Introduction
Cohesinopathies are a group of rare diseases caused
by defects in the cohesin complex. These disorders involve multiple
organ systems, including the brain, heart and skeleton, and stem
from impairments in fundamental cellular processes such as
chromosome segregation, DNA repair, DNA replication,
heterochromatin formation and gene transcription (1). The cohesin complex, an evolutionarily
conserved large functional unit, consists of four core proteins,
structural maintenance of chromosomes (SMC) protein 1A, SMC2, RAD21
and stromal antigen (STAG)1/2. Additionally, several regulatory
proteins associated with this complex have been implicated in a
wide range of human diseases (2).
The STAG2 gene (Online Mendelian Inheritance in Man 300826,
NM_001042751), located on chromosome Xq25, comprises 34 exons and
encodes the STAG2 cohesin complex component, which is involved in
gene expression, DNA repair and genomic integrity (3). Variants in STAG2 have been
identified as the causative factor for neurodevelopmental
disorders, such as X-linked holoprosencephaly 13 (HPE13) and
Mullegama-Klein-Martinez syndrome (MKMS) (4). Patients with STAG2 variants
typically exhibit a wide range of phenotypic abnormalities,
including intellectual disability, developmental delay,
microcephaly, dysmorphic features, short stature, growth
restriction, language impairment, delayed puberty, microtia,
hearing loss and congenital heart and skeletal defects (5). At present, 19 STAG2 variants
have been reported in patients with HPE13 or MKMS (4-13).
These include five missense variants, eight nonsense variants, five
frameshift variants and one splice variant. Additional research is
needed to elucidate the genotype-phenotype associations and
underlying mechanisms of STAG2-related disorders.
Polycystic kidney disease (PKD) is a genetic
disorder characterized by the formation of numerous cysts in the
kidneys, leading to progressive kidney damage and eventual renal
failure (14). The PKD1
gene, located on chromosome 16, encodes polycystin-1, a large
transmembrane protein involved in cell signaling, calcium ion
regulation and the maintenance of normal renal tubular epithelial
cell function (15). Variants in
the PKD1 gene disrupt polycystin-1 function, resulting in
abnormal cell proliferation and the formation of fluid-filled cysts
(16). These cysts gradually
enlarge, compressing and replacing normal kidney tissue, thereby
impairing kidney function (17).
PKD1 variants are responsible for most cases of autosomal
dominant PKD (ADPKD), which is the most common form of PKD
(18).
In the present report, a novel de novo
heterozygous STAG2 variant [NM_001042750.2:c.1775_1777del,
p.(Pro592del)] and a novel heterozygous frameshift PKD1
variant [NM_001009944.3:c.8985delC, p.(Ser2996fs*78)] were
identified in a Chinese infant diagnosed with MKMS and familial
PKD. Furthermore, through a comprehensive review of the literature,
the genotypic, phenotypic and clinical features of STAG2-related
disorders were summarized.
Case report
Patients and methods. Patients
In June 2024, a Chinese family with a member
presenting seizures and developmental delays was referred to the
Department of Pediatric Neurology of Maternal and Child Health
Hospital of Guangxi Zhuang Autonomous Region (Nanning, China) for
genetic evaluation. The study protocol was approved by the Ethics
Committee of Maternal and Child Health Hospital of Guangxi Zhuang
Autonomous Region (approval no. METc 2017-2-11) and conducted in
accordance with the principles of The Declaration of Helsinki.
Written informed consent was obtained from the patients and/or the
parents of the affected individual for the publication of clinical
data and images.
Whole-exome sequencing (WES) and Sanger
validation. WES and Sanger validation were performed on genomic
DNA extracted from 2 ml peripheral blood lymphocytes of the proband
and available family members with the Lab-Aid DNA kit (cat. no.
604016; Xiamen Zeesan Biotech Co., Ltd.). DNA integrity was
verified on a 1% agarose gel (≥20 kb high-molecular-weight band)
and quantified by a Qubit dsDNA BR Assay (cat. no. Q32853; Thermo
Fisher Scientific, Inc.). Trio-WES was carried out for the proband
and parents: 3 µg of each DNA sample were sheared to 180-250 bp,
end-repaired, A-tailed and ligated to Illumina-compatible adapters.
Exome enrichment was performed with the Agilent SureSelect Human
All Exon V5 capture kit (cat. no. 5190-6210; Agilent Technologies,
Inc.). Captured libraries were pooled equimolarly, and the 280-320
bp insert size was confirmed on an Agilent High-Sensitivity DNA
chip (Agilent Technologies, Inc.) run on the 2100 Bioanalyzer (cat.
no. 5067-4626; Agilent Technologies, Inc.). Post-ligation Illumina
NGS libraries served as the DNA source; their concentration was
measured by SYBR Green I qPCR with the KAPA Library Quantification
kit (Roche Diagnostics) using primers P5,
5'-AATGATACGGCGACCACCGA-3', and P7, 5'-CAAGCAGAAGACGGCATACGA-3',
and a six-point kit-supplied linearized standard series (20-0.0002
pM). Reactions (20 µl) were run on a CFX96 (Bio-Rad Laboratories,
Inc.) at 95˚C for 5 min, followed by 35 cycles of 95˚C for 30 sec
and 60˚C for 45 sec, and finally a 65-97˚C melt curve. Unknown
libraries were quantified by interpolation from the standard curve,
diluted to 2 nM, denatured with 0.2 N NaOH and loaded at 10 pM onto
an Illumina HiSeq 2000 flow-cell together with 1% PhiX control
(cat. no. FC-110-3001; Illumina, Inc.). Sequencing was performed
with the HiSeq 2000 Rapid SBS Kit v2, 100-cycle paired-end (cat.
no. FC-402-4021; Illumina, Inc.) to generate 100-bp paired-end
reads. Raw reads were aligned to the hg19/GRCh38 human reference
genome with BWA-MEM (v0.7.15; https://github.com/lh3/bwa), and variant calling was
completed using the Genome Analysis Toolkit (GATK v3.4; Broad
Institute) following the best-practice workflow. Variant calling
and annotation were performed using LifeMap TGex Version 3.0
(https://auth.shanyint.com/; customised
website), with a focus on variants exhibiting a minor allele
frequency of ≤0.001 in public databases, including the 1000 Genomes
Project (https://www.internationalgenome.org/data), Exome
Sequencing Project (http://evs.gs.washington.edu/EVS/) and Exome
Aggregation Consortium (http://exac.broadinstitute.org).
