Introduction
A recent large-scale study reported that fracture
risk was markedly increased in patients with type 1 diabetes, with
an approximately threefold higher risk, and modestly but
significantly increased in those with type 2 diabetes (20-30%
higher risk), compared with non-diabetic individuals (1). In addition, recommendations for bone
health management in diabetic patients have been added to the
American Diabetes Association guidelines published in 2024(2). Low bone mineral density in type 1
diabetes mellitus (3), low bone
turnover, defects in microarchitecture, alterations in vitamin D
regulation and diabetes-related complications, such as neuropathy
and retinopathy, are associated with an increased risk of fractures
in diabetic patients (4-6).
Oxidative stress, advanced glycation end products (AGEs),
homocysteine, and the reduction in insulin and insulin-like growth
factor 1 activity can contribute to diabetes-related bone fragility
(7). Research on osteoporosis
treatments specifically for patients with diabetes is lacking, with
the same osteoporosis treatment being recommended for both the
general population and diabetic patients (8).
The environment of osteoblasts under hyperglycemic
conditions can be experimentally reproduced using 2-deoxy-D-ribose
(dRib), a deoxy-sugar derived from the pentose sugar ribose. dRib
has a high reactivity with proteins and readily generates reactive
oxygen species (ROS) through autoxidation and glycation reactions,
thereby mimicking oxidative and glycative stress under diabetic
conditions (9). Previous study has
demonstrated that dRib induces mitochondrial dysfunction,
endoplasmic reticulum (ER) stress and apoptosis in MC3T3-E1
osteoblastic cells (10),
suggesting that dRib is a reliable in vitro model for
investigating diabetes-related oxidative damage during bone
metabolism.
Spironolactone, a well-known mineralocorticoid
receptor (MR) antagonist used to treat hypertension, heart failure
and primary aldosteronism, has attracted attention due to the
diverse biological actions it exhibits, extending beyond
cardiovascular effects (11).
Beyond its mineralocorticoid receptor-blocking properties,
spironolactone exerts a range of additional biological actions,
such as attenuating fibrotic and inflammatory processes, reducing
thrombotic activity and tissue congestion, and enhancing vascular
function (11). Our previous study
demonstrated that spironolactone mitigates methylglyoxal
(MG)-induced oxidative injury in osteoblasts by reducing
intracellular ROS levels (12).
Considering that both MG and dRib are reactive carbonyl compounds
that contribute to AGE formation and oxidative stress, it was
hypothesized that spironolactone could also protect osteoblasts
from dRib-induced cytotoxicity.
Therefore, the present study aimed to investigate
the protective effects of spironolactone on bone metabolism by
mitigating oxidative and glycative stress in osteoblasts, thereby
providing mechanistic insight into its potential as a therapeutic
option to prevent diabetes-related bone fragility.
Materials and methods
Materials and reagents
Spironolactone was obtained from MilliporeSigma.
Culture media and antibiotics were supplied by Gibco (Thermo Fisher
Scientific, Inc.), whereas all other reagents were sourced from
MilliporeSigma, unless otherwise stated.
Cell culture
The osteoblastic MC3T3-E1 subclone 4 cell line was
purchased from the American Type Culture Collection. Cells were
grown in α-modified minimal essential medium (α-MEM; Gibco; Thermo
Fisher Scientific, Inc) at 37˚C in a humidified incubator with 5%
CO2. The medium was supplemented with 10% FBS,
penicillin (100 U/ml), streptomycin (100 µg/ml) and amphotericin B
(25 µg/ml). Once the cultures reached 100% confluence, they were
switched to an osteogenic differentiation α-MEM containing 10% FBS,
penicillin (100 U/ml), streptomycin (100 µg/ml), amphotericin B (25
µg/ml), 5 mM β-glycerophosphate and 50 µg/ml ascorbic acid
containing 5 mM β-glycerophosphate and 50 µg/ml ascorbic acid.
Spironolactone treatment was administered for 48 h at 37˚C in a
humidified incubator with 5% CO2 after 6 days [for
collagen measurement and alkaline phosphatase (ALP) activity] or 14
days (for mineralization analysis).
Cell viability
A preliminary study was conducted prior to the main
experiments to assess the effects of a number of spironolactone and
dRib concentrations on the viability of MC3T3-E1 osteoblastic
cells. MC3T3-E1 cells were plated in 24-well plates at a density of
2x104 cells/well. After 48 h, the cells were exposed at
37˚C for 1 h to α-MEM containing 0.1% FBS and spironolactone (0,
10, 20, 50, 100, 200, 300 and 500 µM) and then treated with dRib
(0, 5, 10, 15, 20 and 30 mM) for an additional 48 h. Cell viability
was determined using the water-soluble tetrazolium (WST) assay
(Dojindo Molecular Technologies, Inc.). Briefly, WST reagent was
added to each well and incubated at 37˚C for 2 h before
measurement. Absorbance was read at 570 nm with a Multiskan
microplate reader (Thermo Fisher Scientific, Inc.). Untreated
control cells cultured in medium alone were set as 100% viable and
the percentage survival of treated cells was calculated relative to
this control.
Lactate dehydrogenase (LDH)
cytotoxicity assay
MC3T3-E1 cells were plated in 24-well dishes at a
density of 2x104 cells/well. After 48 h, the cells were
pretreated at 37˚C with varying spironolactone concentrations (0,
10, 20, 50, 70 and 100 µM) for 1 h, followed by exposure to dRib 15
mM for an additional 48 h. Cell injury was assessed by measuring
plasma membrane disruption. As LDH is a stable enzyme released upon
membrane damage, its leakage into the culture medium was quantified
using the LDH Cytotoxicity Assay Kit (BioVision, Inc.; Abcam)
according to the manufacturer's protocol. Results were normalized
against both positive controls (cells treated with 1% Triton X-100)
and negative controls, with the Triton-treated samples defined as
representing 100% cytotoxicity.
Collagen content assay
Osteoblast cultures were first rinsed with
Dulbecco's PBS and subsequently fixed in Bouin's solution at room
temperature for 1 h. After fixation, the dishes were washed under
running tap water for 15 min, air-dried and then stained at room
temperature with Sirius red for 1 h with gentle agitation. Excess
dye was removed with 0.01N HCl and the bound stain was solubilized
in 0.1N NaOH. Absorbance was recorded at 550 nm and collagen
content was quantified using a calibration curve prepared from
known concentrations of commercial collagen (MilliporeSigma).
Absorbance was measured at 540 nm with a Zenyth 3100 multimode
detector (Anthos Labtec Instruments GmbH).
ALP activity assay
ALP activity was determined from cell lysates using
the ALP Activity Assay Kit (BioVision, Inc.; cat. no. K412-500)
according to the supplier's protocol. ALP activity was normalized
to total protein content. Total protein content was quantified
using the Bio-Rad protein assay reagent (Bio-Rad Laboratories,
Inc.). Absorbance was measured with a Zenyth 3100 multimode
detector (Anthos Labtec Instruments GmbH).
Mineralization assay
Mineralization was evaluated by assessing calcium
deposition. After fixation with 70% ethanol, cells were stained
with Alizarin Red S at room temperature for 10 min and the
extracted dye was quantified by measuring absorbance at 561 nm.
Absorbance was measured at 540 nm with a Zenyth 3100 multimode
detector (Anthos Labtec Instruments GmbH).
Measurement of TNF-α and IL-6
levels
TNF-α (cat. no. ELM-IL6) and IL-6 (cat. no. ELM TNF
a) concentrations in the culture supernatants were determined using
enzyme immunoassay kits (R&D Systems, Inc.) according to the
manufacturer's guidelines. Culture supernatants were obtained from
centrifugation and determined. Absorbance was measured with a
Zenyth 3100 multimode detector (Anthos Labtec Instruments
GmbH).
Measurements of glyoxalase I activity
and glutathione (GSH) levels
Glyoxalase I activity was assessed according to a
previously reported protocol (13). The intracellular GSH content from
cell lysates was quantified using a GSH Assay Kit (BioAssay
Systems) according to the manufacturer's instructions. Catalog
numbers were glyoxalase I activity (DGLO-100) and GSH
(EGTT-100).
Determination of the mitochondrial
membrane potential (MMP)
Cells were plated in black 96-well plates at a
density of 1x104 cells/well and cultured for 24 h. The
cells were pretreated with numerous spironolactone concentrations
(0, 50, 70 and 100 µM) at 37˚C for 1 h, followed by exposure to
dRib (15 mM) for 48 h. MMP was assessed using the JC-1 MMP Assay
Kit (Cayman Chemical Company). JC-1 is a cationic, lipophilic dye
that accumulates in intact mitochondria and emits red fluorescence.
In depolarized mitochondria, it remains monomeric and fluoresces
green. Cells were incubated with JC-1 for 20 min at 37˚C, washed
twice with PBS and analyzed with a fluorescence microplate reader
(Molecular Devices, LLC) at 550/600 nm (red) and 485/535 nm
(green). A reduction in the red/green fluorescence ratio indicated
mitochondrial depolarization.
Measurement of ATP levels
Cells were plated in 24-well dishes at a density of
2x104 cells/well. After 48 h of culture, the cells were
pretreated at 37˚C with numerous concentrations of spironolactone
(0, 50, 70 and 100 µM) for 1 h and then exposed to dRib (15 mM) for
48 h. The cells were then lysed using cell lysis buffer II (cat.
no. FNN0021; Thermo Fisher Scientific, Inc.) and homogenized in
PBS. The homogenates were centrifuged at 13,000 x g for 15 min at
4˚C and the resulting supernatants were collected for ATP and
protein analyses. ATP levels were quantified by luciferase-based
bioluminescence using the EnzyLight™ ATP Assay Kit (cat. no.
EATP-100, BioAssay Systems) according to the manufacturers protocol
and total protein content was measured with the Bio-Rad Protein
Assay Kit. Absorbance was measured with a Zenyth 3100 multimode
detector (Anthos Labtec Instruments GmbH). ATP levels were
normalized to total protein content.
Measurement of intracellular ROS
levels
Cells were seeded in black 96-well plates at a
density of 1x104 cells per/well. After 24 h, the cells
were pretreated with spironolactone (50-100 µM) at 37˚C for 1 h,
followed by the addition of dRib and incubation at 37˚C for another
48 h. Subsequently, the cells were incubated with 5 µM
2',7'-dichlorofluorescein diacetate with spironolactone (50-100 µM)
at 37˚C for 1 h. Following three PBS washes, intracellular ROS
levels were quantified by detecting fluorescence at 485 nm
excitation and 530 nm emission using a fluorescence microplate
reader (13).
Measurement of mitochondrial
superoxide levels
Cells were seeded in black 96-well plates at a
density of 1x104 cells per/well. After 24 h, the cells
were pretreated with spironolactone at 37˚C for 1 h, followed by
the addition of dRib and incubation for another 48 h. Mitochondrial
superoxide production was measured with the MitoSOX Red probe
(Invitrogen; Thermo Fisher Scientific, Inc.). This fluorogenic dye
specifically targets the mitochondria and exhibits fluorescence
(excitation 510 nm; emission 580 nm) upon reacting with superoxide
radicals (14). The cells were
incubated with 2 mM MitoSOX Red at 37˚C for 20 min, in accordance
with the manufacturer's instructions. After washing the cells, the
levels of MitoSOX Red fluorescence were measured by microplate
reader.
Measurements of inositol-requiring
enzyme 1 (IRE1) and activating transcription factor 6 (ATF-6)
levels
IRE1 and ATF-6 are ER stress markers. Cytosolic
concentrations were quantified using ELISA kits (MyBioSource, Inc.)
following the supplier's protocols. Catalog numbers were IRE1
(MBS728814) and ATF-6 (MBS2533467).
RNA isolation and reverse
transcription-quantitative PCR (qPCR)
MC3T3-E1 osteoblastic cells were plated in 100-mm
culture dishes at a density of 2x105 cells/well and
maintained in growth medium. After 48 h, the cells were pretreated
with spironolactone (100 µM) for at 37˚C for 1 h, followed by
exposure to 100 nM dRib for 24 h. Total RNA was isolated using the
RNeasy Mini Kit (Qiagen GmbH) and cDNA was synthesized with the
PrimeScript First Strand cDNA Synthesis Kit (Takara Bio, Inc.)
according to the manufacturer's protocol. qPCR was performed using
the SYBR® Premix Ex Taq™ Kit (Takara Bio, Inc.) on an
ABI Prism 7500 system (Applied Biosystems; Thermo Fisher
Scientific, Inc.) to evaluate gene expression. The thermal cycling
protocol comprised an initial denaturation at 95˚C for 10 min,
followed by 40 cycles of 94˚C for 10 sec and 60˚C for 30 sec.
Reactions were performed in 20-µl mixtures containing 0.8 µl each
primer (10 µM), 10 µl SYBR Premix, 0.4 µl ROX reference dye, 6 µl
distilled water and 2 µl cDNA template. Table I lists the primer sequences used.
All assays were conducted in quadruplicate and relative expression
levels were analyzed using the 2-ΔΔCq method (15) with glucose-6-phosphate
dehydrogenase as the reference gene, with results expressed as fold
change relative to controls.
 | Table IPrimer sequences used for reverse
transcription-quantitative PCR. |
Table I
Primer sequences used for reverse
transcription-quantitative PCR.
| Genes | Forward primer | Reverse primer |
|---|
| ALP | 5'-GGC CAG CTA CAC
CAC AAC A-3' | 5'-CTG AGC GTT GGT
GTT ATA TGT CTT-3' |
| Collagen | 5'-AGA CAT GTT CAG
CTT TGT GGA C-3' | 5'-CAT CCC TGA AGT
CAG CTG C-3' |
| Osteocalcin | 5'-CAC CAT GAG GAC
CCT CTC TC-3' | 5'-TGG ACA TGA AGG
CTT TGT CA-3' |
| G6PD | 5'-TGC AGC AGC TGT
CCT CTA TG-3' | 5'-ACT TCA GCT TTG
CGC TCA TT-3' |
Statistical analysis
Experiments were carried out in at least three
independent experiments. Data are presented as the mean ± standard
error of the mean. Statistical comparisons were performed using
one-way ANOVA, followed by Dunnett's post-hoc test. Analyses were
conducted using PASW software (version 20.0; IBM Corp.) and
P<0.05 was considered to indicate a statistically significant
difference.
Results
Cytoprotective effects of
spironolactone on dRib-induced cytotoxicity in osteoblasts
After treating MC3T3-E1 osteoblastic cells with
spironolactone alone for 48 h, cell viability was measured. Cell
viability decreased in response to 200-500 µM viability, whereas
concentrations ≤100 µM did not significantly affect cell viability
(Fig. S1A). Therefore, in the
present study, spironolactone was used at concentrations ≤100 µM,
which did not affect cell viability. After treating MC3T3-E1
osteoblastic cells with dRib (0-30 mM) alone for 48 h, cell
viability was reduced in a concentration-dependent manner (Fig. S1B). dRib was then applied to the
cells at a concentration of 15 mM, which reduced the cell viability
to 50%.
Osteoblasts were treated with spironolactone at
concentrations ranging between 0 and 100 µM and cultured 1 h later
with 15 mM dRib for 48 h. Treatment with 15 mM dRib for 48 h
significantly decreased cell viability to 19.4% of control
(P<0.05), whereas pretreatment with spironolactone (70 and 100
µM) restored viability to 32.5±5.6 and 79.2±12.8%, respectively
(Fig. S1C). LDH release increased
to 69.1±3.9% of control after dRib exposure, but 50 and 100 µM
spironolactone reduced LDH leakage to 19.1±1.7 and 21.2±3.9%,
respectively (P<0.05; Fig.
S1D). Morphological changes were photographed under an inverted
microscope. Morphologically, dRib-treated cells showed marked
shrinkage and detachment, whereas spironolactone treatment (100 µM)
largely restored the normal polygonal morphology (Fig. S2).
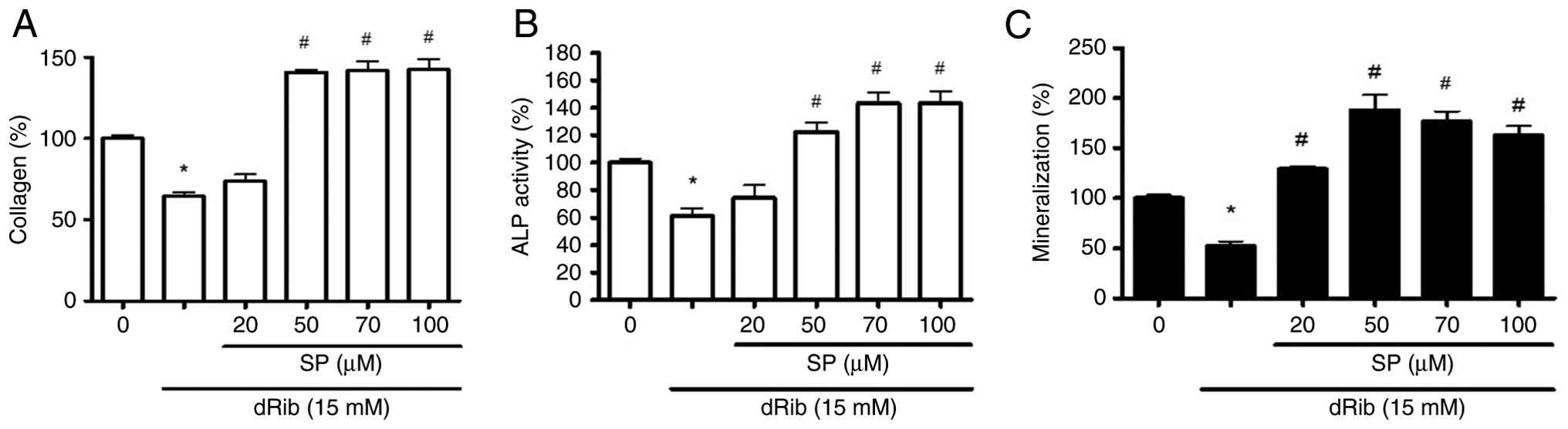
Effect of spironolactone on
dRib-induced osteoblast differentiation
To examine the effect of spironolactone on the
differentiation of the MC3T3-E1 osteoblast cell line, collagen
content, ALP activity and calcium deposition were measured.
Collagen content was decreased to 65.3±2.4% compared with that in
the control group after dRib treatment; however, it increased again
to between 140.6±1.5 and 142.3±6.8% after spironolactone treatment
(50-100 µM; P<0.05; Fig. 1A).
ALP activity showed a similar pattern, declining to 61.3±5.3%
compared with the control group, then recovering to 143.2±8.5% with
100 µM spironolactone treatment (P<0.05; Fig. 1B). Calcium deposition, measured by
Alizarin Red S, fell to 52.4±4.5% after dRib treatment, but was
restored to between 129.1±2.6 and 188.1±15.4% of the control levels
following spironolactone treatment (20-100 µM; P<0.05, Fig. 1C). Considering that collagen
synthesis and ALP activity reflect the early stages of osteoblast
differentiation, whereas mineralization represents the late stage,
it is suggested that spironolactone promotes osteoblast
differentiation, which is suppressed by dRib, from the early to
late phase of differentiation.
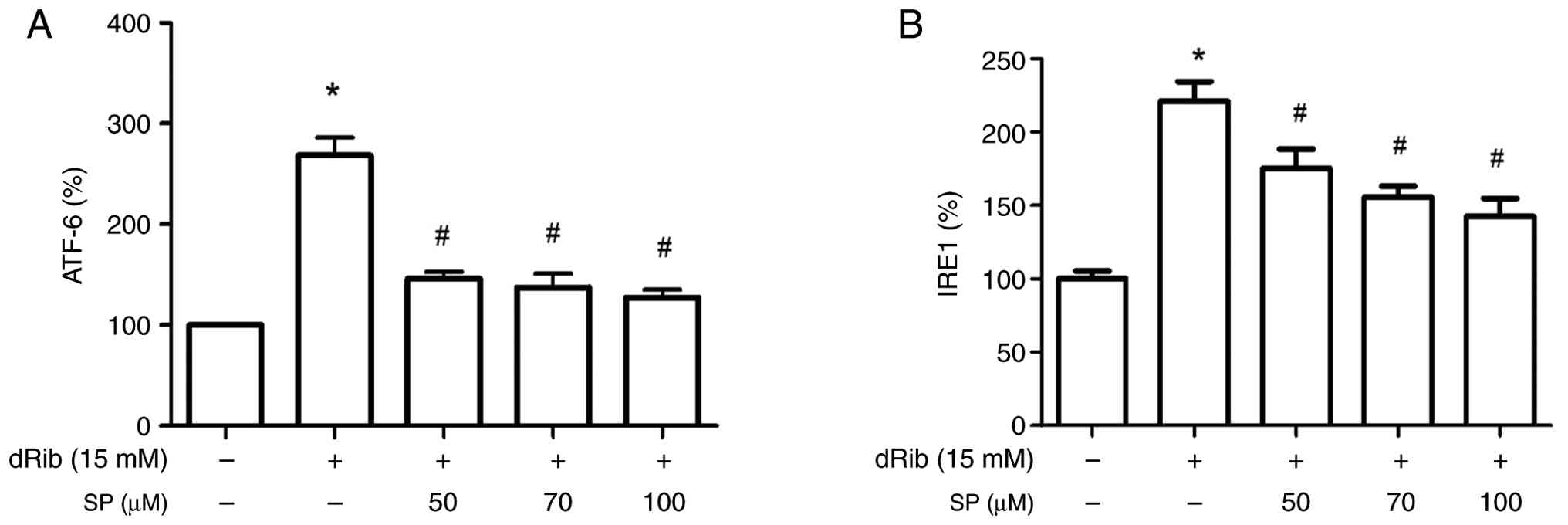
Effect of spironolactone on ER stress
and inflammatory cytokine production in dRib-treated MC3T3-E1
cells
After dRib administration, ATF-6 and IRE1 activities
increased to 268.4±17.8 and 221.1±13.3% of the control levels,
respectively, indicating increased ER stress (P<0.05; Fig. 2A and B). Pretreatment with 100 µM
spironolactone significantly decreased the dRib-induced increase in
ATF-6 and IRE1 activities to 127.4±7.7 and 142.6±12.4%,
respectively (P<0.05; Fig. 2A
and B), suggesting that
spironolactone mitigated ER stress stimulated by dRib.
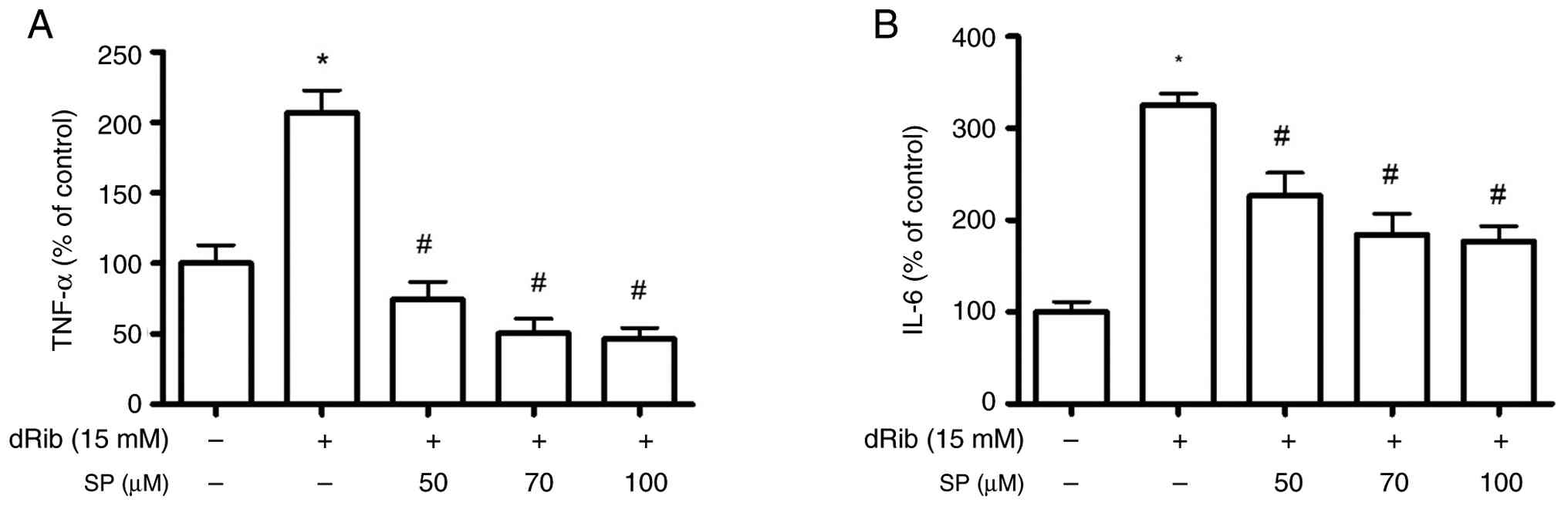
Furthermore, dRib treatment (15 mM) led to a
significant increase in TNF-α and IL-6 production to 206.6±16.3 and
325.5±12.3% of the control levels, respectively (P<0.05;
Fig. 3A and B). Increased TNF-α and IL-6 production
was significantly reduced to 46.3±8.0 and 176.9±16.7%,
respectively, by 100 µM spironolactone pretreatment (P<0.05;
Fig. 3A and B), suggesting that the mechanism by which
spironolactone prevents osteoblast damage caused by dRib involves a
reduction in inflammatory cytokine levels.
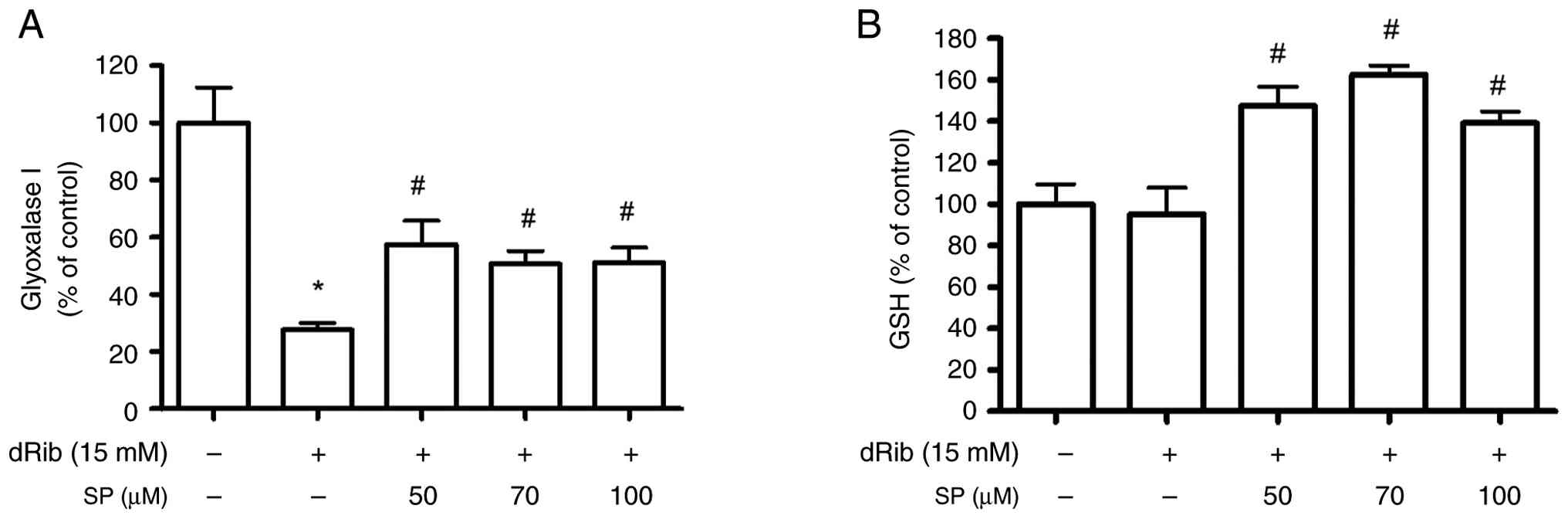
Effect of spironolactone on glyoxalase
I activity and GSH levels in dRib-treated cells
dRib is detoxified by the glyoxalase enzyme system.
Measurement of glyoxalase I activity revealed a significant
decrease to 27.7±2.4% of the control after dRib treatment (15 mM),
whereas glyoxalase I activity was increased to 57.5±8.3% when the
osteoblasts were pretreated with 50 µM spironolactone (P<0.05;
Fig. 4A). Additionally, GSH levels
were increased to 162.5±4.3% in dRib-treated osteoblasts after 70
µM spironolactone treatment (P<0.05; Fig. 4B). These results indicated that
spironolactone may detoxify dRib by increasing GSH and glyoxalase I
activity.
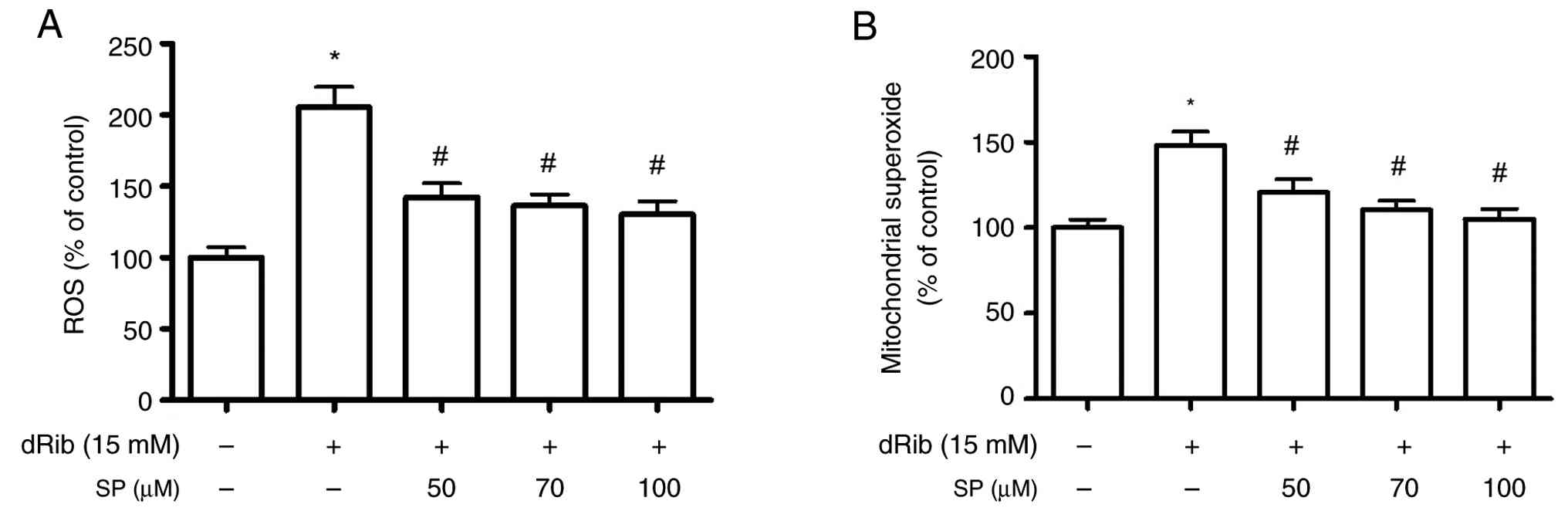
Inhibitory effect of spironolactone on
dRib-induced oxidative stress in cells
dRib exposure markedly increased intracellular ROS
generation to 205.3±14.2% of the control. Spironolactone
pretreatment progressively and dose-dependently reduced ROS levels
to 142.4±9.6% (50 µM), 136.4±7.8% (70 µM) and 130.3±9.3% (100 µM)
(P<0.05; Fig. 5A). Similarly,
mitochondrial superoxide production rose to 148.0±8.2% of the
control after dRib treatment, but was gradually suppressed by
spironolactone to 120.5±7.8, 110.4±5.5 and 105.1±5.8% at 50, 70 and
100 µM, respectively (P<0.05; Fig.
5B). These findings indicated that spironolactone reduced
dRib-induced ROS production and oxidative stress in
mitochondria.
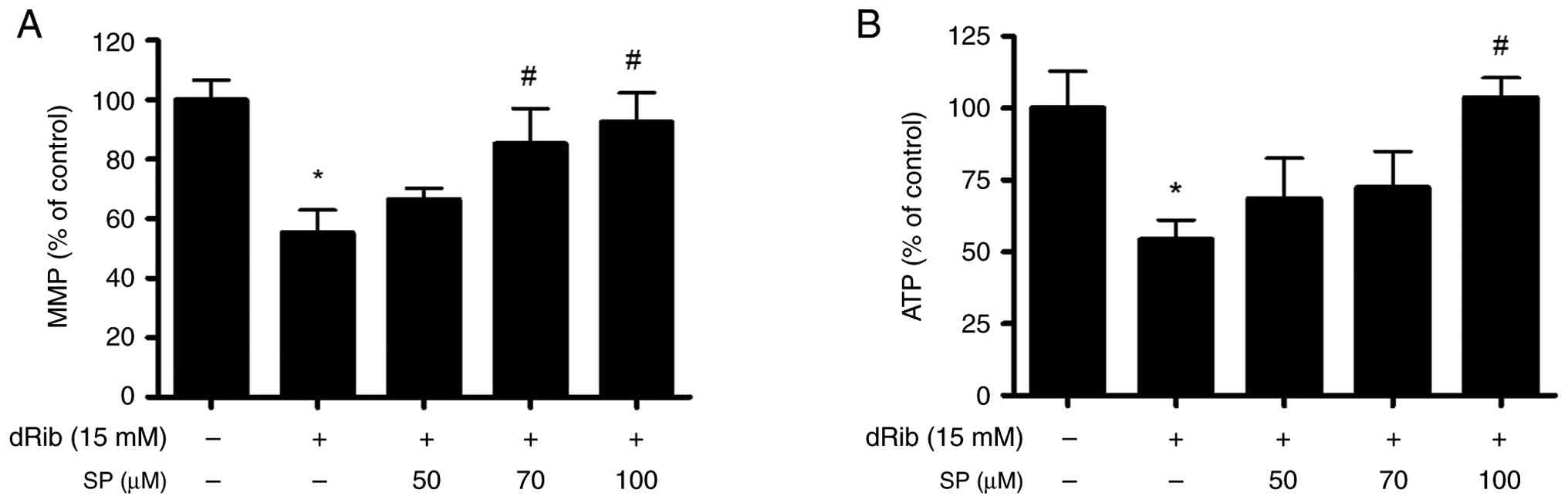
Effect of spironolactone on the
dRib-induced mitochondrial dysfunction in osteoblastic MC3T3-E1
cells
When osteoblasts were treated with dRib, MMP
decreased to 55.3±7.7% of the control, whereas it increased to
66.4±3.8, 85.1±11.9 and 92.5±9.8% in response to 50, 70 and 100 µM
spironolactone pretreatment, respectively. (P<0.05 in 70 and 100
µM spironolactone; Fig. 6A). In
addition, ATP production was reduced to 54.3±6.8% of the control
following dRib treatment, but was restored to 68.4±14.3, 72.4±12.6
and 103.7±6.9% after 50, 70 and 100 µM spironolactone treatment,
respectively (P<0.05 in 100 µM spironolactone; Fig. 6B). These findings suggested that
spironolactone may have reduced dRib toxicity by increasing
mitochondrial biogenesis.
Effect of spironolactone on the mRNA
expression levels of ALP, type I collagen and osteocalcin in
dRib-treated cells
In osteoblasts treated with 15 mM dRib, the mRNA
expression levels of ALP, collagen and osteocalcin were
significantly reduced (0.3±0.1, 0.2±0.1 and 0.42±0.1 fold of
control, respectively; P<0.05), whereas treatment with
spironolactone restored their expression (0.8±0.2, 0.9±0.1 and
1.1±0.1 fold of control, respectively; P<0.05) (Fig. S3).
Discussion
Within the present study, the mechanism by which
spironolactone protects osteoblastic MC3T3-E1 cells from
dRib-induced oxidative and glycative stress was examined. Rather
than reversing cytotoxicity, spironolactone was shown to modulate
numerous interlinked stress-response pathways, including redox
balance, ER stress, mitochondrial function and inflammation,
ultimately preserving osteoblast differentiation capacity.
In our previous study, antioxidants such as
N-acetyl-L-cysteine and α-lipoic acid were revealed to reverse
dRib-induced cytotoxicity (16),
indicating that oxidative stress is a key contributor to this
damage. Consistent with these findings, the present study further
demonstrated this mechanism by exhibiting that dRib exposure
markedly increased intracellular ROS and mitochondrial superoxide
levels, implicating mitochondrial dysfunction as a key source of
oxidative stress. ROS, while being important in normal cellular
processes (17), can cause notable
damage under oxidative stress, leading to DNA, lipid and protein
damage, and enhanced apoptosis (18). The observed reduction in ROS and
mitochondrial superoxide, coupled with the restoration of MMP and
ATP production, implies that spironolactone stabilizes
mitochondrial integrity, likely by limiting ROS-driven damage and
supporting biogenesis.
The ER, which is responsible for protein synthesis
and folding, also serves a role in protein quality surveillance and
degradation (19). Unresolved ER
stress caused by misfolded proteins activates the unfolded protein
response mediated by IRE1 and ATF-6 (20-22).
In the present study, both ATF-6 and IRE1 levels were elevated
following dRib treatment, indicating an increase in ER stress. ER
stress and mitochondrial dysfunction are associated, forming a
cycle that promotes apoptosis and oxidative stress (23). Spironolactone may affect this cycle
by reducing ROS accumulation, restoring MMP and ATP synthesis, and
attenuating ER stress, thereby improving cell survival and
osteogenic capacity. The present findings suggest that
spironolactone acts as an antioxidant.
dRib also increased inflammatory markers TNF-α and
IL-6, contributing to oxidative damage (24). By contrast, spironolactone
suppressed the production of these inflammatory cytokines, thus
reinforcing its role as an anti-inflammatory modulator. A previous
in vitro study demonstrated that spironolactone inhibits the
stimulated release of TNF-α and IL-6 from human peripheral blood
mononuclear cells (25).
Experimental studies have shown that spironolactone protects
myocardial and endothelial cells by reducing oxidative stress and
inflammatory responses, thereby ameliorating diabetic
cardiomyopathy and endothelial dysfunction through mechanisms
involving suppression of reactive oxygen species generation and
inhibition of the AGE/RAGE signaling pathway (26-28).
The enhancement of GSH levels and recovery of
glyoxalase I activity further reinforces the antioxidant potential
of spironolactone. As glyoxalase I detoxifies MG, a precursor of
AGEs (29), this mechanism may
contribute to the prevention of diabetes-related bone
deterioration. Thus, spironolactone not only reduces the oxidative
burden, but may also improve osteoblast redox homeostasis and
matrix formation through both direct and enzymatic pathways.
Patients with primary aldosteronism show increased
urinary calcium excretion, leading to secondary
hyperparathyroidism, which accelerates bone loss and increases
fracture risk (30). Consistent
with these pathophysiological mechanisms, spironolactone treatment
has been shown to reduce bone loss, enhance bone strength and
prevent fractures in this population (30,31).
Besides the systemic effects, the present data suggest that
spironolactone could exert direct osteoblastic protection through
integrated antioxidant and anti-inflammatory mechanisms. This dual
action underscores its therapeutic potential as an adjunct strategy
for treating diabetes-related bone fragility.
The present study has some limitations. First,
although bone metabolism involves osteoblasts, osteoclasts and
osteocytes, only osteoblasts were examined. Nevertheless, the
present study demonstrated meaningful qualitative and quantitative
outcomes in terms of osteogenic activity. Further research using
animal models, osteoblast-osteoclast co-culture systems, and the
evaluation of bone mass and quality is needed. Second,
spironolactone was tested without comparison to other antioxidants
or medications, limiting the ability to evaluate its relative
efficacy. For example, quantitative efficacy analyses through
comparisons with agents such as SGLT2 inhibitors are recommended in
future studies. Third, numerous factors such as hyperglycemia,
insulin resistance and kidney dysfunction influence
diabetes-related bone fragility and should be considered in future
studies. Lastly, dRib may represent a model closer to
stress-induced premature senescence rather than true aging and
therefore may have limitations in fully reflecting the
physiological aging process. However, considering that oxidative
stress is one of the key mechanisms underlying diabetes-related
bone fragility, the present findings remain meaningful.
In conclusion, the present study revealed that
spironolactone protects osteoblasts from dRib-induced oxidative
stress by suppressing ROS and ER stress, enhancing mitochondrial
integrity and activating the endogenous antioxidant system. These
findings provide mechanistic insights into the non-classical
actions of spironolactone and support its potential as an
adjunctive therapeutic strategy for preventing diabetes-related
bone fragility.
Supplementary Material
Cytoprotective effects of SP on
dRib-induced cytotoxicity in osteoblastic cells. (A) Cell viability
in response to various concentrations of SP. (B) Cell viability in
response to various concentrations of dRib. (C) Osteoblasts were
treated with SP in the absence or presence of 15 mM dRib for 48 h
and cell viability was measured. (D) Cells were treated with SP in
the absence or presence of 15 mM dRib for 48 h and LDH levels were
measured. *P<0.05 vs. untreated cells;
#P<0.05 vs. cells treated with dRib alone. SP,
spironolactone; dRib, 2-deoxy-D-ribose; LDH, lactate
dehydrogenase.
Effect of SP on the dRib-induced
morphology of MC3T3-E1 osteoblast cells. (A) Control, (B) SP (100
μM), (C) dRib (15 mM), (D) SP (100 μM) + dRib (15
mM). Inverted microscopy, magnification, x100. SP, spironolactone;
dRib, 2-deoxy-D-ribose.
RNA isolation and reverse
transcription-quantitative PCR. The mRNA expression levels of ALP,
collagen, and osteocalcin were measured in cells treated with dRib
(15 mM) in the presence or absence of spironolactone (100
μM). Gene expression levels are presented as fold changes
relative to the control group. Data are expressed as mean ± SEM
from independent in vitro experiments. #P<0.05
vs. untreated cells; *P<0.05 vs. cells treated with
dRib alone. SP, spironolactone; dRib, 2-deoxy-D-ribose; ALP,
alkaline phosphatase.
Acknowledgements
An abstract based on part of this work was
previously presented and published in the Journal of the
Endocrine Society (2024; 8(Supplement 1): bvae163.410).
Funding
Funding: The present was supported by Korea United Pharm.
Inc.
Availability of data and materials
The data generated in the present study may be
requested from the corresponding author.
Authors' contributions
SYP contributed towards conducting the
investigation, writing the original draft, and reviewing and
editing the manuscript. SYP, SOC and KSS confirm the authenticity
of all the raw data. KSS contributed towards collection of raw
data, formal analysis, conducting the investigation, experimental
methodology, validation, and reviewing and editing the manuscript.
HSK contributed towards collection of raw data, formal analysis,
conducting the investigation, methodology, validation and reviewing
and editing the manuscript. SJY contributed towards conducting the
investigation, validation, and reviewing and editing the
manuscript. HS contributed towards conducting the investigation,
validation, and reviewing and editing the manuscript. SOC
contributed towards conceptualization, funding acquisition,
conducting the investigation, validation, and reviewing and editing
the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Liao CC, Lin CS, Shih CC, Yeh CC, Chang
YC, Lee YW and Chen TL: Increased risk of fracture and postfracture
adverse events in patients with diabetes: Two nationwide
population-based retrospective cohort studies. Diabetes Care.
37:2246–2252. 2014.PubMed/NCBI View Article : Google Scholar
|
|
2
|
American Diabetes Association Professional
Practice Committee. 4. Comprehensive medical evaluation and
assessment of comorbidities: Standards of care in diabetes-2024.
Diabetes Care. 47 (Suppl 1):S52–S76. 2024.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Starup-Linde J, Hygum K, Harslof T and
Langdahl B: Type 1 diabetes and bone fragility: Links and risks.
Diabetes Metab Syndr Obes. 12:2539–2547. 2019.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Carnevale V, Romagnoli E and D'Erasmo E:
Skeletal involvement in patients with diabetes mellitus. Diabetes
Metab Res Rev. 20:196–204. 2004.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Hygum K, Starup-Linde J, Harslof T,
Vestergaard P and Langdahl BL: MECHANISMS IN ENDOCRINOLOGY:
Diabetes mellitus, a state of low bone turnover-a systematic review
and meta-analysis. Eur J Endocrinol. 176:R137–R157. 2017.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Starup-Linde J, Lykkeboe S, Gregersen S,
Hauge EM, Langdahl BL, Handberg A and Vestergaard P: Bone structure
and predictors of fracture in type 1 and type 2 diabetes. J Clin
Endocrinol Metab. 101:928–936. 2016.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kanazawa I and Sugimoto T: Diabetes
Mellitus-induced bone fragility. Intern Med. 57:2773–2785.
2018.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Sheu A, White CP and Center JR: Bone
metabolism in diabetes: A Clinician's guide to understanding the
bone-glucose interplay. Diabetologia. 67:1493–1506. 2024.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Koh G, Suh KS, Chon S, Oh S, Woo JT, Kim
SW, Kim JW and Kim YS: Elevated cAMP level attenuates
2-deoxy-d-ribose-induced oxidative damage in pancreatic beta-cells.
Arch Biochem Biophys. 438:70–79. 2005.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Kim HS, Suh KS, Ko A, Sul D, Choi D, Lee
SK and Jung WW: The flavonoid glabridin attenuates
2-deoxy-D-ribose-induced oxidative damage and cellular dysfunction
in MC3T3-E1 osteoblastic cells. Int J Mol Med. 31:243–251.
2013.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Ferreira JP, Verdonschot J, Wang P, Pizard
A, Collier T, Ahmed FZ, Brunner-La-Rocca HP, Clark AL, Cosmi F,
Cuthbert J, et al: Proteomic and mechanistic analysis of
spironolactone in patients at risk for HF. JACC Heart Fail.
9:268–277. 2021.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Park SY, Suh KS, Jung WW and Chin SO:
Spironolactone attenuates methylglyoxal-induced cellular
dysfunction in MC3T3-E1 osteoblastic cells. J Korean Med Sci.
36(e265)2021.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Suh KS, Chon S and Choi EM: Protective
effects of piceatannol on methylglyoxal-induced cytotoxicity in
MC3T3-E1 osteoblastic cells. Free Radic Res. 52:712–723.
2018.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Piazena H and Kelleher D: Comments on
‘Cellular response to infrared radiation involves retrograde
mitochondrial signaling’. Free Radic Biol Med. 44:1869–1871.
2008.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Suh KS, Choi EM, Kwon M, Chon S, Oh S, Woo
JT, Kim SW, Kim JW and Kim YS: Kaempferol attenuates
2-deoxy-d-ribose-induced oxidative cell damage in MC3T3-E1
osteoblastic cells. Biol Pharm Bull. 32:746–749. 2009.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Rhee SG: Cell signaling. H2O2, a necessary
evil for cell signaling. Science. 312:1882–1883. 2006.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Jacobson MD: Reactive oxygen species and
programmed cell death. