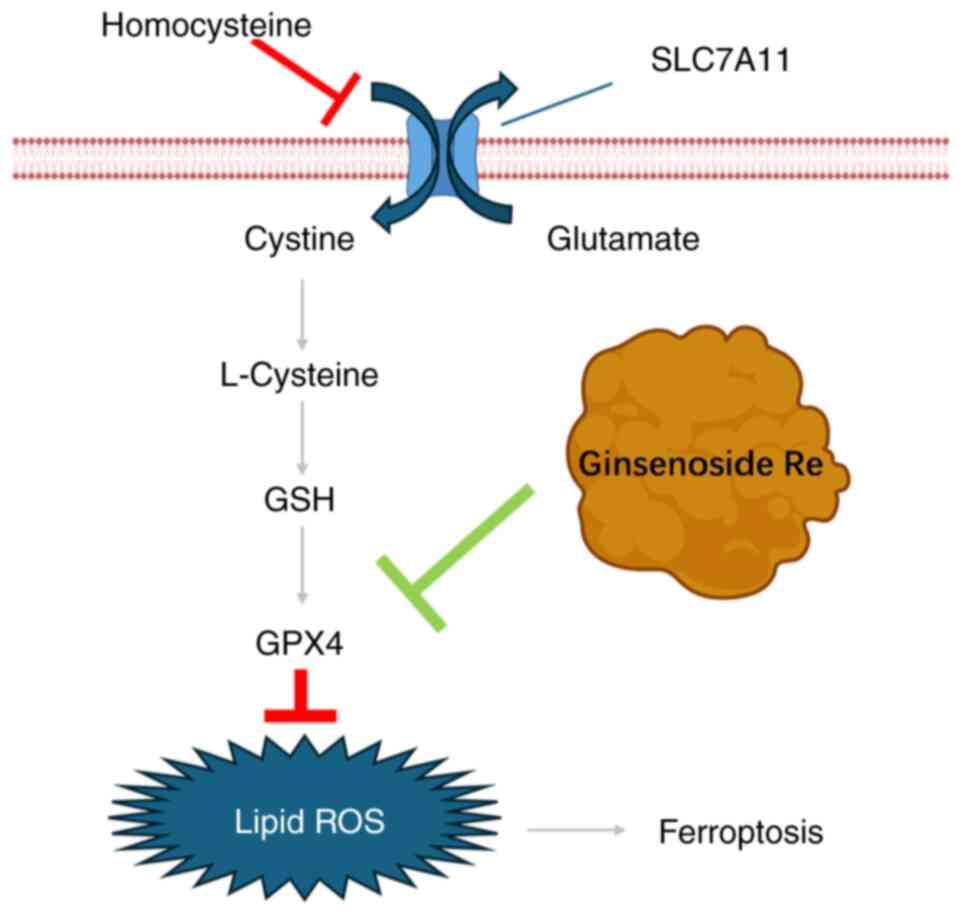
Introduction
Endothelial cell dysfunction serves as a key
foundation for the pathophysiology of atherosclerosis (AS). Normal
endothelial cells regulate the vascular tone, prevent thrombosis
and modulate inflammation, serving key roles in maintaining
vascular homeostasis and inhibiting AS (1). The initiation of AS involves
endothelial apoptosis, ferroptosis and autophagy, among other
programmed cell death mechanisms (2,3).
Ferroptosis, a previously identified form of cell death, is
distinct from apoptosis and autophagy and is characterized by
molecular changes such as glutathione (GSH) depletion or
GSH-peroxidase (GPX) inactivation, increased intracellular free
iron levels and enhanced reactive oxygen species (ROS) generation,
ultimately resulting in the accumulation of toxic lipid peroxides
within cells (4-9).
During ferroptosis, mitochondrial abnormalities such as
condensation or swelling, increased membrane density, decreased or
absent cristae and outer membrane rupture are commonly observed
(10). Previous studies have
indicated a close association between ferroptosis and various
cardiovascular diseases, including AS (11), acute myocardial infarction
(12), ischemia/reperfusion injury
(13), cardiomyopathy and heart
failure (14,15).
GPX4, one of the eight GPX proteins in mammals, is
considered to be the only enzyme within cells capable of directly
reducing phospholipid hydroperoxides. GPX4 converts lipid
hydroperoxides into lipid alcohols, and the inhibition of GPX4
synthesis leads to exacerbated lipid peroxidation and subsequent
ferroptosis (16,17). GSH serves as a key antioxidant in
the body and acts as a co-factor for GPX4, participating in the
reduction of lipid hydroperoxides (18). The depletion of GSH can result in
GPX4 inactivation and increased production of intracellular lipid
peroxides, ultimately triggering ferroptosis. Extracellular cystine
is transported into cells through solute carrier family 7 member 11
(SLC7A11) with intracellular glutamate, which is then converted to
the cysteine required for GSH synthesis. Inhibition of cystine
uptake leads to reduced GSH synthesis, resulting in the
accumulation of lipid peroxidation products and subsequent
ferroptosis (19).
Homocysteine (Hcy) is a sulfur-containing amino
acid, and high levels of homocysteine in the body can be attributed
to factors such as the deficiency of methionine, folic acid,
vitamin B12, B6 and B2 in the diet, and abnormal Hcy metabolic
pathways. High levels of Hcy can lead to AS, hypertension and
congestive heart failure, among other conditions (20,21).
Hcy weakens vascular repair function through mechanisms such as
oxidative stress, damage to the NO system and mitochondrial
destruction, leading to vascular injury (22). A previous study indicated that Hcy
can promote ferroptosis mediated by GPX4 methylation in nucleus
pulposus cells and that folic acid intervention can reduce
ferroptosis-related indicators induced by Hcy (23). However, studies on Hcy-induced
endothelial cell ferroptosis are limited.
Ginsenoside Re, a dietary phytochemical (24), possesses advantages such as easy
accessibility, low cost, efficient and simple purification
techniques as well as low toxicity (25). Notably, ginsenoside Re exhibits
various pharmacological effects, including antidiabetic (26), neuroregulatory (27), anti-infective (28), cardioprotective (29) and antitumor activities (30) and can alleviate the cellular
oxidative stress response (31).
However, studies on whether ginsenoside Re can inhibit ferroptosis
are scarce. The present study aimed to establish a model of
Hcy-induced endothelial cell damage and to investigate whether
ginsenoside Re can suppress Hcy-induced endothelial cell
ferroptosis by upregulating the expression of GPX4, thus providing
a basis for the application of ginsenoside Re in anti-AS therapy
(Fig. 1).
Materials and methods
Cell culture
The EA.hy926 cell line was purchased (cat. no.
CL-0272; Procell®) and cultured in DMEM/F12 (cat. no.
11330032; Gibco; Thermo Fisher Scientific, Inc.) at 37˚C in a
humidified incubator with 5% CO2. All media contained
10% FBS (cat. no. LV-FBSCN500S; Ausgenex Pvt Ltd.), 100 U/ml
penicillin and 100 µg/ml streptomycin (cat. no. C0222; Beyotime
Biotechnology).
Cell toxicity assay
The cytotoxic effect of Hcy (cat. no. H4628;
MilliporeSigma), ferroptosis inducers erastin (cat. no. HY-15763;
MedChemExpress) and ginsenoside Re (cat. no. SG8310; Beijing
Solarbio Science & Technology Co., Ltd.) on EA.hy926 cells was
evaluated using an MTT assay at 37˚C. The cells were seeded at
1x105 cells per well in a 96-well plate and allowed to
adhere overnight. Subsequently, the cells were treated with
different concentrations of Hcy (0-5 mM), erastin (0-20 µM) and
ginsenoside Re (0-200 µM) in the culture medium for 48 h.
Afterwards, 50 ml MTT was added and the resulting formazan crystals
were dissolved in 150 µl DMSO. The absorbance was measured at 570
nm per well with a 96-well plate reader, and cell viability was
expressed as the percentage of untreated controls. Our previous
study demonstrated that exposure to Hcy (0-8 mM) resulted in a
progressive reduction in cell viability from 100 to 30% (32). Consequently, in the present study,
Hcy concentrations of 0-5 mM were selected.
Flow cytometry (FCM) to assess the
cellular levels of lipid ROS (Lip-ROS)
EA.hy926 cells were cultured overnight in a 6-well
culture plate (4x105 cells/well). After discarding the
culture medium from each well, cells were washed three times with
PBS. Subsequently, the cells were treated with Hcy and ginsenoside
Re and divided into the following four groups: The control group
(untreated), the Hcy (2 mM) group, the Hcy (2 mM) + ginsenoside Re
(12 µM) group and the Hcy (2 mM) + ginsenoside Re (24 µM) group.
All groups were cocultured at 37˚C for 24 h. After 24 h of drug
treatment, cell staining was performed. First, the culture medium
was discarded from each well and 1 ml/well sterile PBS solution was
added to remove any residual drugs. Subsequently, the pre-prepared
BODIPY™ 581/591 C11 probe solution (cat. no. D3861;
Thermo Fisher Scientific, Inc.) was diluted to 10 µM and 2 ml
diluted probe solution was slowly added to each well of the 6-well
plate. The plate was covered with aluminum foil and incubated in a
37˚C cell culture incubator for 30 min.
