-label, phase 3 trial. Lancet.
390:849–860. 2017.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Jiang L, Zhang H, Dizhoor AM, Boye SE,
Hauswirth WW, Frederick JM and Baehr W: Long-term RNA interference
gene therapy in a dominant retinitis pigmentosa mouse model. Proc
Natl Acad Sci USA. 108:18476–18481. 2011.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Jiang L, Frederick JM and Baehr W: RNA
interference gene therapy in dominant retinitis pigmentosa and
cone-rod dystrophy mouse models caused by GCAP1 mutations. Front
Mol Neurosci. 7(25)2014.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Jiang L, Li TZ, Boye SE, Hauswirth WW,
Frederick JM and Baehr W: RNAi-mediated gene suppression in a
GCAP1(L151F) cone-rod dystrophy mouse model. PLoS One.
8(e57676)2013.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Ortín-Martínez A, Valiente-Soriano FJ,
García-Ayuso D, Alarcón-Martínez L, Jiménez-López M, Bernal-Garro
JM, Nieto-López L, Nadal-Nicolás FM, Villegas-Pérez MP, Wheeler LA
and Vidal-Sanz M: A novel in vivo model of focal light emitting
diode-induced cone-photoreceptor phototoxicity: neuroprotection
afforded by brimonidine, BDNF, PEDF or bFGF. PLoS One.
9(e113798)2014.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Punzo C, Kornacker K and Cepko CL:
Stimulation of the insulin/mTOR pathway delays cone death in a
mouse model of retinitis pigmentosa. Nat Neurosci. 12:44–52.
2009.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Aït-Ali N, Fridlich R, Millet-Puel G,
Clérin E, Delalande F, Jaillard C, Blond F, Perrocheau L, Reichman
S, Byrne LC, et al: Rod-derived cone viability factor promotes cone
survival by stimulating aerobic glycolysis. Cell. 161:817–832.
2015.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Yang Y, Mohand-Said S, Danan A, Simonutti
M, Fontaine V, Clerin E, Picaud S, Léveillard T and Sahel JA:
Functional cone rescue by RdCVF protein in a dominant model of
retinitis pigmentosa. Mol Ther. 17:787–795. 2009.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Johnson S, Michaelides M, Aligianis IA,
Ainsworth JR, Mollon JD, Maher ER, Moore AT and Hunt DM:
Achromatopsia caused by novel mutations in both CNGA3 and CNGB3. J
Med Genet. 41(e20)2004.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Ellingford JM, Barton S, Bhaskar S,
O'Sullivan J, Williams SG, Lamb JA, Panda B, Sergouniotis PI,
Gillespie RL, Daiger SP, et al: Molecular findings from 537
individuals with inherited retinal disease. J Med Genet.
53:761–767. 2016.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Reuter P, Koeppen K, Ladewig T, Kohl S,
Baumann B and Wissinger B: Achromatopsia Clinical Study Group.
Mutations in CNGA3 impair trafficking or function of cone cyclic
nucleotide-gated channels, resulting in achromatopsia. Hum Mutat.
29:1228–1236. 2008.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Carss KJ, Arno G, Erwood M, Stephens J,
Sanchis-Juan A, Hull S, Megy K, Grozeva D, Dewhurst E, Malka S, et
al: Comprehensive rare variant analysis via whole-genome sequencing
to determine the molecular pathology of inherited retinal disease.
Am J Hum Genet. 100:75–90. 2017.PubMed/NCBI View Article : Google Scholar
|