Introduction
Hypogonadotropic hypogonadism (HH) is a rare
disorder caused by the decreased secretion or dysfunction of
gonadotropin-releasing hormone (GnRH). In addition to the acquired
causes of HH, congenital etiologies have also been established,
which are less frequently observed (1). Congenital HH (CHH) has an estimated
prevalence of 1 in 15,000 to 50,000 and shows a significant male
predominance with a male-to-female ratio of ~3.6:1. Although the
majority of the cases are associated with anosmia, about one-third
of the patients have a normosmic presentation. Among the >40
mutations identified in CHH, the most common is the KAL1
mutation, which typically presents with symptoms of anosmia
(2,3). Among the rarer causes, mutations in
the kisspeptin-1 receptor gene (KISS1R/GPR54), which follow
an autosomal recessive inheritance pattern, have been identified.
The KISS1R gene encodes a G protein-coupled receptor
expressed in the hypothalamus that plays a critical role in
kisspeptin signaling. Mutations of the KISS1R gene impair
kisspeptin-mediated signaling, resulting in a reduction in GnRH
secretion. Thus, the pituitary gland receives insufficient GnRH
stimulation, resulting in inadequate secretion of
follicle-stimulating hormone (FSH) and luteinizing hormone (LH).
Unlike many other mutations associated with CHH, KISS1R
mutations may not be accompanied by symptoms of anosmia (4,5). CHH
is characterized by low or inappropriately normal levels of FSH and
LH, accompanied by decreased serum testosterone concentrations. In
patients who are diagnosed during adulthood, the clinical findings
may include reduced testicular volume (<4 ml), underdeveloped
secondary sexual characteristics, micropenis, erectile dysfunction,
decreased libido, impaired sperm quality, gynecomastia and various
psychiatric symptoms (6). One
treatment option for CHH is pulsatile GnRH therapy. Furthermore,
human chorionic gonadotropin (hCG) alpha, which is purified from
the urine of pregnant women, can be used as a substitute for LH.
This treatment has a success rate of ~75%. The combination therapy
of hCG alpha and FSH represents another therapeutic approach
(7). This study aimed to
investigate a 21-year-old male diagnosed with CHH due to a rare
mutation in the KISS1R gene.
Case report
This study included a 21-year-old, single male
patient who presented to the Endocrinology and Metabolic Diseases
Clinic of Ankara Etlik City Hospital (Ankara, Turkey) in July 2024
with complaints of micropenis, decreased muscle strength, sparse
body hair and erectile dysfunction. The patient had no history of
chronic illness, trauma or previous surgery. The patient's parents
were first-degree cousins. The patient had a 14-year-old sister who
presented with delayed puberty, in whom a genetic analysis revealed
a homozygous KISS1R mutation (c.505+2T>G variant). The
patient's sister is currently under follow-up and treatment at
Ankara Etlik City Hospital Pediatric Endocrinology Clinic (Ankara,
Turkey). The patient's other sibling, an 18-year-old sister, is
healthy and 20 weeks pregnant at the time of writing the
manuscript. Physical examination of the patient revealed sparse
facial and pubic hair, increased subcutaneous adiposity,
gynecomastia, micropenis and small testes. The patient's body mass
index was 27.3 kg/m² (Normal range: 18.5-24.9 kg/m2)
(height, 176 cm; weight, 84.6 kg). Olfactory and auditory functions
were intact.