After incubation, the probe solution was aspirated
and the cells were washed 2-3 times with sterile PBS. After
washing, 0.5 ml EDTA-free trypsin was added to digest the cells and
the plate was centrifuged at 415 x g at 37˚C for 5 min. After
centrifugation, the supernatant was removed and 1 ml PBS solution
was added to resuspend the cells. The resuspended cells were
centrifuged at 415 x g at 37˚C for 5 min to remove as much residual
trypsin digestion solution from the cell surface as possible. The
residual solution in the tube was then removed and an appropriate
amount of PBS buffer was added to resuspend the cells. The stained
cells were transferred to a FCM tube and labeled, with all
procedures conducted in the dark. Cell lipid peroxidation levels
were measured using FCM with the BD FACSCalibur™ flow
cytometer (BD Biosciences). A total of 10,000 gated events were
recorded for each sample and analysis was performed using the BD
FACSDiva™ software (version 8.0.1; BD Biosciences).
FCM to assess the intracellular levels
of ROS
Experimental grouping and cell culture methods were
performed as aforementioned. After 24 h of drug treatment, cell
staining was performed. First, the culture medium was removed from
each well and 1 ml/well sterile PBS solution was added to remove
any residual drugs. Subsequently, the pre-prepared
2',7'-dichlorodihydrofluorescein diacetate probe solution (cat. no.
S0033S; Beyotime Institute of Biotechnology) was diluted to 10 µM
and 2 ml diluted probe solution was slowly added to each well of
the 6-well plate. The plate was covered with aluminum foil and
incubate in a 37˚C cell incubator for 30 min. Stained cells were
then transferred to FCM tubes and labeled, with all procedures
conducted in the dark. FCM was performed using the BD FACSCalibur
flow cytometer to measure cellular ROS levels. A total of 10,000
gated events were recorded for each sample and analysis was
performed using the BD FACSDiva™ software (version
8.0.1).
Fluorescence detection of the levels
of cellular Lip-ROS
After 24 h of drug treatment as aforementioned, cell
staining was conducted. The initial steps of cell staining were the
same as those for ROS detection. Subsequently, the cells were
stained with Antifade Mounting Medium with DAPI (cat. no. P0131;
Beyotime Institute of Biotechnology). Images were captured using a
fluorescence microscope (x10 magnification; Olympus Corporation) in
the dark. Quantitative analysis was performed using ImageJ software
(version 1.54; National Institutes of Health).
Determination of intracellular total
iron ion content
EA.hy926 cells were cultured overnight in 6-well
plates (4x105 cells/well) and divided into four
treatment groups as aforementioned, namely the control group, the
Hcy (2 mM) group, the Hcy (2 mM) + ginsenoside Re (12 µM) group and
the Hcy (2 mM) + ginsenoside Re (24 µM) group, with each group
incubated for 24 h. After treatment, cells were collected and
washed twice with cold sterile PBS, followed by low-speed
centrifugation (37˚C, 415 x g, 5 min). PBS was then aspirated and
100-200 µl lysis buffer in the Total Iron Content Colorimetric
Assay Kit (cat. no. E1042; Applygen Technologies, Inc.) was added.
The cells were vigorously shaken or vortexed for 20-30 sec, then
lysis was conducted at 4˚C for 2 h, before centrifugation at 4˚C at
12,000 x g for 5 min to collect the supernatant for subsequent
determination of iron ion concentration and protein concentration
was measured using a BCA assay. Preparation of the standard
solution was then performed. A 3 mM standard solution was diluted
with the dilution solution provided in the Total Iron Content
Colorimetric Assay Kit to concentrations of 300, 150, 75, 37.5,
18.75, 9.38 and 4.69 µM. A mixture of reagent 2 and 4.5% potassium
permanganate solution was prepared in a 1:1 ratio to form solution
A. The blank control group, standard group and sample group were
set up and mixed with solution A before being incubated in a water
bath at 60˚C for 1 h. After the tubes cooled to room temperature,
the liquid remaining on the wall and cap of the tubes was
centrifuged at low speed (37˚C, 415 x g, 5 min) to the bottom of
the tubes. Subsequently, 30 µl iron ion detection reagent, included
in the aforementioned kit, was added and the mixture was incubated
at room temperature for 30 min. After centrifugation at 4˚C at
12,000 x g for 5 min, the supernatant was collected. Finally, 200
µl sample was added to each well of a 96-well plate and the
absorbance of the samples was measured at 550 nm using a microplate
reader. The relative iron ion levels were checked in the cells.
Determination of the intracellular GSH
concentration
The cells were ultrasonically crushed using PBS as
the homogenizing medium and then centrifuged to take the
supernatant for determination. A total of 0.1 ml cell supernatant
was removed, and then 0.1 ml reagent 1 (precipitating agent) was
added to the tube and thorough mixing was performed. The mixture
was centrifuged at 37˚C at 205 x g for 10 min and the supernatant
was collected for measurement. After the control group, Hcy group,
low-dose ginsenoside Re + Hcy group and high-dose ginsenoside Re +
Hcy group were treated with reagent 1, 100 µl reagent 2 (buffer
solution) and 25 µl reagent 3 (color developer) were added to the
supernatant, mixed and allowed to stand for 5 min. The absorbance
of the samples was measured at 405 nm using a microplate reader.
GSH standard solutions were prepared by diluting a GSH standard
stock solution to concentrations of 100, 50, 20, 10, 5 and 0 µM.
All reagents used were included in the GSH Assay Kit (cat. no.
A006-2-1; Nanjing Jiancheng Bioengineering Institute). The GSH
content was analyzed based on the measured absorbance values using
a standard curve. Based on the absorbance measurement value, the
relative GSH level was checked in the cells.
Determination of the intracellular
malondialdehyde (MDA) content
To measure MDA content using an MDA kit (cat. no.
A003-4-1; Nanjing Jiancheng Bioengineering Institute), 100 µl
anhydrous ethanol, standard solution and test samples were mixed
separately with 1,000 µl working solution to prepare control tubes,
standard tubes and sample tubes, respectively. After thorough
mixing, the tubes were heated in a water bath at >95˚C for 40
min. The solutions were then removed from the water bath and cooled
under running water, before being centrifuged at 37˚C at 268 x g
for 10 min. The optical density (OD) of the blank plate was
measured at 530 nm using a microplate reader. Subsequently, 0.25 ml
of each sample was transferred to a new 96-well plate and the OD
value of each well was measured using a microplate reader (the OD
value of the blank plate was subtracted from the sample OD value).
The results were normalized to the percentage of the control
group.
Molecular docking
Molecular docking evaluation of ginsenoside Re
combined with different proteins was performed. Briefly, the 3D
molecular structure of ginsenoside Re was retrieved from the
PubChem database (https://pubchem.ncbi.nlm.nih.gov/). In addition, the
X-ray crystal structures of the GPX4, SLC7A11 and acyl-CoA
synthetase long-chain family member 4 (ACSL4) proteins were
obtained from Protein Data Bank (http://www.rcsb.org/). For protein preparation, PyMOL
(https://github.com/schrodinger/pymol-open-source)
(version 2.5.2) was used to remove water molecules and heteroatoms
and the proteins were saved in ‘pdb format’. AutoDock (https://autodock.scripps.edu/download-autodock4/)
(version 4.2.6) and PyMOL software were used to perform docking
studies between ginsenoside Re and GPX4, SLC7A11 and ACSL4
proteins. The grid-box function of AutoDock Tools was used to
define specific pockets of active ingredients for protein-protein
interaction binding to proteins. Subsequently, molecular docking
analysis was performed using the command prompt and the results
were displayed using PyMOL. The default settings of the software
were used.
Western blotting
Cells were seeded on a 6-well plate at
2.5x105 cells/well and allowed to adhere overnight.
Subsequently, the cells were incubated with Hcy (2 mM), Hcy (2 mM)
+ ginsenoside Re (12 µM) or Hcy (2 mM) + ginsenoside Re (24 µM) for
24 h, after which, the medium in the 6-well plate was discarded.
Precooled PBS was then added to rinse the cells twice. A prepared
protein lysis solution RIPA (cat. no. P0038; Beyotime Institute of
Biotechnology) + PMSF (cat. no. ST507; Beyotime Institute of
Biotechnology) (RIPA:PMSF, 100:1) was added to lyse the cells for
30 min at 4˚C (shaking the 6-well plate every 10 min), the cells
were quickly collected with a cell scraper and centrifuged using a
high-speed refrigerated centrifuge at 950 x g for 15 min at 4˚C.
The supernatant was aspirated and a BCA protein assay kit was used
to determine the total protein concentration. Subsequently, western
blotting of the lysates was performed. Equal amounts of protein (20
µg per lane) were electrophoresed on 10% SDS-polyacrylamide gels
and transferred onto 0.45 µm PVDF membranes. The membranes were
then blocked with 5% skimmed milk for 1 h at room temperature,
followed by an overnight incubation with the primary antibody at
4˚C. The primary antibodies used included anti-GPX4 (1;2,000; cat.
no. ab125066; Abcam), anti- SLC7A11 (1;2,000; cat. no. ab300667;
Abcam), anti-ACSL4 (1;2,000; cat. no. ab155282; Abcam) and
anti-GAPDH (1:2,000; cat. no. ab8245; Abcam). Following washes with
TBST buffer (0.05% Tween-20), the membranes were incubated with
horseradish peroxidase-conjugated secondary antibodies (1;5,000;
cat. no. XD345904; Thermo Fisher Scientific, Inc.) at 37˚C for 60
min. The immunoreactive bands were then detected using an Odyssey
CLX imaging system (LI-COR, Inc.), and Image Studio™
software (LI-COR, Inc.) was used to measure the OD of the
bands.
