1. Introduction
Metabolic dysfunction-associated steatohepatitis
(MASH) is characterized by hepatic steatosis, inflammation,
hepatocellular injury and progressive fibrosis, becoming a major
cause of cirrhosis and hepatocellular carcinoma (HCC) (1,2).
Despite the growing global prevalence of MASH, effective
pharmacological therapies remain elusive, underscoring the need for
a comprehensive understanding of the cellular mechanisms that drive
disease progression (3). Among the
diverse hepatic cell types involved in MASH, hepatic stellate cells
(HSCs) represent the central fibrogenic population, with
accumulating evidence highlighting cellular senescence as a
critical and context-dependent regulator of HSC behavior (4-6).
Senescent HSCs demonstrate a dual nature. Transient
senescence may be protective during acute injury by arresting
proliferation, upregulating matrix-degrading enzymes and enhancing
immune-mediated clearance (6).
However, under the chronic metabolic and inflammatory stress
conditions typically associated with MASH, senescent HSCs persist
and ultimately acquire an expanded senescence-associated secretory
phenotype (SASP). The SASP promotes inflammation, stimulates
extracellular matrix (ECM) deposition, disrupts immune surveillance
and contributes to malignant transformation, thereby accelerating
disease progression.
This duality reflects the dynamic and multifactorial
regulation of HSC senescence, influenced by factors such as
lipotoxicity, oxidative stress, mitochondrial dysfunction (5,7),
impaired autophagy and alterations in immune surveillance (6,8,9).
However, the timing, triggers and downstream consequences of HSC
senescence remain incompletely characterized (10), as do the interactions between
senescent HSCs, hepatocytes, macrophages and other immune cell
populations (11).
The present review summarizes current knowledge
regarding both the beneficial and detrimental roles of senescent
HSCs in MASH, alongside the molecular pathways leading to HSC
senescence and SASP formation. In addition, the mechanisms by which
senescent HSCs alter the hepatic inflammatory and fibrotic
microenvironment are delineated. Finally, emerging therapeutic
strategies targeting cellular senescence, including senolytics,
senomorphics and biomarker-based approaches are highlighted, which
may offer promising avenues for modulating disease progression and
improving outcomes in MASH.
2. HSC activation and dual role in MASH
pathogenesis
Dichotomy of senescent HSCs in liver
fibrosis
Senescent HSCs function both as a protective
mechanism to limit tissue damage and as a deleterious force that
drives chronic pathological progression (7,12).
Research has shown that HSCs exhibit both an aging-related
secretory phenotype that promotes fibrosis and an anti-fibrotic
effect through cell cycle arrest (6,13).
Therefore, senescent HSCs play a dual role in MASH progression.
Certain cytokines and inflammatory factors, including TGF-β,
platelet-derived growth factor (PDGF), TNF-α, IL-1β and IL-6, serve
key roles in promoting fibrosis (14).
Related studies propose that TNF-α is a key
inflammatory factor regulated by lipopolysaccharide-induced tumor
necrosis factor (LITAF) (15-17).
Reduced nuclear translocation of LITAF diminishes TNF-α production,
thereby inhibiting HSC activation and fibrosis. In addition, TNF-α
and IL-17 exhibit a synergistic effect on HSC activity (18). A clinical experiment indicated that
IL-17 amplifies the effects of TNF-α on IL-1β and IL-6 in HSCs and
the interaction between hepatocytes and HSCs may modulate the
effects of IL-17 and TNF-α on fibrosis-related genes (19).
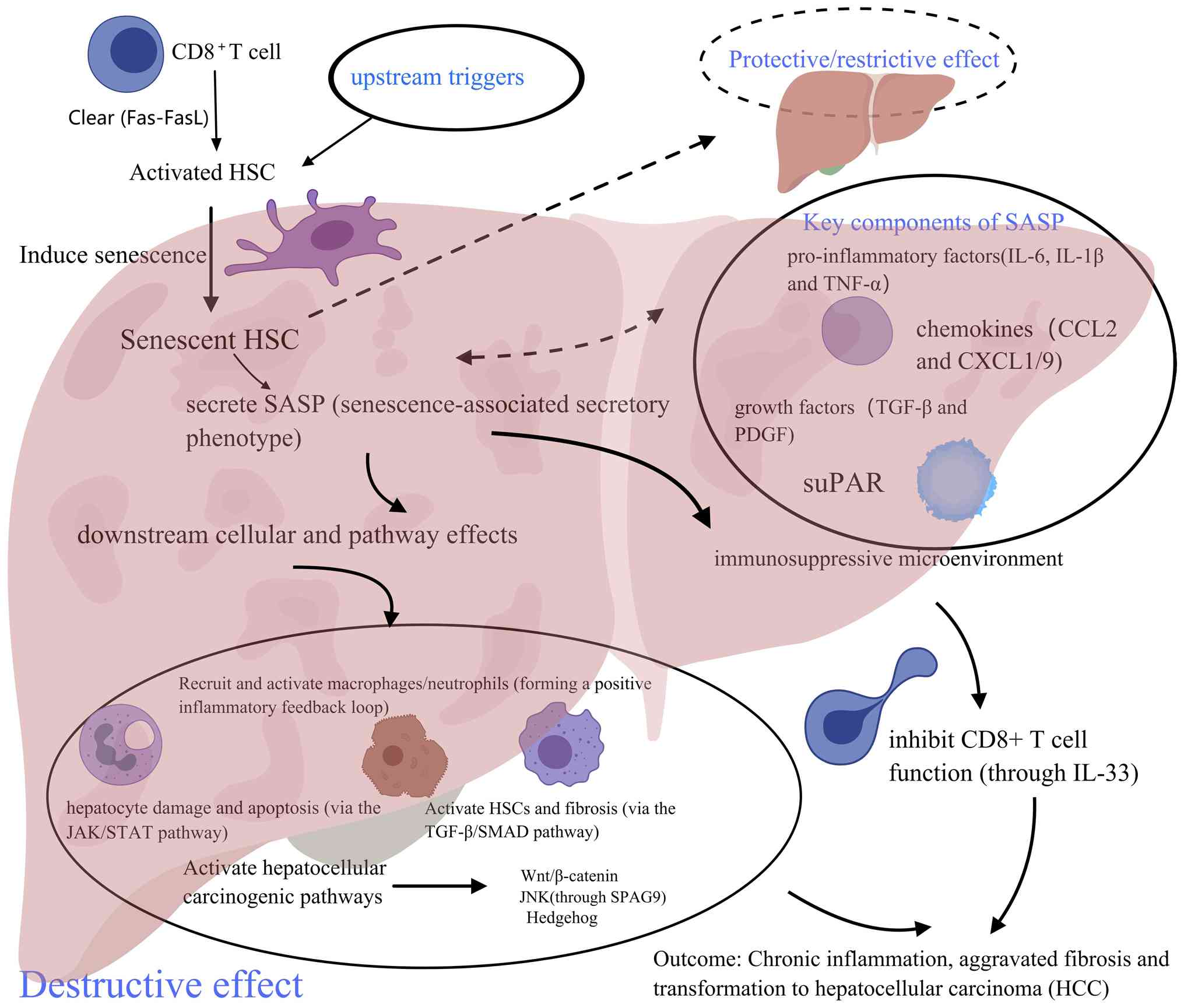
Senescent HSCs promote inflammation,
fibrosis and malignant transformation. SASP is an effective driver
of chronic inflammation and fibrosis in MASH
Senescent HSCs are potent drivers of chronic
inflammation in MASH through their SASP. The present section
examines the mechanisms by which SASP components recruit immune
cells, maintain inflammatory signaling and amplify tissue damage.
During hepatocyte fibrosis, aged HSCs may secrete a series of
specific pro-inflammatory cytokines (such as IL-1β, IL-6 and
TNF-α), chemokines [such as C-X-C motif chemokine ligand (CXCL)-1,
CXCL9 and C-C motif chemokine ligand (CCL)-2], growth factors (such
as PDGF and TGF-β) and mechanism remodeling factors, identified as
the SASP, leading to chronic inflammation (20-29).
IL-1β and CCL2 are SASP factors that can recruit and
activate Kupffer cells and monocyte-derived macrophages from the
bloodstream (30), prompting them
to secrete TNF-α and IL-6, thus generating an inflammatory positive
feedback loop. Concurrently, chemokines such as CXCL1 promote
neutrophil and monocyte infiltration, exacerbating liver
inflammation. For example, combined in vitro experiments in
a mouse model and human MASH-HCC samples revealed notably elevated
mRNA levels of IL-1β, IL-6, CXCL1 and CXCL9 within the secretome of
senescent HSCs. These factors amplified the effects of internal and
external environmental factors, exacerbating the inflammatory
microenvironment of MASH (20).
TGF-β is a core component of the SASP. Within the
liver, TGF-β induces cell senescence in both acute and chronic
liver injury models. Senescent HSCs release a number of profibrotic
factors through the SASP, with TGF-β1 serving as the core
regulatory molecule (31,32). Recent studies indicate that TGF-β1
markedly inhibits cytoglobin expression through the SMAD2/SMAD3-M1
signaling pathway, thus activating HSCs and promoting collagen
deposition, a process positively associated with advanced fibrosis
in patients with MASH. Furthermore, TGF-β initiates a positive
feedback pathway (oxidative stress-fibrosis) by regulating the
antioxidant defense system (33,34).
Soluble urokinase-type plasminogen activator
receptor (suPAR), secreted by senescent HSCs as part of the SASP
(10), has been shown to
co-localize with IL-6 in a CCL4-induced liver fibrosis model,
forming a chronic inflammatory microenvironment that activates
neighboring HSCs and recruits immune cells. In a MASH mouse model,
suPAR secretion has been shown to promote macrophage infiltration
and collagen deposition, further aggravating liver fibrosis
(35).
SASP induces hepatocyte damage and apoptosis.
