Introduction
Hearing loss is a common congenital sensory
impairment, affecting 2-3 out of every 1,000 newborns in China
(1). Genetic factors are a primary
cause, accounting for ≥50% of cases of severe bilateral neonatal
hearing loss. Among these cases, 70% are non-syndromic hearing loss
(NSHL), while the remainder are syndromic, presenting with
additional abnormalities and clinical features, such as visual
impairment and developmental delays. Furthermore, genetic factors
are also implicated in delayed childhood hearing loss (2). The majority of NSHL cases follow
monogenic hereditary patterns; however, the high degree of clinical
and genetic heterogenicity often complicates the identification of
a definitive molecular etiology for numerous individuals (3). The advent of next-generation
sequencing has revolutionized the genetic diagnosis of hearing
loss; its high-throughput capacity has facilitated the
identification of numerous pathogenic variants and novel hearing
loss genes. To date, >120 genes have been associated with NSHL
(4).
The ability of the auditory system to process sound
relies on precise coordination among proteins. Within the inner
ear, hair cells function as sensory receptors, converting
mechanical sound vibrations into electrical signals (5). The apical surface of hair cells is
adorned with stereocilia, which are microvillar projections,
including static and motile cilia (6,7).
Plastin-1 (PLS1) is characterized by four calponin-homology domains
and an EF-hand calcium-binding motif for linking filamentous
(F)-actin (8). The rigidity and
structural integrity of these stereocilia bundles are key in
mechanotransduction and are maintained by actin-binding proteins,
including PLS1(9). The existence
of numerous F-actin crosslinkers regulates the rigidity of the
stereocilia, thereby modulating the sensitivity of the
sound-to-mechanotransduction process (9). Studies on plastin-1-deficient mice
have demonstrated that this protein is key for the proper bundling
of actin filaments within stereocilia, with its absence leading to
stereocilia pathology and hearing impairment (10,11).
The key role of PLS1 in human hearing was first established
in a study by Schrauwen et al (12), which identified it as a pathogenic
gene in an NSHL family. Subsequent reports have further
demonstrated that PLS1 mutations may be a cause of deafness
(13-15).
In the present study, the identification of a novel
splicing mutation of the PLS1 gene (c.981+5G>A) in a
family with NSHL through whole-exome sequencing is reported. IThe
mutation was investigated to elucidate the molecular mechanism
underlying the deafness phenotype in this family. The findings
expand the mutation spectrum of PLS1 and reinforce its key
role in hearing, providing valuable insights for improving the
clinical and molecular diagnosis, as well as prenatal
screening.
Materials and methods
Clinical characteristics
At the Northwest Women's and Children's Hospital
(Xi'an, China), an affected family was identified in December 2024
when the mother came for advice on having a second child. A
7-year-old girl and her mother, aged 36 years, exhibited congenital
hearing loss within this family. Subsequently, clinical information
was comprehensively collected from the affected individuals and
peripheral blood samples (6 ml each) were obtained from the
proband, and her parents and grandparents, in EDTA-anticoagulant
tubes. All patients or their legal guardians provided written
informed consent for both genetic counseling and molecular genetic
testing. The present study was approved by the Research Ethics
Committees of Northwest Women's and Children's Hospital and was
conducted in accordance with the ethical principles of the Helsinki
Declaration for medical research.
Molecular genetics analysis
Genomic DNA was extracted from peripheral blood
samples. Library preparation, sequencing and data analysis were
performed as previously described (16). Briefly, the standard procedure
included DNA fragmentation, end repair, amplification purification
and library quality control. Target regions were captured and
enriched to ensure sufficient coverage. Multiplexed sequencing was
performed on the AmCareSeq 2000 sequencer (Guangzhou Jiajian
Medical Testing Co., Ltd.) using a 2x150 bp paired-end sequencing
kit (Guangzhou Jiajian Medical Testing Co., Ltd.). The sequencing
covered all coding exons and flanking 20-base pair introns,
generating raw FASTQ data with an average depth of ~200x. Raw FASTQ
data were processed using a standard bioinformatics pipeline
whereby: i) Trimmomatic software (version 0.39) (17)was used to remove low-quality reads
(Phred quality score <20; read length <50 bp) and
adapter-contaminated reads; ii) clean reads were aligned to the
human reference genome (GRCh37/hg19) using the Burrows-Wheeler
Aligner software package (version 0.7.15) (18) under default parameters; iii)
aligned Binary Alignment/Map files were processed with the Genome
Analysis Toolkit (version 4.4.0; Broad Institute) for duplicate
marking (‘MarkDuplicates’), base quality score recalibration,
variant calling (‘HaplotypeCaller’) and outputting variants in
Variant Call Format files; and iv) variants were filtered by
excluding those with a minor allele frequency (MAF) >1% in
public databases [Exome Aggregation Consortium (https://avillach-lab.hms.harvard.ed-u/access-data/open-data);
Genome Aggregation Database (https://gnomad.broadinstitute.org)] low-quality copy
number variation (CNV) and synonymous or deep intronic variants not
near splicing sites. Furthermore, pathogenicity and evolutionary
conservation analyses were analyzed using the PolyPhen tool
(version 2) and VarCards database (version 2.0) (19). The final pathogenicity assessment
of variants and CNVs was conducted according to the guidelines of
the American College of Medical Genetics and Genomics (ACMG)
(20). Primer pairs were then
designed to validate these candidate pathogenic variants using
Sanger sequencing (Table I).