Statistical analysis
All data are presented as the mean ± SD of at least
three independent experiments, and the data were analyzed with
GraphPad Prism (version 5.0; GraphPad; Dotmatics). For comparisons
involving three or more groups, a one-way ANOVA was conducted,
followed by Tukey's post-hoc test to assess significance. P<0.05
was considered to indicate a statistically significant
difference.
Results
Hcy may induce ferroptosis in EA.hy926
endothelial cells
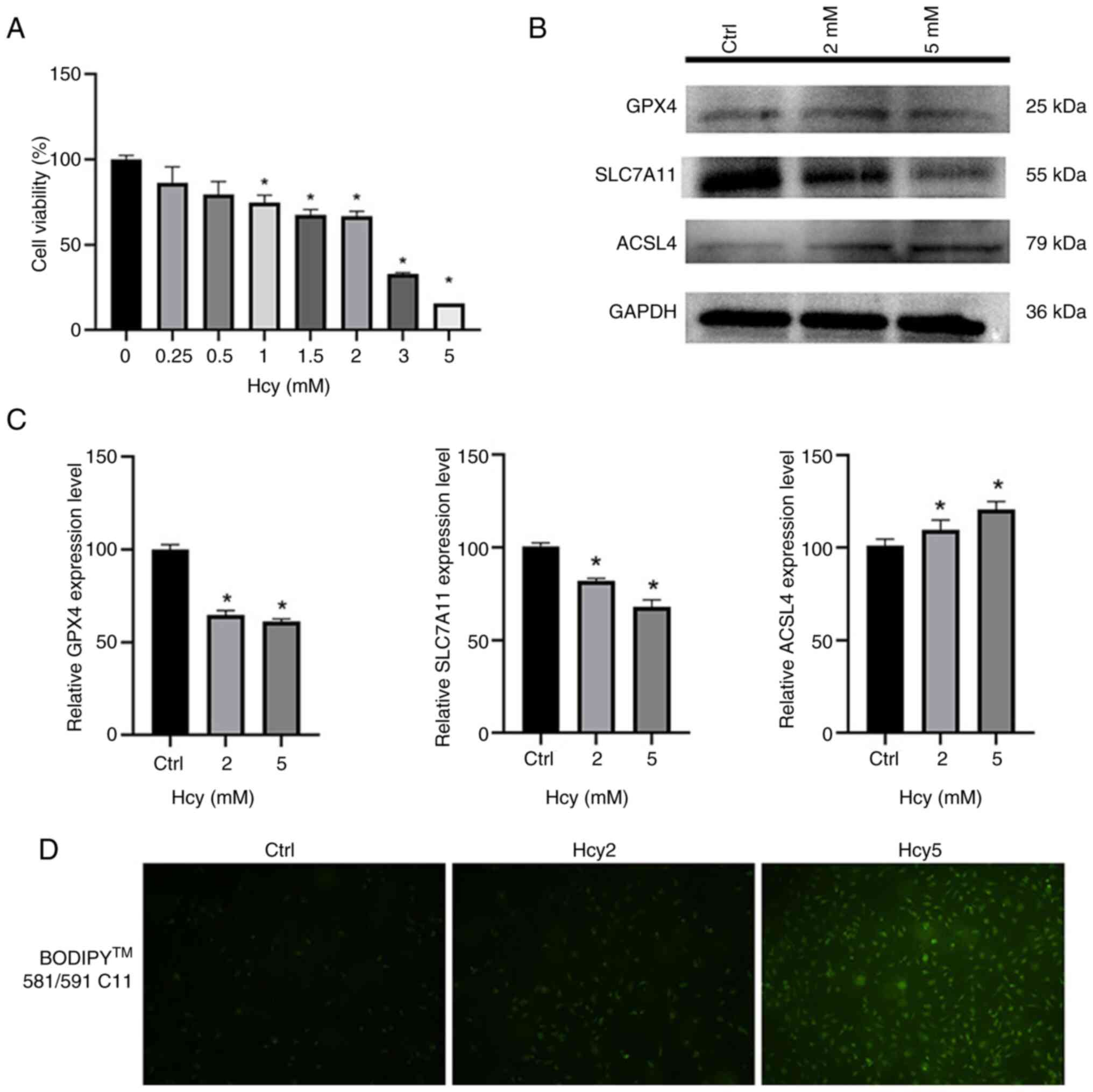
EA.hy926 cells were seeded into a 96-well plate and
treated with different concentrations of Hcy for 24 h. Results
indicated a gradual decrease in cell viability with increasing
concentrations of Hcy (Fig. 2A).
Subsequently, HCY were selected with a cell viability close to the
IC50 at a concentration of 2 mmol (cell viability, 60%)
and the lowest cell viability at 5 mmol (cell viability, 20%) for
the subsequent western blotting experiment. Western blotting was
conducted on cells treated with 2 or 5 mM Hcy for 24 h to detect
expression of the ferroptosis-related proteins GPX4, SLC7A11 and
ACSL4 (Fig. 2B) (32). Results demonstrated that the
expression levels of GPX4 and SLC7A11 decreased with increasing Hcy
concentration, while the expression levels of ACSL4 increased with
increasing Hcy concentration, potentially indicating an increase in
ferroptosis in EA.hy926 cells in response to increasing Hcy
concentration (Fig. 2C).
Furthermore, EA.hy926 cells treated with 2 or 5 mM Hcy for 24 h
were stained with the diluted BODIPY 581/591 C11 probe for lipid
peroxidation detection through inverted fluorescence microscopy
(Fig. 2D). With increasing Hcy
concentration, the intensity of green fluorescence within the cell
markedly increased, further suggesting that Hcy induces ferroptosis
in EA.hy926 cells.
Ginsenoside Re may mitigate
erastin-induced ferroptosis in endothelial cells
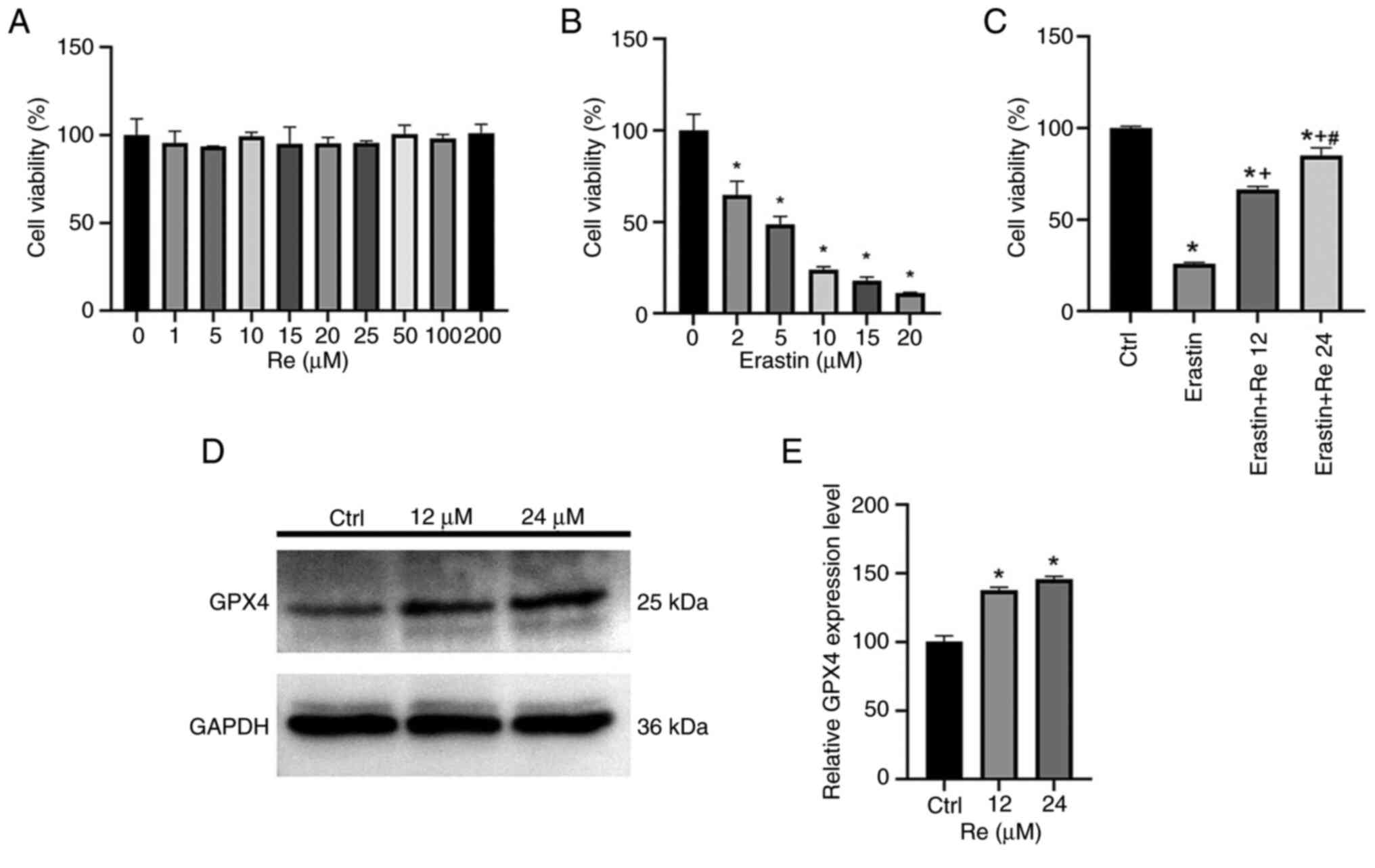
To validate the impact of ginsenoside Re on cell
viability, different concentrations of ginsenoside Re were applied
to EA.hy926 cells for 24 h (Fig.