The functional impact of candidate variants was
predicted using in silico tools, including REVEL (https://sites.google.com/site/revelgenomics/),
PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), Sorting
Intolerant From Tolerant (https://sift.bii.a-star.edu.sg/), Combined Annotation
Dependent Depletion (https://cadd.gs.washington.edu/snv), MutationTaster
(http://www.mutationtaster.org/) and
NMDEscPredictor (https://fursham-h.github.io/factR/reference/predictNMD.html).
SWISS-MODEL (https://swissmodel.expasy.org/) was used to construct
a 3D model of the STAG2 protein. Co-segregation analysis of the
STAG2 and PKD1 variants was performed among family
members using Sanger sequencing, with primers designed to amplify
the STAG2 variant [NM_001042750.2:c.1775_1777del,
p.(Pro592del)] and the PKD1 variant
[NM_001009944.3:c.8985delC, p.(Ser2996fs*78)]. PCR amplification
was performed with Takara PrimeSTAR Max DNA Polymerase (Takara
Biotechnology Co., Ltd.) using the following thermocycling
conditions: Initial denaturation at 95˚C for 5 min; 35 cycles of
denaturation at 95˚C for 3 sec, annealing at 60˚C for 30 sec and
extension at 72˚C for 30 sec; and a final extension at 72˚C for 5
min. The primer sequences were as follows: STAG2 forward,
5'-TTTCCCTAAATGCCTCACAGAA-3' and reverse,
5'-AGGTACAGTTGTGGGCATGA-3'; and PKD1 forward,
5'-CTCTGAGACTGCGACATCCA-3' and reverse, 5'-CACAGGAAACACAAAGCGGA-3'.
The pathogenicity of candidate variants was assessed according to
the guidelines of the American College of Medical Genetics and
Genomics (ACMG)/Association for Molecular Pathology (AMP) and the
ClinGen Sequence Variant Interpretation Working Group (19,20).
Case presentation. Patient
characteristics
The proband (III.1), a 4-month-old female infant,
was the first child of unrelated non-consanguineous Chinese parents
(Fig. 1A). The patient was born
full-term at 40 weeks of gestation by vaginal delivery with a
normal birth weight (3.19 kg). The Apgar scores of the infant at 1,
5 and 10 min after birth were 9, 9 and 10, respectively (21). At the age of 4 months, the patient
was admitted to the Department of Pediatric Neurology of Maternal
and Child Health Hospital of Guangxi Zhuang Autonomous Region for
seizures. The proband experienced the first generalized
tonic-clonic seizure at 3 months and 21 days old, with a frequency
of 1-2 seizures per day during the first 9 days. A 24-h ambulatory
electroencephalogram was performed, and the results showed
bilateral slow waves and spike-like slow waves in the frontal pole,
occipital and temporal areas (data not shown due to the large size
of the video). Global developmental delay was observed in the first
4 months of life, and the patient was unable to hold their head up
or roll over. A physical examination revealed that the patient had
mild short stature (60 cm, <-2 SD), hypotonia and dysmorphic
features, including microcephaly (head circumference of 39 cm,
<-1 SD), a narrow forehead, saddle nose, large ears,
micrognathia, incomplete cleft palate and microstomia (Fig. 1B). Additional findings included
spina bifida occulta and a duplication of the middle phalanx of the
third finger on the left hand. The neurodevelopmental parameters of
the infant were assessed using the Bayley Scales of Infant and
Toddler Development, Third Edition at ~6 months of age, and the
cognitive, motor and language developmental ages were equivalent to
those at 3, 4 and 2 months, respectively (22). Therefore, the patient was diagnosed
with MKMS. Renal ultrasound demonstrated increased renal
echogenicity (Fig. S1A). Notably,
the mother, aunt and grandmother of the patient were all diagnosed
with PKD, although no other malformations were reported in the
family history. The proband did not exhibit other symptoms of ADPKD
beyond the increased echogenicity observed in the renal ultrasound.
Additionally, the patient did not have hypertension, and both
urinalysis and serum creatinine levels were within normal ranges.
Brain magnetic resonance imaging was performed at 4 months of age,
and the results were normal (Fig.
S1B). Audiological evaluation revealed no hearing deficits, but
a middle ear infection was detected. The 4-month-old infant's ear
infection was managed with a 10-day course of high-dose amoxicillin
at 80-90 mg/kg per day, divided into two oral doses (~40 mg/kg
twice daily). The last follow-up of the child was in November 2025,
at which time the patient was 1.5 years old and developing
normally.
