Introduction
Lung adenocarcinoma (LUAD) is the most prevalent
subtype of lung cancer and according to statistics reported in
2024, LUAD continues to rank among the leading malignancies in both
incidence and mortality worldwide. In the United States, LUAD
accounted for ~45% of lung cancers in 2021, with an age-adjusted
incidence of ~22 per 100,000 population (1). For example, recent advances include
low-dose computed tomography screening that reduces lung-cancer
mortality, and precision therapies such as EGFR-targeted
osimertinib (with or without chemotherapy), RET inhibitors
(selpercatinib), KRAS p.G12C inhibitors (sotorasib), and immune
checkpoint blockade (pembrolizumab-based chemoimmunotherapy).
Despite notable progress in diagnostic and therapeutic strategies,
such as low-dose CT screening, EGFR-targeted therapy and immune
checkpoint inhibitors, the 5-year survival rate for LUAD remains
poor at ~15-20% (2,3). With the rapid development of
high-throughput sequencing and bioinformatics, researchers can now
systematically identify key molecular targets related to tumor
initiation and progression at the genomic level (4-6);
however, precise therapeutic targets for LUAD are still
lacking.
In addition to genetic and transcriptomic
characterization, advances in nanotechnology and biomolecular
engineering have introduced new opportunities for improving LUAD
diagnosis and treatment. For example, macrophage-mediated
sulfate-based nanomedicine has demonstrated enhanced drug delivery
efficacy within the tumor microenvironment of lung cancer (7), whilst nano-assisted radiotherapy
strategies have shown potential in increasing treatment precision
and efficacy in non-small cell lung cancer (8). Furthermore, self-assembled DNA-based
biosensors have enabled rapid and portable detection of circulating
tumor cells in lung cancer patient blood samples (n=46), achieving
100% specificity and 86.5% sensitivity, supporting early diagnosis
and disease monitoring in lung cancer (9). These technological innovations
underscore the need to explore molecular-level biomarkers that
could complement or integrate with such novel platforms to improve
clinical outcomes in LUAD.
The present study employed integrated bioinformatics
tools and machine learning algorithms and conducted in
silico validation using independent GEO (GSE31210 discovery;
GSE68465 validation) and TCGA-LUAD cohorts, as well as molecular
validation by reverse transcription-quantitative (RT-q) PCR on
paired LUAD and adjacent normal tissues (n=50) to identify and
validate potential therapeutic targets for LUAD, with the aim to
providing a foundation for future targeted treatment
strategies.
Materials and methods
Identification of differentially
expressed genes (DEGs)
A total of two LUAD-related datasets, GSE31210 and
GSE68465(10), were retrieved from
the GEO database (11), in which
the control samples comprised non-tumorous normal lung tissues
rather than patient-matched adjacent tissues (GSE31210: 20 normal,
226 LUAD; GSE68465: 19 normal, 443 LUAD); these datasets were
chosen for their large sample sizes, availability of normal
controls, and rich clinical annotation (including overall
survival), making them widely used benchmark cohorts for discovery
(GSE31210) and external validation (GSE68465). DEGs were identified
using the ‘limma’ R package (v3.60.6, Bioconductor), implemented in
R v4.4.1 (www.r-project.org/foundation/) (12), with screening criteria set as
|log2fold change|>1 and P<0.05. Volcano plots were
generated using the ‘EnhancedVolcano’ package for visualization
(13).
Weighted gene co-expression network
analysis (WGCNA)
WGCNA was performed on the GSE31210 dataset using
the ‘WGCNA’ R package (14),
because this dataset contains both tumor and normal samples with
comprehensive clinical annotation and a sufficiently large sample
size, making it suitable for constructing reliable co-expression
networks to identify key gene modules associated with LUAD. A
soft-thresholding power β=6 was chosen based on the scale-free
topology criterion (scale-free R²>0.85). The minimum module size
was set to 30 to ensure module robustness. The resulting sample
dendrogram and trait heatmap are presented in Fig. S1. Modules with a Pearson
correlation coefficient of r>0.92 and P<0.05 were considered
statistically significant, with correlations evaluated between
module eigengenes and clinical traits (such as tumor vs. normal
status, stage), measured from the sample annotation of the GSE31210
dataset.
Gene set enrichment analysis
(GSEA)
GSEA was performed using GSEA software (v4.3.2)
(15) on the entire set of DEGs,
whereas Gene Ontology (GO) and Kyoto Encyclopedia of Genes and
Genomes (KEGG) enrichment analyses were performed using the
‘clusterProfiler’ R package (v4.12.0; www.r-project.org/foundation/) (16,17)
on the same DEG set. GO analysis included biological process,
cellular components and molecular functions. KEGG analysis was used
to identify significantly enriched signaling pathways. P<0.05
and q<0.05 were considered to indicate a statistically
significant difference.
Construction and analysis of the
protein-protein interaction (PPI) network
Identified DEGs were submitted to the STRING
database to construct a PPI network (18), with a confidence score threshold of
>0.9 (high confidence). Interaction data were imported into
Cytoscape (v3.10.0; https://cytoscape.org/) for visualization. Network
topology was analyzed using two centrality algorithms: Degree
Centrality and Betweenness Centrality (19).
Machine learning analysis
A total of two machine learning algorithms were
applied: Random Forest (RF) and Support Vector Machine-Recursive
Feature Elimination (SVM-RFE) (20,21).
RF analysis was performed using the ‘randomForest’ package
(v4.7-1.2) in R v4.4.1 (www.r-project.org/foundation/), and feature importance
was ranked accordingly according to the default feature importance
measures implemented in the ‘randomForest’ R package (Mean Decrease
Accuracy and Mean Decrease Gini, as recommended by the package
developers). SVM-RFE analysis was performed using the ‘e1071’
package (v1.7-14) in R v4.4.1 (www.r-project.org/foundation/) to further select
optimal features based on gene importance. A linear kernel was
applied with the default cost parameter (C=1), and feature ranking
was performed using 10-fold cross-validation. The gene subset
corresponding to the lowest cross-validated classification error
was selected as the optimal feature set. P<0.05 was considered
to indicate a statistically significant difference.
RT-qPCR
RT-qPCR (22) was
used to validate the expression levels of caveolin-1 (CAV1) and
cadherin 5 (CDH5) in tumor and adjacent normal tissues from 50
patients with LUAD, with adjacent tissues collected at least 5 mm
away from the tumor margin. All tissue specimens were collected
during surgical resection at Hebei Provincial Hospital of
Traditional Chinese Medicine (Shijiazhuang, China) between May 2021
and June 2023, immediately snap-frozen in liquid nitrogen, and
stored at -80˚C until RNA extraction. Among the patients, 28 were
male and 22 were female. Inclusion criteria were
histopathologically confirmed LUAD and availability of paired tumor
and adjacent normal tissues; exclusion criteria included prior
chemotherapy, radiotherapy, or other malignancies. The median age
was 63 years (range, 45-78 years). The study protocol was approved
by the Ethics Committee of Hebei Provincial Hospital of Traditional
Chinese Medicine (Shijiazhuang, China; approval no.