3A). Results indicated that ginsenoside Re had no significant
effect on EA.hy926 cell viability. Erastin, an activator of
ferroptosis, was used at various concentrations to treat EA.hy926
cells for 24 h, resulting in decreased cell viability with
increasing concentrations of erastin (Fig. 3B). Erastin at a drug concentration
of 5 µM with a cell viability of 50% was selected for subsequent
experiments. For the convenience of concentration calculation,
cells were treated with 12 µM or 24 µM of ginsenoside Re (Fig. 3C), with increasing concentrations
of ginsenoside Re notably improving cell viability. These findings
suggested that ginsenoside Re alleviated erastin-induced
endothelial cell ferroptosis in a dose-dependent manner. After
treatment with 12 or 24 µM ginsenoside Re for 24 h, western
blotting was performed (Fig. 3D
and E) and the results showed that
the expression level of GPX4 increased after the addition of
ginsenoside Re.
Ginsenoside Re may alleviate
Hcy-induced endothelial cell ferroptosis
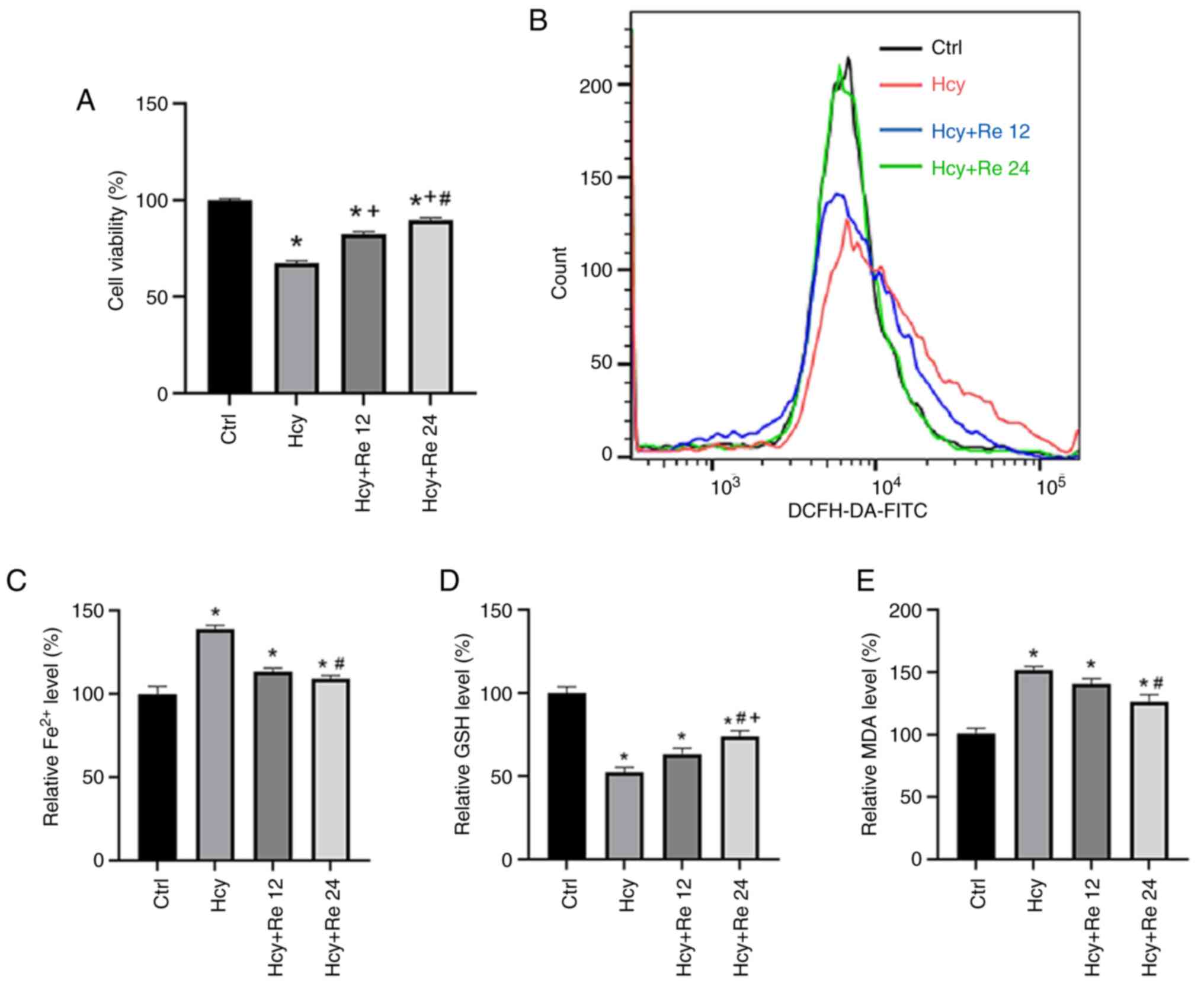
To validate the impact of ginsenoside Re on the cell
damage caused by Hcy, EA.hy926 cells were treated with 2 mM Hcy and
with 12 or 24 µM ginsenoside Re. Results demonstrated that the
addition of ginsenoside Re could mitigate the cell damage caused by
Hcy, as the reduction in cell viability caused by Hcy was reversed
with increasing concentrations of ginsenoside Re (Fig. 4A). Intracellular ROS levels were
measured using FCM, which revealed an increase in ROS levels in the
Hcy-treated groups. Ginsenoside Re markedly reduced the increase in
ROS concentration induced by Hcy (Fig.
4B). Using a microplate reader, intracellular iron ion, GSH and
MDA levels were measured. Results revealed that Hcy could increase
the levels of Fe2+ and MDA, while decreasing GSH levels
in cells compared with those in the control group. By contrast,
ginsenoside Re decreased the levels of Fe2+ and MDA,
while increasing GSH levels compared with those in Hcy-treated
cells (Fig. 4C-E). These findings
suggested that ginsenoside Re alleviates Hcy-induced endothelial
cell ferroptosis.
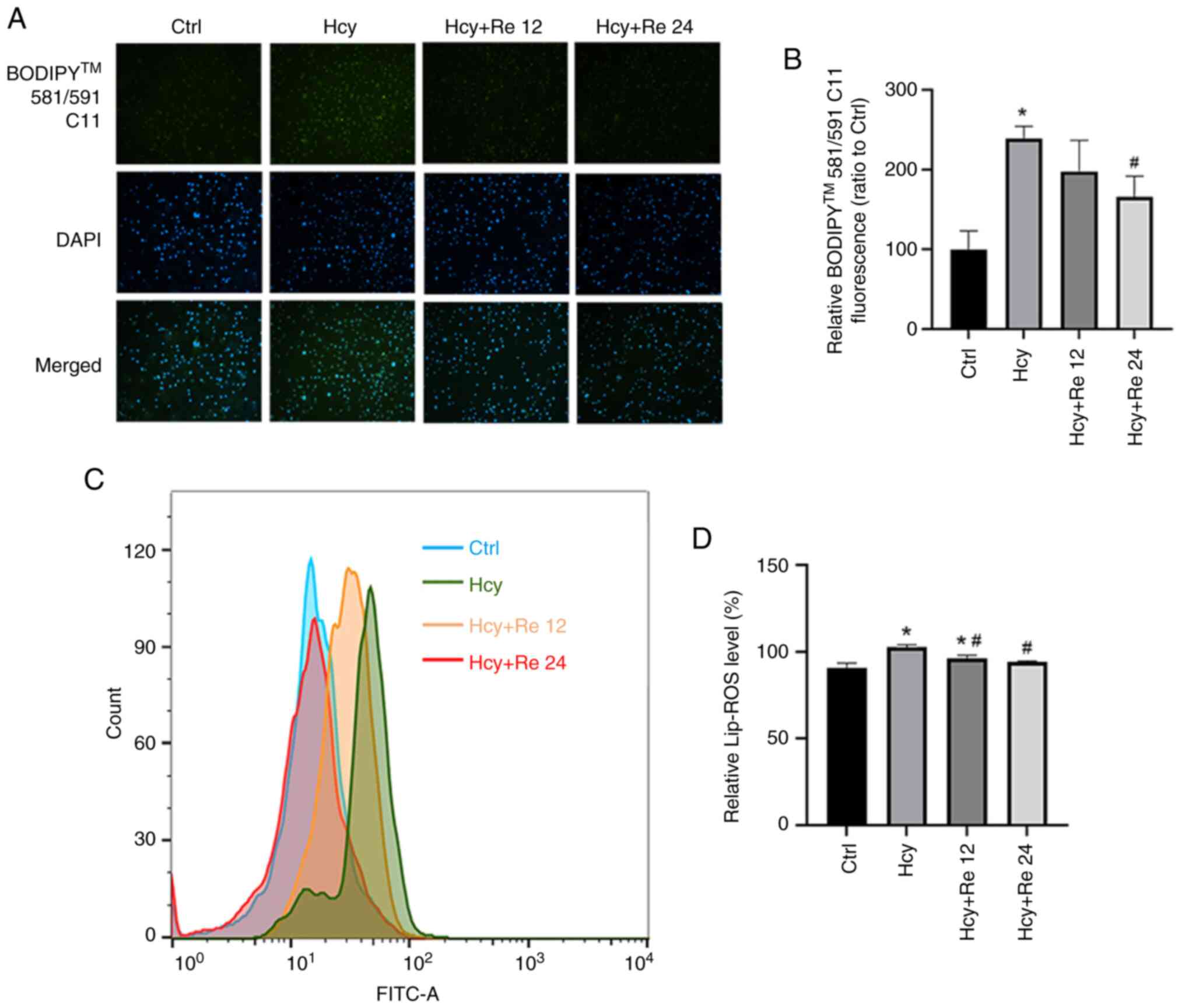
Effect of ginsenoside Re on Lip-ROS
levels in EA.hy926 cells
After EA.hy926 cells were treated with 2 mM Hcy and
either 12 or 24 µM ginsenoside Re, the BODIPY 581/591 C11 probe was
added for staining. Fluorescence microscopy and FCM were used to
measure the level of lipid peroxidation in cells. After Hcy
treatment, the fluorescence intensity of the cells increased,
whereas the fluorescence intensity decreased in the groups treated
with ginsenoside Re compared with that treated with Hcy alone.