Studies have shown that reactive oxygen species (ROS) and IL-6
secreted by a SASP induce mitochondrial dysfunction and DNA damage
[evidenced by increased γ-H2A.X variant histone-b (γ-H2AXb) marker]
by activating the hepatocyte janus kinase (JAK)/STAT3 pathway,
indicating that the SASP can induce hepatocyte damage and apoptosis
(36-38).
This phenomenon may serve a role in the progression of MASH.
Concurrently, senescent HSCs may alter the microenvironment and
induce DNA damage in hepatocytes by secreting IL-6 and matrix
metalloproteinases (MMPs) in the SASP. This interaction aggravates
inflammation and fosters an immune-privileged microenvironment
conducive to the development of MASH into HCC by inhibiting the
function of CD8+ T cells (39). This effect was effectively
demonstrated in MASH mouse models induced by a choline-deficient
high-fat diet or methionine-choline diet alongside programmed
death-ligand-1 knockout mouse models (29).
Senescent HSCs promote the malignant
transformation of MASH to HCC. MASH and non-alcoholic fatty
liver disease (NAFLD) are recognized as precursors to HCC (38,40).
The SASP of senescent HSCs can affect adjacent hepatocytes, which
may be in states of fatty degeneration or dysplasia, through
paracrine signaling, thereby promoting their malignant
transformation (20). A key
mechanism underlying this process is the activation of oncogenic
signaling pathways in hepatocytes by a SASP, specifically the
Wnt/β-catenin and Hedgehog pathways. In addition, proteomic
analysis indicates that sperm-associated antigen-9, secreted by
senescent HSCs, influences the growth and metastasis of liver
cancer cells through the JNK pathway, while fatty acid binding
protein-5 enhances angiogenesis, collectively facilitating the
proliferation of cancer cells (20).
In addition to directly promoting cell
transformation, a SASP also promotes conditions favorable for tumor
growth by reshaping the immune microenvironment (20). In human MASH-related HCC samples, a
recent study demonstrated that IL-33, released from senescent HSCs
and mediated by gasdermin D, inhibits the anti-fibrotic function of
CD8+ T cells by binding to ST2 receptors to activate
regulatory T cells (41). This
immunosuppressive microenvironment may hinder the clearance of
activated HSCs, providing a fertile ground for tumor cells to evade
the immune response.
SASP factors such as IL-6 and CXCL9, secreted by
senescent HSCs driven by obesity-induced intestinal flora
metabolites including deoxycholic acid, can recruit macrophages and
neutrophils, amplifying liver inflammation and activating
fibroblasts to promote collagen deposition and fibrosis. This
ultimately leads to liver cancer development (42). Senescent HSCs may also reduce the
secretion of ECM and stimulate the SASP to attract natural killer
(NK) cells for cleaning, preventing HSCs from excessive
proliferation or transdifferention into fibroblasts (43,44),
thereby preventing liver fibrosis (7). The co-localization of MMP2/9 and
fibrotic areas in liver tissue of patients with MASH, indicates
that MMP2/9 degrades normal ECM, while tissue inhibitor of
metalloproteinases-1 expression inhibits ECM clearance, leading to
an imbalance in fibrotic remodeling (Fig. 1) (38).
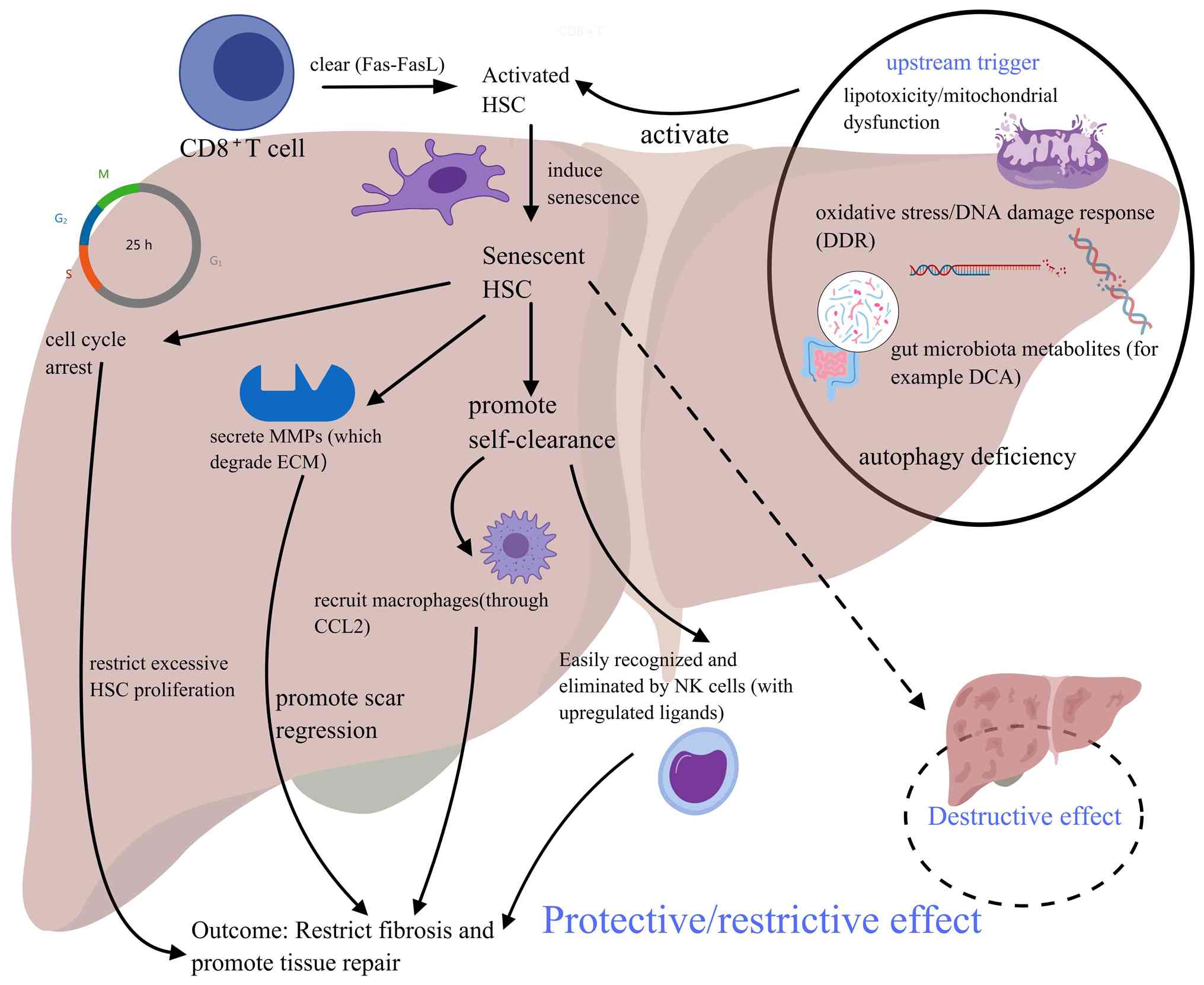
Senescent HSCs limit fibrosis and
promote tissue repair
A defining characteristic of aging is irreversible
cell cycle arrest, predominantly occurring during G1
phase arrest (45). When activated
HSCs enter senescence, they lose their ability to proliferate
(25). Gene expression profiling
analysis has demonstrated that senescent HSCs downregulate genes
associated with cell cycle progression and secretion of ECM
components (such as collagen), thus limiting both their
proliferation and fibrogenic potential. In addition, senescent HSCs
markedly change their secretory properties, increasing the
secretion of ECM-degrading enzymes such as MMPs (6). These enzymes exhibit fibrinolytic
activity and can actively degrade the deposited fibrotic matrix,
thereby promoting the regression of scars and normal tissue
remodeling. Concurrently, gene expression profiling indicates that
monocyte chemoattractant protein-1 (MCP-1), secreted by senescent
HSCs, recruits macrophages to fibrotic areas, promoting the
clearance of senescent HSCs and inhibiting inflammation (7). Aging HSCs actively recruit
macrophages, including monocyte-derived macrophages, to fibrotic
areas by secreting chemokines such as MCP-1 (CCL2) (46). These recruited macrophages perform
two primary anti-fibrotic functions, including timely clearance of
senescent HSCs, limiting inflammation and chronic accumulation of a
SASP (46). Additionally, these
macrophages cause active matrix degradation and tissue remodeling.
Macrophages involved in fibrosis resolution, typically M2-like or
scar-associated macrophages (SAMacs) degrade the ECM by secreting
multiple MMPs (such as, MMP-9, -12 and -13) and releasing
anti-fibrotic cytokines (such as IL-10), thereby promoting scar
tissue remodeling (21).
Senescent HSCs also upregulate immune surveillance
molecules on their surfaces, particularly ligands for the NK cell
receptor NK group 2 member D (NKG2D), such as MHC class I-related
chain A. This allows for their efficient recognition and
preferential elimination by NK cells. Such clearance processes,
actively initiated by senescent HSCs, ensure the prompt removal of
potentially harmful cells from tissue, ultimately promoting the
regression of fibrosis (Fig. 2)
(7,47).
Mechanisms underlying the coexistence
of two opposing effects in aging HSCs
Senescent HSCs exhibit a dual role, which may be
attributed to the decoupling between intrinsic cell cycle arrest
mechanisms and extrinsic, environmentally regulated SASP functions,
alongside variations in immune clearance efficiency. Current
research suggests that early senescence primarily drives fibrosis,
while in later stages, it may promote the reversal or stabilization
of fibrosis within the context of injury resolution and repair
(5,6). This process is also influenced by
cytokines such as IL-6, TGF-β and IL-10 at a mechanistic level.
Assessment of the ‘age’ of HSCs
A variety of methodological approaches have been
used to determine the age of HSCs in previous studies (6,48).