The laboratory examination revealed a FSH level of
5.58 IU/l (reference: 1.5-12.4 IU/l), LH of 4.19 IU/l (reference:
1.7-8.6 IU/l) and total testosterone of 54.9 ng/dl (reference:
249-836 ng/dl), which is consistent with HH (8). Other anterior pituitary hormone
levels and routine biochemical parameters were within normal
limits. A 0.1 mg GnRH stimulation test was performed (9). The response shown by the LH and FSH
levels confirmed the hypothalamic origin of HH (Table I).
 | Table IGonadotropin-releasing hormone
stimulation test results. |
Table I
Gonadotropin-releasing hormone
stimulation test results.
| Time-point, min | FSH, IU/l | LH, IU/l |
|---|
| 0 | 5.81 | 4.2 |
| 30 | 8.8 | 17.6 |
| 60 | 9.91 | 17.1 |
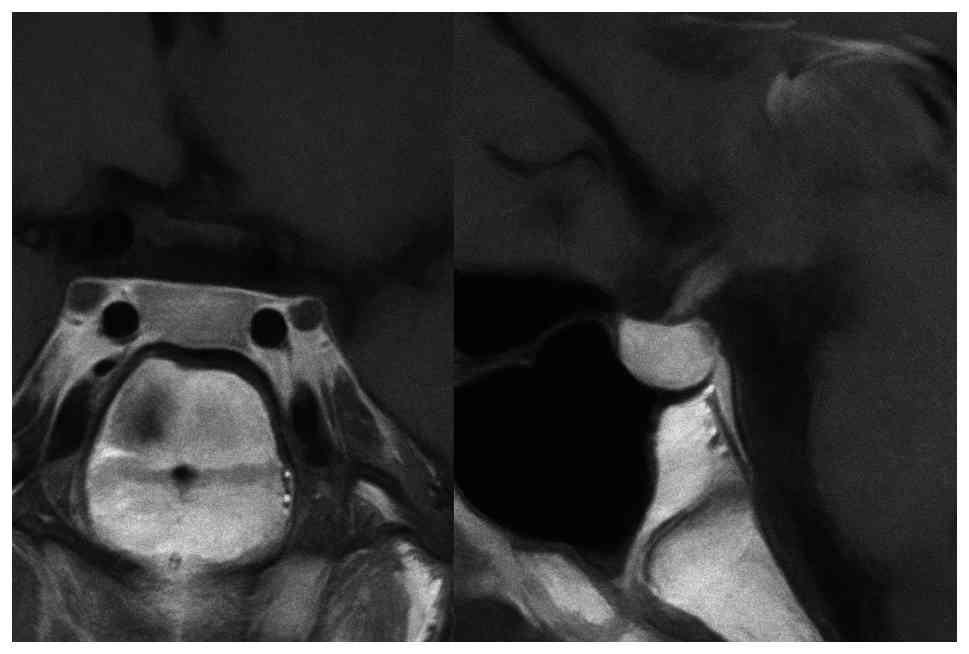
The pituitary magnetic resonance imaging (MRI) with
intravenous contrast revealed a normal pituitary gland measuring 6
mm in height, with no evidence of mass lesions (Fig. 1). Although semen analysis was
planned, the patient was unable to complete the procedure. The bone
mineral density assessment by dual-energy X-ray absorptiometry
showed a Z-score of -2.0 at the femoral neck and -2.5 at the lumbar