These results indicate that Hcy increased the Lip-ROS level in the
cells, while a low dose of ginsenoside Re had no significant effect
on the Lip-ROS level in Hcy group cells. High-dose ginsenoside Re
reduced Lip-ROS levels (Fig. 5A
and B). Subsequently, it was shown
through flow cytometry that the Hcy group could increase the
intracellular Lip-ROS level, and the low-dose ginsenoside Re
(12) intervention could reduce
the reactive oxygen species level, but the rate was still higher
than that of the control group. The high-dose ginsenoside re group
(24) could reduce Lip-ROS levels
to normal levels (Fig. 5C and
D).
Association between ginsenoside Re and
the GPX4, SLC7A11 and ACSL4 antioxidant axis
To elucidate the mechanism by which ginsenoside Re
reduces ferroptosis in EA.hy926 cells, the effect of ginsenoside Re
on the GPX4, SLC7A11 and ACSL4 antioxidant axis in ferroptosis was
evaluated. Previous studies have reported that protein binding to
ligands is influenced by binding energies, with binding energies
<-5.0 kcal/mol indicating good binding and those <-7.0
kcal/mol indicating very firm binding (33,34).
Docking results showed that the binding energies of ginsenoside Re
with GPX4, SLC7A11 and ACSL4 were all <-7.0 kcal/mol (Table I). To more intuitively reflect the
combination of active ingredients and key targets, PyMOL was used
to visualize the results, revealing amino acid residues and the
combination of hydrogen bonding and active compounds. Therefore,
active components that bind strongly to target proteins were
selected for visualization (Fig.
6). The results revealed that ginsenoside Re binds to GPX4 on
amino acid residues methionine (MET)-102 and lysine (LYS)-99
(Fig. 6A), to ACSL4 on amino acid
residues aspartate (ASP)-66, arginine (ARG)-60, glycine (GLY)-523,
glutamine (GLN)-524 and histidine (HIS)-291 (Fig. 6B) and to SLC7A11 on amino acid
residues GLN-438, ASP-350 and ARG-440 (Fig. 6C). These findings suggested a
direct association between ginsenoside Re and the GPX4, SLC7A11 and
ACSL4 antioxidant axis.
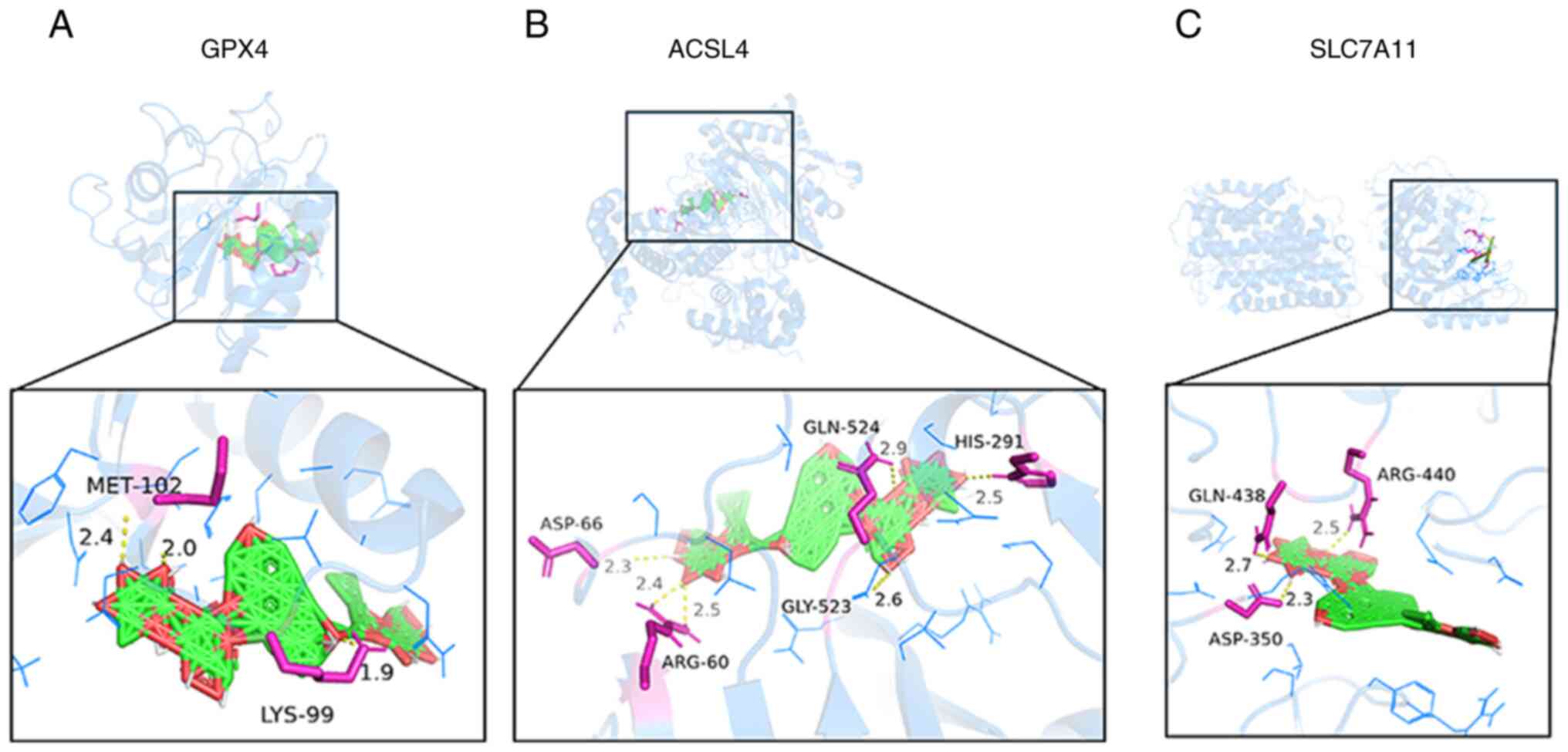 | Figure 6Optimal docking map of ginsenoside Re
and key gene molecules. Ginsenoside Re can bind to (A) GPX4, (B)
ACSL4 and (C) SLC7A11. The yellow dotted line represents hydrogen
bonds, and the number beside the dotted line indicates the length
of the hydrogen bond in Ångströms. GPX4, glutathione peroxidase 4;
SLC7A11; solute carrier family 7 member 11; ACSL4, acyl-CoA
synthetase long-chain family member 4; MET, methionine; LYS,
lysine; ASP, aspartate; ARG, arginine; GLY, glycine; GLN,
glutamine; HIS, histidine. |
 | Table IDocking analysis between ginsenoside
Re and target proteins. |
Table I
Docking analysis between ginsenoside
Re and target proteins.