![Clinical and genetic features. (A)
Pedigree chart of the family of the proband with
Mullegama-Klein-Martinez syndrome and familial polycystic kidney
disease. The pedigree depicts the segregation of both the STAG2
(shown in black) and PKD1 (shown in blue) variants within the
family. (B) Facial appearance of the proband (Ⅲ-1) at the age of 4
months, showing microcephaly (head circumference of 39 cm, <-1
SD), narrow forehead, saddle nose, large ears, micrognathia and
microstomia. (C) The Sanger sequencing chromatograms illustrate the
presence of a novel heterozygous STAG2 variant
[NM_001042750.2:c.1775_1777del, p.(Pro592del)] and a novel
heterozygous frameshift PKD1 variant [NM_001009944.3:
c.8985delC, p.(Ser2996fs*78)] in the proband. (D) The spectrum of
STAG2 pathogenic variants. The red variant is the novel
variant identified in the present study. STAG, stromal antigen;
SCD, stromalin conservative domain; HEAT_SCC3-SA, cohesin subunit
SCC3/SA, HEAT-repeats domain.](/article_images/etm/31/3/etm-31-03-13057-g00.jpg) | Figure 1Clinical and genetic features. (A)
Pedigree chart of the family of the proband with
Mullegama-Klein-Martinez syndrome and familial polycystic kidney
disease. The pedigree depicts the segregation of both the STAG2
(shown in black) and PKD1 (shown in blue) variants within the
family. (B) Facial appearance of the proband (Ⅲ-1) at the age of 4
months, showing microcephaly (head circumference of 39 cm, <-1
SD), narrow forehead, saddle nose, large ears, micrognathia and
microstomia. (C) The Sanger sequencing chromatograms illustrate the
presence of a novel heterozygous STAG2 variant
[NM_001042750.2:c.1775_1777del, p.(Pro592del)] and a novel
heterozygous frameshift PKD1 variant [NM_001009944.3:
c.8985delC, p.(Ser2996fs*78)] in the proband. (D) The spectrum of
STAG2 pathogenic variants. The red variant is the novel
variant identified in the present study. STAG, stromal antigen;
SCD, stromalin conservative domain; HEAT_SCC3-SA, cohesin subunit
SCC3/SA, HEAT-repeats domain. |
Genetic analysis. Trio-WES identified a novel
de novo heterozygous in-frame variant in STAG2
[NM_001042750.2:c.1775_1777del, p.(Pro592del)] and a novel
heterozygous frameshift variant in PKD1
[NM_001009944.3:c.8985delC, p.(Ser2996fs*78)] (Fig. 1C). Both variants were confirmed in
the proband and the family members by Sanger sequencing. The
STAG2 variant [c.1775_1777del, p.(Pro592del)] was absent in
the parents, aunt and grandmother, confirming its de novo
origin. By contrast, the PKD1 variant [c.8985delC,
p.(Ser2996fs*78)] was identified in the mother, aunt and
grandmother of the proband (Fig.
1C). To the best of our knowledge, these variants have not been
reported previously and are not identified in public databases,
including the Exome Sequencing Project (https://evs.gs.washington.edu/EVS/), Genome
Aggregation Database (https://gnomad.broadinstitute.org/), 1000 Genomes
Project (https://www.internationalgenome.org/data) and the
Single Nucleotide Polymorphism database (http://www.ncbi.nlm.nih.gov/SNP/), as well as
disease-related databases such as ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/) and the Human
Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/). To date, a total of 21
distinct STAG2 variants, including the one reported here, have been
identified in patients with HPE13 or MKMS, distributed across the
entire gene (Fig. 1D). The variant
described in the present report falls within the HEAT_SCC3-SA
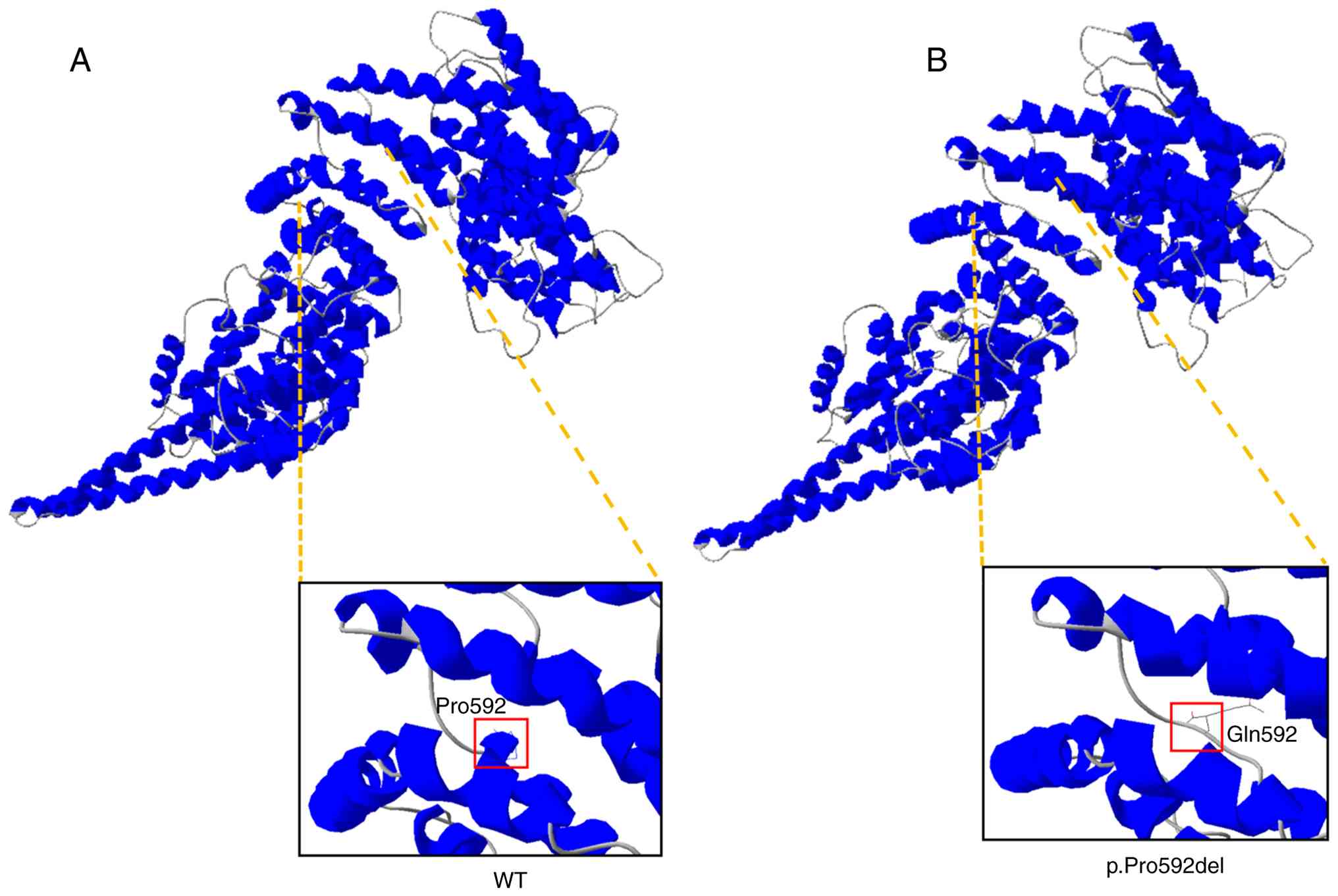
domain. To further analyze the effect of the amino acid changes
caused by the c.1775_1777del, p.(Pro592del) variant on the
structure of the protein, the variant was modelled using the
wild-type STAG2 crystal structure. Pro592 is located at a junction
between multiple α-helices, where it may influence their stability
and packing (Fig. 2A); however,