spine (L1-L4) (data not shown; expected normal Z score for age,
>-2.0), indicating decreased bone density (10). Genomic DNA was isolated from
peripheral blood samples using the QIAamp DNA Blood Kit (Qiagen
GmbH) on an automated extraction system. Library preparation was
performed using the SOPHiA DDM™ Clinical Exome Solution v3 (SOPHiA
Genetics) and sequencing was carried out on the NextSeq 2000
platform (Illumina, Inc.) in accordance with the manufacturer's
standard protocols. Raw sequencing data were analyzed using the
SOPHiA DDM™ platform (SOPHiA Genetics) and sequence reads were
aligned to the human reference genome GRCh37. A clinical gene panel
associated with hypogonadism was analyzed. Variants were filtered
based on a minimum read depth of 30x and a minor allele frequency
of <1%. Splice-site variants were evaluated within ±20 base
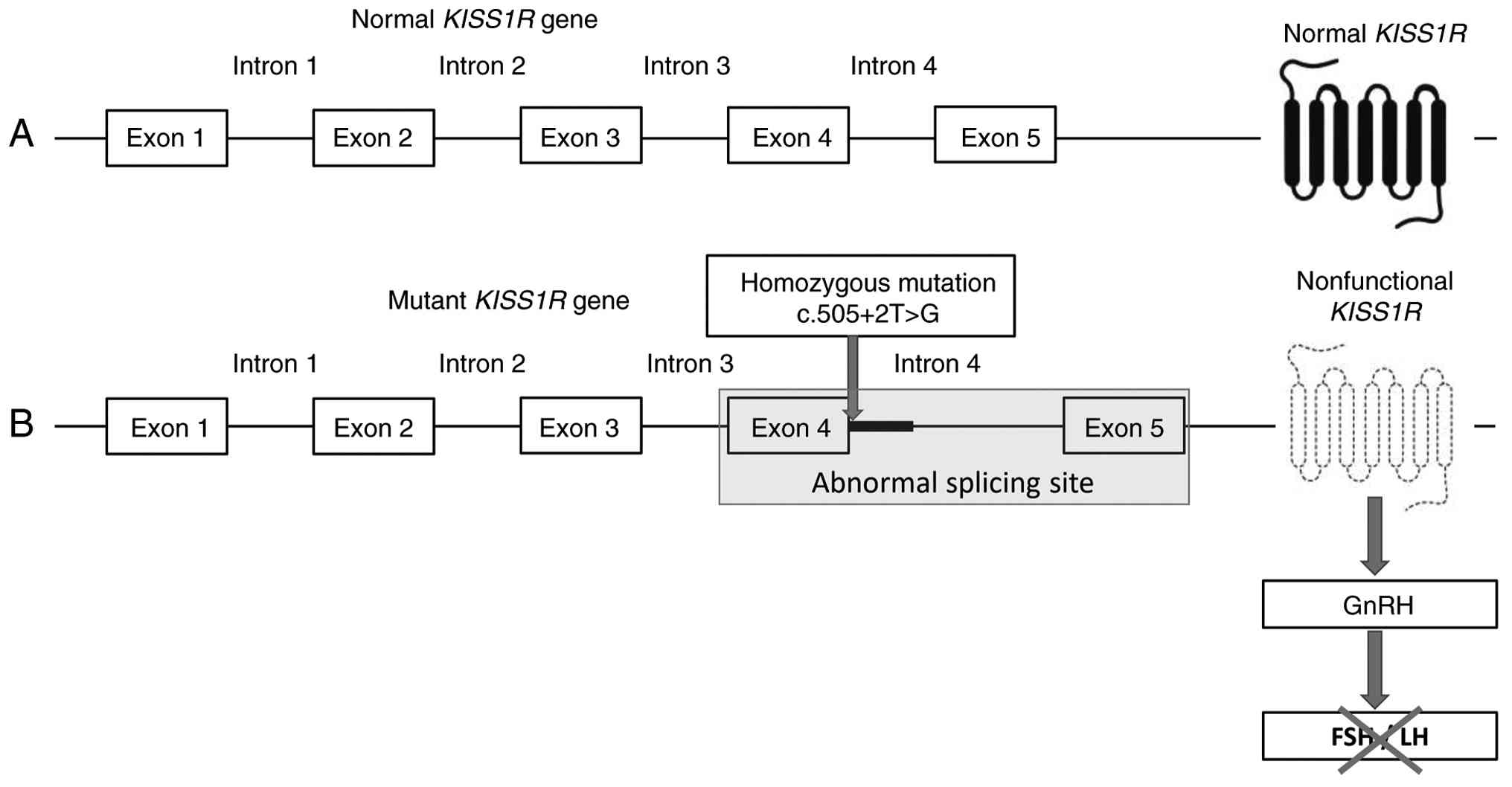
pairs of exon-intron boundaries. Genetic analysis identified a
homozygous c.505+2T>G variant, which was classified as likely
pathogenic according to the American College of Medical Genetics
and Genomics guidelines (Fig. 2).