HBZY2023-KY-076-01) and followed the ethical principles of The
Declaration of Helsinki. Written informed consent was obtained from
all participants.
Total RNA was extracted using TRIzol®
reagent (Thermo Fisher Scientific, Inc.), and RNA purity and
concentration were assessed using a NanoDrop 2000
spectrophotometer. High-quality RNA was reverse transcribed into
cDNA, and qPCR was performed using SYBR Green PCR Master Mix on a
QuantStudio 6 Real-Time PCR System (Thermo Fisher Scientific,
Inc.). Relative gene expression was calculated using the
2-ΔΔCq method (22),
with GAPDH used as the internal control. Primers were designed and
validated using Primer-BLAST. The following primer sequences were
used: GAPDH forward, 5'-AATGGACAACTGGTCGTGGAC-3' and reverse,
5'-CCCTCCAGGGGATCTGTTTG-3'; CAV1 forward,
5'-GCAGAACCAGAAGGGACACAG-3' and reverse,
5'-CCAAAGAGGGCAGACAGCAAGC-3'; and CDH5 forward,
5'-AGGTGCTAACCCTGCCCAAC-3' and reverse,
5'-CGGAAGACCTTGCCCACATA-3'.
Receiver operating characteristic
(ROC) curve analysis
ROC curve analysis was performed using the ‘pROC’
package (v1.18.5) in R v4.4.1 (www.r-project.org/foundation/) (23). An area under the curve (AUC) of
>0.9 was considered to indicate notable diagnostic performance.
P<0.05 was considered to indicate a statistically significant
difference.
Single-cell RNA sequencing (scRNA-seq)
analysis
scRNA-seq data related to LUAD were obtained from
the Tumor Immune Single Cell Hub (TISCH) database (http://tisch.comp-genomics.org) (24). The expression levels of key genes
across different cell types were visualized using heatmaps and
feature plots, which were generated directly from the TISCH
database portal based on preprocessed single-cell RNA-seq data
provided by the database.
Statistical analysis
Statistical analysis was performed using GraphPad
Prism 9.0 (Dotmatics). Differences between tumor and normal tissues
in The Cancer Genome Atlas (TCGA) and GEO datasets were assessed
using unpaired Student's t-test, while differences between matched
tumor and adjacent normal tissues in the clinical validation cohort
and paired comparisons within the TCGA cohort (Fig. 3D) were assessed using paired
Student's t-test. For comparisons among multiple subgroups, one-way
ANOVA followed by Tukey's post hoc test was applied. Kaplan-Meier
survival analysis was performed using the Kaplan-Meier Plotter
database (http://kmplot.com/analysis/) based on
TCGA-LUAD data; patients were stratified into high- and
low-expression groups using the median expression value as the
cut-off, and statistical significance was evaluated using the
log-rank test. P<0.05 was considered to indicate a statistically
significant difference.
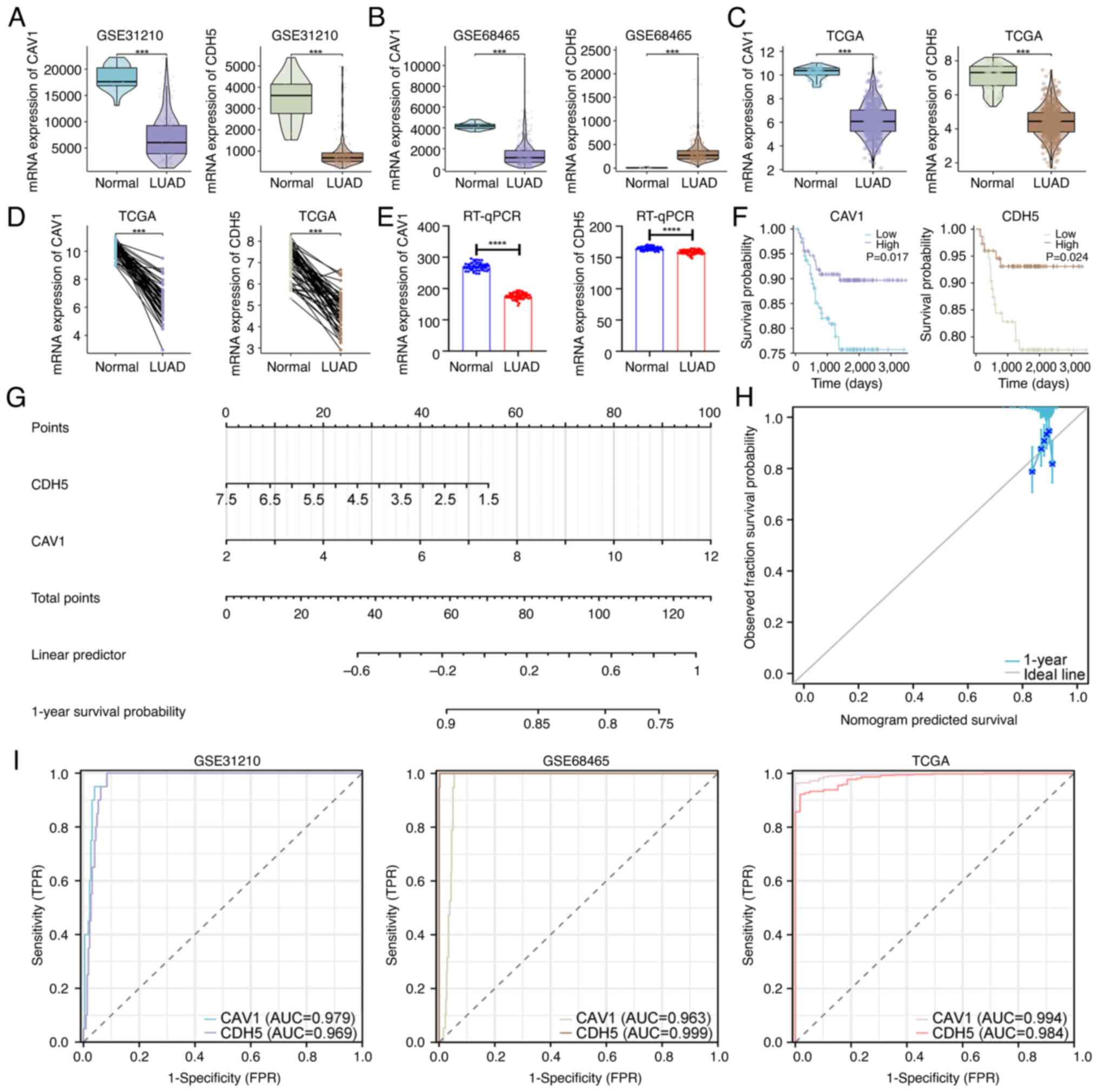 | Figure 3Expression and prognostic analysis of
key genes. Expression of CAV1 and CDH5 in the (A) GSE31210, (B)
GSE68465 and (C) TCGA datasets. (D) Paired expression analysis of
CAV1 and CDH5 in TCGA samples. (E) RT-qPCR validation in tissues
from patients with LUAD (n=50). (F) Kaplan-Meier survival curves
for CAV1 and CDH5, comparing patients with high expression and low
expression (grouped according to the median expression level).
P-values were calculated using the log-rank test. (G) Diagnostic
nomogram based on CAV1 and CDH5. (H) Calibration curve evaluating
nomogram accuracy. (I) Receiver operating characteristic curve
analysis in the GSE31210, GSE68465 and TCGA datasets.
***P<0.001 and ****P<0.0001. CAV1,
caveolin-1; CDH5, cadherin 5; TCGA, The Cancer Genome Atlas;
RT-qPCR, reverse transcription-quantitative PCR; LUAD, lung
adenocarcinoma; AUC, area under the curve; TRP, true positive rate;
FRP, false positive rate. |
Results
Identification of overlapping genes
between DEGs and key WGCNA modules
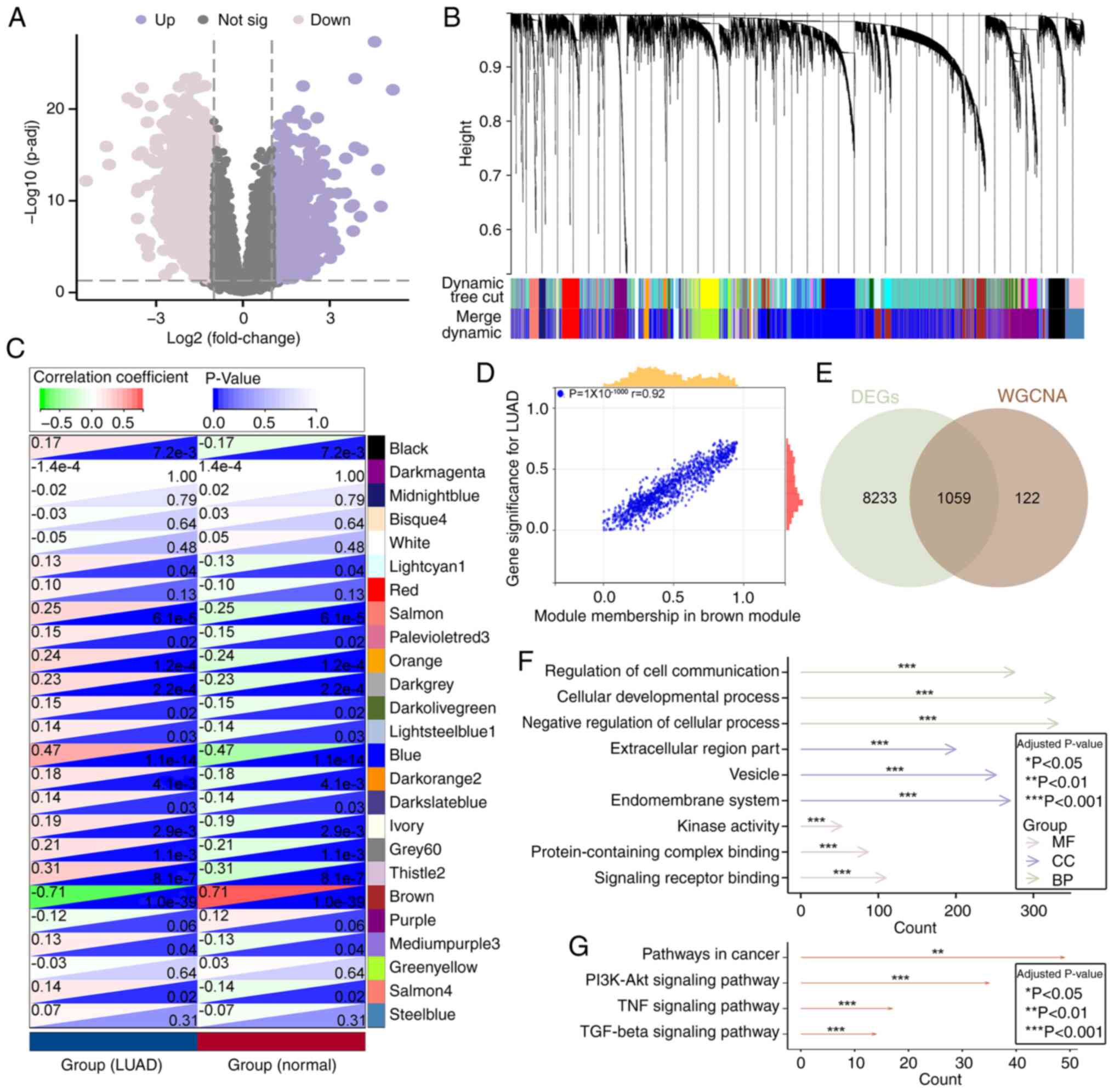
Based on the GSE31210 dataset, a total of 9,292 DEGs
were identified, including 5,399 upregulated and 3,893
downregulated genes in LUAD tumor tissues compared with controls,
as this dataset contains both tumor and non-tumor lung tissues with
comprehensive clinical annotation, making it suitable for DEG
identification. A volcano plot was generated to visualize these
DEGs (Fig. 1A). To further
identify key genes, a WGCNA was performed using the same dataset,
resulting in the identification of 25 co-expression modules
(Fig. 1B).
Correlation analysis between these modules and LUAD
phenotypes revealed that 17 modules were significantly associated
with LUAD, among which the brown module showed the strongest
correlation (Fig. 1C). This
suggested that this module may serve a critical role in the
pathogenesis and progression of LUAD. Further scatter plot analysis
of module membership and gene significance demonstrated a strong
positive correlation within the brown module (Pearson's r=0.92,
P=1x10-1,000), indicating its relevance as a key module
(Fig. 1D).