Pro592del leads to structural defects, disrupting the formation of
α-helices (Fig. 2B). Therefore, we
hypothesize that the c.1775_1777del, p.(Pro592del) variant may lead
to defective protein folding and ultimately reduced stability of
STAG2. According to the ACMG/AMP guidelines, the variant
c.1775_1777del is classified as likely pathogenic (PS2 +
PM2_Supporting + PP3), while the c.8985delC variant is classified
as pathogenic (PVS1 + PM2_Supporting + PP1 + PP4) (Table I).
 | Table IPredicted pathogenicity of
STAG2 and PKD1 variants. |
Table I
Predicted pathogenicity of
STAG2 and PKD1 variants.
| Gene | Reference allele
(GRCh38) | Variant | Inheritance | Variant Taster |
NMDEscPredictor | PolyPhen-2 | SIFT | CADD | ACMG/AMP
guidelines |
|---|
| STAG2
(NM_001042750.2) | ChrX:124063159_
124063161 | c.1775_1777del,
p.(Pro592del) | DNV | D | NA | NA | NA | NA | LP (PS2 + PM2_
Supporting + PP3) |
| PKD1
(NM_001009944.3) | Chr16:2102597 | c.8985delC,
p.(Ser2996fs*78) | Maternal | D | Causes NMD | NA | NA | NA | P (PVS1 +
PM2_Supporting + PP1 + PP4) |
Discussion
STAG2 has been identified as a causative gene
associated with a spectrum of neurodevelopmental disorders,
including microcephaly, microphthalmia, hearing loss, developmental
delay, dysmorphic features, congenital heart defects and digital
anomalies (4,5). The association between STAG2
and neurodevelopmental disorders was initially established through
the identification of copy number variants affecting this gene. In
addition, this association has been corroborated by the
identification of novel single nucleotide variants, which have
provided deeper insights into the genetic architecture of
STAG2-related disorders (4,5). At
present, 19 STAG2 variants have been identified in 25
patients with HPE13 or MKMS (4-13).
In the current report, trio-based WES was performed, which
identified a novel de novo heterozygous variant in the
STAG2 gene in a female Chinese infant. The patient exhibited
a clinical phenotype consistent with STAG2-related
disorders, including epilepsy, global developmental delay, short
stature, hypotonia, dysmorphic features, incomplete cleft palate,
micrognathia, spina bifida occulta and a duplication of the middle
phalanx of the third finger on her left hand; therefore, the
patient was diagnosed with MKMS.
The clinical features of the 26 reported patients
with HPE13 or MKMS, including the patient in the current report,
are summarized in Table II.
Phenotypic analysis of these patients revealed marked heterogeneity
in the clinical features associated with pathogenic or likely
pathogenic variants of the STAG2 gene; however, certain
common features were identified in >50% of cases. All patients
with available data exhibited developmental abnormalities of
varying severity across multiple domains. Intellectual disability
or developmental delay was observed in all patients with available
data (20/20), ranging from mild to severe. Pathogenic variants in
the STAG2 gene are associated with a spectrum of brain
abnormalities. Brain anomalies were observed in almost all patients
(15/17), and included microcephaly, delayed or incomplete
myelination, myelin hypotrophy, agenesis or dysgenesis of the
corpus callosum, white matter hypoplasia, holoprosencephaly and
atelencephaly. Among these, holoprosencephaly (9/19) was the most
frequently reported, whereas alobar holoprosencephaly represented
the most severe manifestation. In the present patient, however, no
brain malformations were observed at 4 months of age. Additionally,
dysmorphic facial features were present in nearly all cases
(19/21); individuals with MKMS typically exhibit characteristic
facial dysmorphisms, including a broad forehead, low-set ears, a
short and broad nose, a shallow philtrum, cleft lip/palate, a small
mouth with thin lips and a small, receding chin (4,13).