| Compound | Key target | Protein Data Bank
ID | Binding energy
(kcal/mol) |
|---|
| Ginsenoside Re | GPX4 | 6HN3 | -12.8 |
| | SLC7A11 | 7EPZ | -13.1 |
| | ACSL4 | AF_AFO60488F1 | -12.8 |
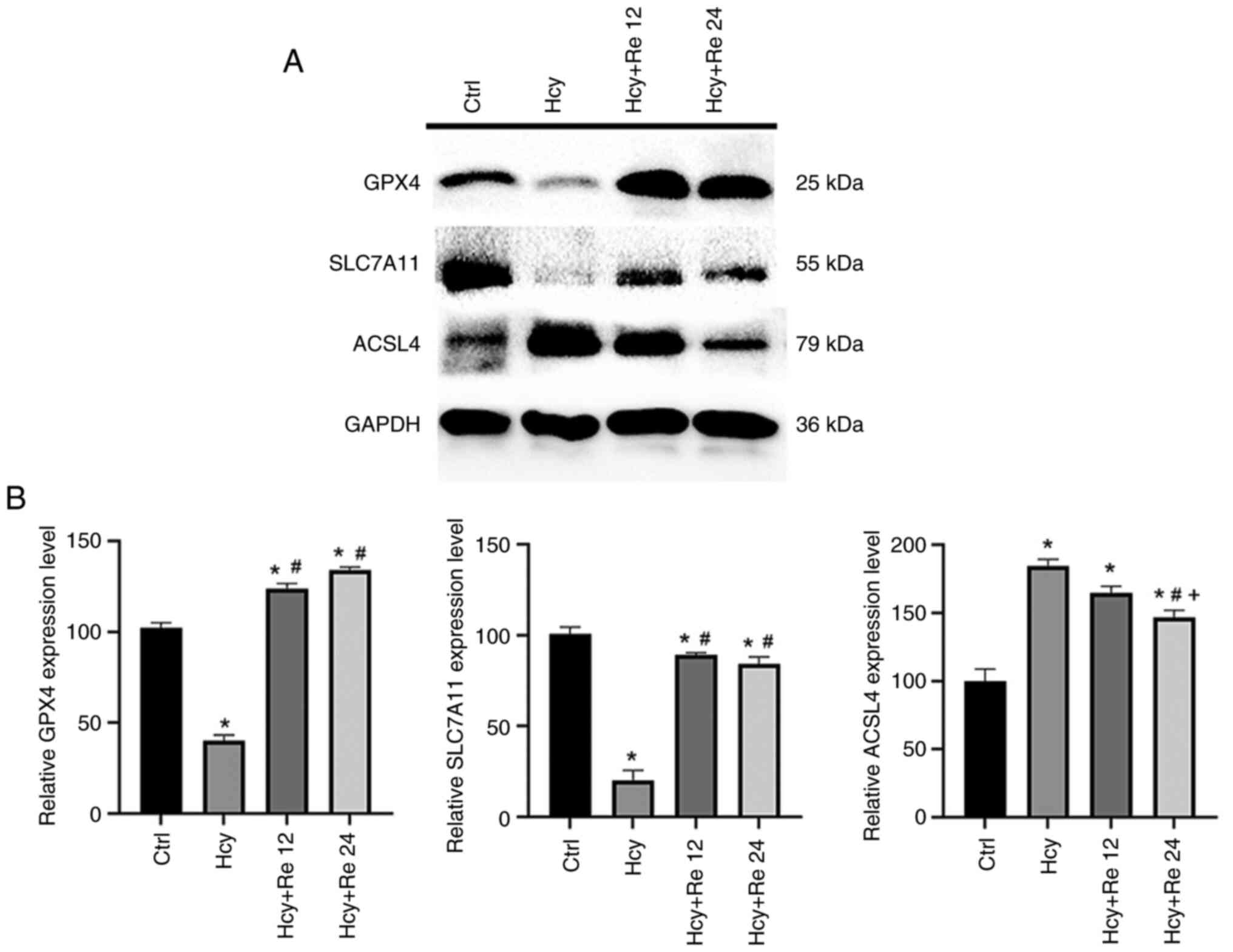
Impact of ginsenoside Re on the
expression of ferroptosis-related proteins induced by Hcy in
EA.hy926 cells
The present study indicated that after Hcy treatment
of cells, the expression levels of GPX4 and SLC7A11 proteins
decreased. After treating EA.hy926 cells with 12 and 24 µM
ginsenoside Re, it was found that the expression levels of GPX4 and
SLC7A11 increased compared with the Hcy alone group, indicating
that ginsenoside Re treatment could significantly increase the
expression level of ferroptosis protein induced by Hcy. The
ferroptosis protein marker ACSL4 was significantly increased in the
Hcy group, but significantly decreased in the ginsenoside Re
groups, thus suggesting that ginsenoside Re may alleviate
Hcy-induced endothelial cell ferroptosis (Fig. 7A and B). The present study demonstrated that
ginsenoside Re significantly increased the expression levels of
GPX4 in Hcy-treated EA.hy926 cells and therefore may mitigate
Hcy-induced ferroptosis in EA.hy926 cells. The results of this
study have clinical application value for the treatment of AS
caused by endothelial cell injury resulting from ferroptosis.
Discussion
Ferroptosis serves a key role in the progression of
AS, participating in various pathological processes, such as
endothelial cell damage, macrophage inflammation, foam cell
formation and vascular smooth muscle cell proliferation and
migration (35-38),
making ferroptosis a novel target for AS research. Therefore,
exploring the relevant mechanisms of endothelial cell ferroptosis
and identifying effective intervention methods are key in the
treatment of AS.
Hcy is an independent risk factor for cardiovascular
disease. Increased levels of Hcy can damage endothelial cells,
leading to local infiltration of lipids and inflammatory cells and
the formation of foam cells and lipid streaks, which gradually
progress to atherosclerotic plaques (20,39).
In recent years, a number of studies have demonstrated the
involvement of ferroptosis in cell damage caused by Hcy; for
example, a study found that the protective properties of Fer-1
against KGN cell Hcy-induced injury were mediated by TET activity
and DNA demethylation (40). Wang
et al (41) reported that
Hcy may promote pulmonary microvascular endothelial cell
ferroptosis by upregulating ACSL4 and downregulating GPX4 and
ferroptosis suppressor protein 1 expression. The present study
showed that the expression levels of GPX4 and SLC7A11 were lower in
Hcy-treated EA.hy926 cells than those in the control group, while
the expression levels of ACSL4 and the levels of Lip-ROS were
greater than those in the control group. These results indicated
that Hcy could decrease the expression of GPX4 and SLC7A11,
potentially leading to increased Lip-ROS generation and therefore
endothelial cell ferroptosis.
It has been shown that ginsenoside Re can upregulate
the expression of GPX4 in cells through a process mediated by
phosphoinositide 3-kinase and extracellular signal-regulated
kinase, thereby alleviating oxidative stress-induced neuronal
damage (31). In a previous study,
10-50 µM ginsenoside Re alone was applied to SH-SY5Y cells for 9 h,
which markedly increased the expression levels of GPX4 in a
concentration-dependent manner (31). The present study showed that within
a certain concentration range (0-200 µM), ginsenoside Re had no
significant effect on cell viability. After treating EA.hy926 cells
with ginsenoside Re at concentrations of 12 and 24 µM for 24 h, the
expression levels of GPX4 were increased compared with those in the
control group. Ye et al (42) pretreated rats with 150 mg/kg
ginsenoside Re for 5 days, after which a myocardial
ischemia/reperfusion injury model was generated. This previous
study revealed that ginsenoside Re alleviated myocardial cell
ferroptosis induced by ischemia/reperfusion injury through microRNA
(miR)-144-3p and SLC7A11. These findings suggest that ginsenoside
Re may increase the expression levels of GPX4 and exert a
protective effect on ferroptosis in various cell types at
appropriate concentrations and treatment durations.
Since the discovery of ferroptosis, there has been a
focus on its two major regulatory mechanisms, namely the
disturbance in iron metabolism and lipid peroxidation. The Fenton
reaction refers to the process where Fe2+ reacts with
hydrogen peroxide to generate Fe3+ and oxygen radicals.
Excessive iron within cells can generate oxygen radicals and ROS
through the iron-dependent Fenton reaction, activating
iron-containing enzymes such as lipoxygenase, and leading to
oxidative stress and lipid peroxidation, thereby triggering
ferroptosis (43). In 2012, Dixon
et al (10) stimulated
HT1080 cells with erastin, which resulted in a marked increase in
intracellular iron ion and Lip-ROS levels. Notably, intervention
with iron chelators and lipid peroxidation inhibitors markedly
improved cell viability. Chen et al (44) established a ferroptosis model in
H9C2 cells by stimulating them with artemisinin for 24 h.
Fluorescence microscopy revealed marked increases in intracellular
ROS and Fe2+ levels as well as lipid peroxidation. By
contrast, SLC7A11 overexpression or miR-16-5p knockdown inhibits
ferroptosis and exhibits cardioprotective effects in vitro
and in vivo. The present study revealed that Hcy can lead to
a significant increase in the levels of ferroptosis related factors
Fe2+, MDA and ROS. However, after the intervention with
ginsenoside Re, the levels of Fe2+, ROS, Lip-ROS and MDA
were all lower than those in the Hcy group. These findings
suggested that ginsenoside Re treatment potentially reduced the
occurrence of the Fenton reaction, decreased cellular lipid
peroxidation levels and thereby alleviated Hcy-induced endothelial
cell ferroptosis.
Cystine/glutamate antiporter (system
xc-)/GPX4 is an important antioxidant system and one of
the central mechanisms underlying ferroptosis (45). System xc- consists of a
light chain subunit (xCT, also known as SLC7A11) and a heavy chain
(hc) subunit (CD98hc, also known as solute carrier family 3 member
2) located on the cell membrane (46). Extracellular cystine is absorbed
into cells in equimolar amounts and rapidly reduced to cysteine,
participating in the synthesis of the important intracellular free
radical scavenger GSH (47). GSH
is an essential antioxidant in cells that is synthesized by
cysteine synthase and GSH synthetase and is also a key
neurotransmitter and endogenous antioxidant in the body (48). System xc- participates
in the biosynthesis of GSH, and GSH, as an important cofactor of
GPX4, reduces intracellular oxygen levels. When system
xc- is inhibited by certain compounds (such as erastin),
GSH synthesis is decreased, leading to the inability of GPX4 to use
GSH to reduce lipid peroxides, resulting in ferroptosis. To
evaluate the extent to which ginsenoside Re binds
ferroptosis-related proteins, the present study performed molecular
docking experiments with GPX4, ACSL4, SLC7A11 and ginsenoside Re.
Results showed that ginsenoside Re tightly bound to the three
target proteins. Notably, the binding energies of ginsenoside Re to
GPX4, ACSL4 and SLC7A11 were all <-7.0 kcal/mol, indicating a
strong binding affinity. Molecular docking analysis of ginsenoside
Re with the ferroptosis-related proteins GPX4, ACSL4 and SLC7A11
revealed key interactions, suggesting their potential therapeutic
relevance. The interaction between ginsenoside Re and GPX4 at the
MET-102 and LYS-99 residues is particularly notable, as this
binding may stabilize GPX4, thereby enhancing its antioxidant
capacity and reducing iron-dependent lipid peroxidation (49). Similarly, the binding of
ginsenoside Re to ACSL4 at the residues ASP-66, ARG-60, GLY-523,
GLN-524 and HIS-291 suggests that ginsenoside Re may interfere with
ACSL4 function and inhibit its ferroptosis-promoting activity
(50). Moreover, the interaction
between ginsenoside Re and SLC7A11 at residues GLN-438, ASP-350 and
ARG-440 may enhance the stability of SLC7A11, which is a key
component of the system xc- pathway and serves a central
role in regulating ferroptosis (51). This hypothesis was verified by
western blotting and cellular GSH assays. GPX4 and SLC7A11
expression levels and intracellular GSH levels were significantly
decreased in the Hcy group. However, when compared with those in
the Hcy group, GPX4 and SLC7A11 expression levels and intracellular
GSH levels were significantly increased in the ginsenoside Re
intervention groups. The present study suggested that Hcy impaired
system xc-function and reduced GSH synthesis by decreasing SLC7A11
expression. By contrast, ginsenoside Re upregulated the expression
of GPX4, increased the synthesis of GSH and reduced
ferroptosis.
Ginsenoside Re has been shown to alleviate the
inhibitory effect of miR-144-3p on SLC7A11 expression, resulting in
the upregulation of SLC7A11. It was further confirmed that the
solute carrier family 7 member 11 (SLC7A11) was the target gene of
miR-144-3p by database analysis and western blotting (42). Specifically, miR-144-3p targets
SLC7A11 mRNA, suppressing its translation and stability. Through
modulation of miR-144-3p, ginsenoside Re indirectly enhances
SLC7A11 expression, promoting cystine uptake and GSH synthesis
(42). This cascade inhibits lipid
peroxidation associated with ferroptosis. Elevated SLC7A11 levels
increase GSH concentrations, which are key for the function of GPX4
as an antioxidant. Consequently, ginsenoside Re may indirectly
augment GPX4 activity through SLC7A11-dependent mechanisms
(42).
The pharmacokinetics and bioavailability of
ginsenoside Re are the key limiting factors to its clinical
application. The oral bioavailability of ginsenoside Re is
generally low, mainly due to its macromolecular structure and
hydrophilic properties, which lead to intestinal malabsorption,
rapid first-pass metabolism and enzyme-mediated degradation
(52). A study showed that the use
of ginsenoside Re complexes with polysaccharide protects
ginsenoside Re from degradation through hydrogen bonding and
regulates the expression of intestinal efflux proteins and tight
junction proteins, thereby enhancing absorption and bioavailability
(53). In animal experiments,
ginsenoside Re has demonstrated rapid absorption and a relatively
long elimination half-life. For example, after oral administration
of red ginseng extract, ginsenoside Re has been detected as a
20(S)-protopanaxatriol saponin in mouse plasma. The high plasma
protein binding and low hepatic distribution contribute to the
prolonged plasma circulation time observed with highly glycosylated
ginsenoside structures such as ginsenoside Re, which exhibits high
plasma exposure; however, tetra- or tri-glycosylated saponins
demonstrate greater accumulation susceptibility than ginsenoside
Re, highlighting its metabolic uniqueness (54).
In conclusion, the present study indicated that
ginsenoside Re may improve Hcy-induced ferroptosis in EA.hy926
cells by upregulating the expression of GPX4. Nevertheless, this
established cell model may not fully replicate the dynamic
functional adaptations characteristic of native endothelial cells
within atherosclerotic microenvironments. Future in vivo
studies are warranted to elucidate the therapeutic efficacy of
ginsenoside Re in inhibiting endothelial ferroptosis.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Clinical
Research Fund project of Guangdong Medical Association (grant no.
A202302018), the Medical Science and Technology Research Foundation
of Guangdong Province (grant no. B2025385), the
‘Hundred-Thousand-Million Project’ for Science and Technology
Support in 2025 (grant no. 250623211606897) and the Social
Development Science and Technology Plan Project of Heyuan City
(grant no. 250609171604668).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
ShL, CZ, SY, SiL and SZ contributed to the
conception and design of the present study. SL, CZ and SL wrote the
manuscript and collected and analyzed the data. SL and SZ
critically revised the final manuscript. KY and SY interpreted the
data, and SL was responsible for capturing and organizing the
images. SL, CZ, SY, KY, SZ and SL confirm the authenticity of all
the raw data. All authors read and approved the final version of
the manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Duan H, Zhang Q, Liu J, Li R, Wang D, Peng
W and Wu C: Suppression of apoptosis in vascular endothelial cell,
the promising way for natural medicines to treat atherosclerosis.
Pharmacol Res. 168(105599)2021.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Qin X, Zhang J, Wang B, Xu G, Yang X, Zou
Z and Yu C: Ferritinophagy is involved in the zinc oxide
nanoparticles-induced ferroptosis of vascular endothelial cells.
Autophagy. 17:4266–4285. 2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Yang Z, Shi J, Chen L, Fu C, Shi D and Qu
H: Role of pyroptosis and ferroptosis in the progression of
atherosclerotic plaques. Front Cell Dev Biol.
10(811196)2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Carlson BA, Tobe R, Yefremova E, Tsuji PA,
Hoffmann VJ, Schweizer U, Gladyshev VN, Hatfield DL and Conrad M:
Glutathione peroxidase 4 and vitamin E cooperatively prevent
hepatocellular degeneration. Redox Biol. 9:22–31. 2016.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Imai H and Nakagawa Y: Biological
significance of phospholipid hydroperoxide glutathione peroxidase
(PHGPx, GPx4) in mammalian cells. Free Radic Biol Med. 34:145–169.
2003.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Tang D, Chen X, Kang R and Kroemer G:
Ferroptosis: Molecular mechanisms and health implications. Cell
Res. 31:107–125. 2021.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Illés E, Patra SG, Marks V, Mizrahi A and
Meyerstein D: The Fe(II)(citrate) Fenton reaction under
physiological conditions. J Inorg Biochem.
206(111018)2020.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Kist M and Vucic D: Cell death pathways:
Intricate connections and disease implications. EMBO J.
40(e106700)2021.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Kagan VE, Mao G, Qu F, Angeli JP, Doll S,
Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al: Oxidized
arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem
Biol. 13:81–90. 2017.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta
R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS,
et al: Ferroptosis: An iron-dependent form of nonapoptotic cell
death. Cell. 149:1060–1072. 2012.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Bai T, Li M, Liu Y, Qiao Z and Wang Z:
Inhibition of ferroptosis alleviates atherosclerosis through
attenuating lipid peroxidation and endothelial dysfunction in mouse
aortic endothelial cell. Free Radic Biol Med. 160:92–102.
2020.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Huang D, Zheng S, Liu Z, Zhu K, Zhi H and
Ma G: Machine learning revealed ferroptosis features and a novel
ferroptosis-based classification for diagnosis in acute myocardial
infarction. Front Genet. 13(813438)2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Tang LJ, Zhou YJ, Xiong XM, Li NS, Zhang
JJ, Luo XJ and Peng J: Ubiquitin-specific protease 7 promotes
ferroptosis via activation of the p53/TfR1 pathway in the rat
hearts after ischemia/reperfusion. Free Radic Biol Med.
162:339–352. 2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Guo Y, Zhang W, Zhou X, Zhao S, Wang J,
Guo Y, Liao Y, Lu H, Liu J, Cai Y, et al: Roles of ferroptosis in
cardiovascular diseases. Front Cardiovasc Med.
9(911564)2022.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Chen X, Xu S, Zhao C and Liu B: Role of
TLR4/NADPH oxidase 4 pathway in promoting cell death through
autophagy and ferroptosis during heart failure. Biochem Biophys Res
Commun. 516:37–43. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Stockwell BR, Friedmann Angeli JP, Bayir
H, Bush AI, Conrad M, Dixon SJ, Fulda S, Gascón S, Hatzios SK,
Kagan VE, et al: Ferroptosis: A regulated cell death nexus linking
metabolism, redox biology, and disease. Cell. 171:273–285.
2017.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Forcina GC and Dixon SJ: GPX4 at the
crossroads of lipid homeostasis and ferroptosis. Proteomics.
19(e1800311)2019.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Shimada K, Skouta R, Kaplan A, Yang WS,
Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ and
Stockwell BR: Global survey of cell death mechanisms reveals
metabolic regulation of ferroptosis. Nat Chem Biol. 12:497–503.
2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Dixon SJ, Patel DN, Welsch M, Skouta R,
Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS
and Stockwell BR: Pharmacological inhibition of cystine-glutamate
exchange induces endoplasmic reticulum stress and ferroptosis.