Telomeres, repetitive DNA sequences at chromosome ends,
progressively shorten with each cell division and are regarded as a
‘mitotic clock’ for gauging cellular replicative senescence and
biological age. One study found that in the normal human liver,
patterns of telomere shortening differ among cell types, whereby
the telomere length of cholangiocytes do not markedly shorten with
age, whereas Kupffer cells and HSCs exhibit notable age-related
telomere shortening, underscoring the utility of telomere length as
an effective metric for assessing chronological age-related changes
in HSCs (49).
Primary detection markers and methods. i)
Senescence-associated β-galactosidase (SA-β-gal) activity. As a
marker, SA-β-gal is most widely used for senescent cells. The
detection principle involves a histochemical stain at a
non-physiological pH of 6.0, where the activity of β-gal results in
a distinct blue coloration in senescent cells. Optimized protocols
for SA-β-gal staining in frozen liver tissue sections are available
(50), indicating that this
technique is fully applicable for identifying senescent cells,
including HSCs.
ii) p16INK4a protein expression. As a
cyclin-dependent kinase inhibitor, p16INK4a is notably
upregulated in senescent cells, where it maintains the senescent
state by inhibiting the cell cycle (51). The expression of
p16INK4a can be detected using methods including
immunohistochemistry and western blotting. For example, in liver
tissue samples from children with end-stage liver disease,
p16INK4a expression was found to be elevated, along with
SA-β-gal activity, demonstrating its validity as a marker of liver
senescence (6).
Expression profiles of associated cytokines.
At the cytokine level, pro-inflammatory cytokines constitute a
prominent component of the SASP (52). Identified factors include IL-6,
TNF-α, IL-1β, IL-1α, IL-12 and IFN-γ. Collectively, these factors
promote a potent pro-inflammatory microenvironment capable of
recruiting and activating immune cells.
Chemokines. Chemokine molecules recruit
immune cells to sites of injury or aging, with key examples
including MCP-1/CCL2, IL-8, CXCR2 ligands and neutrophil
chemotactic proteins (53).
Immunomodulatory cytokines (54),
specifically IL-10, is not only secreted by senescent HSCs but has
also functions as a signal that induces activated HSCs to enter a
senescent state, thereby establishing a complex feedback regulatory
loop. In addition to the aforementioned cytokines, the SASP of
senescent HSCs includes multiple growth factors (including
epidermal growth factors and insulin-like growth factor) and tissue
remodeling-associated proteins. Collectively, these influence the
actions of senescent HSCs.
The onset of HSC senescence and the establishment of
the SASP are dynamic processes rather than instantaneous events.
Through in vivo models, such as those involving partial
hepatectomy, HSCs have been shown to exhibit signs of senescence
and begin IL-6 secretion to promote liver regeneration within just
2 days (55). This observation
indicates that senescence programs can be initiated quite rapidly
during acute repair responses. By contrast, in vitro
cultures treated with inducers (such as etoposide) typically
require 7-10 days for full SASP establishment, during which complex
intracellular signaling and gene expression reprogramming occur,
ultimately leading to the sustained release of secreted proteins
(56).
Theoretically, SASP can persist as long as senescent
cells survive and are not cleared by the immune system. Immune
surveillance, including NK cell activity, represents a key
clearance mechanism for senescent cells, thereby terminating their
SASP. A previous study has indicated that IL-10 mRNA expression in
human HSCs can persist for <120 days during long-term culture.
However, mRNA levels are not fully associated with sustained
protein secretion (57). Overall,
precisely quantifying the kinetic profiles (onset, peak and
duration) of specific cytokines secreted by senescent HSCs under
numerous stimuli remains an unresolved issue requiring further
research.
3. Molecular mechanisms regulating HSC aging
and SASP formation
Causes of aging: Cellular stress in
the MASH environment
A recent study has shown that small extracellular
vesicles (sEVs) enriched with LIM domain and actin binding 1
(LIMA1) released from lipotoxic hepatocytes serve a key role in
promoting HSC activation in NAFLD-related liver fibrosis by
negatively regulating PINK1-mediated mitochondrial autophagy
(58). In this study, a high-fat
diet (HFD)-induced mouse model was constructed to demonstrate LIMA1
expression. Furthermore, sEV injection was used to assess whether
LIMA1 could accelerate HFD-induced liver fibrosis in mice (58). The results showed that LIMA1 was
upregulated in sEVs produced by HFD-induced fatty liver and
lipotoxic hepatocytes and that these LIMA1-enriched sEVs increased
LIMA1 protein expression in HSCs, consequently inducing HSC
activation. Moreover, this study further elucidated that LIMA1
further promoted the activation of HSCs by inhibiting mitochondrial
autophagy.
ROS and reactive nitrogen species (RNS) are
byproducts of mitochondrial dysfunction and inflammatory responses
and may cause extensive oxidative damage to cellular
macromolecules, especially DNA damage. This damage activates the
DNA damage response (DDR) of the cell, marked by the formation of
γH2AXb phosphorylation sites. Sustained or severe DDR is a key
mechanism underlying cellular senescence (26). Damaged mitochondria function as the
main source of ROS and their functional defects, including energy
metabolism disorders and can exacerbate cellular stress,
perpetuating a continuous cycle (25).
Interweaving with liver
disease-related signaling pathways
An example of a key pathway implicated in liver
cancer development is Wnt signaling, with β-catenin as a key
protein. β-catenin demonstrates notable interaction potential
within the network of senescent HSC secretory proteins. In a
previous study, ELISA experiments, immunofluorescence and
immunohistochemical staining demonstrated that Wnt signaling
pathway activation in tumor tissues was associated with an elevated
expression of related genes β-catenin and cellular-Myc, thus
demonstrating the role of senescent HSCs in promoting the malignant
transformation of individual cells by activating the Wnt signaling
pathway (20,59,60).
This study also identified the Hedgehog signaling
pathway. The Hedgehog pathway is abnormally activated in chronic
liver injury and MASH and is an important pathway driving fibrosis
and tumorigenesis (61). SASP
components from senescent HSCs act as potent activators of this
pathway (62). Immunofluorescence
staining showed activation levels of the pathway's key protein, GLI
family zinc finger 1 (Gli-1), initially increased and subsequently
decreased throughout MASH-HCC progression, consistent with changes
in the number of senescent HSCs (63). Among senescent HSCs, expression
levels of key molecules of the Hedgehog signaling pathway
(including Gli-1, Patched, cyclin D1 and B cell leukemia-1), were
markedly elevated, indicating that the pathway is continuously
activated in senescent HSCs. Therefore, the amount of main Hedgehog
signaling pathway ligand, sonic hedgehog SHh, also exhibited an
upward trend. SHh can directly interact with fibrotic cytokine
TGF-β and ECM components (including fibronectin), thereby
activating the Hedgehog signaling pathway and promoting the
malignant transformation of hepatocytes (20,64,65).
The JAK/STAT signaling pathway is a key downstream
mediator of the SASP-induced bystander effect. Key components of
the SASP, particularly IL-6, bind to receptors on the surface of
adjacent hepatocytes, activating the JAK/STAT3 signaling pathway.
Continuous activation of this pathway can lead to mitochondrial
dysfunction and DNA damage in hepatocytes (characterized by
increased γH2AX and 53BP1 markers), inducing hepatocyte damage and
apoptosis. This process not only exacerbates the inflammatory
response but also promotes a continuing cycle of liver damage
(26).
Interactions between autophagy and
aging
Cellular autophagy degrades damaged or redundant
organelles, protein aggregates, pathogen and other cellular
components in the cell, breaking them down into smaller molecules
for recycling. Autophagy serves a core role in maintaining cell
homeostasis, responding to environmental stress (such as nutrient
deficiency or oxidative stress) and regulating cell fate. Within
hepatocytes, autophagy helps to remove damaged mitochondria and
control the intracellular lipidome (66,67),
while selective autophagy of mitochondria usually involves distinct
signaling pathways, such as the PINK1/Parkin-dependent pathway and
receptor-mediated mechanisms involving BNIP3 and NIX (68). By preserving a functional pool of
mitochondria, autophagy minimizes ROS production and thus inhibits
the activation of intracellular pro-inflammatory factors (69-71).
Furthermore, autophagy is key for lipid metabolism as autophagy
defects are associated with increased triglyceride accumulation
both in vivo and in vitro (20). Impaired autophagy leads to
dysfunctional mitochondria, which reduces the production of ROS and
activates the DNA damage response, thereby inducing
p53/p21-dependent cell cycle arrest and cell senescence (72). Senescent HSCs further exacerbate
MASH-induced fibrosis and promote the progression of MASH. In
addition, ROS-dependent alterations in mitochondrial metabolism
have been associated with metabolic disorders, particularly insulin
resistance (73). Similar to ROS,
nitric oxide (NO) and RNS exhibit diverse biological effects,
ranging from physiological signaling to pathological nitrosative
stress under inflammatory conditions (74). Excessive NO and RNS induce similar
impairments in energy metabolism, covalently modifying a large
group of proteins and enzymes involved in mitochondrial respiration
including mitochondrial complex IV, ultimately leading to cell
death (64-79).
Bidirectional interactions between
lipid metabolism and HSC aging
In the early stages of MASH, lipid accumulation
(steatosis) in hepatocytes leads to lipotoxicity. This metabolic
stress serves as a key initial signal for inducing HSC senescence.
Lipotoxic hepatocytes release extracellular vesicles rich in
specific proteins such as LIMA1. Upon uptake by HSCs, these
vesicles induce HSC senescence and activation through mechanisms
that inhibit PINK1-Parkin-mediated mitochondrial autophagy
(58,80). Fatty acid oxidation (FAO) is a key
metabolic pathway responsible for breaking down fatty acids into
acetyl-CoA for energy production, primarily occurring within
mitochondria. FAO is important in maintaining the long-term
function and quiescent state of HSCs.
Within one notable study, it was demonstrated that
the promyelocytic leukemia protein (PML) regulates FAO through the
peroxisome proliferator-activated receptor (PPAR)-δ, which is key
for sustaining HSC quiescence and functionality (81). In this study, inactivation of the
PML-PPAR-δ-FAO axis led to HSC functional exhaustion, demonstrating
that FAO is a key metabolic pathway involved in sustaining HSC
stemness.
In aging cells, the dynamic equilibrium of lipid
droplets is often disrupted. A recent study conducted in 2024
revealed that during cellular senescence, glycerol-3-phosphate
accumulation triggers lipid metabolic reprogramming, leading to
marked triglyceride accumulation within lipid droplets. Such
abnormal lipid storage not only modifies the cellular metabolic
state but also activates senescence-associated gene expression
programs (82). The formation,
growth and degradation of lipid droplets is precisely regulated by
a series of lipid droplet-associated proteins, with perilipin
(PLIN) family proteins serving as core members. PLIN2, one of the
most extensively studied members, is widely expressed in
non-adipose cells and regulates lipid droplet stability and
lipolysis processes (83-86).
Certain aging models exhibit notable alterations in PLIN2
expression. Studies indicate that PLIN2 expression is downregulated
in aged mesenchymal stem cells, which is further associated with
changes in lipid droplet content and alterations in β-oxidation
capacity (87-89).
4. Interactions between senescent HSCs and
other cells
Interactions between senescent HSC and
macrophages
Interactions between HSCs and macrophages can
promote inflammation and fibrosis in MASH (30,90,91).
The SASP of senescent HSCs is rich in chemokines, such as CCL2
(MCP-1) and CXCL1, which recruit a number of circulating monocytes
to the liver (25). Once within
the liver, these monocytes differentiate into macrophages
exhibiting pro-inflammatory and pro-fibrotic phenotypes,
facilitated by SASP factors such as IL-1β and IL-6(30). In recent years, single-cell RNA
sequencing has identified MASH-specific macrophage subsets, such as
MASH-associated macrophages or SAMacs, whose formation may be
affected by senescent HSCs (90).
In return, activated macrophages secrete a number of potent HSC
activators, including TGF-β, PDGF and TNF-α. These factors not only
activate the remaining quiescent HSCs but also promote the survival
of both activated and senescent HSCs, thus forming a robust,
self-amplifying pro-fibrotic positive feedback loop (30).
Interactions between senescent HSCs
and NK cells
NK cells selectively eliminate early activated HSCs
by recognizing activated ligands on the surface of HSCs, such as
NKG2D and Raf-1 proto-oncogene serine/threonine kinase, thereby
inhibiting fibrosis (43).
Senescent HSCs are more easily cleared by NK cells due to the
upregulation of NKG2D ligands and tumor necrosis factor-related
apoptosis-inducing ligand receptors, thereby limiting the
development of fibrosis (43,44).
In addition, NK cells secrete IFN-γ, which induces HSC apoptosis
and cell cycle arrest, impeding their capacity to participate in
liver repair and proliferation. Disease progression is further
aggravated by persistent MASH-related damaging factors (7,43).
Gene expression profiling analysis has identified that MCP-1,
secreted by senescent HSCs, recruits macrophages to fibrotic areas,
promoting the clearance of senescent HSCs and inhibiting
inflammation (7). After
macrophages are recruited to the liver, Kupffer cells may inhibit
NK cell activity by secreting TGF-β, thereby reducing the ability
of NK cells to eliminate HSCs. However, TGF-β secreted by senescent
HSCs may attenuate NK cell inhibition (7,21,43,92).
Dual role of T cells in MASH
Within MASH, the role of T cells is also dualistic,
with senescent HSCs serving an important regulatory role. T cells
inhibit HSC fibrosis during the regression of MASH fibrosis.
CD8+ memory T cells have been shown to attract HSCs
through the C-C chemokine receptor type 5 (CCR5) and induce HSC
apoptosis through Fas-FasL, promoting the reversal of fibrosis
(30). In addition, the
persistence of senescent HSCs can effectively orchestrate an immune
escape strategy, inhibit the activity of CD8+ T cells,
hinder the clearance of senescent HSCs, promote collagen deposition
and promote an immune-exempt microenvironment for subsequent
tumorigenesis.
Anti-fibrotic T cell subsets: HSC
clearance mechanism based on the Fas-FasL pathway
Anti-fibrotic T cell subsets mainly promote HSC
apoptosis through specific molecular pathways. CD8+
memory T cells are directed to the vicinity of HSCs through
increased expression of the chemokine receptor CCR5, directly
triggering HSC apoptosis through the Fas-FasL death signaling
pathway. Simultaneously, liver-enriched γδ T cells also induce
programmed death of HSCs through the same Fas-FasL molecular
mechanism. This synergistic effect based on specific molecular
markers (including CCR5) and signaling pathways (including
Fas-FasL) constitutes a key immune mechanism that promotes fibrosis
reversal (93).
Pro-fibrotic T cell subsets:
IL-33-suppression of tumorigenicity 2 (ST2)-mediated
immunosuppressive mechanism. Pro-fibrotic regulatory T cells
hinder HSC clearance by establishing an immunosuppressive
microenvironment. This activation depends on the specific binding
of IL-33 released by senescent HSCs through the gasdermin D protein
pore to the ST2 receptor. Activated regulatory T cells form an
immunosuppressive circuit based on the IL-33-ST2 molecular axis by
inhibiting the antifibrotic function of CD8+ T cells,
ultimately leading to the accumulation of senescent HSCs, increased
collagen deposition and therefore an increased risk of
tumorigenesis (94,95).
Factors that can reverse the aging
process of hematopoietic stem cells. Inducing senescence to
terminate fibrosis
i) Pharmacological induction. Doxazosin, the α-1
adrenergic receptor antagonist, has been shown to reverse the
activation of HSCs by inducing senescence (96). Experiments have demonstrated that
doxazosin treatment upregulates senescence markers p53 and p21,
thereby inhibiting HSC proliferation and the expression of
pro-fibrotic genes.
ii) Modulation of the FoxO3a/S-phase kinase
associated protein 2 (SKP2)/p27 pathway. Notably, the soluble egg
antigen of Schistosoma japonicum, has been demonstrated to
induce HSC senescence by activating the transcription factor
FoxO3a, which subsequently inhibits SKP2 (an E3 ubiquitin ligase)
expression, leading to the accumulation of the cell cycle inhibitor
p27(97).
iii) Inhibition of endogenous hydrogen sulfide
(H2S). Inhibition of endogenous H2S
production induces HSC senescence and reduces HSC activation
primarily through the PI3K-Akt signaling pathway. This inhibition
leads to cell cycle arrest via upregulation of p53 and p21, and
promotes the SASP, thereby reversing the fibrogenic activity of
HSCs (89,94).
Promoting phenotypic reversion to a quiescent
state. i) Genetic regulation. In HSCs, specific knockout of
Delta-like 1 homolog, which is highly expressed upon HSC activation
(98), has been shown to markedly
reverse the activated phenotype, restoring these cells to a
quiescent or differentiated state. This effect is achieved by
inhibiting the Wnt signaling pathway and upregulating PPAR-γ
activity, thereby providing evidence that targeting DLK1 via
genetic engineering could be a potential therapeutic strategy for
reversing HSC activation (98).
ii) Signaling pathways and small-molecule compounds.
PPAR-γ is a key transcription factor for maintaining the quiescent
state of HSCs (99). The
synergistic interactions between retinoic acid (a derivative of
vitamin A) and PPAR-γ agonists can be considered effective
strategies for promoting the reversal of liver fibrosis. Their
combined action may suppress HSC activation markers and restore
their quiescent phenotypes.
Selective clearance of senescent HSCs. Given
the potential risks associated with the long-term presence of
senescent cells, senotherapy, the use of senolytic drugs to
selectively induce senescent cell death, has emerged as a new
research avenue (100).
Effects of aging HSCs on liver
regenerative capacity. A pro-regenerative role
Notably, a previous study reshaped the understanding
of the role of senescent HSCs in liver regeneration. Using a mouse
model of 2/3 partial hepatectomy, this study demonstrated that in
the early phase of regeneration following acute liver injury (~2
days post-surgery), HSCs rapidly enter a transient state of
senescence (101). This
phenomenon is not a pathological accumulation, but a programmed
physiological response key for regeneration.
The underlying mechanism can be attributed to the
SASP of these senescent HSCs, which secrete IL-6 and ligands for
the chemokine receptor CXCR2 (including CXCL1 and CXCL2). These
secreted factors exert a marked effect on adjacent hepatocytes,
driving their proliferation by activating downstream STAT3 and
YES-associated protein signaling pathways, thereby promoting the
restoration of liver mass. Experimental evidence shows that
specific elimination of these senescent HSCs during the early
regenerative phase notably impairs the regenerative capacity of the
liver, thereby demonstrating the positive role of transient
senescence in the acute repair process (102).
An inhibitory role. By contrast with their
beneficial role in acute injury, the persistent presence and
accumulation of senescent HSCs in chronic liver injuries, or the
naturally aged liver, is often associated with decreased
regenerative capacity and pathological progression.
First, in the aged liver, the quantity of quiescent
HSCs increases, coupled with phenotypic changes, such as increased
accumulation of lipid droplets. This alteration in the basal state
may compromise the ability of HSCs to maintain homeostasis of the
sinusoidal microenvironment (103).
Second, during chronic injury (for example, chronic
hepatitis and NAFLD), HSCs are continuously activated and may enter
senescent states. The SASP exhibited by long-existing senescent
HSCs is more complex, including pro-inflammatory factors and
numerous pro-fibrotic factors such as TGF-β. This chronic,
low-grade inflammatory and fibrotic environment inhibits normal
hepatocyte proliferation. A number of studies have demonstrated
that factors secreted by senescent cells (including HSCs and
neutrophils) can suppress hepatic progenitor cell activation and
proliferation, thereby obstructing compensatory regenerative
pathways (104-106).
5. Future directions and outlook
Development of biomarkers
Within aging-related therapies, the primary
challenge remaining is the lack of reliable, non-invasive
biomarkers. This gap makes patient selection, efficacy tracking and
dose optimization during human trials challenging, meaning diseases
cannot be accurately diagnosed, the burden of senescent HSCs cannot
be quantified and challenges remain regarding treatment response
evaluation, subsequently hindering clinical translation (107). Although liver biopsy is the
established standard for diagnosing MASH, the invasive nature of
this procedure imposes limitations (108). Therefore, the development of
biomarkers is key in addressing these challenges (109).
Identifying molecules in the blood that reflect the
aging state of the liver remains an active area of research. While
the SASP can be detected in plasma, studies reveal that its
association with frailty in liver transplant patients is marginally
notable, often lacking specificity as it is also associated with
systemic inflammation (110,111). The expression of
senescence-related genes, including p16INK4a and
p21CIP1, in circulating T cells is associated with
frailty and increased duration of hospitalization in patients
undergoing liver transplants, rendering them viable replacement
markers for systemic aging burdens (112). The anti-aging effects of
acyl-CoA-binding protein (ACBP) neutralization extend across a
number of cell types. Elevated plasma ACBP levels have been
documented in aging and liver injury models induced by a Western
diet and repeated CCl4 injections. Neutralization of ACBP prevents
cellular senescence, indicating its potential as a valuable
biomarker (113).
Ongoing research continues to explore non-invasive
imaging biomarkers. Elastic imaging techniques, such as transient
elastography and magnetic resonance elastography, are used to
assess the degree of fibrosis by quantifying the shear wave
velocity or tissue displacement generated by ultrasound or physical
pulses. However, their application is limited by an unclear optimal
cutoff point, inability to assess obese patients, poor diagnostic
accuracy in the early stages of fibrosis and the non-specificity
exhibited in senescent HSC detection (114). In addition, positron emission
tomography (PET) technology is highly promising due to its high
sensitivity and specificity. PET tracers targeting aging-related
SA-β-gal have demonstrated successful applications in preclinical
models including liver cancer, having also entered preliminary
human trial stages (115).
Potential of anti-aging drugs
Given the integral role of senescent HSCs in MASH
pathogenesis, senescent cell scavengers and phenotype regulators
have arisen as new therapeutic strategies. Senescent cell
scavengers are a class of drugs that selectively induce apoptosis
in senescent HSCs. Targeting the pro-survival pathways, these cells
upregulate to resist their own SASP toxicity (116).
The combination of dasatinib and quercetin (D + Q)
is a widely studied senescent cell scavenger regimen. Studies
demonstrate that D + Q effectively clears senescent HSCs in the
liver, reduces liver fat deposition and markedly lowers expression
levels of key pro-fibrotic factor TGF-β1 (116,117), with validation in a metabolic
dysfunction-associated fatty liver disease (a reclassified
terminology for MASH) model established using medaka fish (118). Additionally, ABT-263, an
inhibitor of BCL-2 family proteins, has been shown to effectively
eliminate senescent HSCs and hepatocytes, reduce expression of SASP
factors and improve mitochondrial function in a mouse model of
liver injury. Furthermore, it can facilitate the clearance of
senescent HSCs in liver regeneration models (119). However, the thrombocytopenia
associated with senescent cell removal and the tumor-promoting
effects of D + Q in HCC models reflect the limitations of this
particular therapy (119-124).
Senescence phenotype regulators constitute an
alternative therapeutic strategy that does not directly eliminate
senescent HSCs, instead regulating their phenotype primarily
through inhibition of harmful SASP production and secretion of SASP
factors (125). Potential targets
for senescence phenotype regulators include transcription factors
such as GATA binding protein 4 (an upstream regulator of NF-κB) as
well as mTOR and p38/MAPK signaling pathways. Analogs including
rapamycin, ruxolitinib, glucocorticoids and metformin are also
considered to exhibit senescence phenotype regulatory effects
(126). However, since these
signaling pathways are also essential for the physiological
function of normal cells, their inhibition may lead to significant
off-target effects and toxicity (127,128).
Other treatment and prevention
methods
Lifestyle interventions constitute a foundational
strategy for preventing and managing MASH (129). An unhealthy diet rich in fat and
sugar can promote intestinal flora imbalance, leading to the
translocation of lipopolysaccharides and other pathogenic molecular
patterns to the liver, triggering liver inflammation and
aggravating MASH. Healthy dietary interventions primarily target
obesity reduction, improve dyslipidemia and attenuate MASH
progression (130).
Supplementation with obeticholic acid may also provide additional
benefits in inhibiting liver inflammation and preventing disease
progression (131). Therefore,
treatments that restore healthy intestinal microecology, such as
probiotics, prebiotics, synbiotics and fecal microbiota
transplantation, may serve as innovative treatment strategies
(132).
6. Summary
Senescent HSCs occupy a central and multifaceted
role in the pathophysiology of MASH. The most notable feature of
senescent HSCs is their dual role, as during acute injury and
repair, they protect the body by arresting the cell cycle and
facilitating immune-mediated clearance. However, in the chronic and
persistent pathological state of MASH, a reduction in immune
clearance mechanisms allows long-term persistence of senescent
HSCs, continually secreting harmful SASP factors that drive chronic
inflammation, fibrogenesis and malignant transformation towards
liver cancer in MASH. Impaired autophagy induces senescence, while
SASP promotes pathological processes through key signaling
pathways, such as Wnt and Hedgehog. Simultaneously, the senescent
SASP promotes a microenvironment conducive to its survival and
disease development through interactions with immune cells. It is
recommended that future anti-aging therapeutic strategies should
evolve from broad-spectrum pathway inhibitors to target the
clearance or inhibition of pathogenic senescent HSCs. Focus should
remain on the specific identification and clearance of pathogenic
senescent HSCs phenotypes or the fine-tuning of their SASP
components. In the future, development of refined treatment
strategies based on a deeper understanding of the complex
interaction networks of senescent HSCs should be prioritized.
Acknowledgements
Not applicable.
Funding
Funding: The present review was supported by the National
Natural Science Foundation of China (Youth Fund; grant no.
82300666) and the College Students' Innovation and Entrepreneurship
Training Program of Nanjing Medical University (grant no.
202510312047).
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
ZH, YS and DW contributed to research design,
literature review, core content drafting and analysis within the
present study. DW acquired the funding. NZ, ZL and ZW assisted with
literature retrieval, checking citations of the key signaling
pathways and participated in writing the first draft. XZ guided
research direction, reviewed mechanism analysis logic and
coordinated author division of labor. XZ also participated in
writing and proofreading the manuscript. SL led the review design,
determined the framework, guided key issue analysis, finalized the
manuscript, served as the data contact and also acquired funding
for the present study. All authors read and approved the final
version of the manuscript. Data authentication is not
applicable.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Rinella ME, Lazarus JV, Ratziu V, Francque
SM, Sanyal AJ, Kanwal F, Romero D, Abdelmalek MF, Anstee QM, Arab
JP, et al: A multi-society Delphi consensus statement on new fatty
liver disease nomenclature. J Hepatol. 79:1542–1556.
2023.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Huang DQ, El-Serag HB and Loomba R: Global
epidemiology of NAFLD-related HCC: Trends, predictions, risk
factors and prevention. Nat Rev Gastroenterol Hepatol. 18:223–238.
2021.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Riazi K, Azhari H, Charette JH, Underwood
FE, King JA, Afshar EE, Swain MG, Congly SE, Kaplan GG and Shaheen
AA: The prevalence and incidence of NAFLD worldwide: A systematic
review and meta-analysis. Lancet Gastroenterol Hepatol. 7:851–861.
2022.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Tsuchida T and Friedman SL: Mechanisms of
hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol.
14:397–411. 2017.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Ferreira-Gonzalez S, Rodrigo-Torres D,
Gadd VL and Forbes SJ: Cellular senescence in liver disease and
regeneration. Semin Liver Dis. 41:50–66. 2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Krizhanovsky V, Yon M, Dickins RA, Hearn
S, Simon J, Miething C, Yee H, Zender L and Lowe SW: Senescence of
activated stellate cells limits liver fibrosis. Cell. 134:657–667.
2008.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Aravinthan A, Scarpini C, Tachtatzis P,
Verma S, Penrhyn-Lowe S, Harvey R, Davies SE, Allison M, Coleman N
and Alexander G: Hepatocyte senescence predicts progression in
non-alcohol-related fatty liver disease. J Hepatol. 58:549–556.
2013.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Hernández-Gea V, Ghiassi-Nejad Z,
Rozenfeld R, Gordon R, Fiel MI, Yue Z, Czaja MJ and Friedman SL:
Autophagy releases lipid that promotes fibrogenesis by activated
hepatic stellate cells in mice and in human tissues.
Gastroenterology. 142:938–946. 2012.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Allaire M, Rautou PE, Codogno P and
Lotersztajn S: Autophagy in liver diseases: Time for translation? J
Hepatol. 70:985–998. 2019.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kale A, et al: Senescent cells and
therapeutic targeting in liver fibrosis. Gut. 2020.
|
|
11
|
Friedman SL, Neuschwander-Tetri BA,
Rinella M and Sanyal AJ: Mechanisms of NAFLD development and
therapeutic strategies. Nat Med. 24:908–922. 2018.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Campisi J and d'Adda di Fagagna F:
Cellular senescence: When bad things happen to good cells. Nat Rev
Mol Cell Biol. 8:729–740. 2007.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Tchkonia T, Zhu Y, van Deursen J, Campisi
J and Kirkland JL: Cellular senescence and the senescent secretory
phenotype: Therapeutic opportunities. J Clin Invest. 123:966–972.