Ocular anomalies such as hypertelorism, hypotelorism, ptosis and
epicanthal folds, as well as facial asymmetry, are also commonly
observed. By contrast, patients with X-linked HPE13 often present
with midline facial defects, such as a single central incisor, a
flat nasal bridge and a proboscis (10). Ocular and eyelid abnormalities,
including microphthalmia, anophthalmia, colobomas, ptosis and
ankyloblepharon, are frequently noted, alongside oral and
mandibular anomalies such as microstomia, macroglossia, cleft
lip/palate and micrognathia. Dysmorphic features were shared in
both conditions. The present patient also exhibited mild dysmorphic
features including mild microcephaly, narrow forehead, saddle nose,
large ears, micrognathia, incomplete cleft palate and microstomia.
Thoracic vertebra abnormalities were observed in more than
two-thirds (11/14) of the patients, primarily affecting the
thoracic spine; these included hemivertebrae, butterfly vertebrae,
scoliosis, spina bifida and fused ribs. Other skeletal anomalies
involved left hip dysplasia, broad hands and feet and
hyperextensibility of the hand and foot joints. The present patient
also presented with spina bifida occulta accompanied by a novel
clinical manifestation of middle phalanx duplication in the third
digit of the left hand. Congenital cardiac malformations were also
common (10/15), with a spectrum of anomalies ranging from patent
foramen ovale to severe dextroposition of the heart and aortic
valve atresia; notably, the present patient had no cardiac
problems. Additional dysmorphic features included seizures, left
facial palsy, mild left pelviectasis, sacral dimple, congenital
dislocation of the hip, gastroesophageal reflux, hypotonia,
pulmonary hypoplasia, single kidney, hearing loss, polycystic
kidney, duodenal atresia and left-sided diaphragmatic hernia. These
findings highlight the broad phenotypic spectrum associated with
STAG2 gene variants and underscore the importance of a
comprehensive clinical evaluation in patients with these
conditions. Further studies are needed to elucidate the underlying
mechanisms and improve diagnostic and therapeutic approaches.
| Table IIClinical features of patients with
STAG2 variants. |
Table II
Clinical features of patients with
STAG2 variants.
|
Patienta | Sex | STAG2
variants | Inheritance | ID/DD | Brain
abnormalities | Micro-cephaly | Dysmorphic features
and malformations | Cleft
lip/palate | Congenital heart
defects | Thoracic vertebra
anomalies | Seizures | Other features | (Refs.) |
|---|
| 1 | M | c.3027A>T,
p.(Lys1009Asn) | De novo | + | - | + | + | - | - | - | - | Polycystic
kidney | (4) |
| One family (5
patients) | All M | c.980G>A,
p.(Ser327Asn) | Maternally | 5/5 | NA | 0/5 | 5/5 | 1/5 | NA | NA | 0/5 | NA | (5) |
| 7 | F | c.3097C>T,
p.(Arg1033*) | De novo | NA | HPE | NA | NA | + | Left heart
hypoplasia | NA | - | NA | (6) |
| 8 | F | c.2229G>A,
p.(Trp743*) | De novo | + | White matter
hypoplasia | - | + | + | - | Hemivertebra | + | Amblyopia | |
| 9 | F | c.205C>T,
p.(Arg69*) | De novo | + | HCC, subarachnoid
cyst, subgaleal | + | + | + | VSD | Scoliosis,
hemivertebra, butterfly vertebra | - | Left facial palsy,
mild left pelviectasis | (7) |
| 10 | F | c.1913_1922del,
p.(Ala638Valfs*10) | De novo | NA | NA | NA | NA | NA | NA | NA | NA | NA | |
| 11 | F | c.1840C>T,
p.(Arg614*) | De novo | + | Cystic pituitary
lesion | + | NA | NA | NA | Scoliosis | - | Sacral dimple,
CDH | (8) |
| 12 | F | c.418C>T,
p.(Gln140*) | De novo | + | NA | - | + | - | Left heart
hypoplasia, VSD, CA | Scoliosis, rib
fusion, vertebral clefts | + | Gastroesophageal
reflux, CDH | |
| 13 | F | c.1605T>A,
p.(Cys535*) | De novo | + | NA | + | + | - | NA | Scoliosis, rib
fusion | + | Hypotonia | |
| 14 | F | c.1658_1660delinsT,
p.(Lys553Ilefs*6) | De novo | + | HPE
(microform) | + | + | - | NA | Scoliosis, rib
fusion | + | Hypotonia | (9) |
| 15 | F | c.1811G>A,
p.(Arg604Gln) | De novo | + | NA | + | + | NA | - | Vertebral
clefts | - | Gastroesophageal
reflux, CDH, pulmonary hypoplasia | |
| 16 | M | c.476A>G,
p.(Tyr159Cys) | De novo | + | Ectopic posterior
pituitary, short pituitary stalk | - | + | + | Minimal PFO | Scoliosis | - | Single kidney,
hypotonia | |
| 17 | F | c.205C>T,
p.(Arg69*) | De novo | + | HPE
(semi-lobar) | + | + | + | PFO, PDA | - | - | NA | (10) |
| 18 | F | c.436C>T,
p.(Arg146*) | NA | NA | HPE (alobar) | + | + | - | VSD | Hemivertebra | - | Duodenal
atresia | |
| 19 | F | c.775C>T,
p.(Arg259*) | De novo | + | HPE (septo-optic
dysplasia) | - | - | - | VSD | NA | - | Left hip dysplasia,
bilateral optic nerve hypoplasia | |
| 20 | F | c.3034C>T,
p.(Arg1012*) | De novo | NA | HPE (alobar) | + | + | + | NA | Spina bifida | - | Gastroesophageal
reflux | |
| 21 | F | c.2898_2899del,
p.(Glu968Serfs*15) | De novo | + | HPE
(microform) | + | NA | NA | - | NA | - | NA | |
| 22 | F | c.2533+1G>A | Maternally | NA | HPE
(semi-lobar) | + | - | - | Left heart
hypoplasia, DORV | NA | - | Hypotonia | |
| 23 | F | c.1639delG,
p.(Val547Cysfs*29) | De novo | + | + | NA | + | NA | NA | NA | + | Hearing loss | (11) |
| 24 | F | c. 3458_3459delTT,
p.(Leu1153Argfs*3) | De novo | NA | Dandy-Walker
malformation | - | NA | - | Dextrocardia, CA,
VSD | NA | - | CDH | (12) |
| 25 | M | c.475T>C,
p.(Tyr159His) | Maternally | + | HPE, PMG, HCC | - | + | - | PFO | - | + | Hands and feet as
well as fingers and toes were broad, with soft dorsal surfaces; the
nails were deeply inserted and the joints of the hands and feet
were hyperextensible | (13) |
| 26 | F | c.1775_1777delCTC,
p.(Pro592del) | De novo | + | - | + | + | + | - | Spina bifida | + | Polycystic kidney,
hypotonia | Present case |
| Total | M=8, F=18 | Frameshift=5,
Splicing=1, Nonsense=8, In-frame deletion=1, Missense=5 | | 20/20 | 15/17 | 12/23 | 19/21 | 8/21 | 10/15 | 11/14 | 7/25 | | |
To date, only 26 affected individuals (including the
present case) have been reported with STAG2 variants,
including five missense variants, one in-frame deletion (present
case), eight nonsense variants, five frameshift variants and one
splice variant (4-13).