Subsequently, a Venn diagram was used to identify
overlapping genes between DEGs and those in the key module
identified by WGCNA, resulting in 1,059 common genes (Fig. 1E). GO and KEGG enrichment analyses
were then performed on these overlapping genes. GO analysis
revealed enrichment in signaling-related molecular functions such
as ‘signaling receptor binding’, ‘kinase activity’ and
‘protein-containing complex binding’, cellular components such as
‘vesicle’, ‘endomembrane system’ and ‘extracellular region part’,
as well as biological processes such as ‘regulation of cell
communication’, ‘cellular developmental process’ and ‘negative
regulation of cellular process’ (Fig.
1F). KEGG analysis revealed significant enrichment in ‘TGF-β
signaling pathway’, ‘TNF signaling pathway’, ‘PI3K-Akt signaling
pathway’ and ‘pathways in cancer’, suggesting that these genes may
serve regulatory roles in LUAD initiation and progression (Fig. 1G).
Identification of key LUAD genes based
on PPI network and machine learning
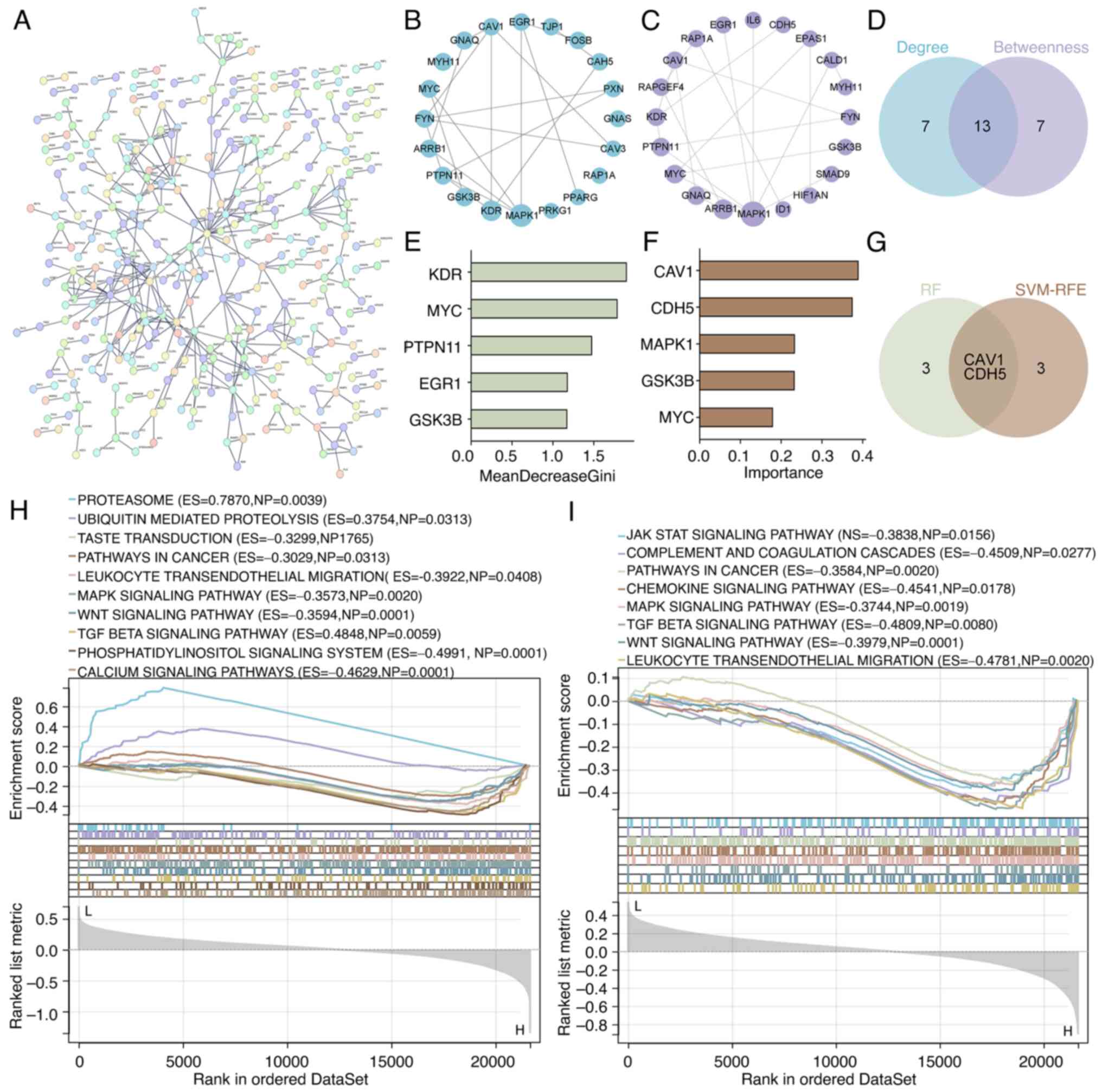
A PPI network was constructed using the 1,059
overlapping genes (Fig. 2A).
Topological analysis in Cytoscape using the Degree Centrality and
Betweenness Centrality algorithms identified the top 20 hub genes
(Fig. 2B and C, respectively). The intersection of
these two analyses yielded 13 shared hub genes (Fig. 2D).
Subsequently, two machine learning approaches, RF
and SVM-RFE, were employed to further refine key gene selection.
The RF algorithm identified the top five genes based on feature
importance scores (Fig. 2E),
whereas SVM-RFE selected another top five genes through iterative
optimization (Fig. 2F). A Venn
diagram analysis of both methods identified two overlapping
candidate genes: CAV1 and CDH5 (Fig.
2G).
Furthermore, GSEA indicated that both CAV1 and CDH5
were significantly enriched in signaling pathways such as MAPK, Wnt
and TGF-β, suggesting their involvement in LUAD development,
progression and modulation of the tumor immune microenvironment
(Fig. 2H and I).
Expression analysis and prognostic
model construction of key genes
To explore the roles of CAV1 and CDH5 in LUAD, their
expression levels were analyzed in the GSE31210 dataset. Both genes
were significantly downregulated in LUAD tissues compared with
those in normal tissues (Fig. 3A).
Moreover, consistent downregulation trends were observed in the
GSE68465 and TCGA datasets for CAV1 (Fig. 3B and C). Although CDH5 expression was
upregulated in the LUAD tissues in the GSE68465 dataset, it was
downregulated in the TCGA cohort. To further investigate whether
tumor stage contributes to this inter-dataset discrepancy, a
subgroup analysis was performed using the GSE68465 dataset,
stratifying patients by N stage (N0, N1 and N2). As shown in
Fig. S2, CDH5 expression was
significantly higher in N0-stage samples compared with that in N1
(P<0.001) and a significant difference was also observed between
N1 and N2 (P<0.05). This stage-associated decline supports a
biological trend of progressive CDH5 downregulation during LUAD
advancement.
Pairwise analyses in the TCGA dataset demonstrated
significantly lower expression of CAV1 and CDH5 in LUAD tissues
compared with available adjacent normal lung tissues, eliminating
potential interference from inter-individual biological variation
(Fig. 3D). Similarly, RT-qPCR
results from 50 LUAD samples demonstrated significantly decreased
expression levels of CAV1 and CDH5 in tumor tissues compared with
those in matched adjacent normal tissues (P<0.0001; Fig. 3E). Notably, although the difference
in CDH5 expression between tumor and control tissues was relatively
small in magnitude, it remained statistically significant. This
expanded validation supports the reliability and reproducibility of
the bioinformatics-driven identification of these genes.
Furthermore, Kaplan-Meier survival analysis revealed
that high expression of CAV1 and CDH5 was significantly associated
with improved overall survival, suggesting that CAV1 and CDH5 have
potential tumor suppressor functions (Fig. 3F). A diagnostic nomogram model
based on the expression of CAV1 and CDH5 was then constructed
(Fig. 3G), with calibration curves
indicating strong agreement between predicted and actual outcomes,
as the calibration curves closely overlapped with the ideal 45˚
reference line (Fig. 3H). In
addition, ROC curve analysis demonstrated notable diagnostic
performance for CAV1 (AUC=0.979) and CDH5 (AUC=0.969) in GSE31210,
and this was validated in both GSE68465 and TCGA datasets (Fig. 3I), supporting the potential of CAV1
and CDH5 as diagnostic biomarkers for LUAD.
Expression features of key genes in
single-cell transcriptomics
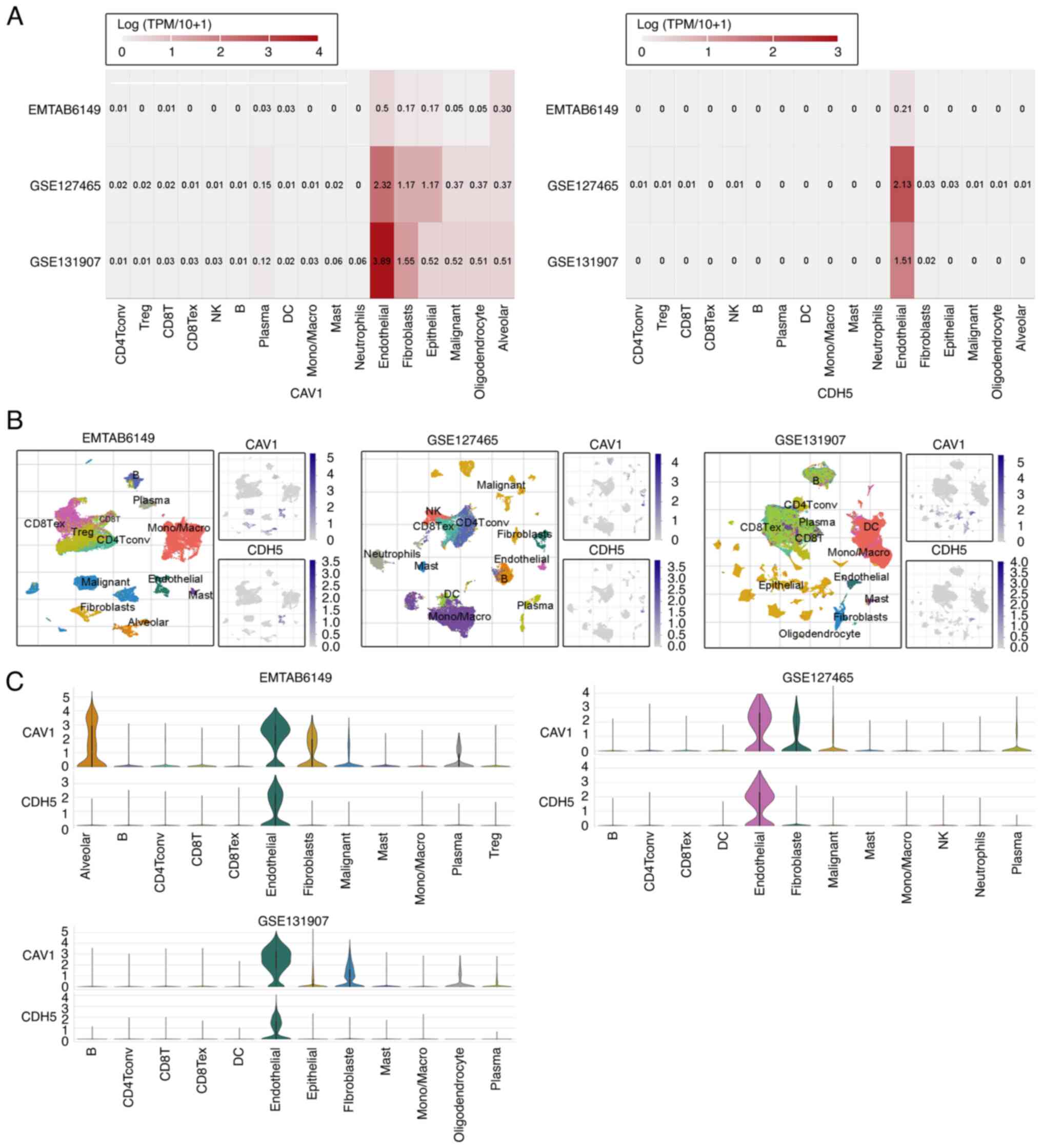
To precisely characterize the cell-type-specific
expression and potential functions of CAV1 and CDH5, three
LUAD-related scRNA-seq datasets (EMTAB6149, GSE127465 and
GSE131907) from the TISCH database were analyzed: EMTAB6149
includes ~20,000 cells in total from the primary LUAD tissues of 4
patients; GSE127465 comprises ~14,000 cells in total from
surgically resected LUAD tumors and adjacent tissues from 7
patients; and GSE131907 includes ~208,506 cells in total derived
from 11 patients with LUAD. These datasets represent a diverse
range of LUAD microenvironments and provide robust resolution of
immune and stromal cell populations. Both genes were revealed to be
highly expressed in endothelial cells (Fig. 4A), suggesting potential roles in
tumor-associated angiogenesis, vascular barrier maintenance or
microenvironment regulation. To visualize their spatial expression
across immune cell subpopulations, t-distributed stochastic
neighbor embedding plots were generated for CAV1 and CDH5, which
showed that both genes were predominantly expressed in endothelial
cell clusters, with lower expression observed in other immune cell
subtypes (Fig. 4B). Additionally,
violin plots were used to quantify expression levels across major
cell types, revealing marked heterogeneity in gene expression
patterns, with CAV1 and CDH5 showing higher expression in
endothelial cells but minimal expression in most immune cell
subsets, indicating substantial variability across cell populations
(Fig. 4C). These findings not only
corroborate the high expression observed in heatmaps, particularly
in endothelial cells, but also highlight the complexity of gene
regulation in the tumor microenvironment, providing a theoretical
basis for further functional studies.