The variant was subsequently confirmed by Sanger sequencing using
the BigDye™ Terminator v3.1 Cycle Sequencing Kit (Thermo Fisher
Scientific, Inc.) on an Applied Biosystems 3130 Genetic Analyzer
(Thermo Fisher Scientific, Inc.) (11). Based on this result, a definitive
diagnosis of CHH due to KISS1R mutation was established.
The patient was started on subcutaneous hCG alpha
treatment at a dose of 6,500 IU once a week. By the 12th week of
treatment, a significant increase in the total testosterone levels
was observed, along with significant improvements in muscle
strength, body hair growth and erectile function. Furthermore, the
semen analysis performed during this period revealed an adequate
sperm reserve and function (12).
Clinical and laboratory improvements were maintained through to the
24th week of treatment (Table
II). At that time, hCG alpha therapy was continued at the same
dose, and the patient was scheduled for regular endocrinological
follow-up every 3-6 months. As hCG alpha provides an exogenous
LH-like stimulus; its beneficial effects on testosterone production
and spermatogenesis are expected to be dependent on ongoing
treatment and to diminish if therapy is discontinued (13). At the time of writing, the patient
remains on hCG alpha with sustained clinical and biochemical
response.
| Table IIBiochemical, semen analysis and
scrotal USG results before and after treatment. |
Table II
Biochemical, semen analysis and
scrotal USG results before and after treatment.
| Parameters | Normal values | Week 0 | Week 12 | Week 24 |
|---|
| FSH, IU/l | 1.5-12.4 | 5.58 | 0.39 | 1.24 |
| LH, IU/l | 1.7-8.6 | 4.19 | 0.45 | 1.41 |
| Total testosterone,
ng/dl | 249-836 | 54.9 | 628 | 474 |
| TSH, mIU/l | 0.4-4.2 | 2.12 | 1.82 | 1.08 |
| Free T4, ng/dl | 0.9-1.7 | 1.28 | 0.91 | 1.15 |
| Free T3, ng/l | 2-4.4 | 3.38 | 3.98 | 3.98 |
| Spermiogram | | | | |
|
Sperm
concentration, million/ml | ≥15 | - | 15.6 | 74.6 |
|
Total
mobility, % | ≥40 | - | 12 | 19 |
|
Motile sperm
concentration, million/ml | ≥5 | - | 1.2 | 14.2 |
|
Concentration
of progressively motile sperm, million/ml | ≥5 | - | 0 | 11.2 |
|
Total sperm
count, million | ≥39 | - | 25 | 37.3 |
| Testicular volumes on
USG, right/left, ml | ≥10 | 7/6 | 11/6 | 11/6 |
Discussion
This study assessed a 21-year-old male presenting
with decreased body hair, micropenis and erectile dysfunction for
HH. After a thorough evaluation to exclude acquired causes of HH
(including chronic systemic disease, hyperprolactinemia, previous
cranial irradiation or trauma, and pituitary or hypothalamic
lesions on MRI), a congenital etiology was suspected. The genetic
testing results revealed a homozygous mutation in the KISS1R
gene. To our knowledge, this specific variant, c.505+2T>G, has
not been previously reported in the literature. Thus, this case
represents the first documented instance of this novel
KISS1R mutation in a patient with CHH, expanding the known
genetic spectrum of the disorder.
CHH is a rare hypothalamic-pituitary disorder
characterized by absent or delayed pubertal development. This case
shows a typical clinical presentation of CHH in a young male with
absent pubertal progression, low serum gonadotropin levels and a
normal pituitary gland on contrast-enhanced MRI. CHH results from
defects in the migration or secretion of GnRH neurons. Both the
sporadic and familial forms of CHH have been described (2). The absence of anosmia in this patient
helped distinguish the condition from Kallmann syndrome (14). The genetic analysis results
revealed a KISS1R mutation, consistent with normosmic CHH
(15). The KISS1R gene
encodes a G protein-coupled receptor for the KISS1 peptide.