 | Table IPrimers of the Sanger sequencing
performed in the present study. |
Table I
Primers of the Sanger sequencing
performed in the present study.
| Primer | Sequence |
|---|
| PLS1-E9F |
5'-GAATCCAGGGAAGGCATACG-3' |
| PLS1-E9R |
5'-AGGGCTGAGGGCTCTACTGG-3' |
|
ELMOD3-12F |
5'-GGAAAGTTACTAAAGGTCACTGA-3' |
|
ELMOD3-12R |
5'-AGCCAGCAGGTGGAGTAGAG-3' |
| TNC-3F |
5'-GTCTGCGAACCTGGCTGGAA-3' |
| TNC-3R |
5'-CATGTGCCGTGCTCCTCACT-3' |
In silico analysis of novel splicing
mutations and protein structure
To predict the effect of the novel variant on mRNA
transcripts of PLS1, the online software AUGUSTUS
(https://bioinf.uni-greifswald.de/webaugustus/) was
used with scripts and parameters set according to the official
manual (21). Prediction results
were visualized using the Integrative Genomics Viewer (version
2.19.7) (22). Homology modeling
was employed to construct structural models of the PLS1 and β-actin
(ACTB) complex using SWISS-MODEL (https://swissmodel.expasy.org) and MODELLER (version
10.8) (23). The model was
constructed using a multi-template alignment strategy based on the
crystal structure of human PLS1 [Protein Data Bank (PDB): 1AOA], a
plant fimbrin structure resolved by cryo-electron microscopy and
ACTB (PDB: 3BYH). The final structural model was visualized
using PyMOL (version 3.1; Schrödinger).
Identification of splice transcripts
by reverse transcription PCR
Total RNA was extracted from the peripheral blood of
all patients and controls using the TRIzol® (Thermo
Fisher Scientific, Inc.) method. Furthermore, 1,000 ng of mRNA was
reverse-transcribed into cDNA using the PrimeScript™ RT
reagent kit according to the manufacturer's instructions (Takara
Bio Inc.). Primers were designed (forward,
5'-CTTGGTCTTGGGACTTCTCTG-3'; and reverse,
5'-TGTTCTTCCCAAGTTCCACT-3') to amplify the PLS1 exons of
interest in affected family members. Thermocycling conditions were
an initial denaturation at 98˚C, followed by annealing at 58˚C and
extension at 72˚C for 35 cycles. The amplification products were
verified through 1% agarose gel electrophoresis, and visualized by
gel imaging system (ChemiDoc™XRS; Bio-Rad Inc.). and
then subjected to Sanger sequencing (conducted by Beijing Tsingke
Biotech Co., Ltd.).
Results
Clinical characteristics of
patients
Within the present study, the two affected
individuals exhibiting hearing loss, were members of a
three-generation Chinese family from Shanxi (Fig. 1A). The proband (III-1), a
7-year-old girl, was diagnosed with congenital hearing loss,
failing newborn hearing screening, which included otoacoustic
emissions and automatic auditory brainstem response (Fig. 1B). The patient also exhibited an
abnormal V wave response in the right ear. At age 6, pure-tone
audiometry revealed bilateral symmetric severe hearing loss at
medium-to-high frequencies (250-8000 Hz), with abnormal air and
bone conduction thresholds (Fig.
1C). Tympanometry showed a ‘type As’ tympanogram in the right
ear and a ‘type B’ in the left. Typical inflammatory symptoms were
absent, ruling out conditions such as otitis media. The probands
mother, aged 36 years, also exhibited congenital hearing loss, with
pure-tone audiometry demonstrating levels markedly above the normal
threshold (≤25 dB HL) (Fig. 1D).
At present, both mother and daughter require hearing aids for daily
life. Furthermore, the father and maternal grandparents of the
proband currently exhibit no hearing abnormalities.