Discussion
LUAD exhibits notable heterogeneity in terms of its
molecular characteristics, cellular composition, and clinical
behavior, posing ongoing challenges for both diagnosis and targeted
therapy (25). The present study
systematically identified key genes participating in LUAD, namely
CAV1 and CDH5, through integrated differential expression analysis
across multiple GEO and TCGA datasets, WGCNA, PPI network topology
screening and dual machine learning algorithms. The expression
profiles and potential functions of these genes in LUAD were then
further explored.
CAV1 is a scaffolding protein involved in membrane
signal integration, cytoskeletal remodeling and tumor-suppressive
signal transduction, and has been implicated in LUAD proliferation,
migration and mechanisms of drug resistance (26). CDH5 (coding for VE-cadherin) is an
endothelial cell-specific adhesion molecule that maintains vascular
integrity and participates in tumor angiogenesis and immune evasion
(27). In the context of LUAD
progression, the downregulation of CAV1 and CDH5 may actively
contribute to tumor aggressiveness through multiple mechanisms.
CAV1, a structural component of caveolae, is known to regulate
membrane-associated signaling and actin cytoskeleton dynamics
(28). Its loss has been
associated with the activation of the MAPK/ERK and Wnt/β-catenin
pathways, both of which are implicated in promoting
epithelial-mesenchymal transition (EMT), increased motility and the
metastatic capacity of LUAD cells (29). CAV1 downregulation also facilitates
invadopodia formation, enabling extracellular matrix degradation
and local invasion (30).
Furthermore, loss of CDH5 expression has been associated with
enhanced invasiveness in several cancers, including breast, gastric
and colorectal cancers, as demonstrated in a recent pan-cancer
analysis (31). In the present
study, survival analysis demonstrated that high expression of both
CAV1 and CDH5 was significantly associated with improved overall
survival, suggesting the tumor-suppressive roles of these genes in
LUAD.
CDH5/VE-cadherin serves a critical role in
maintaining endothelial junctional integrity (32), and reduced CDH5 expression in LUAD
may disrupt the vascular endothelial barrier, thereby increasing
vascular permeability and promoting tumor cell intravasation into
the bloodstream. This mechanism supports distant metastasis and has
been associated with enhanced extravasation and colonization at
secondary sites (33).
Furthermore, loss of CDH5 expression may impair immune surveillance
by altering leukocyte transmigration, thereby contributing to the
formation of an immune-privileged tumor niche, characterized by
reduced immune cell infiltration and evasion from host immune
responses (34). These effects
highlight the functional importance of CAV1 and CDH5 beyond their
diagnostic value, further supporting their role as potential
therapeutic targets in LUAD. Additionally, a diagnostic nomogram
model constructed based on these two genes exhibited strong
predictive accuracy. ROC analysis across multiple independent
datasets also demonstrated robust diagnostic potential of CAV1 and
CDH5, supporting their reliability as candidate molecular
biomarkers for LUAD. While CDH5 expression was found to be
upregulated in the GSE68265 dataset, this trend was not replicated
in other independent datasets. To explore whether clinical staging
may account for this discrepancy, a subgroup analysis stratified by
N stage was performed using the GSE68465 cohort. The results
demonstrated a significant stepwise decline in CDH5 expression from
N0 to N2 stages, supporting a stage-dependent downregulation
pattern. This finding aligns with the trends observed in the TCGA
dataset and the RT-qPCR validation cohort conducted in the present
study. Taken together, these data suggest that CDH5 downregulation
is a consistent feature in LUAD progression, and that isolated
inter-dataset variability may be attributable to sample
composition, stromal content or platform-specific differences.
These observations underscore the importance of integrating
stratified analysis in biomarker evaluation to mitigate confounding
effects and enhance biological interpretability.
In terms of underlying mechanisms, GSEA revealed
that CAV1 and CDH5 were significantly enriched in key signaling
pathways, including MAPK, Wnt and TGF-β. These pathways are known
to be involved in cell proliferation, migration, metastasis and
immune regulation in LUAD (35-37).
CAV1 generally acts as an inhibitor of the MAPK/ERK and
Wnt/β-catenin pathways, while loss of CDH5 function disrupts
endothelial junctions and facilitates pro-tumorigenic signaling,
thereby highlighting their tumor-suppressive roles. Notably, the
activation of MAPK and Wnt signaling has been reported to promote
tumor cell growth and maintain cancer stemness (38), whereas the TGF-β pathway in LUAD
has shown stage-dependent effects, exhibiting tumor-suppressive
roles in early stages but promoting EMT and metastasis in later
stages (39).
Further analysis of single-cell transcriptomic data
revealed that CAV1 and CDH5 are predominantly expressed in
endothelial cells, suggesting the hypothesis that they may be
involved in maintaining endothelial function (40), regulating tumor-associated
angiogenesis and contributing to the formation of the immune
barrier. A recent study has reported that dysfunction of tumor
endothelial cells is associated with abnormal vascularization,
immune escape and distant metastasis, which is consistent with the
findings of the present study (41). Although these processes were not
directly investigated in the present study, such associations can
be hypothesized based on the enriched terms and pathways identified
in the bioinformatics analysis, which are consistent with the
findings of the previous report. Whilst this endothelial-specific
expression pattern suggests a possible involvement in angiogenic
regulation, this inference is based on gene localization and prior
biological evidence rather than direct functional association. In
the current study, the proportion of endothelial subclusters
expressing CAV1 or CDH5 were not quantified, and associations with
angiogenesis-related signatures or histological vessel density were
not assessed. Therefore, future studies integrating these analyses
will be necessary to confirm the functional roles of these genes in
LUAD-associated angiogenesis.
From a clinical standpoint, the incorporation of
CAV1 and CDH5 into diagnostic workflows may hold promising
translational potential. Given their consistent differential
expression between LUAD and normal tissues, both genes could be
evaluated using immunohistochemistry on formalin-fixed
paraffin-embedded lung biopsy specimens, allowing pathologists to
assess protein levels directly in tumor samples. Alternatively,
emerging liquid biopsy techniques, such as detection of circulating
tumor RNA or protein in plasma, could potentially enable the
non-invasive, dynamic monitoring of these biomarkers during disease
progression or treatment response. Although CAV1 and CDH5 are
cytoskeleton-associated proteins and are unlikely to be secreted
directly, their dysregulation may still be detected indirectly
through circulating tumor RNA or extracellular vesicles. A study
demonstrated the feasibility of liquid biopsy for monitoring
molecular alterations in patients with LUAD (42). Although further validation is
necessary, particularly at the protein and clinical implementation
level, the findings of the present study provide a rationale for
the future development of CAV1 and CDH5 as clinically useful
biomarkers in LUAD diagnosis and patient management.