2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Xiong J, Dong L, Lv Q, Yin Y, Zhao J, Ke
Y, Wang S, Zhang W and Wu M: Targeting senescence-associated
secretory phenotypes to remodel the tumour microenvironment and
modulate tumour outcomes. Clin Transl Med. 14(1772)2024.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Myung PK, Kugel JF and Goodrich JA: The
Borrelia burgdorferi p66 protein interacts with the human
lipopolysaccharide-induced tumor necrosis factor-alpha factor. J
Bacteriol. 184:1350–1357. 2002.
|
|
16
|
Tang X, Metzger D, Leeman S and Amar S:
LPS-induced TNF-alpha factor (LITAF)-deficient mice show delayed
LPS-induced cytokine expression. Proc Natl Acad Sci USA.
103:13777–13782. 2006.
|
|
17
|
Ceccarelli S, Panera N, Mina M, Gnani D,
De Stefanis C, Crudele A, Rychlicki C, Petrini S, Bruscalupi G,
Agostinelli L, et al: LPS-induced TNF-α factor mediates
pro-inflammatory and pro-fibrogenic pattern in non-alcoholic fatty
liver disease. Oncotarget. 6:41434–41452. 2015.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Beringer A and Miossec P: IL-17 and TNF-α
co-operation contributes to the proinflammatory response of hepatic
stellate cells. Clin Exp Immunol. 198:111–120. 2019.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Lemmers A, Moreno C, Gustot T, Maréchal R,
Degré D, Demetter P, de Nadai P, Geerts A, Quertinmont E,
Vercruysse V, et al: The interleukin-17 pathway is involved in
human alcoholic liver disease. Hepatology. 49:646–657.
2009.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Zhou Y, Zhang L, Ma Y, Xie L, Yang YY, Jin
C, Chen H, Zhou Y, Song GQ, Ding J and Wu J: Secretome of senescent
hepatic stellate cells favors malignant transformation from
nonalcoholic steatohepatitis-fibrotic progression to hepatocellular
carcinoma. Theranostics. 13:4430–4448. 2023.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Kuilman T and Peeper DS:
Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev
Cancer. 9:81–94. 2009.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Gorgoulis V, Adams PD, Alimonti A, Bennett
DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K,
Ferbeyre G, et al: Cellular senescence: Defining a path forward.
Cell. 179:813–827. 2019.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Faget DV, Ren Q and Stewart SA: Unmasking
senescence: Context-dependent effects of SASP in cancer. Nat Rev
Cancer. 19:439–453. 2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz
DP, Goldstein J, Nelson PS, Desprez PY and Campisi J:
Senescence-associated secretory phenotypes reveal
cell-nonautonomous functions of oncogenic RAS and the p53 tumor
suppressor. PLoS Biol. 6:2853–2868. 2008.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Engelmann C and Tacke F: The potential
role of cellular senescence in non-alcoholic fatty liver disease.
Int J Mol Sci. 23(652)2022.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Li Q and Wang L: Navigating the complex
role of senescence in liver disease. Chin Med J (Engl).
137:3061–3072. 2024.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Rosenthal SB, Liu X, Ganguly S, Dhar D,
Pasillas MP, Ricciardelli E, Li RZ, Troutman TD, Kisseleva T, Glass
CK and Brenner DA: Heterogeneity of HSCs in a mouse model of NASH.
Hepatology. 74:667–685. 2021.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Du K, Jun JH, Dutta RK and Diehl AM:
Plasticity, heterogeneity and multifunctionality of hepatic
stellate cells in liver pathophysiology. Hepatol Commun.
8(e0411)2024.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Kumar P, Hassan M, Tacke F and Engelmann
C: Delineating the heterogeneity of senescence-induced-functional
alterations in hepatocytes. Cell Mol Life Sci.
81(200)2024.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Carter JK and Friedman SL: Hepatic
stellate cell-immune interactions in NASH. Front Endocrinol
(Lausanne). 13(867940)2022.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Razdan N, Vasilopoulos T and Herbig U:
Telomere dysfunction promotes transdifferentiation of human
fibroblasts into myofibroblasts. Aging Cell.
17(e12838)2018.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Akkız H, Gieseler RK and Canbay A: Liver
fibrosis: From basic science towards clinical progress, focusing on
the central role of hepatic stellate cells. Int J Mol Sci.
25(7873)2024.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Okina Y, Sato-Matsubara M, Matsubara T,
Daikoku A, Longato L, Rombouts K, Thanh Thuy LT, Ichikawa H,
Minamiyama Y, Kadota M, et al: TGF-β1-driven reduction of
cytoglobin leads to oxidative DNA damage in stellate cells during
non-alcoholic steatohepatitis. J Hepatol. 73:882–895.
2020.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Frippiat C, Chen QM, Zdanov S, Magalhaes
JP, Remacle J and Toussaint O: Subcytotoxic H2O2 stress triggers a
release of transforming growth factor-beta1, which induces
biomarkers of cellular senescence of human diploid fibroblasts. J
Biol Chem. 276:2531–2537. 2001.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Amor C, Feucht J, Leibold J, Ho YJ, Zhu C,
Alonso-Curbelo D, Mansilla-Soto J, Boyer JA, Li X, Giavridis T, et
al: Senolytic CAR T cells reverse senescence-associated
pathologies. Nature. 583:127–132. 2020.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Nelson G, Wordsworth J, Wang C, Jurk D,
Lawless C, Martin-Ruiz C and von Zglinicki T: A senescent cell
bystander effect: Senescence-induced senescence. Aging Cell.
11:345–349. 2012.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Grohmann M, Wiede F, Dodd GT, Gurzov EN,
Ooi GJ, Butt T, Rasmiena AA, Kaur S, Gulati T, Goh PK, et al:
Obesity drives STAT-1-Dependent NASH and STAT-3-Dependent HCC.
Cell. 175:1289–1306.e20. 2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Anstee QM, Reeves HL, Kotsiliti E, Govaere
O and Heikenwalder M: From NASH to HCC: Current concepts and future
challenges. Nat Rev Gastroenterol Hepatol. 16:411–428.
2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Loo TM, Kamachi F, Watanabe Y, Yoshimoto
S, Kanda H, Arai Y, Nakajima-Takagi Y, Iwama A, Koga T, Sugimoto Y,
et al: Gut Microbiota Promotes Obesity-Associated Liver Cancer
through PGE2-Mediated Suppression of Antitumor Immunity. Cancer
Discov. 7:522–538. 2017.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Brennan PN, Elsharkawy AM, Kendall TJ,
Loomba R, Mann DA and Fallowfield JA: Antifibrotic therapy in
nonalcoholic steatohepatitis: Time for a human-centric approach.
Nat Rev Gastroenterol Hepatol. 20:679–688. 2023.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Yamagishi R, Kamachi F, Nakamura M,
Yamazaki S, Kamiya T, Takasugi M, Cheng Y, Nonaka Y, Yukawa-Muto Y,
Thuy LTT, et al: Gasdermin D-mediated release of IL-33 from
senescent hepatic stellate cells promotes obesity-associated
hepatocellular carcinoma. Sci Immunol. 7(eabl7209)2022.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Yoshimoto S, Loo TM, Atarashi K, Kanda H,
Sato S, Oyadomari S, Iwakura Y, Oshima K, Morita H, Hattori M, et
al: Obesity-induced gut microbial metabolite promotes liver cancer
through senescence secretome. Nature. 499:97–101. 2013.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Gao B and Radaeva S: Natural killer and
natural killer T cells in liver fibrosis. Biochim Biophys Acta.
1832:1061–1069. 2013.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Park O, Jeong WI, Wang L, Wang H, Lian ZX,
Gershwin ME and Gao B: Diverse roles of invariant natural killer T
cells in liver injury and fibrosis induced by carbon tetrachloride.
Hepatology. 49:1683–1694. 2009.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Wagner KD and Wagner N: The senescence
markers p16INK4A, p14ARF/p19ARF and p21 in organ development and
homeostasis. Cells. 11(1966)2022.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Jiang Z, Qian B, Xu T, Bai J and Fu W:
Immune microenvironment on the molecular mechanisms and therapeutic
targets of MAFLD. Immunotargets Ther. 14:719–733. 2025.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Carter J, Wang S and Friedman SL: Ten
thousand points of light: Heterogeneity among the stars of NASH
Fibrosis. Hepatology. 74:543–546. 2021.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Wiemann SU, Satyanarayana A, Tsahuridu M,
Tillmann HL, Zender L, Klempnauer J, Flemming P, Franco S, Blasco
MA, Manns MP and Rudolph KL: Hepatocyte telomere shortening and
senescence are general markers of human liver cirrhosis. FASEB J.
16:935–942. 2002.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Verma S, Tachtatzis P, Penrhyn-Lowe S,
Scarpini C, Jurk D, Von Zglinicki T, Coleman N and Alexander GJ:
Sustained telomere length in hepatocytes and cholangiocytes with
increasing age in normal liver. Hepatology. 56:1510–1520.
2012.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Jannone G, Rozzi M, Najimi M, Decottignies
A and Sokal EM: An optimized protocol for histochemical detection
of senescence-associated beta-galactosidase activity in
cryopreserved liver tissue. J Histochem Cytochem. 68:269–278.
2020.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Serrano M, Lin AW, McCurrach ME, Beach D
and Lowe SW: Oncogenic RAS provokes premature cell senescence
associated with accumulation of p53 and p16INK4a. Cell. 88:593–602.
1997.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Guo J, Huang X, Dou L, Yan M, Shen T, Tang
W and Li J: Aging and aging-related diseases: From molecular
mechanisms to interventions and treatments. Signal Transduct Target
Ther. 7(391)2022.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Friedman SL: Hepatic stellate cells:
Protean, multifunctional and enigmatic cells of the liver. Physiol
Rev. 88:125–172. 2008.PubMed/NCBI View Article : Google Scholar
|
|
54
|
De Waal Malefyt R, Abrams J, Bennett B,
Figdor CG and de Vries JE: Interleukin 10(IL-10) inhibits cytokine
synthesis by human monocytes: An autoregulatory role of IL-10
produced by monocytes. J Exp Med. 174:1209–1220. 1991.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Cheng N, Kim KH and Lau LF: Senescent
hepatic stellate cells promote liver regeneration through IL-6 and
ligands of CXCR2. JCI Insight. 7(158207)2022.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Irvine KM, Skoien R, Bokil NJ, Melino M,
Thomas GP, Loo D, Gabrielli B, Hill MM, Sweet MJ, Clouston AD and
Powell EE: Senescent human hepatocytes express a unique secretory
phenotype and promote macrophage migration. World J Gastroenterol.