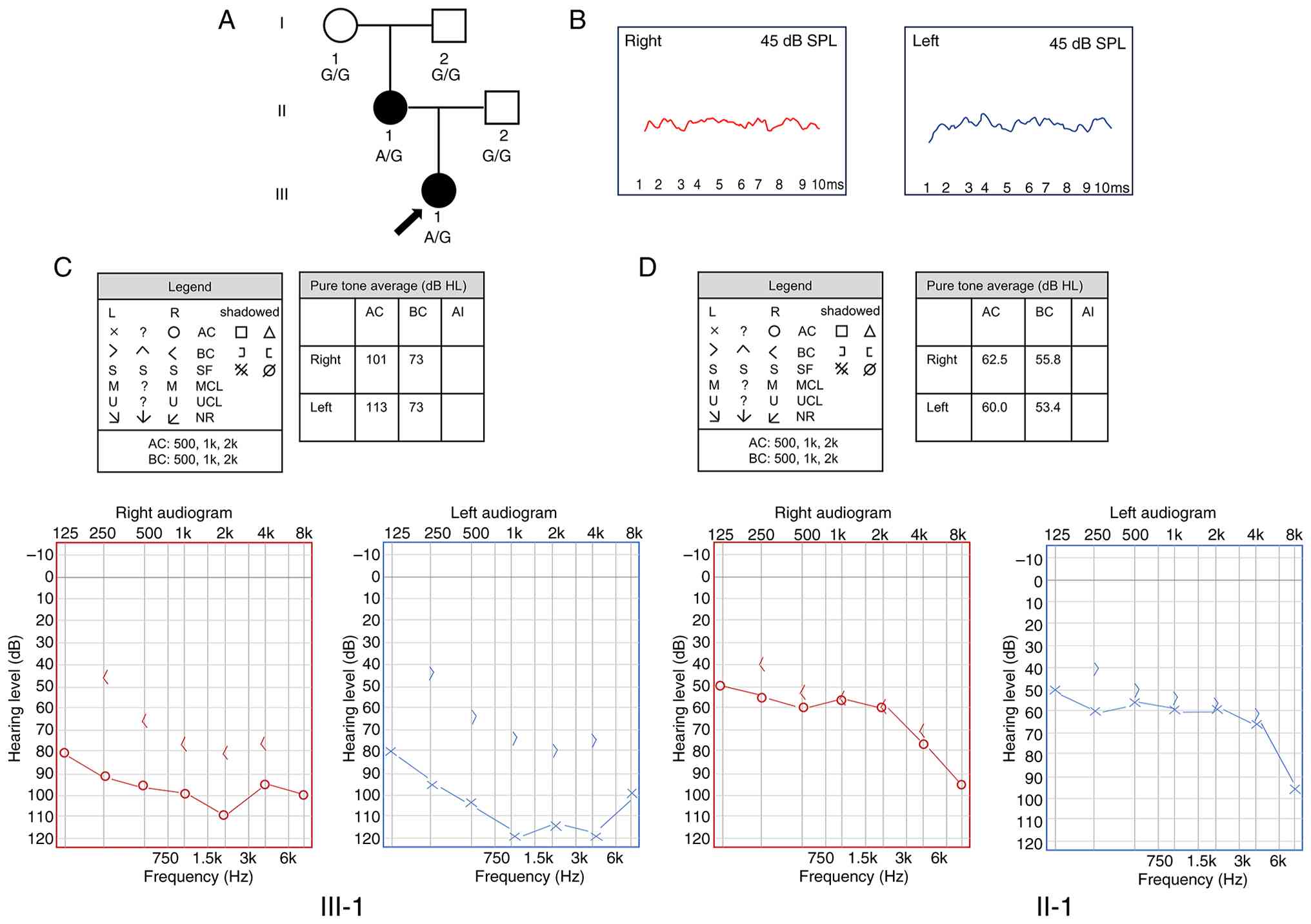 | Figure 1Identification of a family with
non-syndromic hearing loss. (A) Pedigree of the family. The arrow
indicates the proband. Affected family members are shown in black.
(B) Results of auditory brainstem response testing at the proband's
newborn hearing screening, showing an abnormal wave in the right
ear. (C) Pure-tone hearing testing results of the proband at 250
Hz-8k Hz, showing flat or declining curves in the bilateral ears.
Pure tone averages indicate severe-to-profound hearing loss. (D)
Pure-tone hearing testing results of the proband's mother at 250
Hz-8k Hz, showing flat or declining curves in the bilateral ears.
Pure tone averages indicate severe-to-profound hearing loss. AC,
air conduction; BC, bone conduction; SF, sound field; MCL, most
comfortable level; UCL, uncomfortable level; NR, no response; AI,
asymmetry index; HL, hearing level; SPL, sound pressure level. |
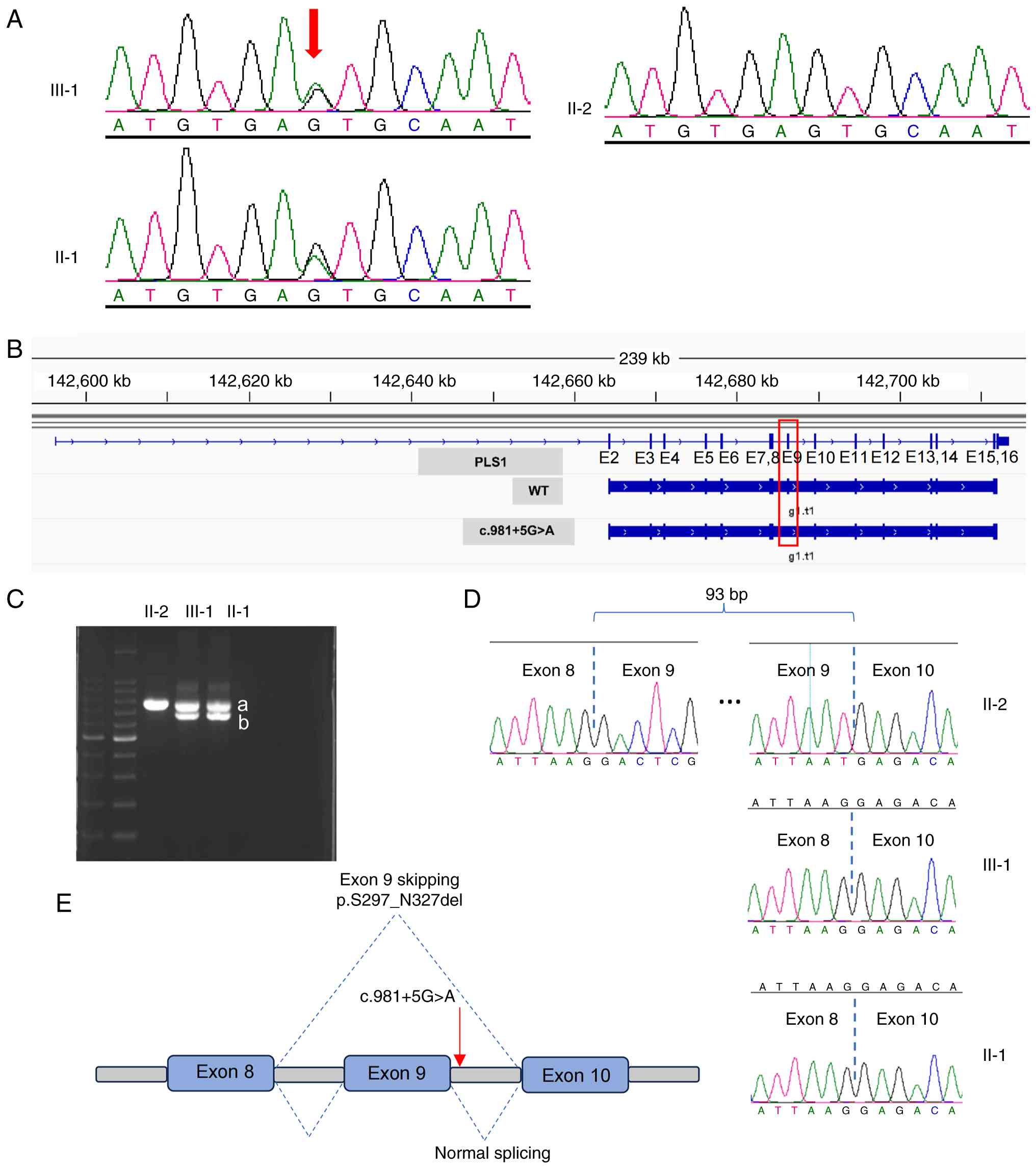
Identification of a novel PLS1
mutation through whole-exome sequencing
To determine the genetic etiology of the family's
congenital hearing loss, whole-exome sequencing was performed on
the proband. After filtering out variants with an MAF >0.01 in
public databases, the hearing loss-related genes matching the
patient phenotype were focused on. Subsequent bioinformatics
analysis identified a novel mutation, PLS1 c.981+5G>A
(reference sequence: NM_001145319.2), as the most likely candidate
variant. Sanger sequencing was applied to further demonstrate
segregation of this mutation within the disease presented by the
family (Fig. 2A). The mutation was
found to be present in the affected mother but absent in the
healthy grandparents (data not shown), indicating the de
novo nature of the mutation within the mother.
According to the ACMG guidelines and specialized
criteria for hereditary hearing loss (24), the PLS1 c.981+5G>A
variant is classified as a Variant of Uncertain Significance (VUS)
based on the following evidence codes: i) PM2 (absent or very low
frequency in normal population databases); ii) PM6-P (assumed de
novo without confirmed maternity and paternity. A phenotype
consistent with the genotype but not highly specific and with a
high degree of genetic heterogeneity); and iii) PP1 (co-segregation
with disease). Supporting its potential pathogenicity, the mutation
was predicted to alter PLS1 splicing by SpliceAI (Illumina,
Inc.).
PLS1 c.981+5G>A mutation generates
a novel transcript through exon 9 skipping
The PLS1 c.981+5G>A variant is located
near a splicing site. Therefore, using the AUGUSTUS program, this
mutation was predicted to cause the skipping of exon 9 (Fig. 2B). To validate this, total RNA was
extracted from the lymphocytes of all patients, including the
control (the father) and fragments were analyzed through reverse
transcription PCR and agarose gel electrophoresis. In addition to
the normal band, the proband and her affected mother exhibited an
unexpected, lower band that was absent in the control (Fig. 2C). Sanger sequencing demonstrated
that this extra band corresponded to a transcript lacking the
entire exon 9 (Fig. 2D). These
findings demonstrate that the PLS1 c.981+5G>A mutation
causes exon 9 skipping, generating an aberrant splicing
product.
