Cardiomyopathy is a heterogeneous group of
conditions with structural and/or functional myocardial lesions,
usually in the form of cardiac dilatation, arrhythmias or heart
failure (1). Incidence and
mortality rates continue to rise globally, making it one of the
most notable causes of cardiovascular-related mortality, with the
incidence in China reaching 11.76 per 100,000 person-years and an
overall mortality rate of 3.38% (2). Cardiomyopathies, based on etiology
and clinical presentation, are categorized into dilated,
hypertrophic, restrictive and arrhythmogenic right ventricular
types (3). The pathological
substrate of cardiomyopathy is multifactorial, and comprises
genetic mutation, metabolic derangement, inflammatory activation
and oxidative stress. Present evidence has highlighted that
impaired cellular energy metabolism, calcium dysregulation and
apoptosis are the principal molecular processes underlying the
onset and progression of the disease (4). Intracellular homeostasis disruption
thereby leads to systemic abnormalities in cardiomyocyte energy
metabolism, protein quality control and signal transduction and
eventually culminates in cardiac remodeling and contractile
dysfunction (5). Elucidation of
these pathogenic mechanisms is important for deciphering the
molecular pathogenesis of cardiomyopathy and identifying new
therapeutic targets.
The pathological alterations of cardiomyopathy are
primarily at the level of cardiomyocytes, whose dysfunction is
responsible for the development of the disease. Cardiomyocytes
contain different membranous and non-membranous organelles such as
mitochondria, endoplasmic reticulum (ER), Golgi apparatus,
lysosomes and ribosomes that collectively create a complex
intracellular network of organelles to keep cardiac function
intact. Prior research has focused on myofibrillar (6,7) and
ion channel abnormalities (8,9);
however, recent advances in cell biology and high-resolution
microscopy have demonstrated that organelle homeostasis serves a
major role in the pathogenesis of cardiomyopathy (10-12).
Since mitochondrial dysfunction was originally associated with
cardiomyopathy during the second half of the twentieth century
(13), more evidence has uncovered
other mechanisms, including ER stress (14), Golgi apparatus fragmentation
(15), lysosomal-autophagic
dysfunction (16) and defects in
ribosomal quality control (17),
which have provided novel insights into the molecular
pathophysiology of cardiomyopathy. Organelles not only regulate
cardiomyocyte metabolism and survival, but also orchestrate
global-cardiac reprogramming of metabolism and stress adaptation,
suggesting that the targeting of organelle biology represents a
valuable direction for mechanistic exploration and pharmacological
identification of novel drugs.
The present review summarized the advances in
understanding the functions of several organelles in
cardiomyopathy. Mitochondrial energy metabolism disorders, calcium
overload and imbalanced dynamics have been reported to be marked
contributors to cardiomyocyte dysfunction; ER stress and disturbed
ER-mitochondria coupling serve a key role in protein homeostasis
and calcium signaling; Golgi fragmentation and aberrant
glycosylation are detrimental to membrane protein traffic and
signaling pathways; lysosomal dysfunction and impaired autophagic
flux compromise intracellular degradation; and ribosome biogenesis
and translational regulation defects reveal pathological
alterations at the protein synthesis level. Disruption of nuclear
envelope integrity and intercalated disc architecture perturbs
essential mechanical signaling and gene regulatory programs.
Together, these findings indicate that cardiomyopathy is a systemic
illness of deranged multi-organelle homeostasis. In the future, the
integration of single-cell omics, spatial transcriptomics and
high-resolution imaging technologies will enable dynamic dissection
of inter-organelle communication and spatiotemporal coordination,
thereby redirecting cardiomyopathy research from ‘single-organelle
pathology’ to a ‘global organelle network pathology’ and paving the
way to precision therapeutic strategies.
A total of two independent reviewers conducted a
three-stage screening process: i) Title screening; ii) abstract
screening; and iii) full-text evaluation. Studies were included if
they met the following criteria: i) Investigated organelle
structure, function or homeostasis in the context of
cardiomyopathy; ii) provided mechanistic evidence at the cellular,
animal or human level; and iii) reported molecular, biochemical,
ultrastructural or signaling-based insights relevant to cardiac
pathophysiology. Studies were excluded if they: i) Lacked relevance
to organelle biology or cardiomyopathy mechanisms; ii) did not
present mechanistic data (such as commentary, purely descriptive
reports); and iii) focused on non-cardiac tissues without
cardiovascular implications. The reference lists of key articles
were also manually screened to identify additional eligible
studies. Any disagreements were resolved through discussion until
consensus was reached.
For each included study, data were extracted on the
type of organelle investigated, experimental model and mechanistic
pathways. Evidence was categorized into three levels: i) Cellular
evidence, including in vitro experiments, molecular
signaling and organelle function assays; ii) animal evidence,
including knockout/knock-in models, disease phenotyping,
mechanistic validation; and iii) human evidence, including patient
tissue, clinical correlations and genetic variants.
A total of 166 peer-reviewed articles met the
inclusion criteria and were synthesized narratively, with emphasis
on mechanistic pathways and organelle-specific contributions to
cardiomyopathy.
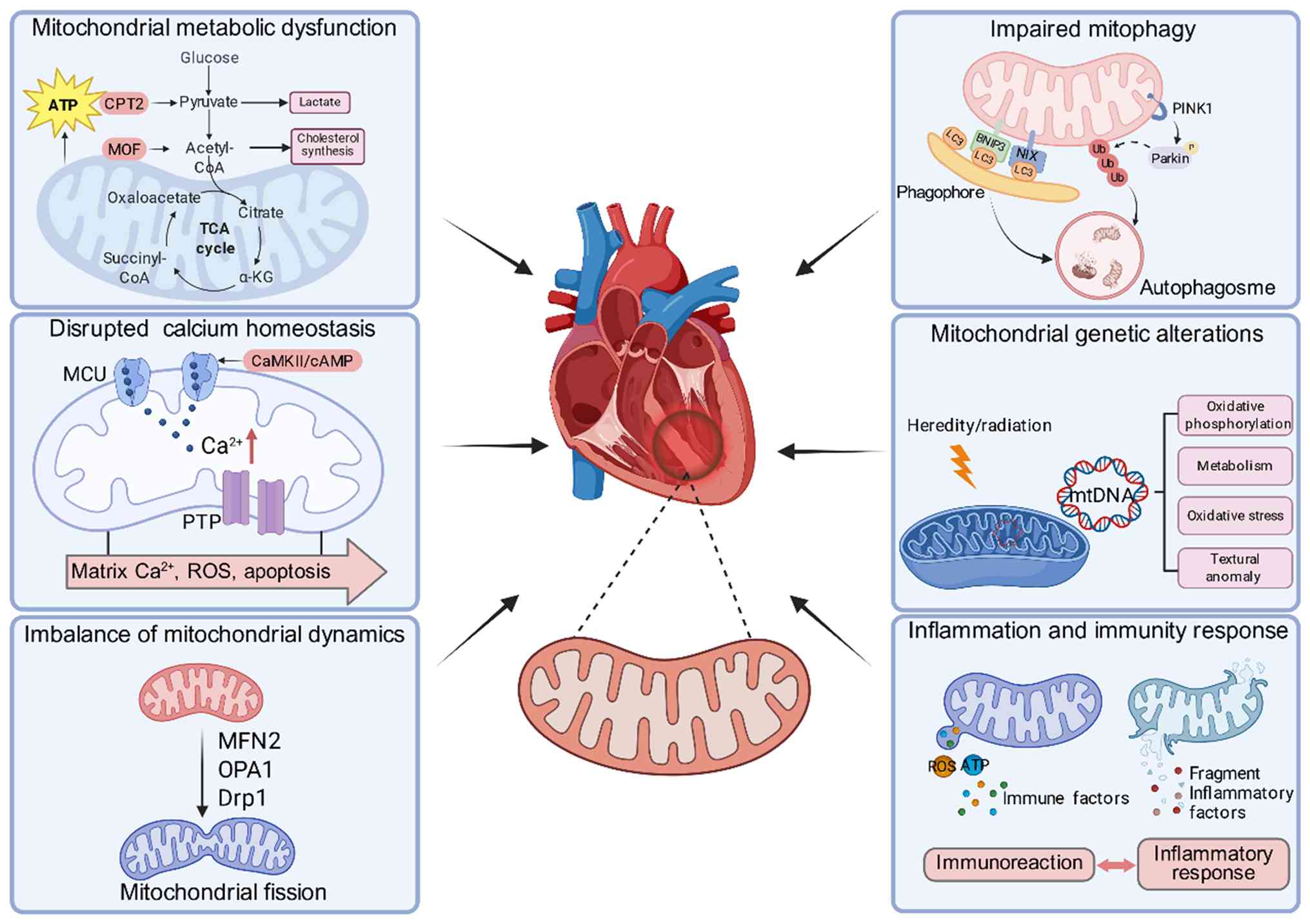
Over the years, accumulating evidence has shed light
on the pivotal position occupied by mitochondria in the
pathogenesis and progression of cardiomyopathies. Mitochondria not
only act as the primary source of cellular energy, but also
participate in various biological processes, including calcium
homeostasis, control of oxidative stress and apoptosis (18,19).
Mitochondrial dysfunction has been shown to induce myocardial
disturbances in energy metabolism (20), cellular injury (21) and cardiac failure (22). Therefore, understanding the
mechanisms through which mitochondria are implicated in
cardiomyopathy is required in order to study its pathogenesis at a
deeper level and for the identification of new therapeutic
targets.