Elife. 3(e02523)2014.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Kim J, Kim H, Roh H and Kwon Y: Causes of
hyperhomocysteinemia and its pathological significance. Arch Pharm
Res. 41:372–383. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Qin X, Xu M, Zhang Y, Li J, Xu X, Wang X,
Xu X and Huo Y: Effect of folic acid supplementation on the
progression of carotid intima-media thickness: A meta-analysis of
randomized controlled trials. Atherosclerosis. 222:307–313.
2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Esse R, Barroso M, Tavares de Almeida I
and Castro R: The contribution of homocysteine metabolism
disruption to endothelial dysfunction: State-of-the-art. Int J Mol
Sci. 20(867)2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Zhang X, Huang Z, Xie Z, Chen Y, Zheng Z,
Wei X, Huang B, Shan Z, Liu J, Fan S, et al: Homocysteine induces
oxidative stress and ferroptosis of nucleus pulposus via enhancing
methylation of GPX4. Free Radic Biol Med. 160:552–565.
2020.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Mancuso C and Santangelo R: Panax ginseng
and Panax quinquefolius: From pharmacology to toxicology. Food Chem
Toxicol. 107:362–372. 2017.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Quan HY, Yuan HD, Jung MS, Ko SK, Park YG
and Chung SH: Ginsenoside Re lowers blood glucose and lipid levels
via activation of AMP-activated protein kinase in HepG2 cells and
high-fat diet fed mice. Int J Mol Med. 29:73–80. 2012.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Kim JM, Park CH, Park SK, Seung TW, Kang
JY, Ha JS, Lee DS, Lee U, Kim DO and Heo HJ: Ginsenoside Re
ameliorates brain insulin resistance and cognitive dysfunction in
high fat diet-induced C57BL/6 mice. J Agric Food Chem.
65:2719–2729. 2017.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Wang H, Lv J, Jiang N, Huang H, Wang Q and
Liu X: Ginsenoside Re protects against chronic restraint
stress-induced cognitive deficits through regulation of NLRP3 and
Nrf2 pathways in mice. Phytother Res. 35:2523–2535. 2021.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Su F, Xue Y, Wang Y, Zhang L, Chen W and
Hu S: Protective effect of ginsenosides Rg1 and Re on
lipopolysaccharide-induced sepsis by competitive binding to
Toll-like receptor 4. Antimicrob Agents Chemother. 59:5654–5663.
2015.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Wang QW, Yu XF, Xu HL, Zhao XZ and Sui DY:
Ginsenoside Re improves isoproterenol-induced myocardial fibrosis
and heart failure in rats. Evid Based Complement Alternat Med.
2019(3714508)2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Yao H, Li J, Song Y, Zhao H, Wei Z, Li X,
Jin Y, Yang B and Jiang J: Synthesis of ginsenoside Re-based carbon
dots applied for bioimaging and effective inhibition of cancer
cells. Int J Nanomedicine. 13:6249–6264. 2018.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Lee GH, Lee WJ, Hur J, Kim E, Lee HG and
Seo HG: Ginsenoside Re mitigates 6-hydroxydopamine-induced
oxidative stress through upregulation of GPX4. Molecules.
25(188)2020.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Shi J, Chen D, Wang Z, Li S and Zhang S:
Homocysteine induces ferroptosis in endothelial cells through the
systemXc-/GPX4 signaling pathway. BMC Cardiovasc Disord.
23(316)2023.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Yang R, Gao W, Wang Z, Jian H, Peng L, Yu
X, Xue P, Peng W, Li K and Zeng P: Polyphyllin I induced
ferroptosis to suppress the progression of hepatocellular carcinoma
through activation of the mitochondrial dysfunction via
Nrf2/HO-1/GPX4 axis. Phytomedicine. 122(155135)2024.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Wu B, Lan X, Chen X, Wu Q, Yang Y and Wang
Y: Researching the molecular mechanisms of Taohong Siwu Decoction
in the treatment of varicocele-associated male infertility using
network pharmacology and molecular docking: A review. Medicine
(Baltimore). 102(e34476)2023.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Gimbrone MA Jr and García-Cardeña G:
Endothelial cell dysfunction and the pathobiology of
atherosclerosis. Circ Res. 118:620–636. 2016.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Botts SR, Fish JE and Howe KL:
Dysfunctional vascular endothelium as a driver of atherosclerosis:
Emerging insights into pathogenesis and treatment. Front Pharmacol.
12(787541)2021.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Mou Y, Wang J, Wu J, He D, Zhang C, Duan C
and Li B: Ferroptosis, a new form of cell death: Opportunities and
challenges in cancer. J Hematol Oncol. 12(34)2019.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Ouyang S, You J, Zhi C, Li P, Lin X, Tan
X, Ma W, Li L and Xie W: Ferroptosis: The potential value target in
atherosclerosis. Cell Death Dis. 12(782)2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Azad MAK, Huang P, Liu G, Ren W, Teklebrh
T, Yan W, Zhou X and Yin Y: Hyperhomocysteinemia and cardiovascular
disease in animal model. Amino Acids. 50:3–9. 2018.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Shi Q, Liu R and Chen L: Ferroptosis
inhibitor ferrostatin-1 alleviates homocysteine-induced ovarian
granulosa cell injury by regulating TET activity and DNA
methylation. Mol Med Rep. 25(130)2022.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Wang Y, Kuang X, Yin Y, Han N, Chang L,
Wang H, Hou Y, Li H, Li Z, Liu Y, et al: Tongxinluo prevents
chronic obstructive pulmonary disease complicated with
atherosclerosis by inhibiting ferroptosis and protecting against
pulmonary microvascular barrier dysfunction. Biomed Pharmacother.
145(112367)2022.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Ye J, Lyu TJ, Li LY, Liu Y, Zhang H, Wang
X, Xi X, Liu ZJ and Gao JQ: Ginsenoside Re attenuates myocardial
ischemia/reperfusion induced ferroptosis via miR-144-3p/SLC7A11.
Phytomedicine. 113(154681)2023.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Yang WS, Kim KJ, Gaschler MM, Patel M,
Shchepinov MS and Stockwell BR: Peroxidation of polyunsaturated
fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci
USA. 113:E4966–E4975. 2016.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Chen Y, Deng Y, Chen L and Huang Z, Yan Y
and Huang Z: miR-16-5p regulates ferroptosis by targeting SLC7A11
in adriamycin-induced ferroptosis in cardiomyocytes. J Inflamm Res.
16:1077–1089. 2023.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Xu S, Wu B, Zhong B, Lin L, Ding Y, Jin X,
Huang Z, Lin M, Wu H and Xu D: Naringenin alleviates myocardial
ischemia/reperfusion injury by regulating the nuclear
factor-erythroid factor 2-related factor 2 (Nrf2)/System
xc-/glutathione peroxidase 4 (GPX4) axis to inhibit ferroptosis.
Bioengineered. 12:10924–10934. 2021.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Zhang L, Liu W, Liu F, Wang Q, Song M, Yu
Q, Tang K, Teng T, Wu D, Wang X, et al: IMCA induces ferroptosis
mediated by SLC7A11 through the AMPK/mTOR pathway in colorectal
cancer. Oxid Med Cell Longev. 2020(1675613)2020.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Wang L, Liu Y, Du T, Yang H, Lei L, Guo M,
Ding HF, Zhang J, Wang H, Chen X and Yan C: ATF3 promotes
erastin-induced ferroptosis by suppressing system Xc. Cell Death
Differ. 27:662–675. 2020.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Parker JL, Deme JC, Kolokouris D, Kuteyi
G, Biggin PC, Lea SM and Newstead S: Molecular basis for redox
control by the human cystine/glutamate antiporter system xc. Nat
Commun. 12(7147)2021.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Qiao O, Zhang L, Han L, Wang X, Li Z, Bao
F, Hao H, Hou Y, Duan X, Li N and Gong Y: Rosmarinic acid plus
deferasirox inhibits ferroptosis to alleviate crush
syndrome-related AKI via Nrf2/Keap1 pathway. Phytomedicine.
129(155700)2024.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Ren S, Wang J, Dong Z, Li J, Ma Y, Yang Y,
Zhou T, Qiu T, Jiang L, Li Q, et al: Perfluorooctane sulfonate
induces ferroptosis-dependent non-alcoholic steatohepatitis via
autophagy-MCU-caused mitochondrial calcium overload and MCU-ACSL4
interaction. Ecotoxicol Environ Saf. 280(116553)2024.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Wei XL, Yu AG, Zhang X, Pantopoulos K, Liu
ZB, Xu PC, Zheng H and Luo Z: Fe2O3
nanoparticles induce ferroptosis by binding to the N-terminus of
ACSL4 and suppressing its ubiquitin-mediated degradation. Free
Radic Biol Med. 238:123–136. 2025.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Cai J, Huang K, Han S, Chen R, Li Z, Chen
Y, Chen B, Li S, Xinhua L and Yao H: A comprehensive system review
of pharmacological effects and relative mechanisms of Ginsenoside
Re: Recent advances and future perspectives. Phytomedicine.
102(154119)2022.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Zhu Y, Yue J, Yan R, Xia W, Li T and Fu X:
Enhancement in the intestinal absorption of ginsenoside Re by
ginseng polysaccharides and its mechanisms. Food Chem.
488(144914)2025.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Jeon JH, Lee J, Choi MK and Song IS:
Pharmacokinetics of ginsenosides following repeated oral
administration of red ginseng extract significantly differ between
species of experimental animals. Arch Pharm Res. 43:1335–1346.
2020.PubMed/NCBI View Article : Google Scholar
|