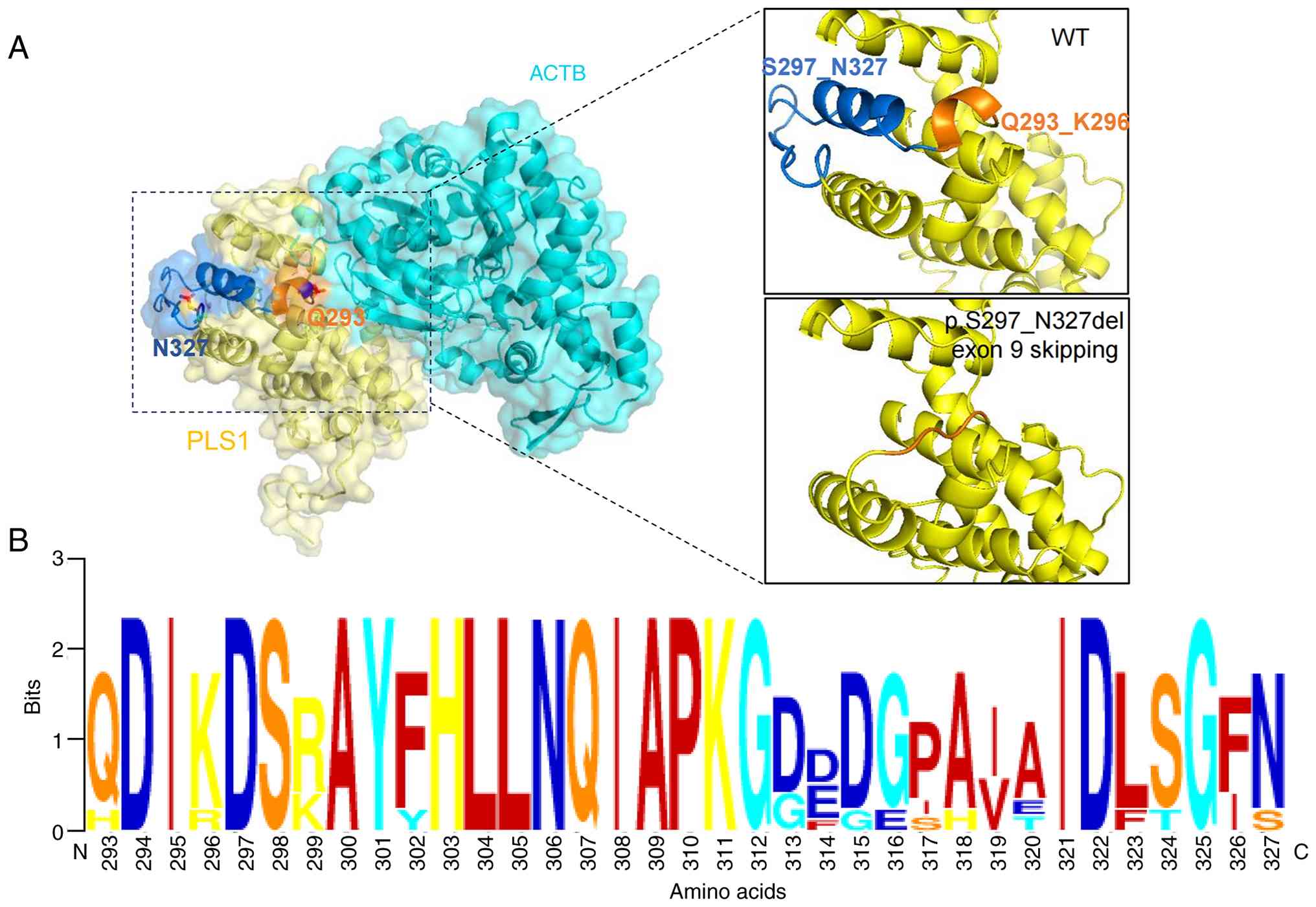
Mutant PLS1 protein impairs the
PLS1-ACTB interaction
To understand the physiological impact of the exon 9
skipping, a structural model of the actin-binding domain 1 (ABD1)
of PLS1 complexed with ACTB was constructed based on homology
modeling (Fig. 3A) using the
SWISS-MODEL program. Structural analysis revealed that the 31 amino
acid residues (p.S297_N327) encoded by exon 9 were part of the
calponin-homology (CH)-2 domain within ABD1. The deletion of this
region results in the loss of an α-helix (residues 293-296), which
directly faces the ACTB (Fig. 3A).
Therefore, the present study proposes that this structural
alteration disrupts the PLS1-ACTB interaction. Furthermore,
residues 293-327 are evolutionarily conserved across species,
underscoring their functional importance (Fig. 3B). Collectively, these data suggest
that the PLS1 c.981+5G>A mutation impairs the binding
between PLS1 and ACTB.
Discussion
Within the present study, two additional VUSs in the
probands mother were also identified by deafness gene panel, namely
ELMO domain containing 3 (ELMOD3) c.815+1G>C and
tenascin-C (TNC) c.625G>A. While both genes are
associated with hearing loss [Online Mendelian Inheritance in Man
(OMIM)_619500 and OMIM_615629, respectively], they are linked to
postlingual, progressive, high-frequency hearing impairment with an
onset of 8-30 years of age (25,26).
By contrast, both affected individuals in the present family
exhibited prelingual deafness, consistent with other reported NSHL
cases caused by PLS1 mutations (12-15).
Furthermore, neither of these mutations were inherited by the
proband (Fig. S1) and two primer
pairs were designed to rule them out (Table I). Consequently, ELMOD3
c.815+1G>C and TNC c.625G>A were excluded as
candidates. This underscores that the clinical features,
particularly the age of hearing loss onset, are key to an accurate
molecular diagnosis, especially given the large number of genes
associated with hearing loss. Auditory brainstem response is the
most commonly employed test to identify post-cochlear lesions and
central nervous system disorders. In the present study, since the
auditory brainstem response data originate from the newborn hearing
screening of the proband when the patient was born, only waveforms
less than a sound pressure level of 45 dB could be obtained.
Although the lack of additional waveform analysis under multiple
stimulation is a limitation of the present study, we consider that
integrating all hearing test findings provides an adequate basis
for diagnosing the congenital hearing loss of the proband.
PLS1 encodes plastin-1 (also known as
fimbrin), a 629-amino acid protein containing two EF-hand domains
and two actin-binding domains (ABD1 and ABD2), each composed of CH
domains (27). Reported cases of
hearing loss due to PLS1 mutations are scarce. To the best
of our knowledge, including the present study, only 12 families
with NSHL caused by PLS1 mutations have been described
(12-15),
eight of which are from Europe. The remaining three originate from
Asia, two of which belong to the Han Chinese population. While this
may suggest a higher incidence in Europe, more data is needed to
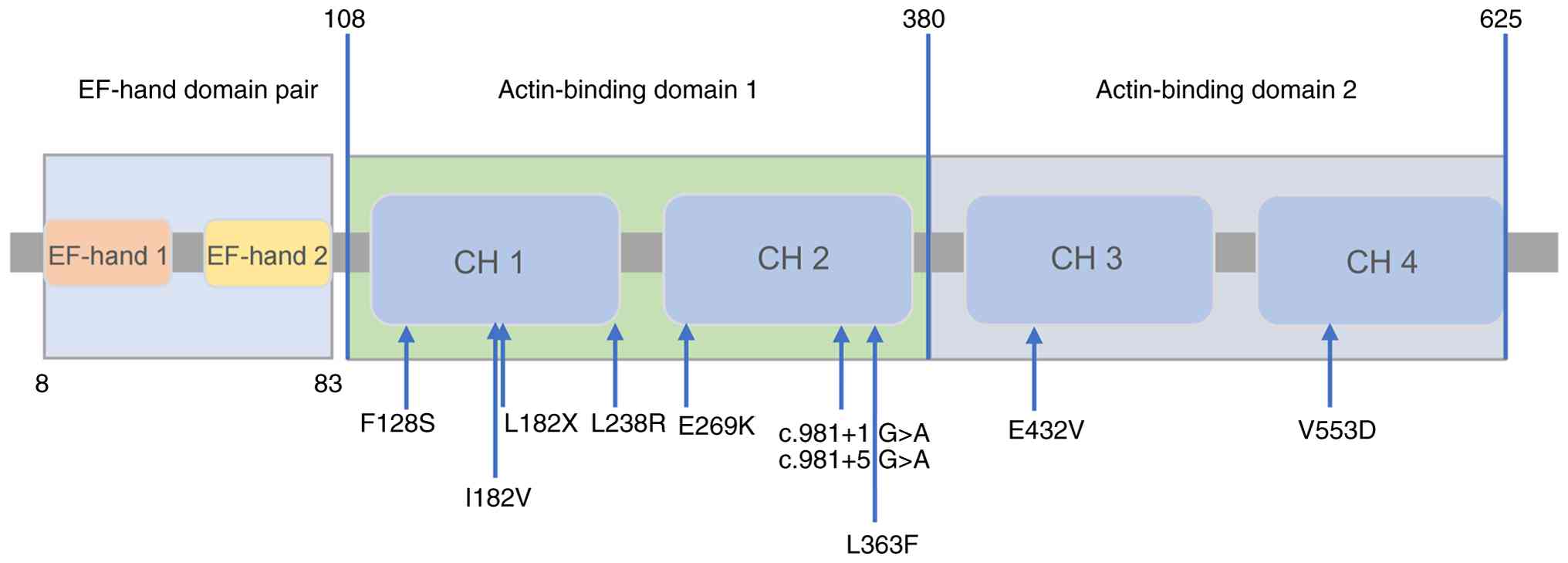
validate this. To date, 10 PLS1 mutations causing autosomal
dominant deafness have been identified, including seven missenses,
two splice sites and one frameshift. Notably, both reported
splicing mutations were identified in Chinese families. As
summarized in Fig. 4, the majority
of variants are located within the CH domains of ABD1. PLS1
crosslinks actin filaments by using its two ABDs to recruit and
bind ACTB molecules on opposite faces (28). Therefore, structural defects in
ABD1 or ABD2, whether caused by missense or splicing variants,
likely impair actin binding, ultimately disrupting stereocilia
formation and causing hearing loss.