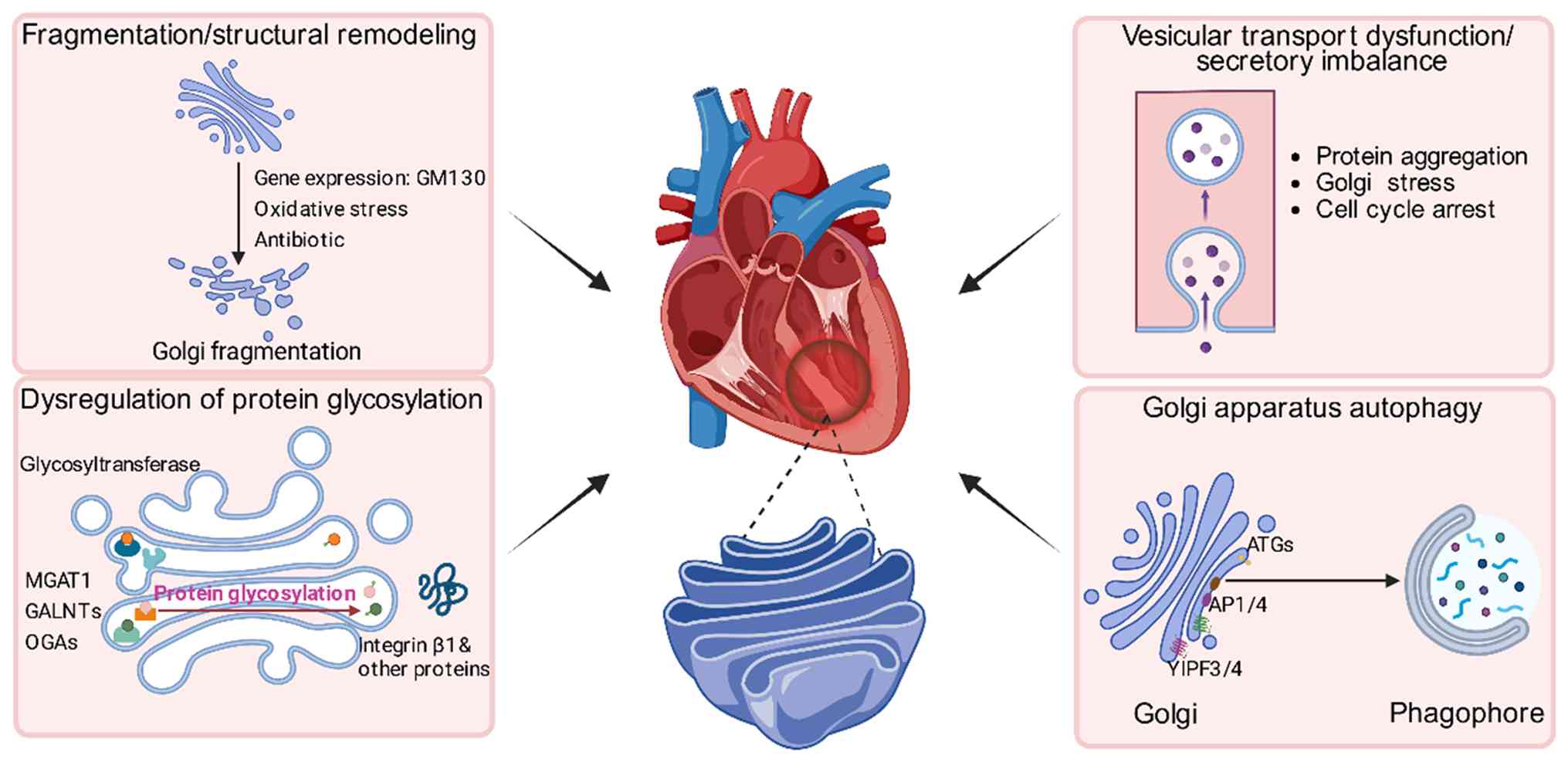
In complex network of organelles, the Golgi
apparatus serves a notable regulatory role in maintaining
post-translational modification, protein sorting and vesicular
trafficking. Increased evidence supports the Golgi apparatus
structural and functional dysfunctions as key to the development
and progression of cardiomyopathy. The subsequent section provides
brief summaries of research advances in the role of the Golgi
apparatus in cardiomyopathies from various pathological process
viewpoints.
In summary, the Golgi apparatus contributes to
cardiomyopathies pathogenesis by various molecular pathways,
including structural deficiency, defect in protein modification,
dysfunction of autophagy, disruption of the metabolism pathways of
extracellular matrix and failure in stress response (Fig. 2). These findings further provide
insights into cardiomyopathy pathogenesis and potential therapeutic
intervention.
Highly differentiated terminal cardiomyocytes rely
on ER homeostasis for structure and function (73). Recent research has shown that ER
stress and the resulting unfolded protein response (UPR) are
central to the pathogenesis of numerous cardiomyopathies, including
dilated, hypertrophic and ischemic cardiomyopathies (74,75).
Failure of the ER drives pathological processes such as
cardiomyocyte apoptosis, fibrosis and contractile dysfunction
through different mechanisms, including impaired protein quality
control, disrupted calcium homeostasis and dysregulated
inter-organelle communication (76).
Lysosomes play roles in the maintenance of
cardiomyocyte homeostasis through processes such as regulation of
autophagy, protein homeostasis, energy metabolism and regulation of
calcium signaling. Lysosomal disease is strongly linked to various
forms of cardiomyopathy, most prominently hypertrophic and
restrictive cardiomyopathy observed in lysosomal storage disorders.
Beyond these inherited conditions, impaired lysosomal degradation
and autophagic flux have also been linked to maladaptive cardiac
remodeling in pressure overload-induced hypertrophy, ischemic
cardiomyopathy and metabolic cardiomyopathy, although the strongest
and most direct association is observed in lysosomal storage
disease-associated cardiomyopathies. Mutations in lysosome-related
genes may potentially influence cardiac structure and function
directly. The present section is aimed at critically discussing the
advances in elucidating the role of lysosomes in cardiomyopathy
pathogenesis and reporting new information regarding their
underlying molecular and pathophysiological mechanisms.
A previous study demonstrated that downregulation of
the lysosome-associated membrane protein LAMP2 in hypertrophic
cardiomyopathy models leads to autophagosome-lysosome fusion
defects and increases cardiomyocyte apoptosis (110). Conversely, exogenous LAMP2B
expression has been shown to normalize autophagic processing in
myocardial ischemia-reperfusion injury (111). Danon disease, an X-linked
dominant disorder caused by mutations in the LAMP2 gene, is
characterized by giant lysosomes in cardiomyocytes and severe
myocardial hypertrophy (112).
Pan et al (113) also
established a cardiac proteinopathy mouse model and showed that
TFEB activation enhances autophagic flux by activating the
autophagy-lysosome axis, thereby promoting the lysosomal breakdown
of ubiquitinated proteins and minimizing cardiomyocyte
proteotoxicity. In myocardial ischemia-reperfusion injury,
lysosomal inhibition and depletion are also reported, whereas a
rise in TFEB expression or an increase in its nuclear translocation
augments lysosomal biogenesis and recovers lysosomal function
(114). Overall, these findings
highlight the pivotal role played by the lysosomal-autophagy system
in securing cardiac homeostasis, and place a focus on its potential
as an active therapeutic target in the prevention and treatment of
cardiomyopathies.
In summary, lysosomes maintain cardiomyocyte
homeostasis not only through degradation but also as key signaling
hubs coordinating diverse pathophysiological processes. Lysosomal
deficiency impinges upon autophagic flux, energy metabolism,
calcium homeostasis and signal transduction, all synergistically
acting to initiate and advance cardiomyopathies. With continuous
research progresses in this field, lysosome-targeted therapy has
the potential to be a novel treatment method for patients with
cardiomyopathy.
The ribosome, a central molecular machinery
orchestrating cellular protein synthesis, is a notable regulator of
cardiomyocyte homeostasis and cardiac integrity. Previous studies
have indicated that ribosomes not only perform the primary activity
of protein translation but are also involved in cellular stress
adaptation through mechanisms such as ribosome quality control
(RQC) and regulation of translational selection (126,127). The following section summarizes
advances on how ribosomes contribute to cardiomyopathy, providing
new insights into cardiac disease mechanisms.
Overall, cardiomyocyte ribosomes not only synthesize
proteins but also form a central regulatory network governing
proteome homeostasis and cardiac function, where disruptions in
structure, biogenesis, elongation dynamics or quality control may
drive cardiomyopathy. Advances in ribosome profiling and functional
genomics will improve molecular subtyping and mechanistic insights,
guiding ribosome-targeted therapies for cardiomyopathy.
The fine structure of cardiomyocytes is the
foundation of their normal contraction and electrophysiological
activity. Studies have revealed that the nucleus/nuclear envelope
and the intercalated discs are not merely static cellular
components, but dynamic signaling hubs, and their dysfunction
directly drives the occurrence and progression of various
cardiomyopathies.
The nuclear envelope is composed of the inner
nuclear membrane, the outer nuclear membrane and the perinuclear
space between them and provides mechanical support through a
network of nuclear lamina proteins. The LMNA gene, which
encodes nuclear lamina proteins, is one of the most common causes
of autosomal DCM and conduction system diseases, accounting for
5-10% of familial DCM cases and a notably higher proportion
(30-40%) in patients with concomitant conduction defects. Mutations
in this gene lead to disease primarily by disrupting nuclear
membrane stability and function (140). Under pathological conditions,
mutated nuclear membrane proteins can trigger nuclear membrane
rupture, leading to sustained activation of the DNA damage response
and apoptosis of cardiomyocytes, accelerating the progression of
heart failure (141). In
addition, the nuclear membrane acts as a mechanical sensor,
transmitting extracellular mechanical stress to the chromatin and
regulating gene expression; LMNA mutations disrupt this function,
leading to the activation of pathological gene programs (142). Abnormalities in nuclear
membrane-associated proteins such as Emerin are also closely
related to Emery-Dreifuss muscular dystrophy, as well as syndromic
DCM or arrhythmogenic cardiomyopathy (143,144), highlighting the central role of
the nuclear membrane complex in maintaining myocardial
integrity.