Furthermore, whilst the nomogram and ROC analyses in
the present study demonstrated notable diagnostic performance for
CAV1 and CDH5, these results were derived from datasets with
limited clinical covariates and without multivariate survival
adjustment. In particular, the Kaplan-Meier survival curves were
based on univariate analysis and did not account for potential
confounders, such as tumor stage, patient age or treatment
modalities. Similarly, although multiple datasets were used for
validation, the high AUC values may partially reflect
dataset-specific characteristics or overfitting. Therefore, future
studies incorporating multivariate Cox regression analysis, larger
prospective cohorts and bootstrap or external validation frameworks
will be necessary to confirm the independent clinical utility of
these biomarkers.
In addition, whilst the expression levels of CAV1
and CDH5 were validated at the mRNA level in an expanded clinical
cohort, there was a lack of protein-level validation and
histological confirmation of cell-type specificity within LUAD
tissues. Future studies involving immunohistochemical staining and
spatial transcriptomic profiling will be required to fully
elucidate the functional roles and cellular localization of CAV1
and CDH5.
In summary, CAV1 and CDH5 appear to have notable
biological roles in LUAD and may serve as promising targets for
diagnosis and therapy. A limitation of the present study is that it
focused on bioinformatics integration and clinical validation and
did not include in vitro or in vivo functional
experiments. However, the consistent expression patterns of CAV1
and CDH5 across multiple datasets and RT-qPCR analysis support
their tumor-suppressive potential. Mechanistic validation, such as
gene knockdown or overexpression studies in LUAD models, should be
performed in future research to further elucidate the functional
roles and therapeutic implications of these genes.
In conclusion, the present study systematically
identified CAV1 and CDH5 as potential key tumor-suppressor genes in
LUAD. Both genes were significantly downregulated in tumor tissues
and associated with a favorable patient prognosis. The diagnostic
model based on their expression demonstrated a notable predictive
performance. Moreover, single-cell transcriptomic analysis
highlighted the specific expression of CAV1 and CDH5 in endothelial
cells, suggesting their potential involvement in the regulation of
the tumor microenvironment. These findings provide a theoretical
basis and novel molecular targets for the diagnosis and targeted
treatment of LUAD.
Supplementary Material
Sample clustering dendrogram and
phenotype heatmap for the GSE31210 dataset. Hierarchical clustering
was performed using average linkage and Euclidean distance. The
lower color bar represents clinical phenotype classification
(purple, LUAD; blue, normal tissue). The dendrogram and heatmap
confirm consistency between sample grouping and phenotype labels
and indicate no obvious outliers. This visualization supports the
reliability of subsequent module-trait correlation analyses. LUAD,
lung adenocarcinoma.
CDH5 mRNA expression in patients with
lung adenocarcinoma from the GSE68465 dataset, stratified by lymph
node metastasis status (N stage). CDH5 expression was significantly
higher in N0-stage samples compared with that in N1 (P<0.001)
and a significant difference was also observed between N1 and N2
(P<0.05). *P<0.05 and ***P<0.001.
CDH5, cadherin 5.
Acknowledgements
Not applicable.
Funding
Funding: The present study was funded by the Research Project of
the Hebei Provincial Administration of Traditional Chinese Medicine
(grant nos. 2020103 and 2022065).
Availability of data and materials
The datasets generated in the present study may be
requested from the corresponding author.
Authors' contributions
LZ conceived and designed the study, developed the
methodology and drafted the manuscript. YW contributed to software
implementation, validation and project administration. YuL
performed formal analysis, participated in data acquisition and
interpretation and contributed to securing funding. YaL conducted
experimental investigation and provided essential resources
(clinical samples and reagents). LW curated the data, performed
statistical analysis and visualization. CW contributed to the study
conception and manuscript writing, reviewing and editing. LZ and CW
confirm the authenticity of all the raw data. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
The study was approved by the Ethics Committee of
Hebei Provincial Hospital of Traditional Chinese Medicine (approval
no. HBZY2023-KY-076-01). Written informed consent was obtained from
all participants.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Siegel RL, Giaquinto AN and Jemal A:
Cancer statistics, 2024. CA Cancer J Clin. 74:12–49.
2024.PubMed/NCBI View Article : Google Scholar
|
|
2
|
He T, Li J, Wang P and Zhang Z: Artificial
intelligence predictive system of individual survival rate for lung
adenocarcinoma. Comput Struct Biotechnol J. 20:2352–2359.
2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lin JJ, Cardarella S, Lydon CA, Dahlberg
SE, Jackman DM, Jänne PA and Johnson BE: Five-year survival in
EGFR-mutant metastatic lung adenocarcinoma treated with EGFR-TKIs.
J Thorac Oncol. 11:556–565. 2016.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Sogbe M, Aliseda D, Sangro P, de la
Torre-Aláez M, Sangro B and Argemi J: Prognostic value of
circulating tumor DNA in different cancer types detected by
ultra-low-pass whole-genome sequencing: A systematic review and
patient-level survival data meta-analysis. Carcinogenesis.
16(bgae073)2025.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Leong IUS, Cabrera CP, Cipriani V, Ross
PJ, Turner RM, Stuckey A, Sanghvi S, Pasko D, Moutsianas L, Odhams
CA, et al: Large-scale pharmacogenomics analysis of patients with
cancer within the 100,000 genomes project combining whole-genome
sequencing and medical records to inform clinical practice. J Clin
Oncol. 43:682–693. 2025.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Larsson L, Corbett C, Kalmambetova G,
Utpatel C, Ahmedov S, Antonenka U, Iskakova A, Kadyrov A, Kohl TA,
Barilar V, et al: Whole-genome sequencing drug susceptibility
testing is associated with positive MDR-TB treatment response. Int
J Tuberc Lung Dis. 28:494–499. 2024.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Liu C, Chen Y, Xu X, Yin M, Zhang H and Su
W: Utilizing macrophages missile for sulfate-based nanomedicine
delivery in lung cancer therapy. Research (Wash D C).