In the present study, results suggested that the
novel c.981+5G>A variant caused the skipping of exon 9, which
aligns with the effect of a previously reported mutation at another
position, namely c.981+1G>A (15). It has been demonstrated that the
deletion of 31 amino acids (p.S297_N327) causes abnormal inner ear
phenotypes in zebrafish through mRNA microinjection experiments
(15), supporting the deleterious
nature of the variant identified in the present study. This
functional evidence meets the ‘PS3_Moderate’ criterion under ACMG
guidelines. Therefore, the c.981+5G>A variant was reclassified
from a VUS to ‘likely pathogenic’. Moreover, RNA-sequencing
analysis in HEI-OC1 cells suggests that PLS1 contributes to hearing
loss by modulating the PI3K-AKT signaling pathway and focal
adhesion (15). However, the
precise molecular mechanisms require further investigation.
In conclusion, the present study reports on a family
with autosomal dominant NSHL caused by a novel PLS1 splicing
variant, demonstrating that the variant causes exon 9 skipping. The
resulting truncation of the protein causes the loss of key amino
acids, which is predicted to disrupt the actin-binding function of
PLS1 and impair stereocilia formation. The present study therefore
provides a conclusive genetic diagnosis for this family.
Supplementary Material
Sanger sequencing. The results of the
Sanger sequencing confirm the presence of mutations TNC c.625G>A
and ELMOD3 c.815+1G>C in the affected mother (II1) but not the
proband (III1). The red arrows indicate the mutation site.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by the Natural Science
Basic Research Program of Shaanxi (grant nos. 2025JC-YBQN-1137 and
2024JC-YBMS-677) and the Incubation Research Project of Northwest
Women's and Children's Hospital (grant no. 2024FH06).
Availability of data and materials
The data generated in the present study may be found
in the China National GeneBank database under accession number
CNP0008613 or at the following URL: http://db.cngb.org/cnsa/project/CNP0008613_283e261b/reviewlink/.
Authors' contributions
CY, YX and DW performed the bioinformatics analysis
and experiments. CY and JS designed the present study. CY wrote the
manuscript. JS supervised the present study. CY and YX confirm the
authenticity of all the raw data. All authors have read and
approved the final version of the manuscript.
Ethics approval and consent to
participate
The present study was approved by the Research
Ethics Committees of Northwest Women's and Children's Hospital
(Xi'an, China; approval no. 202500802). All patients provided
written informed consent for both genetic counseling and molecular
genetic testing. The written informed consent of minors/children in
the present study was provided by their legal guardian.
Patient consent for publication
The proband, her father (II-2), mother (II-1),
grandmother (I-1) and grandfather (I-2) provided written informed
consent for the publication of all associated data.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Wang Q, Xiang J, Sun J, Yang Y, Guan J,
Wang D, Song C, Guo L, Wang H, Chen Y, et al: Nationwide population
genetic screening improves outcomes of newborn screening for
hearing loss in China. Genet Med. 21:2231–2238. 2019.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Shearer AE, Eppsteiner RW, Booth KT,
Ephraim SS, Gurrola J II, Simpson A, Black-Ziegelbein EA, Joshi S,
Ravi H, Giuffre AC, et al: Utilizing ethnic-specific differences in
minor allele frequency to recategorize reported pathogenic deafness
variants. Am J Hum Genet. 95:445–453. 2014.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Lieu JEC, Kenna M, Anne S and Davidson L:
Hearing loss in children: A review. JAMA. 324:2195–2205.
2020.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Walls WD, Azaiez H and Smith RJH:
Hereditary Hearing Loss. Homepage. Available: https://hereditaryhearingloss.org. Accessed on 19
February 2025.
|
|
5
|
Peng AW, Salles FT, Pan B and Ricci AJ:
Integrating the biophysical and molecular mechanisms of auditory
hair cell mechanotransduction. Nat Commun. 2(523)2011.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Mogensen MM, Rzadzinska A and Steel KP:
The deaf mouse mutant whirler suggests a role for whirlin in actin
filament dynamics and stereocilia development. Cell Motil
Cytoskeleton. 64:496–508. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
7
|
DeRosier DJ and Tilney LG: F-actin bundles
are derivatives of microvilli: What does this tell us about how
bundles might form? J Cell Biol. 148:1–6. 2000.PubMed/NCBI
|
|
8
|
Lin CS, Shen W, Chen ZP, Tu YH and
Matsudaira P: Identification of I-plastin, a human fimbrin isoform
expressed in intestine and kidney. Mol Cell Biol. 14:2457–2467.
1994.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Richardson GP, de Monvel JB and Petit C:
How the genetics of deafness illuminates auditory physiology. Annu
Rev Physiol. 73:311–334. 2011.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Taylor R, Bullen A, Johnson SL,
Grimm-Günter EM, Rivero F, Marcotti W, Forge A and Daudet N:
Absence of plastin 1 causes abnormal maintenance of hair cell
stereocilia and a moderate form of hearing loss in mice. Hum Mol
Genet. 24:37–49. 2015.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Krey JF, Krystofiak ES, Dumont RA,
Vijayakumar S, Choi D, Rivero F, Kachar B, Jones SM and
Barr-Gillespie PG: Plastin 1 widens stereocilia by transforming
actin filament packing from hexagonal to liquid. J Cell Biol.
215:467–482. 2016.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Schrauwen I, Melegh BI, Chakchouk I,
Acharya A, Nasir A, Poston A, Cornejo-Sanchez DM, Szabo Z, Karosi
T, Bene J, et al: Hearing impairment locus heterogeneity and
identification of PLS1 as a new autosomal dominant gene in
Hungarian Roma. Eur J Hum Genet. 27:869–878. 2019.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Morgan A, Koboldt DC, Barrie ES, Crist ER,
García García G, Mezzavilla M, Faletra F, Mihalic Mosher T, Wilson
RK, Blanchet C, et al: Mutations in PLS1, encoding fimbrin, cause
autosomal dominant nonsyndromic hearing loss. Hum Mutat.
40:2286–2295. 2019.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Diaz-Horta O, Bademci G, Tokgoz-Yilmaz S,
Guo S, Zafeer F, Sineni CJ, Duman D, Farooq A and Tekin M: Novel
variant p.E269K confirms causative role of PLS1 mutations in
autosomal dominant hearing loss. Clin Genet. 96:575–578.
2019.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Xu L, Wang X, Li J, Chen L, Wang H, Xu S,
Zhang Y, Li W, Yao P, Tan M, et al: A novel PLS1 c.981+1G>A
variant causes autosomal-dominant hereditary hearing loss in a
family. Clin Genet. 103:413–423. 2023.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Cai H, Bai H, Qiao S, Xue X, Shi W and Shi
J: Clinical exome sequencing for carrier screening in assisted
reproductive technology and sperm donation. J Assist Reprod Genet.
42:1247–1256. 2025.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Bolger AM, Lohse M and Usadel B:
Trimmomatic: A flexible trimmer for illumina sequence data.
Bioinformatics. 30:2114–2119. 2014.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Li H and Durbin R: Fast and accurate
long-read alignment with Burrows-Wheeler transform. Bioinformatics.
26:589–595. 2010.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Wang Z, Zhao G, Zhu Z, Wang Y, Xiang X,
Zhang S, Luo T, Zhou Q, Qiu J, Tang B, et al: VarCards2: An
integrated genetic and clinical database for ACMG-AMP
variant-interpretation guidelines in the human whole genome.
Nucleic Acids Res. 52:D1478–D1489. 2024.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Miller DT, Lee K, Abul-Husn NS, Amendola
LM, Brothers K, Chung WK, Gollob MH, Gordon AS, Harrison SM,
Hershberger RE, et al: ACMG SF v3.2 list for reporting of secondary
findings in clinical exome and genome sequencing: A policy
statement of the American College of Medical Genetics and Genomics
(ACMG). Genet Med. 25(100866)2023.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Stanke M, Diekhans M, Baertsch R and
Haussler D: Using native and syntenically mapped cDNA alignments to
improve de novo gene finding. Bioinformatics. 24:637–644.
2008.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Robinson JT, Thorvaldsdóttir H, Turner D
and Mesirov JP: igv.js: An embeddable JavaScript implementation of
the integrative genomics viewer (IGV). Bioinformatics.
39(btac830)2023.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Marti-Renom MA, Stuart AC, Fiser A,
Sánchez R, Melo F and Sali A: Comparative protein structure
modeling of genes and genomes. Annu RevBiophys Biomol Struct.
29:291–325. 2000.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Oza AM, DiStefano MT, Hemphill SE, Cushman
BJ, Grant AR, Siegert RK, Shen J, Chapin A, Boczek NJ, Schimmenti
LA, et al: Expert specification of the ACMG/AMP variant
interpretation guidelines for genetic hearing loss. Hum Mutat.
39:1593–1613. 2018.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Li W, Sun J, Ling J, Li J, He C, Liu Y,
Chen H, Men M, Niu Z, Deng Y, et al: ELMOD3, a novel causative
gene, associated with human autosomal dominant nonsyndromic and
progressive hearing loss. Hum Genet. 137:329–342. 2018.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhao Y, Zhao F, Zong L, Zhang P, Guan L,
Zhang J, Wang D, Wang J, Chai W, Lan L, et al: Exome sequencing and
linkage analysis identified tenascin-C (TNC) as a novel causative
gene in nonsyndromic hearing loss. PLoS One.
8(e69549)2013.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Bañuelos S, Saraste M and Djinović Carugo
K: Structural comparisons of calponin homology domains:
Implications for actin binding. Structure. 6:1419–1431.
1998.PubMed/NCBI View Article : Google Scholar
|
|
28
|
de Arruda MV, Watson S, Lin CS, Leavitt J
and Matsudaira P: Fimbrin is a homologue of the cytoplasmic
phosphoprotein plastin and has domains homologous with calmodulin
and actin gelation proteins. J Cell Biol. 111:1069–1079.
1990.PubMed/NCBI View Article : Google Scholar
|