20:17851–17862. 2014.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Thompson KC, Trowern A, Fowell A, Marathe
M, Haycock C, Arthur MJ and Sheron N: Primary rat and mouse hepatic
stellate cells express the macrophage inhibitor cytokine
interleukin-10 during the course of activation in vitro.
Hepatology. 28:1518–1524. 1998.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Li S, Yang F, Cheng F, Zhu L and Yan Y:
Lipotoxic hepatocyte derived LIMA1 enriched small extracellular
vesicles promote hepatic stellate cells activation via inhibiting
mitophagy. Cell Mol Biol Lett. 29(82)2024.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Wang Q, Lv Q, Bian H, Yang L, Guo KL, Ye
SS, Dong XF and Tao LL: A novel tumor suppressor SPINK5 targets
Wnt/β-catenin signaling pathway in esophageal cancer. Cancer Med.
8:2360–2371. 2019.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Kordes C, Sawitza I and Häussinger D:
Canonical Wnt signaling maintains the quiescent stage of hepatic
stellate cells. Biochem Biophys Res Commun. 367:116–123.
2008.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Verdelho Machado M and Diehl A: Role of
hedgehog signaling pathway in NASH. Int J Mol Sci.
17(857)2016.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Ding J, Li HY, Zhang L, Zhou Y and Wu J:
Hedgehog signaling, a critical pathway governing the development
and progression of hepatocellular carcinoma. Cells.
10(123)2021.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Chen Y, Gao WK, Shu YY and Ye J:
Mechanisms of ductular reaction in non-alcoholic steatohepatitis.
World J Gastroenterol. 28:2088–2099. 2022.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Shen X, Peng Y and Li H: The
injury-related activation of hedgehog signaling pathway modulates
the repair-associated inflammation in liver fibrosis. Front
Immunol. 8(1450)2017.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Hu Y, Peng L, Zhuo X, Yang C and Zhang Y:
Hedgehog signaling pathway in fibrosis and targeted therapies.
Biomolecules. 14(1485)2024.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Brenner C, Galluzzi L, Kepp O and Kroemer
G: Decoding cell death signals in liver inflammation. J Hepatol.
59:583–594. 2013.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Singh R, Kaushik S, Wang Y, Xiang Y, Novak
I, Komatsu M, Tanaka K, Cuervo AM and Czaja MJ: Autophagy regulates
lipid metabolism. Nature. 458:1131–1135. 2009.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Pickles S, Vigié P and Youle RJ: Mitophagy
and quality control mechanisms in mitochondrial maintenance. Curr
Biol. 28:R170–R185. 2018.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Cui X, Zhou Z, Tu H, Wu J, Zhou J, Yi Q,
Liu O and Dai X: Mitophagy in fibrotic diseases: Molecular
mechanisms and therapeutic applications. Front Physiol.
15(1430230)2024.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Liu S, Wang L, Zhu L, Zhao T, Han P, Yan
F, Wang X, Li C, Wang Z and Yang BF: Mechanism and regulation of
mitophagy in liver diseases: A review. Front Cell Dev Biol.
13(1614940)2025.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Du Y, Zhu S, Zeng H, Wang Z, Huang Y, Zhou
Y, Zhang W, Zhu J and Yang C: Research progress on the effect of
autophagy and exosomes on liver fibrosis. Curr Stem Cell Res Ther.
19:785–797. 2024.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Passos JF, Nelson G, Wang C, Richter T,
Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat
A, et al: Feedback between p21 and reactive oxygen production is
necessary for cell senescence. Mol Syst Biol. 6(347)2010.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Vial G, Dubouchaud H and Leverve XM: Liver
mitochondria and insulin resistance. Acta Biochim Pol. 57:389–392.
2010.PubMed/NCBI
|
|
74
|
Pacher P, Beckman JS and Liaudet L: Nitric
oxide and peroxynitrite in health and disease. Physiol Rev.
87:315–424. 2007.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Beltrán B, Mathur A, Duchen MR,
Erusalimsky JD and Moncada S: The effect of nitric oxide on cell
respiration: A key to understanding its role in cell survival or
death. Proc Natl Acad Sci USA. 97:14602–14607. 2000.PubMed/NCBI View Article : Google Scholar
|
|
76
|
d'Adda di Fagagna F: Living on a break:
Cellular senescence as a DNA-damage response. Nat Rev Cancer.
8:512–522. 2008.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Kim KS, Seu YB, Baek SH, Kim MJ, Kim KJ,
Kim JH and Kim JR: Induction of cellular senescence by insulin-like
growth factor binding protein-5 through a p53-dependent mechanism.
Mol Biol Cell. 18:4543–4552. 2007.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Kortlever RM, Higgins PJ and Bernards R:
Plasminogen activator inhibitor-1 is a critical downstream target
of p53 in the induction of replicative senescence. Nat Cell Biol.
8:877–884. 2006.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Merendino N, Costantini L, Manzi L,
Molinari R, D'Eliseo D and Velotti F: Dietaryω-3
polyunsaturated fatty acid DHA: A potential adjuvant in the
treatment of cancer. Biomed Res Int. 2013(310186)2013.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Wu Z, Xia M, Wang J, Aguilar MM,
Buist-Homan M and Moshage H: Extracellular vesicles originating
from steatotic hepatocytes promote hepatic stellate cell senescence
via AKT/mTOR signaling. Cell Biochem Funct.
42(e4077)2024.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Sun R, Cao M, Zhang J, Yang W, Wei H, Meng
X, Yin L and Pu Y: Benzene exposure alters expression of enzymes
involved in fatty acid β-oxidation in male C3H/He mice. Int J
Environ Res Public Health. 13(1068)2016.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Tighanimine K, Nabuco Leva Ferreira
Freitas JA, Nemazanyy I, Bankolé A, Benarroch-Popivker D, Brodesser
S, Doré G, Robinson L, Benit P, Ladraa S, et al: A homoeostatic
switch causing glycerol-3-phosphate and phosphoethanolamine
accumulation triggers senescence by rewiring lipid metabolism. Nat
Metab. 6:323–342. 2024.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Najt CP, Devarajan M and Mashek DG:
Perilipins at a glance. J Cell Sci. 135(jcs259501)2022.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Sztalryd C and Kimmel AR: Perilipin: Lipid
droplet coat protein adapted for tissue-specific energy storage and
utilization, and lipids cytoprotection. Biochimie. 96:96–101.
2014.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Marschallinger J, Iram T, Zardeneta M, Lee
SE, Lehallier B, Haney MS, Pluvinage JV, Mathur V, Hahn O, Morgens
DW, et al: Lipid-droplet-accumulating microglia represent a
dysfunctional and proinflammatory state in the aging brain. Nat
Neurosci. 23:194–208. 2020.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Mashek DG: Hepatic lipid droplets: A
balancing act between energy storage and metabolic dysfunction in
NAFLD. Mol Metab. 50(101115)2021.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Hosseini MS, Barjesteh F, Azedi F,
Alipourfard I, Rezaei Z and Bahreini E: Comparative analysis of
β-Estradiol and testosterone on lipid droplet accumulation, and
regulatory protein expression in palmitate/oleate-induced fatty
HepG2 cells. BMC Gastroenterol. 25(263)2025.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Li H and Humphreys BD: Targeting de novo
lipogenesis to mitigate kidney disease. J Clin Invest.
134(e178125)2024.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Conte M, Vasuri F, Trisolino G, Bellavista
E, Santoro A, Degiovanni A, Martucci E, D'Errico-Grigioni A,
Caporossi D, Capri M, et al: Increased Plin2 expression in human
skeletal muscle is associated with sarcopenia and muscle weakness.
PLoS One. 8(e73709)2013.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Li H, Zhou Y, Wang H, Zhang M, Qiu P,
Zhang M, Zhang R, Zhao Q and Liu J: Crosstalk between liver
macrophages and surrounding cells in nonalcoholic steatohepatitis.
Front Immunol. 11(1169)2020.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Wang YB, Li T, Wang FY, Yao X, Bai QX, Su
HW, Liu J, Wang L and Tan RZ: The dual role of cellular senescence
in macrophages: Unveiling the hidden driver of age-related
inflammation in kidney disease. Int J Biol Sci. 21:632–657.
2025.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Campisi J: Senescent cells, tumor
suppression and organismal aging: Good citizens, bad neighbors.
Cell. 120:513–522. 2005.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Yue B, Gao Y, Hu Y, Zhan M, Wu Y and Lu L:
Harnessing CD8+ T cell dynamics in hepatitis B virus-associated
liver diseases: Insights, therapies and future directions. Clin
Transl Med. 14(e1731)2024.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Damba T, Zhang M, Serna Salas SA, Wu Z,
van Goor H, Arenas AF, Muñoz-Ortega MH, Ventura-Juárez J,
Buist-Homan M and Moshage H: Inhibition of endogenous hydrogen
sulfide production reduces activation of hepatic stellate cells via
the induction of cellular senescence. Cell Cycle. 23:629–644.