It comprises five exons and four introns (Fig. 2). c.505+2T>G is an intronic
mutation located in intron 4, and intronic mutations are often
polymorphic rather than structural/functional mutations. However,
of note, the c.505+2T>G mutation reported in the present study
was located close to the splicing junctional site (the junction of
exon 4 and intron 4), suggesting the possibility of certain
abnormalities in the receptor. However, further confirmatory
molecular experiments are needed to prove its pathological role
(16,17).
A study by Francou et al (18) assessed 603 patients with normosmic
CHH and identified KISS1R mutations in ~2% of all cases,
emphasizing the rarity of this genetic variant. During the
diagnostic evaluation, significantly low serum levels of LH and FSH
in conjunction with testosterone deficiency were found. This
classic hormonal profile was also observed in the patient of the
present study. The primary differential diagnosis for CHH is a
constitutional delay of puberty; however, unlike CHH, individuals
with constitutional delay typically experience spontaneous pubertal
progression over time, which is not expected in CHH (19).
The primary goals of treatment in CHH include
achieving an age-appropriate virilization, preserving or restoring
fertility, optimizing sexual function, attaining normal adult
height and supporting psychosocial well-being (20). While exogenous testosterone is
effective in promoting the development of secondary sexual
characteristics, it suppresses intratesticular testosterone
production and inhibits spermatogenesis and testicular volume
increase (21). In patients such
as the case of the present study, where both virilization and
fertility are desired, the treatment options include pulsatile GnRH
therapy, hCG alpha monotherapy or combined treatment of hCG alpha
and FSH (22). However, several
studies have shown that in patients with a history of
cryptorchidism and baseline testicular volumes <4 ml, the
likelihood of achieving fertility through such treatments remains
low (23-25).
A 19-year-old male patient with CHH, as reported by Brioude et
al (26), remained azoospermic
after 1 year of testosterone therapy. Given the patient's desire
for fertility, combined gonadotropin therapy was initiated, which
comprised subcutaneous hCG alpha at 1,500 IU three times per week
and FSH at 150 IU three times per week. After 22 months of
treatment, the patient showed a significant increase in testicular
volume and normalization of the sperm count, resulting in a
successful pregnancy in the patient's partner. In this case of CHH,
the treatment objective was to achieve both virilization and
fertility. Thus, subcutaneous hCG alpha therapy was initiated at a
total weekly dose of 6,500 IU. The follow-up assessments at weeks
12 and 24 showed significant improvements in secondary sexual
characteristics and adequate progression of spermatogenesis, as
demonstrated by semen analysis.
In conclusion, the present study reported a novel
KISS1R gene variant (c.505+2T>G) associated with CHH in a
young male patient. The clinical presentation was consistent with
the classical features of CHH and the patient responded favorably
to hCG alpha therapy, which was aimed at achieving both
virilization and fertility. This case emphasizes the importance of
considering CHH in the differential diagnosis of delayed puberty,
as early identification and treatment are essential to prevent
long-term physical and psychosocial consequences. Furthermore,
genetic analysis and counseling play a critical role in confirming
its diagnosis and guiding its management. The present findings
contribute to the expanding body of literature on
KISS1R-related CHH and introduce a previously unreported
mutation, providing valuable insights into the genetic spectrum of
this rare condition.
Acknowledgements
Not applicable.
Funding
Funding: No funding was received.
Availability of data and materials
The data generated in the present study are included
in the figures and/or tables of this article.