7(0448)2024.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zhao L, Li M, Shen C and Luo Y, Hou X, Qi
Y, Huang Z, Li W, Gao L, Wu M and Luo Y: Nano-assisted radiotherapy
strategies: New opportunities for treatment of non-small cell lung
cancer. Research (Wash D C). 7(0429)2024.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Wang Y, Shen C, Wu C, Zhan Z, Qu R, Xie Y
and Chen P: Self-assembled DNA machine and selective complexation
recognition enable rapid homogeneous portable quantification of
lung cancer CTCs. Research (Wash D C). 7(0352)2024.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Zhao Z, Zhao D, Xia J, Wang Y and Wang B:
Immunoscore predicts survival in early-stage lung adenocarcinoma
patients. Front Oncol. 10(691)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Barrett T, Wilhite SE, Ledoux P,
Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH,
Sherman PM, Holko M, et al: NCBI GEO: Archive for functional
genomics data sets-update. Nucleic Acids Res. 41 (Database
Issue):D991–D995. 2013.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43(e47)2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Li W: Volcano plots in analyzing
differential expressions with mRNA microarrays. J Bioinform Comput
Biol. 10(1231003)2012.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9(559)2008.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Subramanian A, Tamayo P, Mootha VK,
Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub
TR, Lander ES and Mesirov JP: Gene set enrichment analysis: A
knowledge-based approach for interpreting genome-wide expression
profiles. Proc Natl Acad Sci USA. 102:15545–15550. 2005.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The gene
ontology consortium. Nat Genet. 25:25–29. 2000.PubMed/NCBI View
Article : Google Scholar
|
|
17
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Stelzl U, Worm U, Lalowski M, Haenig C,
Brembeck FH, Goehler H, Stroedicke M, Zenkner M, Schoenherr A,
Koeppen S, et al: A human protein-protein interaction network: A
resource for annotating the proteome. Cell. 122:957–968.
2005.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT and
Lin CY: cytoHubba: Identifying hub objects and sub-networks from
complex interactome. BMC Syst Biol. 8 (Suppl 4)(S11)2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Breiman L: Random forests. Mach Learn.
45:5–32. 2001.
|
|
21
|
Cortes C and Vapnik V: Support-vector
networks. Mach Learn. 20:273–297. 1995.
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Sedgwick P: How to read a receiver
operating characteristic curve. BMJ. 350(h2464)2015.PubMed/NCBI View Article : Google Scholar
|
|
24
|
González-Silva L, Quevedo L and Varela I:
Tumor Functional heterogeneity unraveled by scRNA-seq technologies.
Trends Cancer. 6:13–19. 2020.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Li X, Lu F, Cao M, Yao Y, Guo J, Zeng G
and Qian J: The pro-tumor activity of INTS7 on lung adenocarcinoma
via inhibiting immune infiltration and activating p38MAPK pathway.
Sci Rep. 14(25636)2024.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kim YJ, Kim JH, Kim O, Ahn EJ, Oh SJ,
Akanda MR, Oh IJ, Jung S, Kim KK, Lee JH, et al: Caveolin-1
enhances brain metastasis of non-small cell lung cancer,
potentially in association with the epithelial-mesenchymal
transition marker SNAIL. Cancer Cell Int. 19(171)2019.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Kakogiannos N, Ferrari L, Giampietro C,
Scalise AA, Maderna C, Ravà M, Taddei A, Lampugnani MG, Pisati F,
Malinverno M, et al: JAM-A acts via C/EBP-α to promote claudin-5
expression and enhance endothelial barrier function. Circ Res.
127:1056–1073. 2016.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Parton RG and del Pozo MA: Caveolae as
plasma membrane sensors, protectors and organizers. Nat Rev Mol
Cell Biol. 14:98–112. 2013.PubMed/NCBI View
Article : Google Scholar
|
|
29
|
Chen D and Che G: Value of caveolin-1 in
cancer progression and prognosis: Emphasis on cancer-associated
fibroblasts, human cancer cells and mechanism of caveolin-1
expression (Review). Oncol Lett. 8:1409–1421. 2014.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Yamaguchi H, Takeo Y, Yoshida S, Kouchi Z,
Nakamura Y and Fukami K: Lipid rafts and caveolin-1 are required
for invadopodia formation and extracellular matrix degradation by
human breast cancer cells. Cancer Res. 69:8594–8602.
2009.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Li Y, Wu Q, Lv J and Gu J: A comprehensive
pan-cancer analysis of CDH5 in immunological response. Front
Immunol. 14(1239875)2023.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Dejana E, Orsenigo F and Lampugnani MG:
The role of adherens junctions and VE-cadherin in the control of
vascular permeability. J Cell Sci. 121:2115–2122. 2008.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Strilic B and Offermanns S: Intravascular
survival and extravasation of tumor cells. Cancer Cell. 32:282–293.
2017.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Vestweber D: VE-cadherin: The major
endothelial adhesion molecule controlling cellular junctions and
blood vessel formation. Arterioscler Thromb Vasc Biol. 28:223–232.
2008.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Sun Y, Liu JQ, Chen WJ, Tang WF, Zhou YL,
Liu BJ, Wei Y and Dong JC: Astragaloside III inhibits MAPK-mediated
M2 tumor-associated macrophages to suppress the progression of lung
cancer cells via Akt/mTOR signaling pathway. Int Immunopharmacol.
154(114546)2025.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Huang F, Xue F, Wang Q, Huang Y, Wan Z,
Cao X and Zhong L: Transcription factor-target gene regulatory
network analysis in human lung adenocarcinoma. J Thorac Dis.
15:6996–7012. 2023.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Zhang J, Yin Y, Wang B, Chen J, Yang H, Li
T and Chen Y: Discovery of novel small molecules targeting TGF-β
signaling for the treatment of non-small cell lung cancer. Eur J
Med Chem. 289(117442)2025.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Guardavaccaro D and Clevers H:
Wnt/β-catenin and MAPK signaling: Allies and enemies in different
battlefields. Sci Signal. 5(pe15)2012.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Derynck R, Turley SJ and Akhurst RJ: TGFβ
biology in cancer progression and immunotherapy. Nat Rev Clin
Oncol. 18:9–34. 2021.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Zhao Y, Li J, Ting KK, Chen J, Coleman P,
Liu K, Wan L, Moller T, Vadas MA and Gamble JR: The
VE-cadherin/β-catenin signalling axis regulates immune cell
infiltration into tumours. Cancer Lett. 496:1–15. 2021.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Zheng F, Chen Z, Jia W and Zhao R:
Editorial: Community series in the role of angiogenesis and immune
response in tumor microenvironment of solid tumor, volume III.
Front Immunol. 15(1495465)2024.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Wan JCM, Massie C, Garcia-Corbacho J,
Mouliere F, Brenton JD, Caldas C, Pacey S, Baird R and Rosenfeld N:
Liquid biopsies come of age: Towards implementation of circulating
tumour DNA. Nat Rev Cancer. 17:223–238. 2017.PubMed/NCBI View Article : Google Scholar
|