2024.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Ducimetière L, Vermeer M and Tugues S: The
interplay between innate lymphoid cells and the tumor
microenvironment. Front Immunol. 10(2895)2019.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Serna-Salas SA, Arroyave-Ospina JC, Zhang
M, Damba T, Buist-Homan M, Muñoz-Ortega MH, Ventura-Juárez J and
Moshage H: α-1 Adrenergic receptor antagonist doxazosin reverses
hepatic stellate cells activation via induction of senescence. Mech
Ageing Dev. 201(111617)2022.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Duan Y, Pan J, Chen J, Zhu D, Wang J, Sun
X, Chen L and Wu L: Soluble egg antigens of schistosoma japonicum
induce senescence of activated hepatic stellate cells by activation
of the FoxO3a/SKP2/P27 pathway. PLoS Negl Trop Dis.
10(e0005268)2016.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Zhu NL, Asahina K, Wang J, Ueno A, Lazaro
R, Miyaoka Y, Miyajima A and Tsukamoto H: Hepatic stellate
cell-derived delta-like homolog 1 (DLK1) protein in liver
regeneration. J Biol Chem. 287:10355–10367. 2012.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Panebianco C, Oben JA, Vinciguerra M and
Pazienza V: Senescence in hepatic stellate cells as a mechanism of
liver fibrosis reversal: A putative synergy between retinoic acid
and PPAR-gamma signalings. Clin Exp Med. 17:269–280.
2017.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Zhu Y, Tchkonia T, Pirtskhalava T, Gower
AC, Ding H, Giorgadze N, Palmer AK, Ikeno Y, Hubbard GB, Lenburg M,
et al: The Achilles' heel of senescent cells: From transcriptome to
senolytic drugs. Aging Cell. 14:644–658. 2015.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Wang MJ, Zhang HL, Chen F, Guo XJ, Liu QG
and Hou J: The double-edged effects of IL-6 in liver regeneration,
aging, inflammation and diseases. Exp Hematol Oncol.
13(62)2024.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Ferreira-Gonzalez S, Lu WY, Raven A, et
al: Paracrine signaling by senescent cells promotes liver
regeneration. EMBO J. 37(e99195)2018.
|
|
103
|
Kordes C, Bock HH, Reichert D, May P and
Häussinger D: Hepatic stellate cells: Current state and open
questions. Biol Chem. 402:1021–1032. 2021.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Bird TG, Müller M, Boulter L, Vincent DF,
Ridgway RA, Lopez-Guadamillas E, Lu WY, Jamieson T, Govaere O,
Campbell AD, et al: TGFβ inhibition restores a regenerative
response in acute liver injury by suppressing paracrine senescence.
Sci Transl Med. 10(eaan1230)2018.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Ritschka B, Storer M, Mas A, Heinzmann F,
Ortells MC, Morton JP, Sansom OJ, Zender L and Keyes WM: The
senescence-associated secretory phenotype induces cellular
plasticity and tissue regeneration. Genes Dev. 31:172–183.
2017.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Cheng Y, Wang X, Wang B, Zhou H, Dang S,
Shi Y, Hao L, Luo Q, Jin M, Zhou Q and Zhang Y: Aging-associated
oxidative stress inhibits liver progenitor cell activation in mice.
Aging (Albany NY). 9:1359–1374. 2017.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Saliev T and Singh PB: Targeting
senescence: A review of senolytics and senomorphics in anti-aging
interventions. Biomolecules. 15(860)2025.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Roeb E: Non-alcoholic fatty liver
diseases: Current challenges and future directions. Ann Transl Med.
9(726)2021.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Geng Y and Schwabe RF: Hepatic stellate
cell heterogeneity: Functional aspects and therapeutic
implications. Hepatology: May 8, 2025 (Epub ahead of print).
|
|
110
|
Schaenman JM, Rossetti M, Sidharthan S, et
al: Increased p16INK4a expression in peripheral blood T cells is
associated with frailty in patients evaluated for liver
transplantation. Am J Transplant. 18:696–703. 2018.
|
|
111
|
Sidharthan S, Wang T, Ying Z, et al:
Sarcopenia, frailty, and immunosenescence in liver transplant
candidates. Transplant Direct. 6(e598)2020.
|
|
112
|
Miller WC, Yousefzadeh MJ, Fisher J,
Sarumi H, Kirchner V, Niedernhofer LJ and Pruett T: A brief report
on biomarkers of cellular senescence associated with liver frailty
and length of stay in liver transplantation. GeroScience.
47:5257–5265. 2025.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Montégut L, Lambertucci F, Moledo-Nodar L,
Fiuza-Luces C, Rodríguez-López C, Serra-Rexach JA, Lachkar S,
Motiño O, Abdellatif M, Durand S, et al: Acyl-CoA-binding protein
as a driver of pathological aging. Proc Natl Acad Sci USA.
122(e2501584122)2025.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Yu JH, Lee HA and Kim SU: Noninvasive
imaging biomarkers for liver fibrosis in nonalcoholic fatty liver
disease: Current and future. Clin Mol Hepatol. 29
(Suppl):S136–S149. 2023.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Xiang X, Dong C, Zhou L, Liu J, Rabinowitz
ZM, Zhang Y, Guo H, He F, Chen X, Wang Y, et al: Novel PET imaging
probe for quantitative detection of senescence in vivo. J Med Chem.
67:5924–5934. 2024.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Birch J and Gil J: Senescence and the
SASP: Many therapeutic avenues. Genes Dev. 34:1565–1576.
2020.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Raffaele M, Kovacovicova K, Frohlich J, Lo
Re O, Giallongo S, Oben JA, Faldyna M, Leva L, Giannone AG, Cabibi
D and Vinciguerra M: Mild exacerbation of obesity- and
age-dependent liver disease progression by senolytic cocktail
dasatinib + quercetin. Cell Commun Signal. 19(44)2021.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Yakubo S, Abe H, Li Y, Kudo M, Kimura A,
Wakabayashi T, Watanabe Y, Kimura N, Setsu T, Yokoo T, et al:
Dasatinib and quercetin as senolytic drugs improve fat deposition
and exhibit antifibrotic effects in the medaka metabolic
dysfunction-associated steatotic liver disease model. Diseases.
12(317)2024.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Luo J, Li L, Chang B, Zhu Z, Deng F, Hu M,
Yu Y, Lu X, Chen Z, Zuo D and Zhou J: Mannan-binding lectin via
interaction with cell surface calreticulin promotes senescence of
activated hepatic stellate cells to limit liver fibrosis
progression. Cell Mol Gastroenterol Hepatol. 14:75–99.
2022.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Trussoni CE, O'Hara SP and LaRusso NF:
Correction to: Cellular senescence in the cholangiopathies: A
driver of immunopathology and a novel therapeutic target. Semin
Immunopathol. 44:527–544. 2022.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Suda M, Paul KH, Tripathi U, Minamino T,
Tchkonia T and Kirkland JL: Targeting cell senescence and
senolytics: Novel interventions for age-related endocrine
dysfunction. Endocr Rev. 45:655–675. 2024.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Watanabe Y, Abe H, Kimura N, Arao Y,
Ishikawa N, Yuichiro M, Setsu T, Sakamaki A, Kamimura H, Yokoo T,
et al: Navitoclax improves acute-on-chronic liver failure by
eliminating senescent cells in mice. Hepatol Res. 53:460–472.
2023.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Mohamad Anuar NN, Nor Hisam NS, Liew SL
and Ugusman A: Clinical review: Navitoclax as a pro-apoptotic and
anti-fibrotic agent. Front Pharmacol. 11(564108)2020.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Sharma AK, Roberts RL, Benson RD Jr,
Pierce JL, Yu K, Hamrick MW and McGee-Lawrence ME: The senolytic
drug navitoclax (ABT-263) causes trabecular bone loss and impaired
osteoprogenitor function in aged mice. Front Cell Dev Biol.
8(354)2020.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Tang Q, Xiao D, Veviorskiy A, Xin Y, Lok
SWY, Pulous FE, Zhang P, Zhu Y, Ma Y, Hu X, et al: AI-driven
robotics laboratory identifies pharmacological TNIK inhibition as a
potent senomorphic agent. Aging Dis. 17:432–51. 2025.PubMed/NCBI View Article : Google Scholar
|
|
126
|
Meijnikman AS, Herrema H, Scheithauer TPM,
Kroon J, Nieuwdorp M and Groen AK: Evaluating causality of cellular
senescence in non-alcoholic fatty liver disease. JHEP Rep.
3(100301)2021.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Ryan P and Lee J: In vitro senescence and
senolytic functional assays. Biomater Sci. 13:3509–3531.
2025.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Fei B, Wang H, Ding Y, Shao N, Wen P, Cao
Y, Li J, Tanaka M, Wang Z and Li S: Reprogramming cellular
senescence of hepatic stellate cells to combat liver fibrosis by
targeted nanodrugs. Mater Today Bio. 33(101996)2025.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Kakde SP, Mushtaq M, Liaqat M, Ali H,
Mushtaq MM, Sarwer MA, Ullah S, Hassan MW, Khalid A and Bokhari
SFH: Emerging Therapies for non-alcoholic steatohepatitis (NASH): A
comprehensive review of pharmacological and non-pharmacological
approaches. Cureus. 16(e69129)2024.PubMed/NCBI View Article : Google Scholar
|
|
130
|
Rinella ME, Neuschwander-Tetri BA,
Siddiqui MS, Abdelmalek MF, Caldwell S, Barb D, Kleiner DE and
Loomba R: AASLD Practice Guidance on the clinical assessment and
management of nonalcoholic fatty liver disease. Hepatology.
77:1797–1835. 2023.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Panyod S, Wu W-K, Hu M-Y, Huang H-S, Chen
R-A, Chen Y-H, Shen T-CD, Ho C-T, Liu C-J, Chuang H-L, Huang C-C,
et al: Healthy diet intervention reverses the progression of NASH
through gut microbiota modulation. Microbiol Spectr.
12(e186823)2024.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Liu Q, Liu S, Chen L, Zhao Z, Du S, Dong
Q, Xin Y and Xuan S: Role and effective therapeutic target of gut
microbiota in NAFLD/NASH. Exp Ther Med. 18:1935–1944.
2019.PubMed/NCBI View Article : Google Scholar
|