Authors' contributions
BM and EU identified and managed the case, curated
the clinical data, performed the literature review and drafted the
manuscript. AB conducted and interpreted the genetic testing
confirming the novel variant and authored the genetics section. SH
and IOU contributed to data acquisition (laboratory, imaging and
follow-up) and assisted in manuscript preparation. MK contributed
to the endocrine assessment and interpretation of biochemical
findings. TA and EC collected the data, performed the statistical
analyses, critically reviewed the article for significant
intellectual content and provided overall supervision. BM and EU
confirm the authenticity of all the raw data. All authors have read
and approved the final manuscript.
Ethics approval and consent to
participate
The study was conducted in accordance with the
ethical standards of institutional and national research
committees, the Declaration of Helsinki and its later amendments,
or comparable ethical standards. This case report does not require
ethics committee approval from our affiliated institution.
Patient consent for publication
The patient was informed of the purpose of the
publication. The patient provided written consent to the
publication of his clinical findings and imaging results in the
journal.
Competing interests
The authors have no competing interests to declare
that are relevant to the content of this article.
References
|
1
|
Bangalore Krishna K, Fuqua JS and Witchel
SF: Hypogonadotropic hypogonadism. Endocrinol Metab Clin North Am.
53:279–292. 2024.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Carriço JN, Gonçalves CI, Al-Naama A, Syed
N, Aragüés JM, Bastos M, Fonseca F, Borges T, Pereira BD,
Pignatelli D, et al: Genetic architecture of congenital
hypogonadotropic hypogonadism: Insights from analysis of a
Portuguese cohort. Hum Reprod Open. 2024(hoae053)2024.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Al Sayed Y and Howard SR: Panel testing
for the molecular genetic diagnosis of congenital hypogonadotropic
hypogonadism-a clinical perspective. Eur J Hum Genet. 31:387–394.
2023.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Szeliga A, Kunicki M, Maciejewska-Jeske M,
Rzewuska N, Kostrzak A, Meczekalski B, Bala G, Smolarczyk R and
Adashi EY: The genetic backdrop of hypogonadotropic hypogonadism.
Int J Mol Sci. 22(13241)2021.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Vezzoli V, Hrvat F, Goggi G, Federici S,
Cangiano B, Quinton R, Persani L and Bonomi M: Genetic architecture
of self-limited delayed puberty and congenital hypogonadotropic
hypogonadism. Front Endocrinol (Lausanne).
13(1069741)2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Young J, Xu C, Papadakis GE, Acierno JS,
Maione L, Hietamäki J, Raivio T and Pitteloud N: Clinical
management of congenital hypogonadotropic hypogonadism. Endocr Rev.
40:669–710. 2019.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Salvio G, Balercia G and Kadioglu A:
Hypogonadotropic hypogonadism as a cause of NOA and its treatment.
Asian J Androl. 27:322–329. 2025.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Dwyer AA, McDonald IR and Quinton R:
Current landscape of fertility induction in males with congenital
hypogonadotropic hypogonadism. Ann NY Acad Sci. 1540:133–146.
2024.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Boehm U, Bouloux PM, Dattani MT, de Roux
N, Dodé C, Dunkel L, Dwyer AA, Giacobini P, Hardelin JP, Juul A, et
al: Expert consensus document: European Consensus Statement on
congenital hypogonadotropic hypogonadism-pathogenesis, diagnosis
and treatment. Nat Rev Endocrinol. 11:547–564. 2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Haseltine KN, Chukir T, Smith PJ, Jacob
JT, Bilezikian JP and Farooki A: Bone mineral density: Clinical
relevance and quantitative assessment. J Nucl Med. 62:446–454.
2021.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Cooper TG, Noonan E, von Eckardstein S,
Auger J, Baker HW, Behre HM, Haugen TB, Kruger T, Wang C, Mbizvo MT
and Vogelsong KM: World Health Organization reference values for
human semen characteristics. Hum Reprod Update. 16:231–245.