Notably, a distinct pattern emerged upon further analysis of these
cases; all male patients (8/8) harbored hemizygous missense
variants, whereas nearly all female patients (16/18) carried
heterozygous null variants. A study by Cheng et al (23) showed that Stag2 knockout
mouse embryos displayed severe developmental defects and underwent
necrosis at day E11.5, whereas conditional knockout mice with
Stag2 deletion in the nervous system exhibited growth
retardation, neurological defects and early death. These results
suggest that a hemizygous deletion of the STAG2 gene may
lead to severe phenotypes and could potentially cause embryonic
lethality, thereby preventing the generation of male individuals
with null variants. In addition, the present findings suggested
that female patients with truncating variants exhibit a higher
incidence of congenital malformations, such as brain malformations,
cleft lip and palate, congenital heart disease, thoracic spine
anomalies and epileptic seizures, compared with male patients with
missense variants (Table III).
These observations are likely driven by the greater pathogenic
potential of truncating variants relative to missense variants,
rather than by sex differences. Specifically, null variants
typically result in a complete loss of function of the STAG2
protein, leading to more profound disruptions in cellular
processes. By contrast, missense variants may retain partial
protein function, potentially explaining the milder phenotypic
manifestations observed in these cases. However, the precise
mechanisms underlying these observations remain unclear and warrant
further investigation through more comprehensive studies.
| Table IIIComparison of the effects of sex and
variant type on clinical manifestations. |
Table III
Comparison of the effects of sex and
variant type on clinical manifestations.
| Clinical
manifestation | Male patients with
missense variants, n (%) | Female patients
with truncating variantsa, n (%) | Total, n (%) |
|---|
| DD/ID | 8/8 (100.00) | 10/10 (100.00) | 18/18 (100.00) |
| Microcephaly | 1/8 (12.50) | 9/13 (69.23) | 10/21 (47.62) |
| Brain
abnormalities | 2/3 (66.67) | 13/13 (100.00) | 15/16 (93.75) |
| Dysmorphic
features | 8/8 (100.00) | 9/11 (81.82) | 17/19 (89.47) |
| Cleft
lip/palate | 2/8 (25.00) | 5/12 (41.67) | 7/20 (35.00) |
| Congenital heart
defects | 2/3 (66.67) | 8/10 (80.00) | 10/13 (76.92) |
| Thoracic vertebra
anomalies | 1/3 (33.33) | 8/9 (88.89) | 9/12 (75.00) |
| Seizures | 1/8 (12.50) | 5/15 (33.33) | 6/23 (26.09) |
In the present study, the proband was also
considered to have PKD. A renal ultrasound conducted at the age of
4 months revealed enhanced renal echogenicity. Furthermore,
whole-exome sequencing in the proband identified pathogenic
variants in the PKD1 gene. A familial investigation further
indicated that the mother, aunt and grandmother of the proband had
previously been diagnosed with PKD. ADPKD is a common genetic renal
disorder with an estimated worldwide incidence of 1:1,000. ADPKD is
frequently associated with progressive renal failure (18), and variants in the PKD1 gene
are responsible for ~85% of ADPKD cases. Trio-WES analysis
identified a heterozygous pathogenic variant [c.8985delC,
p.(Ser2996fs*78)] in exon 25 of the PKD1 gene in both the
proband and the mother. Further validation in other family members
by Sanger sequencing confirmed that this heterozygous variant was
also detected in the affected aunt and grandmother of the patient,
but it was not detected in other unaffected family members. There
was co-segregation of the variant with the disease phenotype in
this family. The novel frameshift variant was predicted to be
disease-causing by MutationTaster, resulting in a premature
termination codon or a translational frameshift, leading to the
production of a truncated protein and markedly reduced mRNA levels
due to nonsense-mediated mRNA decay. Consistent with the ACMG/AMP
guidelines, this frameshift variant was classified as pathogenic,
with the evidence criteria PVS1, PM2_supporting, PP1 and PP4. This
finding confirmed that PKD1 defects are likely to be the
cause of PKD in this family. ADPKD is characterized by
age-dependent and progressive clinical manifestations, usually
becoming apparent in adulthood (18). While the proband, who was only 4
months old, had not shown typical PKD, these symptoms are expected
to become more evident as the proband grows older. Therefore, a
long-term follow-up plan needs to be established for this patient,
with regular monitoring of renal ultrasound, glomerular filtration
rate and urinary protein levels, to detect renal structural or
functional abnormalities at an early stage and intervene in a
timely manner. Notably, studies by Mullegama et al (4) and Yuan et al (9) reported two patients with STAG2
variants who exhibited distinct renal anomalies, one presented with
polycystic kidneys, while the other had a solitary kidney; however,
genetic testing in these cases did not identify variants associated
with PKD or renal dysplasia. Although no definitive studies have
yet explored the specific role of STAG2 in kidney development, its
widespread expression in various tissues, including the kidneys,
and its critical role in cell proliferation, differentiation and
gene regulation suggest that STAG2 may influence kidney development
(5,7-8,10,24).