Intercalated discs are specialized cell-cell
junctions that connect cardiac muscle cells, consisting of
desmosomes, adherens junctions and gap junctions, and are
indispensable for mechanical transmission and electrical coupling
(145). Studies on the
ultrastructure and proteomics of the stratum corneum have revealed
the complexity and plasticity of its composition, particularly
highlighting that the structure and regulation of desmosomes serve
a key role in maintaining cell adhesion (146-148).
Mutations in multiple genes encoding intercalated disc-related
proteins have been confirmed to be closely associated with
arrhythmogenic cardiomyopathy (149), including plakophilin-2,
desmoplakin, desmoglein-2, desmocollin-2 and plakoglobin,
suggesting that intercalated disc instability is one of the core
molecular mechanisms of this disease. The intercalated disc not
only serves a mechanical connection function but also acts as a
‘hub’ for mechanical sensing and signal transduction, participating
in myocardial stress responses and pathological remodeling through
signaling pathways such as Hippo-YAP and Wnt/β-catenin (150). In addition, abnormal localization
and functional changes of intercalated disc proteins have also been
reported in dilated and hypertrophic cardiomyopathy (151). Specifically, key intercalated
disc components such as connexin 43, plakoglobin and N-cadherin are
frequently redistributed from the intercalated discs to the lateral
membranes or cytoplasm of cardiomyocytes, accompanied by impaired
gap junction coupling, weakened mechanical adhesion and altered
electrical conduction. These changes promote electrical
instability, contractile dysfunction and adverse cardiac
remodeling, indicating that intercalated disc pathology plays a
broad disease-promoting role across diverse cardiomyopathy
spectra.
Over the past decade, research on the molecular
pathogenesis of cardiomyopathy has progressed from a
single-organelle dysfunction-driven paradigm to an integrative
system based on systemic homeostatic failure following
inter-organelle crosstalk. The present review synthesizes
increasing evidence pointing towards mitochondria, ER, Golgi
apparatus, lysosomes and ribosomes remodeling their structure and
function markedly during the development and progression of
cardiomyopathy. These membranous and non-membranous compartments
are additionally closely integrated by networks that regulate
intracellular signaling, proteostasis, bioenergetic metabolism,
calcium signaling and vesicular transport. Perturbations in a
single organelle thus cascade through the entire cellular network,
spreading metabolic stress and continuing pathological remodeling.
In contrast to the old paradigm that attributed cardiomyopathy to
abnormalities in isolated metabolic pathways or sarcomeric
proteins, the new paradigm of organelle network disease provides an
improved explanation of the heterogenous and multifactorial
clinical cardiomyopathies.
Mitochondria remain the focal point of remodeling
disease in cardiomyopathy. Their dysfunctional metabolism not only
affects ATP production and contractile efficiency, but also
triggers the endogenous innate immune signaling by releasing DAMPs
such as ROS, mtDNA and cardiolipin (152-154).
Non-normal oxidative phosphorylation and fatty acid oxidation
further exaggerate metabolic stiffness and induce a maladaptive
pathway towards inefficient glucose metabolism. This metabolic
reprogramming causally directs inflammatory signaling cascades
bidirectionally, thus creating a self-perpetuating loop of
metabolic stress and immune activation. For instance, mtDNA release
into the cytosol is found to stimulate type I interferon responses
through cGAS-STING activation (155,156), enhancing the proinflammatory
environment and possibly the kinetics of myocardial fibrosis and
structural remodeling. These findings highlight that therapeutic
approaches aimed specifically at the rehabilitation of
mitochondrial bioenergetics will be likely to provide only
transient benefit unless these therapies also control the spread of
inflammation and ensuing pathologies. The ER, or the intracellular
calcium central reservoir and regulatory site, is functionally
interdependent in a complex way with mitochondria via
mitochondria-associated membranes, thus integrating energy
metabolism and calcium signaling homeostasis (157). In cardiomyopathy, chronic ER
stress deteriorates MAM integrity, leading to dysregulation of
calcium efflux and ensuing mitochondrial calcium overload,
initiating an aberrant positive-feedback cycle of overproduction of
ROS and abnormal opening of the mitochondrial permeability
transition pore (158,159). SERCA2a abnormality also
accelerates the cascade earlier, not only prolonging myocardial
relaxation but also decreasing contractile efficiency and
accelerating heart failure onset. Therapeutically, attempts to
reverse SERCA2a function or to fix MAM structural dynamics are
potential therapies for normalization of calcium equilibrium
(160), However, accumulating
evidence suggests that these interventions are primarily effective
during early or compensatory stages of cardiomyopathy.
The Golgi apparatus, previously considered a passive
accepting organ for protein targeting and post-translational
modification, is now recognized as an active regulatory site
functionally integrated within cellular homeostasis (161,162). In cardiomyopathies, Golgi
apparatus disruption, manifests as structural fragmentation,
defective glycosylation and disruption of vesicular trafficking,
which notably compromises the spatial arrangement of membrane
proteins and intracellular cascades of signals (163,164). These perturbations disrupt proper
receptor and ion channel expression and localization, extracellular
matrix turnover and mechanotransducive responsiveness, thus
remodeling cardiomyocyte biomechanical properties. Additionally,
increasing evidence suggests that Golgi apparatus impairment, ER
stress and ribosomal quality-control failure are temporally and
mechanistically interdependent (165,166). This spectrum suggests that early
dysfunctions of the protein-processing machinery may cause
excessive pressure on subsequent surveillance and degradative
mechanisms, eventually enhancing proteostatic degradation during
cardiac remodeling.
The RQC pathway is a notable protective mechanism
against translational blockage and dysregulated accumulation of
nascent polypeptides in cardiomyocytes (167). During hemodynamic stress or
cardiotoxic injury, dysfunction of integral RQC machinery such as
HOIL-1 and Vms1 eliminates co-translational surveillance, resulting
in dysregulated accumulation of defective stalled ribosomal
complexes and defective proteins (168). This proteostatic deregulation
triggers compensatory hyperactivation of the proteasome and
ultimately the activation of apoptotic cascades. In addition to ER
stress, translational stress in cardiomyocytes is beyond their
scope of adaptability, which results in overall failure during
sarcomeric assembly, membrane channel expression and integrity of
signal transduction. In therapeutic contexts, concerted modulation
of RQC function together with site-specific regulation of protein
degradation machineries has the potential to provide a novel
platform for enhancing proteostasis and alleviating cardiac
dysfunction in the context of precision medicine.
A deep analysis of the causal chain of
multi-organelle network imbalance in the development and
progression of cardiomyopathy will lay the foundation for precise
subtyping and mechanism-guided intervention strategies. First,
advances in single-cell multi-omics and spatial transcriptomics
have allowed spatiotemporal mapping of organelle interactions and
identification of key regulatory nodes, such as mitochondrial-ER
calcium coupling, Golgi apparatus-plasma membrane signaling and
nuclear mechanosensitive structures. These cross-scale datasets
provide a refined framework for molecular subtyping of
cardiomyopathy, and offer a basis for developing network-level
biomarkers capable of predicting disease progression and
therapeutic responsiveness. Second, multi-organelle intervention
strategies will become a core direction for future translational
research. For example, combined regulation of mitochondrial-ER
calcium flux, repair of Golgi glycosylation profiles, enhancement
of nuclear membrane stress resilience or remodeling of the
RQC-autophagy axis may all achieve multidimensional
cardioprotective effects with lower toxicity. Finally, the
development of organelle-penetrating drug delivery systems,
including organelle-targeted nanocarriers, programmable liposomes
and peptides directed to mitochondria or the Golgi apparatus, will
be essential for enabling mechanism-based therapy. In parallel,
platforms such as patient-derived cardiomyocytes, cardiac organoids
and engineered microtissues provide physiologically relevant
readouts of organelle function, allowing more accurate prediction
of individualized therapeutic responses and accelerating the
clinical translation of organelle-targeted interventions.
Not applicable.
Funding: The present review was supported by grants from the
National Natural Science Foundation of China (grant no. 82400312),
Medical and Health Science Program of Zhejiang Province (grant no.
2025KY1879) and Hangzhou Natural Science Foundation of China (grant
no. 2024SZRYBH300002).
Not applicable.
YH, CL, LM, SL and FH were responsible for the
methodology, including literature search strategy design, database
selection, study inclusion and exclusion criteria, and
methodological framework for evidence synthesis. YH, CL, YC, DZ, MC
and QC curated the data. YH and CL wrote the original draft of the
manuscript, and YH, YC and QC reviewed and edited the manuscript.
YH, SL and FH supervised the project, and YH and QC provided
project administration. All authors read and approved the final
manuscript. Data authentication is not applicable.
Not applicable.
Not applicable.
The authors declare that they have no competing
interests.
|
1
|
Parlati ALM, Nardi E, Marzano F, Madaudo
C, Di Santo M, Cotticelli C, Agizza S, Abbellito GM, Perrone
Filardi F, Del Giudice M, et al: Advancing cardiovascular
diagnostics: The expanding role of CMR in heart failure and
cardiomyopathies. J Clin Med. 14(865)2025.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Bai Y, Zheng JP, Lu F, Zhang XL, Sun CP,
Guo WH, Zou YX, Lip GYH and Shi XB: Prevalence, incidence and
mortality of hypertrophic cardiomyopathy based on a population
cohort of 21.9 million in China. Sci Rep. 12(18799)2022.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Wexler RK, Elton T, Pleister A and Feldman
D: Cardiomyopathy: An overview. Am Fam Physician. 79:778–784.
2009.PubMed/NCBI
|
|
4
|
Li T, Wang N, Yi D, Xiao Y, Li X, Shao B,
Wu Z, Bai J, Shi X, Wu C, et al: ROS-mediated ferroptosis and
pyroptosis in cardiomyocytes: An update. Life Sci.
370(123565)2025.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Wang Y, Zhou T, Zhao J, Zhu H, Tan X, Chen
J, Zhang Z, Shen L and Lu S: Calcium handling remodeling in dilated
cardiomyopathy: From molecular mechanisms to targeted therapies.