2010.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Coviello AD, Matsumoto AM, Bremner WJ,
Herbst KL, Amory JK, Anawalt BD, Sutton PR, Wright WW, Brown TR,
Yan X, et al: Low-dose human chorionic gonadotropin maintains
intratesticular testosterone in normal men with
testosterone-induced gonadotropin suppression. J Clin Endocrinol
Metab. 90:2595–2602. 2005.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Kumar Yadav R, Qi B, Wen J, Gang X and
Banerjee S: Kallmann syndrome: Diagnostics and management. Clin
Chim Acta. 565(119994)2025.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Moalla M, Hadj Kacem F, Al-Mutery AF,
Mahfood M, Mejdoub-Rekik N, Abid M, Mnif-Feki M and Hadj Kacem H:
Nonstop mutation in the Kisspeptin 1 receptor (KISS1R) gene causes
normosmic congenital hypogonadotropic hypogonadism. Journal of
assisted reproduction and genetics. J Assist Reprod Genet.
36:1273–1280. 2019.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Hwang JS: The genes associated with
gonadotropin-releasing hormone-dependent precocious puberty. Korean
J Pediatr. 55:6–10. 2012.PubMed/NCBI View Article : Google Scholar
|
|
17
|
de Roux N, Genin E, Carel JC, Matsuda F,
Chaussain JL and Milgrom E: Hypogonadotropic hypogonadism due to
loss of function of the KiSS1-derived peptide receptor GPR54. Proc
Natl Acad Sci USA. 100:10972–10976. 2003.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Francou B, Paul C, Amazit L, Cartes A,
Bouvattier C, Albarel F, Maiter D, Chanson P, Trabado S,
Brailly-Tabard S, et al: Prevalence of KISS1 receptor mutations in
a series of 603 patients with normosmic congenital
hypogonadotrophic hypogonadism and characterization of novel
mutations: A single-centre study. Hum Reprod. 31:1363–1374.
2016.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Gaudino R, De Filippo G, Bozzola E,
Gasparri M, Bozzola M, Villani A and Radetti G: Current clinical
management of constitutional delay of growth and puberty. Ital J
Pediatr. 48(45)2022.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Lambert AS and Bouvattier C: Puberty
induction with recombinant gonadotropin: What impact on future
fertility? Ann Endocrinol (Paris). 83:159–163. 2022.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Naelitz BD, Momtazi-Mar L, Vallabhaneni S,
Cannarella R, Vij SC, Parekh NV, Bole R and Lundy SD: Testosterone
replacement therapy and spermatogenesis in reproductive age men.
Nat Rev Urol. 22:703–719. 2025.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Zheng Y, Bai HZ, Zhao GC, Tian K, Yue JT,
Li DM and Jiang XH: Comparison of outcomes between pulsatile
gonadotropin releasing hormone and combined gonadotropin therapy of
spermatogenesis in patients with congenital hypogonadotropic
hypogonadism. Reprod Biol Endocrinol. 23(46)2025.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Lee HS, Shim YS and Hwang JS: Treatment of
congenital hypogonadotropic hypogonadism in male patients. Ann
Pediatr Endocrinol Metab. 27:176–182. 2022.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Swee DS and Quinton R: Managing congenital
hypogonadotrophic hypogonadism: A contemporary approach directed at
optimizing fertility and long-term outcomes in males. Ther Adv
Endocrinol Metab. 10(2042018819826889)2019.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Liu Z, Mao J, Xu H, Wang X, Huang B, Zheng
J, Nie M, Zhang H and Wu X: Gonadotropin-induced spermatogenesis in
CHH patients with cryptorchidism. Int J Endocrinol.
2019(6743489)2019.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Brioude F, Bouligand J, Francou B, Fagart
J, Roussel R, Viengchareun S, Combettes L, Brailly-Tabard S, Lombès
M, Young J and Guiochon-Mantel A: Two families with normosmic
congenital hypogonadotropic hypogonadism and biallelic mutations in
KISS1R (KISS1 receptor): Clinical evaluation and molecular
characterization of a novel mutation. PLoS One.
8(e53896)2013.PubMed/NCBI View Article : Google Scholar
|