For instance, in renal cancer tissues, the expression level of
STAG2 is markedly reduced, and its function is closely
associated with cell proliferation and migratory capacity,
indicating a potential role in the normal physiological processes
of kidney cells (23).
Additionally, while STAG2 and polycystin-1 are involved in
different biological pathways, their functions may be
interconnected at the cellular level. For instance, abnormalities
in cell division and proliferation caused by STAG2 mutations
may indirectly affect kidney development or tissue homeostasis,
thereby potentially influencing the manifestation of symptoms
associated with polycystin-1; however, there is currently no direct
evidence to suggest a clear molecular interaction between STAG2 and
polycystin-1. In the present report, although pathogenic variants
in the PKD1 gene were detected in the patients, it cannot be
excluded that STAG2 gene variants may be associated with
renal abnormalities. Furthermore, to accurately assess the role of
STAG2 in kidney development, its potential interaction with
polycystin-1 and the impact of STAG2 variants on related
diseases, further functional studies involving larger patient
cohorts are required.
In conclusion, in the present study, a novel de
novo STAG2 variant was identified in a Chinese female infant
with MKMS, expanding the clinical and genetic spectrum of
STAG2-related disorders. Phenotypic analysis of additional
25 patients revealed marked heterogeneity; however, common features
included intellectual disability, brain abnormalities, dysmorphic
features and skeletal anomalies. Notably, loss-of-function variants
were associated with more severe phenotypes compared with missense
or in-frame variants, likely due to the complete loss of
STAG2 protein function. Additionally, a sex-biased
distribution of variant types was observed, in which male patients
predominantly harbored hemizygous missense variants, whereas female
patients typically harbored heterozygous null variants. This
pattern suggests potential sex-biased differences in disease
severity, possibly influenced by X-chromosome inactivation or
residual protein function in male patients. A pathogenic
PKD1 variant was also identified in the present patient,
providing an independent explanation for the observed renal
abnormalities. However, it is noteworthy that renal anomalies,
including cystic dysplasia and renal hypoplasia, have also been
reported in other STAG2 variant carriers without concurrent
PKD1 variants or other identified nephropathy-associated
genetic alterations. This phenotypic overlap suggests a potential
pleiotropic role of STAG2 in renal organogenesis.
Nevertheless, the mechanistic relationship between STAG2
variants and renal pathology remains poorly understood. In summary,
the present findings emphasized the importance of genetic testing
in diagnosing STAG2-related disorders. Future research
should prioritize functional studies in larger cohorts to elucidate
the molecular mechanisms of STAG2 and its potential role in
renal development. Such insights are critical for developing
targeted therapies and improving the clinical management of
patients with STAG2-related conditions.
Supplementary Material
Clinical features of the proband. (A)
An ultrasound examination at the age of 4 months revealed markedly
increased echogenicity in the left kidney. (B) At 4 months of age,
an MRI scan of the brain revealed no abnormalities.
Acknowledgements
Not applicable.
Funding
Funding: The present study was funded by the Health Department
of Guangxi Province (grant nos. Z-A20220256, Z20190311 and
Z20210309).
Availability of data and materials
The sequencing data generated in the present study
may be found in the Sequence Read Archive database under accession
number PRJNA1321735 or at the following URL: https://www.ncbi.nlm.nih.gov/sra/PRJNA1321735.
The other data generated in the present study may be requested from
the corresponding author.
Authors' contributions
QY and JL designed the study and drafted the
manuscript. QiaZ, SheY, XZ, YR, SZ, ShaY, QinZ and ZQ collected the
patients' clinical information and analyzed the WES data. QY and
QiaZ revised the manuscript. All authors contributed to the
coordination of the study and revised the manuscript. QY and JL
confirm the authenticity of all the raw data. All authors read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
The present study, involving the use of genetic
testing for diagnosis, was approved by the Ethics Committee of
Maternal and Child Health Hospital of Guangxi Zhuang Autonomous
Region (approval no. METc 2017-2-11), and adhered to the principles
of The Declaration of Helsinki. Written informed consent for
genetic testing was obtained from the parents of the affected
individual and the other relatives examined.
Patient consent for publication
Written informed consent was obtained from all adult
patients and the parents of the minor individual for the
publication of any potentially identifiable images or data included
in this article.
Competing interests
The authors declare that they have competing
interests.
References
|
1
|
Losada A: Cohesin in cancer: Chromosome
segregation and beyond. Nat Rev Cancer. 14:389–393. 2014.PubMed/NCBI View
Article : Google Scholar
|
|
2
|
Musio A, Selicorni A, Focarelli ML,
Gervasini C, Milani D, Russo S, Vezzoni P and Larizza L: X-linked
cornelia de Lange syndrome owing to SMC1L1 variants. Nat Genet.
38:528–530. 2006.
|
|
3
|
Solomon DA, Kim T, Diaz-Martinez LA, Fair
J, Elkahloun AG, Harris BT, Toretsky JA, Rosenberg SA, Shukla N,
Ladanyi M, et al: Variantal inactivation of STAG2 causes aneuploidy
in human cancer. Science. 333:1039–1043. 2011.
|
|
4
|
Mullegama SV, Klein SD, Mulatinho MV,
Senaratne TN and Singh K: UCLA Clinical Genomics Center. Nguyen DC,
Gallant NM, Strom SP, Ghahremani S, et al: De novo loss-of-function
variants in STAG2 are associated with developmental delay,
microcephaly, and congenital anomalies. Am J Med Genet A.
173:1319–1327. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Soardi FC, Machado-Silva A, Linhares ND,
Zheng G, Qu Q, Pena HB, Martins TMM, Vieira HGS, Pereira NB,
Melo-Minardi RC, et al: Familial aSTAG2 germline variant
defines a new human cohesinopathy. NPJ Genom Med. 2(7)2017.
|
|
6
|
Aoi H, Lei M, Mizuguchi T, Nishioka N,
Goto T, Miyama S, Suzuki T, Iwama K, Uchiyama Y, Mitsuhashi S, et
al: Nonsense variants of STAG2 result in distinct congenital
anomalies. Hum Genome Var. 7(26)2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Mondal G, Stevers M, Goode B, Ashworth A
and Solomon DA: A requirement for STAG2 in replication fork
progression creates a targetable synthetic lethality in
cohesin-mutant cancers. Nat Commun. 10(1686)2019.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Yu L, Sawle AD, Wynn J, Aspelund G, Stolar
CJ, Arkovitz MS, Potoka D, Azarow KS, Mychaliska GB, Shen Y, et al:
Increased burden of de novo predicted deleterious variants in
complex congenital diaphragmatic hernia. Hum Mol Genet.
24:4764–4773. 2015.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Yuan B, Neira J, Pehlivan D, Santiago-Sim
T, Song X, Rosenfeld J, Posey JE, Patel V, Jin W, Adam MP, et al:
Clinical exome sequencing reveals locus heterogeneity and
phenotypic variability of cohesinopathies. Genet Med. 21:663–675.
2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kruszka P, Berger SI, Casa V, Dekker MR,
Gaesser J, Weiss K, Martinez AF, Murdock DR, Louie RJ, Prijoles EJ,
et al: Cohesin complex-associated holoprosencephaly. Brain.
142:2631–2643. 2019.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Epilepsy Genetics Initiative. The epilepsy
genetics initiative: Systematic reanalysis of diagnostic exomes
increases yield. Epilepsia. 60:797–806. 2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Provenzano A, La Barbera A, Lai F, Perra
A, Farina A, Cariati E, Zuffardi O and Giglio S: Non-invasive
detection of a de novo frameshift variant of stag2 in a
female fetus: Escape genes influence the manifestation of X-linked
diseases in females. J Clin Med. 11(4182)2022.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Freyberger F, Kokotović T, Krnjak G,
Frković SH and Nagy V: Expanding the known phenotype of
Mullegama-Klein-Martinez syndrome in male patients. Hum Genome Var.
8(37)2021.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Harris PC and Torres VE: Polycystic kidney
disease. Annu Rev Med. 60:321–337. 2009.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Hughes J, Ward CJ, Peral B, Aspinwall R,
Clark K, San Millán JL, Gamble V and Harris PC: The polycystic
kidney disease 1 (PKD1) gene encodes a novel protein with multiple
cell recognition domains. Nat Genet. 10:151–160. 1995.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Qian F, Germino FJ, Cai Y, Zhang X, Somlo
S and Germino GG: PKD1 interacts with PKD2 through a probable
coiled-coil domain. Nat Genet. 16:179–183. 1997.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Grantham JJ, Mulamalla S and
Swenson-Fields KI: Why kidneys fail in autosomal dominant
polycystic kidney disease. Nat Rev Nephrol. 7:556–566.
2011.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Cornec-Le Gall E, Alam A and Perrone RD:
Autosomal dominant polycystic kidney disease. Lancet. 393:919–935.
2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al: ACMG
Laboratory quality assurance committee. Standards and guidelines
for the interpretation of sequence variants: A joint consensus
recommendation of the American College of Medical genetics and
genomics and the association for molecular pathology. Genet Med.
17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Rivera-Muñoz EA, Milko LV, Harrison SM,
Azzariti DR, Kurtz CL, Lee K, Mester JL, Weaver MA, Currey E,
Craigen W, et al: ClinGen variant curation expert panel experiences
and standardized processes for disease and gene-level specification
of the ACMG/AMP guidelines for sequence variant interpretation. Hum
Mutat. 39:1614–1622. 2018.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Apgar V: A proposal for a new method of
evaluation of the newborn infant. Originally published in July.
1953, volume 32, pages 250-259. Anesth Analg. 120:1056–1059.
2015.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Spencer-Smith MM, Spittle AJ, Lee KJ,
Doyle LW and Anderson PJ: Bayley-III cognitive and language scales
in preterm children. Pediatrics. 135:e1258–e1265. 2015.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Cheng N, Li G, Kanchwala M, Evers BM, Xing
C and Yu H: STAG2 promotes the myelination transcriptional program
in oligodendrocytes. Elife. 12(e77848)2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Yu C, Dai D and Xie J: Molecular subtype
classification of papillary renal cell cancer using miRNA
expression. Onco Targets Ther. 12:2311–2322. 2019.PubMed/NCBI View Article : Google Scholar
|