Channels (Austin). 19(2519545)2025.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Hoskins AC, Jacques A, Bardswell SC,
McKenna WJ, Tsang V, dos Remedios CG, Ehler E, Adams K, Jalilzadeh
S, Avkiran M, et al: Normal passive viscoelasticity but abnormal
myofibrillar force generation in human hypertrophic cardiomyopathy.
J Mol Cell Cardiol. 49:737–745. 2010.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Tanaka A, Yuasa S, Mearini G, Egashira T,
Seki T, Kodaira M, Kusumoto D, Kuroda Y, Okata S, Suzuki T, et al:
Endothelin-1 induces myofibrillar disarray and contractile vector
variability in hypertrophic cardiomyopathy-induced pluripotent stem
cell-derived cardiomyocytes. J Am Heart Assoc.
3(e001263)2014.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Viola H, Johnstone V, Cserne Szappanos H,
Richman T, Tsoutsman T, Filipovska A, Semsarian C and Hool L: The
L-type Ca(2+) channel facilitates abnormal metabolic activity in
the cTnI-G203S mouse model of hypertrophic cardiomyopathy. J
Physiol. 594:4051–4070. 2016.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Castillero E, Akashi H, Pendrak K,
Yerebakan H, Najjar M, Wang C, Naka Y, Mancini D, Sweeney HL, D
Armiento J, et al: Attenuation of the unfolded protein response and
endoplasmic reticulum stress after mechanical unloading in dilated
cardiomyopathy. Am J Physiol Heart Circ Physiol. 309:H459–H470.
2015.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Fan G, Liu T, Chen X, Guo Y, Li X, He Y,
Hua P, Lin X, Lin D, Yan X, et al: Astaxanthin prevented acute
alcoholic cardiomyopathy by maintenance of mitophagy-mediated
mitochondrial homeostasis. J Cell Mol Med.
29(e70714)2025.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Zhao T, Jin K, Wang X, Su X, Wang Y, Gao
M, Luo W, Yang H and Yang Z: GPAT4 sustains endoplasmic reticulum
homeostasis in endocardial cells and safeguards heart development.
Nat Commun. 16(3345)2025.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Wang J, Pu X, Zhuang H, Guo Z, Wang M,
Yang H, Li C and Chang X: Astragaloside IV alleviates septic
myocardial injury through DUSP1-Prohibitin 2 mediated mitochondrial
quality control and ER-autophagy. J Adv Res. 75:561–580.
2025.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Brinkman K, Smeitink JA, Romijn JA and
Reiss P: Mitochondrial toxicity induced by nucleoside-analogue
reverse-transcriptase inhibitors is a key factor in the
pathogenesis of antiretroviral-therapy-related lipodystrophy.
Lancet. 354:1112–1115. 1999.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Liu ZW, Zhu HT, Chen KL, Dong X, Wei J,
Qiu C and Xue JH: Protein kinase RNA-like endoplasmic reticulum
kinase (PERK) signaling pathway plays a major role in reactive
oxygen species (ROS)-mediated endoplasmic reticulum stress-induced
apoptosis in diabetic cardiomyopathy. Cardiovasc Diabetol.
12(158)2013.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Tarazon E, Rosello-Lleti E, Ortega A,
Gil-Cayuela C, González-Juanatey JR, Lago F, Martínez-Dolz L,
Portolés M and Rivera M: Changes in human Golgi apparatus reflect
new left ventricular dimensions and function in dilated
cardiomyopathy patients. Eur J Heart Fail. 19:280–282.
2017.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Zhang J, Cui J, Zhao F, Yang L, Xu X, Shi
Y and Wei B: Cardioprotective effect of MLN4924 on ameliorating
autophagic flux impairment in myocardial ischemia-reperfusion
injury by Sirt1. Redox Biol. 46(102114)2021.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Kaur N, Raja R, Ruiz-Velasco A and Liu W:
Cellular protein quality control in diabetic cardiomyopathy: From
bench to bedside. Front Cardiovasc Med. 7(585309)2020.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Bresilla D, Tawfik I, Hirtl M, Gabrijelčič
S, Ostaku J, Mossegger F, Wurzer L, Lederer S, Kalinova K, Malle E,
et al: Enhancing late-life survival and mobility via mitohormesis
by reducing mitochondrial calcium levels. Aging Cell.
24(e70247)2025.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Xu X, Pang Y and Fan X: Mitochondria in
oxidative stress, inflammation and aging: From mechanisms to
therapeutic advances. Signal Transduct Target Ther.
10(190)2025.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Chen X, Yuan T, Zheng D, Li F, Xu H, Ye M,
Liu S and Li J: Cardiomyocyte mitochondrial mono-ADP-ribosylation
dictates cardiac tolerance to sepsis by configuring bioenergetic
reserve in male mice. Nat Commun. 16(8119)2025.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Huang J, Meng P, Liang Y, Li X, Zhou S, Li
J, Wang X, Miao J, Shen W and Zhou L: Tubular CD44 plays a key role
in aggravating AKI through NF-ĸB p65-mediated mitochondrial
dysfunction. Cell Death Dis. 16(119)2025.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Elbatarny M, Lu YT, Hu M, Coles J, Mital
S, Ross-White A, Honjo O, Barron DJ and Gramolini AO: Systems
biology approaches investigating mitochondrial dysfunction in
cyanotic heart disease: A systematic review. EBioMedicine.
118(105839)2025.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Tran DH and Wang ZV: Glucose metabolism in
cardiac hypertrophy and heart failure. J Am Heart Assoc.
8(e012673)2019.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Xu Y, Zhang X, Tang X, Zhang C, Cahoon JG,
Wang Y, Li H, Lv X, Wang Y, Wang Z, et al: Dexmedetomidine
post-treatment exacerbates metabolic disturbances in septic
cardiomyopathy via α2A-adrenoceptor. Biomed
Pharmacother. 170(115993)2024.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Gallo G, Rubattu S and Volpe M:
Mitochondrial dysfunction in heart failure: From pathophysiological
mechanisms to therapeutic opportunities. Int J Mol Sci.
25(2667)2024.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Wu M, Tan J, Cao Z, Cai Y, Huang Z, Chen
Z, He W, Liu X, Jiang Y, Gao Q, et al: Sirt5 improves
cardiomyocytes fatty acid metabolism and ameliorates cardiac
lipotoxicity in diabetic cardiomyopathy via CPT2 de-succinylation.
Redox Biol. 73(103184)2024.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Guo Y, Zhang Z, Wen Z, Kang X, Wang D,
Zhang L, Cheng M, Yuan G and Ren H: Mitochondrial SIRT2-mediated
CPT2 deacetylation prevents diabetic cardiomyopathy by impeding
cardiac fatty acid oxidation. Int J Biol Sci. 21:725–744.
2025.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Hu Y, Zheng Y, Liu C, You Y, Wu Y, Wang P,
Wu Y, Ba H, Lu J, Yuan Y, et al: Mitochondrial MOF regulates energy
metabolism in heart failure via ATP5B hyperacetylation. Cell Rep.
43(114839)2024.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Bonora M, Wieckowski MR, Sinclair DA,
Kroemer G, Pinton P and Galluzzi L: Targeting mitochondria for
cardiovascular disorders: Therapeutic potential and obstacles. Nat
Rev Cardiol. 16:33–55. 2019.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Wang P, Xu S, Xu J, Xin Y, Lu Y, Zhang H,
Zhou B, Xu H, Sheu SS, Tian R and Wang W: Elevated MCU expression
by CaMKIIdeltaB limits pathological cardiac remodeling.
Circulation. 145:1067–1083. 2022.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Garcia-Pena LM, Abel ED and Pereira RO:
Mitochondrial dynamics, diabetes, and cardiovascular disease.
Diabetes. 73:151–161. 2024.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Alves-Figueiredo H, Silva-Platas C,
Estrada M, Oropeza-Almazán Y, Ramos-González M, Bernal-Ramírez J,
Vázquez-Garza E, Tellez A, Salazar-Ramírez F, Méndez-Fernández A,
et al: Mitochondrial Ca(2+) uniporter-dependent energetic
dysfunction drives hypertrophy in heart failure. JACC Basic Transl
Sci. 9:496–518. 2024.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Niu W, Liu X, Deng B, Hong T, Wang C, Yan
Y, Liu J, Jiang Y and Li J: Piezo1 deletion mitigates diabetic
cardiomyopathy by maintaining mitochondrial dynamics via ERK/Drp1
pathway. Cardiovasc Diabetol. 24(127)2025.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Tokuyama T, Uosaki H, Sugiura A, Nishitai
G, Takeda K, Nagashima S, Shiiba I, Ito N, Amo T, Mohri S, et al:
Protective roles of MITOL against myocardial senescence and
ischemic injury partly via Drp1 regulation. iScience.
25(104582)2022.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Thakkar C, Alikunju S, Venkatasubramanian
A, D'Mello D, Abbas H, Yang Z, Andreas I, Sayed N, Abdellatif M and
Sayed D: Constitutive expression of cardiomyocyte Klf9 precipitates
metabolic dysfunction and spontaneous cardiomyopathy. Cell Signal.
136(112146)2025.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Lazarou M, Sliter DA, Kane LA, Sarraf SA,
Wang C, Burman JL, Sideris DP, Fogel AI and Youle RJ: The ubiquitin
kinase PINK1 recruits autophagy receptors to induce mitophagy.
Nature. 524:309–314. 2015.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Kubli DA, Zhang X, Lee Y, Hanna RA,
Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN and
Gustafsson AB: Parkin protein deficiency exacerbates cardiac injury
and reduces survival following myocardial infarction. J Biol Chem.
288:915–926. 2013.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Vujic A, Koo ANM, Prag HA and Krieg T:
Mitochondrial redox and TCA cycle metabolite signaling in the
heart. Free Radic Biol Med. 166:287–296. 2021.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Wang X, Li J, Zhang Y, Huang M, Yang P,
Huang T and Cheng Q: HIF1A/BNIP3 pathway affects ferroptosis in
sepsis-induced cardiomyopathy through binding to BCL-2. Redox Rep.
30(2544412)2025.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Chen W, Zhao Z, Geng Z, Zhang H and Fu X:
Advances in mitochondria-nucleus crosstalk in septic
cardiomyopathy. Cell Biol Toxicol. 41(136)2025.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Liu H, Liu X, Zhou J and Li T:
Mitochondrial DNA Is a vital driving force in ischemia-reperfusion
injury in cardiovascular diseases. Oxid Med Cell Longev.
2022(6235747)2022.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Lopes LR, Macken WL, Preez SD, Kotwal H,
Savvatis K, Sekhri N, Mohiddin SA, Kabiljo R and Pitceathly RDS: An
analysis of mitochondrial variation in cardiomyopathy patients from
the 100,000 genomes cohort: m.4300A>G as a cause of genetically
elusive hypertrophic cardiomyopathy. Hum Genomics.
18(136)2024.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Constante AD, Abreu SM and Trigo C:
Mitochondrial cardiomyopathy: A puzzle for the final diagnosis.
Cardiol Young. 34:1393–1396. 2024.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Tragni V, Primiano G, Tummolo A, Cafferati
Beltrame L, La Piana G, Sgobba MN, Cavalluzzi MM, Paterno G,
Gorgoglione R, Volpicella M, et al: Personalized medicine in
mitochondrial health and disease: Molecular basis of therapeutic
approaches based on nutritional supplements and their analogs.
Molecules. 27(3494)2022.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Luppi E, De Luise M, Bini C, Pelletti G,
Tioli G, Kurelac I, Iommarini L, Pelotti S and Gasparre G: The
landscape of rare mitochondrial DNA variants in sudden cardiac
death: A potential role for ATP synthase. Heliyon.
11(e41592)2024.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Lee JS, Ko YG, Shin KJ, Kim SK, Park JH,
Hwang KC and Pak HN: Mitochondrial DNA 4977bp deletion mutation in
peripheral blood reflects atrial remodeling in patients with
non-valvular atrial fibrillation. Yonsei Med J. 56:53–61.
2015.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Zhang Y, Sun X, Jin Y, Chen K, Zhang L,
Gao X, Li M, Yuan Z, Jia J, Sun A and Ge J: Mitochondrial
transplantation augments the reparative capacity of macrophages
following myocardial injury. Adv Sci (Weinh).
12(e06337)2025.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Lin H, Xiong W, Fu L, Yi J and Yang J:
Damage-associated molecular patterns (DAMPs) in diseases:
Implications for therapy. Mol Biomed. 6(60)2025.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Yu H, Ren K, Jin Y, Zhang L, Liu H, Huang
Z, Zhang Z, Chen X, Yang Y and Wei Z: Mitochondrial DAMPs: Key
mediators in neuroinflammation and neurodegenerative disease
pathogenesis. Neuropharmacology. 264(110217)2025.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Oka T, Hikoso S, Yamaguchi O, Taneike M,
Takeda T, Tamai T, Oyabu J, Murakawa T, Nakayama H, Nishida K, et
al: Mitochondrial DNA that escapes from autophagy causes
inflammation and heart failure. Nature. 485:251–255.
2012.PubMed/NCBI View Article : Google Scholar
|
|
51
|
He B, Yu H, Liu S, Wan H, Fu S, Liu S,
Yang J, Zhang Z, Huang H, Li Q, et al: Mitochondrial cristae
architecture protects against mtDNA release and inflammation. Cell
Rep. 41(111774)2022.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Mweene BC, Hatwiko H, Povia JP and Masenga
SK: The role of mitochondrial dysfunction and dynamics in
hypertensive heart disease: Mechanisms and recent advances. Biology
(Basel). 14(1212)2025.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Yan J, Cao J, Pan W and Chen L: Insights
into golgi apparatus and centrosome: Implications for ciliogenesis.
Mol Biol Rep. 52(716)2025.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Ferrer MF, Rozes-Salvador V, Tomatis C,
Thomas P, Aquila S, Aguiar MCAM, Carrera Silva EA, Alvarez C and
Gómez RM: Yellow fever virus induces golgi stress and CREB3L1
nuclear translocation in human A549 cells. J Med Virol.
97(e70490)2025.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Cui C, Sun C, Yuan P, Tian S, Xie H, Xu F
and Li H: The golgi apparatus as a strategic target in cancer:
Mechanisms, diagnosis and therapeutic opportunities. J Drug Target.
33:1773–1787. 2025.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Wijaya CS and Xu S: Reevaluating Golgi
fragmentation and its implications in wound repair. Cell Regen.
13(4)2024.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Gonzalez-Torrent I, Gimenez-Escamilla I,
Perez-Carrillo L, Delgado-Arija M, Portolés M, Tarazón E and
Roselló-Lletí E: Alteration in Golgi apparatus fragmentation
related genes in human dilated cardiomyopathy. Sci Rep.
15(7704)2025.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Jungk L, Franke H, Salameh A and Dhein S:
Golgi fragmentation in human patients with chronic atrial
fibrillation: A new aspect of remodeling. Thorac Cardiovasc Surg.
67:98–106. 2019.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Zemet R, Hope KD, Edmondson AC, Shah R,
Patino M, Yesso AM, Berger JH, Sarafoglou K, Larson A, Lam C, et
al: Cardiomyopathy, an uncommon phenotype of congenital disorders
of glycosylation: Recommendations for baseline screening and
follow-up evaluation. Mol Genet Metab. 142(108513)2024.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Raja R, Fonseka O, Ganenthiran H,
Andrea-Ruiz-Velasco and Liu W: The multifaceted roles of ER
and Golgi in metabolic cardiomyopathy. Front Cardiovasc Med.
9(999044)2022.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Chatham JC and Patel RP: Protein
glycosylation in cardiovascular health and disease. Nat Rev
Cardiol. 21:525–544. 2024.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Yu P, Hu L, Xie J, Chen S, Huang L, Xu Z,
Liu X, Zhou Q, Yuan P, Yan X, et al: O-GlcNAcylation of cardiac
Nav1.5 contributes to the development of arrhythmias in diabetic
hearts. Int J Cardiol. 260:74–81. 2018.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Fang R, Jiang Q, Jia X and Jiang Z:
ARMH3-mediated recruitment of PI4KB directs Golgi-to-endosome
trafficking and activation of the antiviral effector STING.
Immunity. 56:500–515 e6. 2023.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Brandizzi F and Barlowe C: Organization of
the ER-Golgi interface for membrane traffic control. Nat Rev Mol
Cell Biol. 14:382–392. 2013.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Sikder K, Phillips E, Zhong Z, Wang N,
Saunders J, Mothy D, Kossenkov A, Schneider T, Nichtova Z, Csordas
G, et al: Perinuclear damage from nuclear envelope deterioration
elicits stress responses that contribute to LMNA cardiomyopathy.
Sci Adv. 10(eadh0798)2024.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Lu LQ, Tang MZ, Qi ZH, Huang SF, He YQ, Li
DK, Li LF and Chen LX: Regulation of the Golgi apparatus via
GOLPH3-mediated new selective autophagy. Life Sci.
253(117700)2020.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Chen Y, Wu Y, Tian X, Shao G, Lin Q and
Sun A: Golgiphagy: A novel selective autophagy to the fore. Cell
Biosci. 14(130)2024.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Kitta S, Kaminishi T, Higashi M, Shima T,
Nishino K, Nakamura N, Kosako H, Yoshimori T and Kuma A: YIPF3 and
YIPF4 regulate autophagic turnover of the Golgi apparatus. EMBO J.
43:2954–2978. 2024.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Huang Y and Klionsky DJ: Identification of
the YIPF3-YIPF4 heterodimer as a novel Golgiphagy receptor.
Autophagy. 20:1211–1212. 2024.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Kang J, Li CM, Kim N, Baek J and Jung YK:
Non-autophagic Golgi-LC3 lipidation facilitates TFE3 stress
response against Golgi dysfunction. EMBO J. 43:5085–5113.
2024.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Li A, Liu Y, Labapuchi Chen Z, Li S, Zhong
R, Cheng D, Chen L and He L: Development of a Golgi-targeted
fluorescent chemosensor for detecting ferrous ions overload under
Golgi stress. Spectrochim Acta A Mol Biomol Spectrosc.
294(122560)2023.PubMed/NCBI View Article : Google Scholar
|
|
72
|
Reiling JH, Olive AJ, Sanyal S, Carette
JE, Brummelkamp TR, Ploegh HL, Starnbach MN and Sabatini DM: A
CREB3-ARF4 signalling pathway mediates the response to Golgi stress
and susceptibility to pathogens. Nat Cell Biol. 15:1473–1485.
2013.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Prola A, Nichtova Z, Pires Da Silva J,
Piquereau J, Monceaux K, Guilbert A, Gressette M, Ventura-Clapier
R, Garnier A, Zahradnik I, et al: Endoplasmic reticulum stress
induces cardiac dysfunction through architectural modifications and
alteration of mitochondrial function in cardiomyocytes. Cardiovasc
Res. 115:328–342. 2019.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Borbein W, Dahmlos L, Saleem U, Reinsch M,
Braren I, Schulze T, Klampe B, Cuello F, Stenzig J, Eschenhagen T
and Hansen A: Altered endoplasmic reticulum calcium loading in
human PLN-R14del cardiomyopathy. Front Cell Dev Biol.
13(1627985)2025.PubMed/NCBI View Article : Google Scholar
|
|
75
|
Tandra V, Zhang L, Lee CM, Wu Y, Yue G, Li
H, Su H and Li J: ufmylation suppresses unfolded protein response
to prevent peripartum cardiomyopathy. JACC Basic Transl Sci.
10(101293)2025.PubMed/NCBI View Article : Google Scholar
|
|
76
|
Chen J, Yang X, Li W, Lin Y, Lin R, Cai X,
Yan B, Xie B and Li J: Endoplasmic reticulum stress-related gene
expression causes the progression of dilated cardiomyopathy by
inducing apoptosis. Front Genet. 15(1366087)2024.PubMed/NCBI View Article : Google Scholar
|
|
77
|
Bhattarai KR, Chaudhary M, Kim HR and Chae
HJ: Endoplasmic reticulum (ER) stress response failure in diseases.
Trends Cell Biol. 30:672–675. 2020.PubMed/NCBI View Article : Google Scholar
|
|
78
|
Bond Newton SE, Shi X, Beratan NR, Perhacs
J, Arya JK, Bond MK, Gidalevitz T, Akay-Espinoza C, Brady DC and
Jordan-Sciutto KL: ER stress tolerance is regulated by
copper-dependent PERK kinase activity. Cell Rep.
44(116318)2025.PubMed/NCBI View Article : Google Scholar
|
|
79
|
Wang S, Binder P, Fang Q, Wang Z, Xiao W,
Liu W and Wang X: Endoplasmic reticulum stress in the heart:
insights into mechanisms and drug targets. Br J Pharmacol.
175:1293–1304. 2018.PubMed/NCBI View Article : Google Scholar
|
|
80
|
Qiu Y, Chen Z, He P and Wang Z:
Endoplasmic reticulum stress in cardiomyopathies: From the unfolded
protein response to therapeutic opportunities. Front Cardiovasc
Med. 12(1577186)2025.PubMed/NCBI View Article : Google Scholar
|
|
81
|
Bertolotti A, Zhang Y, Hendershot LM,
Harding HP and Ron D: Dynamic interaction of BiP and ER stress
transducers in the unfolded-protein response. Nat Cell Biol.
2:326–332. 2000.PubMed/NCBI View Article : Google Scholar
|
|
82
|
Han J, Back SH, Hur J, Lin YH,
Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M,
et al: ER-stress-induced transcriptional regulation increases
protein synthesis leading to cell death. Nat Cell Biol. 15:481–490.
2013.PubMed/NCBI View Article : Google Scholar
|
|
83
|
Fu F and Doroudgar S: IRE1/XBP1 and
endoplasmic reticulum signaling - from basic to translational
research for cardiovascular disease. Curr Opin Physiol.
28(100552)2022.PubMed/NCBI View Article : Google Scholar
|
|
84
|
Nakatsukasa K, Huyer G, Michaelis S and
Brodsky JL: Dissecting the ER-associated degradation of a misfolded
polytopic membrane protein. Cell. 132:101–112. 2008.PubMed/NCBI View Article : Google Scholar
|
|
85
|
Zhu Z, Pu J, Li Y, Chen J, Ding H, Zhou A
and Zhang X: RBM25 regulates hypoxic cardiomyocyte apoptosis
through CHOP-associated endoplasmic reticulum stress. Cell Stress
Chaperones. 28:861–876. 2023.PubMed/NCBI View Article : Google Scholar
|
|
86
|
Schiattarella GG, Altamirano F, Kim SY,
Tong D, Ferdous A, Piristine H, Dasgupta S, Wang X, French KM,
Villalobos E, et al: Xbp1s-FoxO1 axis governs lipid accumulation
and contractile performance in heart failure with preserved
ejection fraction. Nat Commun. 12(1684)2021.PubMed/NCBI View Article : Google Scholar
|
|
87
|
Li C, Qian T, He R, Wan C, Liu Y and Yu H:
Endoplasmic reticulum-plasma membrane contact sites: Regulators,
mechanisms, and physiological functions. Front Cell Dev Biol.
9(627700)2021.PubMed/NCBI View Article : Google Scholar
|
|
88
|
Chen C, Dai G, Fan M, Wang X, Niu K and
Gao W: Mitochondria-associated endoplasmic reticulum membranes and
myocardial ischemia: from molecular mechanisms to therapeutic
strategies. J Transl Med. 23(277)2025.PubMed/NCBI View Article : Google Scholar
|
|
89
|
Feno S, Rizzuto R, Raffaello A and
Vecellio Reane D: The molecular complexity of the mitochondrial
calcium uniporter. Cell Calcium. 93(102322)2021.PubMed/NCBI View Article : Google Scholar
|
|
90
|
Bai X, Zhang Z, Li X, Yang Y and Ding S:
FUNDC1: An emerging mitochondrial and MAMs protein for
mitochondrial quality control in heart diseases. Int J Mol Sci.
24(9151)2023.PubMed/NCBI View Article : Google Scholar
|
|
91
|
Szabadkai G, Bianchi K, Varnai P, De
Stefani D, Wieckowski MR, Cavagna D, Nagy AI, Balla T and Rizzuto
R: Chaperone-mediated coupling of endoplasmic reticulum and
mitochondrial Ca2+ channels. J Cell Biol. 175:901–911.
2006.PubMed/NCBI View Article : Google Scholar
|
|
92
|
Ong SB, Samangouei P, Kalkhoran SB and
Hausenloy DJ: The mitochondrial permeability transition pore and
its role in myocardial ischemia reperfusion injury. J Mol Cell
Cardiol. 78:23–34. 2015.PubMed/NCBI View Article : Google Scholar
|
|
93
|
Zhihao L, Jingyu N, Lan L, Michael S, Rui
G, Xiyun B, Xiaozhi L and Guanwei F: SERCA2a: A key protein in the
Ca(2+) cycle of the heart failure. Heart Fail Rev. 25:523–535.
2020.PubMed/NCBI View Article : Google Scholar
|
|
94
|
Zsebo K, Yaroshinsky A, Rudy JJ, Wagner K,
Greenberg B, Jessup M and Hajjar RJ: Long-term effects of
AAV1/SERCA2a gene transfer in patients with severe heart failure:
analysis of recurrent cardiovascular events and mortality. Circ
Res. 114:101–108. 2014.PubMed/NCBI View Article : Google Scholar
|
|
95
|
Lyon AR, Babalis D, Morley-Smith AC,
Hedger M, Suarez Barrientos A, Foldes G, Couch LS, Chowdhury RA,
Tzortzis KN, Peters NS, et al: Investigation of the safety and
feasibility of AAV1/SERCA2a gene transfer in patients with chronic
heart failure supported with a left ventricular assist device - the
SERCA-LVAD TRIAL. Gene Ther. 27:579–590. 2020.PubMed/NCBI View Article : Google Scholar
|
|
96
|
Nakagama S, Maejima Y, Fan Q,
Shiheido-Watanabe Y, Tamura N, Ihara K and Sasano T: Endoplasmic
reticulum selective autophagy alleviates anthracycline-induced
cardiotoxicity. JACC CardioOncol. 5:656–670. 2023.PubMed/NCBI View Article : Google Scholar
|
|
97
|
Kuijpers M, Kochlamazashvili G, Stumpf A,
Puchkov D, Swaminathan A, Lucht MT, Krause E, Maritzen T, Schmitz D
and Haucke V: Neuronal autophagy regulates presynaptic
neurotransmission by controlling the axonal endoplasmic reticulum.
Neuron. 109:299–313 e9. 2021.PubMed/NCBI View Article : Google Scholar
|
|
98
|
Stolz A, Ernst A and Dikic I: Cargo
recognition and trafficking in selective autophagy. Nat Cell Biol.
16:495–501. 2014.PubMed/NCBI View Article : Google Scholar
|
|
99
|
Wang H, Liu L, Gong H and Li H:
Upregulation of FAM134B inhibits endoplasmic reticulum
stress-related degradation protein expression and promotes
hepatocellular carcinogenesis. J Cell Mol Med.
28(e17964)2024.PubMed/NCBI View Article : Google Scholar
|
|
100
|
Qu Y, Gao R, Wei X, Sun X, Yang K, Shi H,
Gao Y, Hu S, Wang Y, Yang J, et al: Gasdermin D mediates
endoplasmic reticulum stress via FAM134B to regulate cardiomyocyte
autophagy and apoptosis in doxorubicin-induced cardiotoxicity. Cell
Death Dis. 13(901)2022.PubMed/NCBI View Article : Google Scholar
|
|
101
|
Wiersma M, Meijering RAM, Qi XY, Zhang D,
Liu T, Hoogstra-Berends F, Sibon OCM, Henning RH, Nattel S and
Brundel BJJM: Endoplasmic reticulum stress is associated with
autophagy and cardiomyocyte remodeling in experimental and human
atrial fibrillation. J Am Heart Assoc. 6(e006458)2017.PubMed/NCBI View Article : Google Scholar
|
|
102
|
Pires Da Silva J, Monceaux K, Guilbert A,
Gressette M, Piquereau J, Novotova M, Ventura-Clapier R, Garnier A
and Lemaire C: SIRT1 protects the heart from ER stress-induced
injury by promoting eEF2K/eEF2-dependent autophagy. Cells.
9(426)2020.PubMed/NCBI View Article : Google Scholar
|
|
103
|
Karwi QG, Ho KL, Pherwani S, Ketema EB,
Sun Q and Lopaschuk GD: Concurrent diabetes and heart failure:
Interplay and novel therapeutic approaches. Cardiovasc Res.
118:686–715. 2022.PubMed/NCBI View Article : Google Scholar
|
|
104
|
Wang T, Wei Q, Liang L, Tang X, Yao J, Lu
Y, Qu Y, Chen Z, Xing G and Cao X: OSBPL2 is required for the
binding of COPB1 to ATGL and the regulation of lipid droplet
lipolysis. iScience. 23(101252)2020.PubMed/NCBI View Article : Google Scholar
|
|
105
|
Kaur N, Ruiz-Velasco A, Raja R, Howell G,
Miller JM, Abouleisa RRE, Ou Q, Mace K, Hille SS, Frey N, et al:
Paracrine signal emanating from stressed cardiomyocytes aggravates
inflammatory microenvironment in diabetic cardiomyopathy. iScience.
25(103973)2022.PubMed/NCBI View Article : Google Scholar
|
|
106
|
Fonseka O, Raja R, Ross C, Gare SR, Zhang
J, Hille SS, King K, Ruiz-Velasco A, Kaur N, Chen X, et al:
XBP1s-EDEM2 prevents the onset and development of HFpEF by
ameliorating cardiac lipotoxicity. Circulation. 151:1583–1605.
2025.PubMed/NCBI View Article : Google Scholar
|
|
107
|
Yu L, Chen Y and Tooze SA: Autophagy
pathway: Cellular and molecular mechanisms. Autophagy. 14:207–215.
2018.PubMed/NCBI View Article : Google Scholar
|
|
108
|
Zhou R, Zhang Z, Li X, Duan Q, Miao Y,
Zhang T, Wang M, Li J, Zhang W, Wang L, et al: Autophagy in
high-fat diet and streptozotocin-induced metabolic cardiomyopathy:
Mechanisms and therapeutic implications. Int J Mol Sci.
26(1668)2025.PubMed/NCBI View Article : Google Scholar
|
|
109
|
Gu S, Tan J, Li Q, Liu S, Ma J, Zheng Y,
Liu J, Bi W, Sha P, Li X, et al: Downregulation of LAPTM4B
contributes to the impairment of the autophagic flux via unopposed
activation of mTORC1 signaling during myocardial
ischemia/reperfusion injury. Circ Res. 127:e148–e165.
2020.PubMed/NCBI View Article : Google Scholar
|
|
110
|
Qiao L, Hu J, Qiu X, Wang C, Peng J, Zhang
C, Zhang M, Lu H and Chen W: LAMP2A, LAMP2B and LAMP2C: Similar
structures, divergent roles. Autophagy. 19:2837–2852.
2023.PubMed/NCBI View Article : Google Scholar
|
|
111
|
Ma X, Liu H, Foyil SR, Godar RJ,
Weinheimer CJ, Hill JA and Diwan A: Impaired autophagosome
clearance contributes to cardiomyocyte death in
ischemia/reperfusion injury. Circulation. 125:3170–3181.
2012.PubMed/NCBI View Article : Google Scholar
|
|
112
|
Zhao YT, Cao XQ and Mu XL: Hypertrophic
cardiomyopathy secondary to deficiency in lysosome-associated
membrane protein-2: A case report. World J Cardiol. 15:609–614.
2023.PubMed/NCBI View Article : Google Scholar
|
|
113
|
Pan B, Zhang H, Cui T and Wang X: TFEB
activation protects against cardiac proteotoxicity via increasing
autophagic flux. J Mol Cell Cardiol. 113:51–62. 2017.PubMed/NCBI View Article : Google Scholar
|
|
114
|
Ma X, Godar RJ, Liu H and Diwan A:
Enhancing lysosome biogenesis attenuates BNIP3-induced
cardiomyocyte death. Autophagy. 8:297–309. 2012.PubMed/NCBI View Article : Google Scholar
|
|
115
|
Patel S, Radhakrishnan D, Kumari D,
Bhansali P and Setty SRG: Restoration of β-GC trafficking improves
the lysosome function in Gaucher disease. Traffic. 24:489–503.
2023.PubMed/NCBI View Article : Google Scholar
|
|
116
|
Beyer JN, Serebrenik YV, Toy K, Najar MA,
Feierman E, Raniszewski NR, Korb E, Shalem O and Burslem GM:
Intracellular protein editing enables incorporation of noncanonical
residues in endogenous proteins. Science.
388(eadr5499)2025.PubMed/NCBI View Article : Google Scholar
|
|
117
|
Azevedo O, Cordeiro F, Gago MF,
Miltenberger-Miltenyi G, Ferreira C, Sousa N and Cunha D: Fabry
disease and the heart: A comprehensive review. Int J Mol Sci.
22(4434)2021.PubMed/NCBI View Article : Google Scholar
|
|
118
|
Huang W, Zhou R, Jiang C, Wang J, Zhou Y,
Xu X, Wang T, Li A and Zhang Y: Mitochondrial dysfunction is
associated with hypertrophic cardiomyopathy in Pompe
disease-specific induced pluripotent stem cell-derived
cardiomyocytes. Cell Prolif. 57(e13573)2024.PubMed/NCBI View Article : Google Scholar
|
|
119
|
Chen YY, Liu CX, Liu HX and Wen SY: The
emerging roles of vacuolar-type ATPase-dependent lysosomal
acidification in cardiovascular disease. Biomolecules.
15(525)2025.PubMed/NCBI View Article : Google Scholar
|
|
120
|
Kim YC, Park HW, Sciarretta S, Mo JS,
Jewell JL, Russell RC, Wu X, Sadoshima J and Guan KL: Rag GTPases
are cardioprotective by regulating lysosomal function. Nat Commun.
5(4241)2014.PubMed/NCBI View Article : Google Scholar
|
|
121
|
Kinouchi K, Ichihara A, Sano M, Sun-Wada
GH, Wada Y, Kurauchi-Mito A, Bokuda K, Narita T, Oshima Y, Sakoda
M, et al: The (pro)renin receptor/ATP6AP2 is essential for vacuolar
H+-ATPase assembly in murine cardiomyocytes. Circ Res. 107:30–34.
2010.PubMed/NCBI View Article : Google Scholar
|
|
122
|
Qi J, Li Q, Xin T, Lu Q, Lin J, Zhang Y,
Luo H, Zhang F, Xing Y, Wang W, et al: MCOLN1/TRPML1 in the
lysosome: A promising target for autophagy modulation in diverse
diseases. Autophagy. 20:1712–1722. 2024.PubMed/NCBI View Article : Google Scholar
|
|
123
|
Medina DL and Ballabio A: Lysosomal
calcium regulates autophagy. Autophagy. 11:970–971. 2015.PubMed/NCBI View Article : Google Scholar
|
|
124
|
Scotto Rosato A, Montefusco S, Soldati C,
Di Paola S, Capuozzo A, Monfregola J, Polishchuk E, Amabile A,
Grimm C, Lombardo A, et al: TRPML1 links lysosomal calcium to
autophagosome biogenesis through the activation of the CaMKKβ/VPS34
pathway. Nat Commun. 10(5630)2019.PubMed/NCBI View Article : Google Scholar
|
|
125
|
Xing Y, Sui Z, Liu Y, Wang MM, Wei X, Lu
Q, Wang X, Liu N, Lu C, Chen R, et al: Blunting TRPML1 channels
protects myocardial ischemia/reperfusion injury by restoring
impaired cardiomyocyte autophagy. Basic Res Cardiol.
117(20)2022.PubMed/NCBI View Article : Google Scholar
|
|
126
|
McGirr T, Onar O and Jafarnejad SM:
Dysregulated ribosome quality control in human diseases. FEBS J.
292:936–959. 2025.PubMed/NCBI View Article : Google Scholar
|
|
127
|
Gobet C and Naef F: Ribosome profiling and
dynamic regulation of translation in mammals. Curr Opin Genet Dev.
43:120–127. 2017.PubMed/NCBI View Article : Google Scholar
|
|
128
|
Ganapathi M, Argyriou L, Martinez-Azorin
F, Morlot S, Yigit G, Lee TM, Auber B, von Gise A, Petrey DS,
Thiele H, et al: Bi-allelic missense disease-causing variants in
RPL3L associate neonatal dilated cardiomyopathy with
muscle-specific ribosome biogenesis. Hum Genet. 139:1443–1454.
2020.PubMed/NCBI View Article : Google Scholar
|
|
129
|
Liu WY, Wang HQ and Shao ZH: Research
progress on pathogenesis of congenital pure red cell
aplasia-review. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 29:1654–1657.
2021.PubMed/NCBI View Article : Google Scholar : (In Chinese).
|
|
130
|
Desai N, Yang H, Chandrasekaran V, Kazi R,
Minczuk M and Ramakrishnan V: Elongational stalling activates
mitoribosome-associated quality control. Science. 370:1105–1110.
2020.PubMed/NCBI View Article : Google Scholar
|
|
131
|
Balch WE, Morimoto RI, Dillin A and Kelly
JW: Adapting proteostasis for disease intervention. Science.
319:916–919. 2008.PubMed/NCBI View Article : Google Scholar
|
|
132
|
Juszkiewicz S, Chandrasekaran V, Lin Z,
Kraatz S, Ramakrishnan V and Hegde RS: ZNF598 Is a Quality Control
Sensor of Collided Ribosomes. Mol Cell. 72:469–481 e7.
2018.PubMed/NCBI View Article : Google Scholar
|
|
133
|
Douglas T, Zhang J, Wu Z, Abdallah K,
McReynolds M, Gilbert WV, Iwai K, Peng J, Young LH and Crews CM: An
atypical E3 ligase safeguards the ribosome during nutrient stress
bioRxiv [Preprint] 2024.10.10.617692, 2024.
|
|
134
|
Wang Y, Tu J, Wu W, Xu Y, Li Y, Pan X, Liu
B, Lu T, Han Q, Zhang H, et al: The orchestration of cell-cycle
reentry and ribosome biogenesis network is critical for cardiac
repair. Theranostics. 14:3927–3944. 2024.PubMed/NCBI View Article : Google Scholar
|
|
135
|
Qu JH, Tarasov KV, Chakir K, Tarasova YS,
Riordon DR and Lakatta EG: Proteomic landscape and deduced
functions of the cardiac 14-3-3 protein interactome. Cells.
11(3496)2022.PubMed/NCBI View Article : Google Scholar
|
|
136
|
Sciarretta S, Forte M, Frati G and
Sadoshima J: The complex network of mTOR signalling in the heart.
Cardiovasc Res. 118:424–439. 2022.PubMed/NCBI View Article : Google Scholar
|
|
137
|
Yano T, Ferlito M, Aponte A, Kuno A, Miura
T, Murphy E and Steenbergen C: Pivotal role of mTORC2 and
involvement of ribosomal protein S6 in cardioprotective signaling.
Circ Res. 114:1268–1280. 2014.PubMed/NCBI View Article : Google Scholar
|
|
138
|
Ye T, Yan Z, Chen C, Wang D, Wang A, Li T,
Yang B, Ding X and Shen C: Lactoferrin attenuates cardiac fibrosis
and cardiac remodeling after myocardial infarction via inhibiting
mTORC1/S6K signaling pathway. Theranostics. 13:3419–3433.
2023.PubMed/NCBI View Article : Google Scholar
|
|
139
|
Zhang G, Wang X, Li C, Li Q, An YA, Luo X,
Deng Y, Gillette TG, Scherer PE and Wang ZV: Integrated stress
response couples mitochondrial protein translation with oxidative
stress control. Circulation. 144:1500–1515. 2021.PubMed/NCBI View Article : Google Scholar
|
|
140
|
Wang Z, Wu J, Lv Z, Liang P, Li Q, Li Y
and Guo Y: LMNA-related cardiomyopathy: From molecular pathology to
cardiac gene therapy. J Adv Res. 77:443–464. 2025.PubMed/NCBI View Article : Google Scholar
|
|
141
|
Chen SN, Lombardi R, Karmouch J, Tsai JY,
Czernuszewicz G, Taylor MRG, Mestroni L, Coarfa C, Gurha P and
Marian AJ: DNA damage response/TP53 pathway is activated and
contributes to the pathogenesis of dilated cardiomyopathy
associated with LMNA (Lamin A/C) mutations. Circ Res. 124:856–873.
2019.PubMed/NCBI View Article : Google Scholar
|
|
142
|
Veltrop RJA, Kukk MM, Topouzidou K, Didden
L, Muchir A, van Steenbeek FG, Schurgers LJ and Harakalova M: From
gene to mechanics: A comprehensive insight into the mechanobiology
of LMNA mutations in cardiomyopathy. Cell Commun Signal.
22(197)2024.PubMed/NCBI View Article : Google Scholar
|
|
143
|
Cenni V, Evangelisti C, Santi S, Sabatelli
P, Neri S, Cavallo M, Lattanzi G and Mattioli E: Desmin and plectin
recruitment to the nucleus and nuclei orientation are lost in
emery-dreifuss muscular dystrophy myoblasts subjected to mechanical
stimulation. Cells. 13(162)2024.PubMed/NCBI View Article : Google Scholar
|
|
144
|
Bulmer L, Ljungman C, Hallin J, Dahlberg
P, Polte CL, Hedberg-Oldfors C, Oldfors A and Gummesson A: EMD
missense variant causes X-linked isolated dilated cardiomyopathy
with myocardial emerin deficiency. Eur J Hum Genet. 33:775–783.
2025.PubMed/NCBI View Article : Google Scholar
|
|
145
|
Nielsen MS, van Opbergen CJM, van Veen TAB
and Delmar M: The intercalated disc: A unique organelle for
electromechanical synchrony in cardiomyocytes. Physiol Rev.
103:2271–2319. 2023.PubMed/NCBI View Article : Google Scholar
|
|
146
|
Yeruva S and Waschke J: Structure and
regulation of desmosomes in intercalated discs: Lessons from
epithelia. J Anat. 242:81–90. 2023.PubMed/NCBI View Article : Google Scholar
|
|
147
|
Zhao G, Qiu Y, Zhang HM and Yang D:
Intercalated discs: Cellular adhesion and signaling in heart health
and diseases. Heart Fail Rev. 24:115–132. 2019.PubMed/NCBI View Article : Google Scholar
|
|
148
|
Sheikh F, Ross RS and Chen J: Cell-cell
connection to cardiac disease. Trends Cardiovasc Med. 19:182–190.
2009.PubMed/NCBI View Article : Google Scholar
|
|
149
|
Wang L, Liu S, Zhang H, Hu S and Wei Y:
Arrhythmogenic cardiomyopathy: Identification of desmosomal gene
variations and desmosomal protein expression in variation carriers.
Exp Ther Med. 15:2255–2262. 2018.PubMed/NCBI View Article : Google Scholar
|
|
150
|
Pruna M and Ehler E: The intercalated
disc: A mechanosensing signalling node in cardiomyopathy. Biophys
Rev. 12:931–946. 2020.PubMed/NCBI View Article : Google Scholar
|
|
151
|
Rickelt S and Pieperhoff S: Mutations with
pathogenic potential in proteins located in or at the composite
junctions of the intercalated disk connecting mammalian
cardiomyocytes: A reference thesaurus for arrhythmogenic
cardiomyopathies and for Naxos and Carvajal diseases. Cell Tissue
Res. 348:325–333. 2012.PubMed/NCBI View Article : Google Scholar
|
|
152
|
Mishra Y, Kumar A and Kaundal RK:
Mitochondrial Dysfunction is a Crucial Immune Checkpoint for
Neuroinflammation and Neurodegeneration: mtDAMPs in Focus. Mol
Neurobiol. 62:6715–6747. 2025.PubMed/NCBI View Article : Google Scholar
|
|
153
|
Zhang G, Wei H, Zhao A, Yan X, Zhang X,
Gan J, Guo M, Wang J, Zhang F, Jiang Y, et al: Mitochondrial DNA
leakage: underlying mechanisms and therapeutic implications in
neurological disorders. J Neuroinflammation. 22(34)2025.PubMed/NCBI View Article : Google Scholar
|
|
154
|
Guan X, Li H, Zhang L and Zhi H:
Mechanisms of mitochondrial damage-associated molecular patterns
associated with inflammatory response in cardiovascular diseases.
Inflamm Res. 74(18)2025.PubMed/NCBI View Article : Google Scholar
|
|
155
|
Du Q, Ning N, Zhao X, Liu F, Zhang S, Xia
Y, Li F, Yuan S, Xie X, Zhu M, et al: Acylglycerol kinase inhibits
macrophage anti-tumor activity via limiting mtDNA release and
cGAS-STING-type I IFN response. Theranostics. 15:1304–1319.
2025.PubMed/NCBI View Article : Google Scholar
|
|
156
|
Rabinowitz J, Vila IK, Luchsinger C,
Bertelli C, Schüssler M, Taffoni C, Cui B, Dai AZ, Rashid MM,
Cisneros WJ, et al: The ability of SAMHD1-deficient monocytes to
trigger the type I IFN response depends on cGAS and mitochondrial
DNA. J Biol Chem. 301(110430)2025.PubMed/NCBI View Article : Google Scholar
|
|
157
|
Calixto A, Moen KE and Moreno SNJ: The
contribution of the Golgi and the endoplasmic reticulum to calcium
and pH homeostasis in Toxoplasma gondii. J Biol Chem.
301(108372)2025.PubMed/NCBI View Article : Google Scholar
|
|
158
|
Sun W, Zhang J, Li S, Fu W, Liu Y, Liu M,
Dong J, Zhao X and Li X: TAB2 deficiency induces dilated
cardiomyopathy by promoting mitochondrial calcium overload in human
iPSC-derived cardiomyocytes. Mol Med. 31(42)2025.PubMed/NCBI View Article : Google Scholar
|
|
159
|
He P, Chang H, Qiu Y and Wang Z:
Mitochondria associated membranes in dilated cardiomyopathy:
Connecting pathogenesis and cellular dysfunction. Front Cardiovasc
Med. 12(1571998)2025.PubMed/NCBI View Article : Google Scholar
|
|
160
|
Zhao WB and Sheng R: The correlation
between mitochondria-associated endoplasmic reticulum membranes
(MAMs) and Ca(2+) transport in the pathogenesis of diseases. Acta
Pharmacol Sin. 46:271–291. 2025.PubMed/NCBI View Article : Google Scholar
|
|
161
|
Benyair R, Eisenberg-Lerner A and Merbl Y:
Maintaining Golgi homeostasis: A balancing act of two proteolytic
pathways. Cells. 11(780)2022.PubMed/NCBI View Article : Google Scholar
|
|
162
|
Liu J, Huang Y, Li T, Jiang Z, Zeng L and
Hu Z: The role of the Golgi apparatus in disease (Review). Int J
Mol Med. 47(38)2021.PubMed/NCBI View Article : Google Scholar
|
|
163
|
Raval KK, Tao R, White BE, De Lange WJ,
Koonce CH, Yu J, Kishnani PS, Thomson JA, Mosher DF, Ralphe JC and
Kamp TJ: Pompe disease results in a Golgi-based glycosylation
deficit in human induced pluripotent stem cell-derived
cardiomyocytes. J Biol Chem. 290:3121–3136. 2015.PubMed/NCBI View Article : Google Scholar
|
|
164
|
Lu L, Zhou Q, Chen Z and Chen L: The
significant role of the Golgi apparatus in cardiovascular diseases.
J Cell Physiol. 233:2911–2919. 2018.PubMed/NCBI View Article : Google Scholar
|
|
165
|
Sun Z and Brodsky JL: Protein quality
control in the secretory pathway. J Cell Biol. 218:3171–3187.
2019.PubMed/NCBI View Article : Google Scholar
|
|
166
|
Gallagher CM and Walter P: Ceapins inhibit
ATF6alpha signaling by selectively preventing transport of ATF6α to
the Golgi apparatus during ER stress. Elife.
5(e11880)2016.PubMed/NCBI View Article : Google Scholar
|
|
167
|
Wang L, Xu Y, Yun S, Yuan Q,
Satpute-Krishnan P and Ye Y: SAYSD1 senses UFMylated ribosome to
safeguard co-translational protein translocation at the endoplasmic
reticulum. Cell Rep. 42(112028)2023.PubMed/NCBI View Article : Google Scholar
|
|
168
|
Izawa T, Park SH, Zhao L, Hartl FU and
Neupert W: Cytosolic protein vms1 links ribosome quality control to
mitochondrial and cellular homeostasis. Cell. 171:890–903 e18.
2017.PubMed/NCBI View Article : Google Scholar
|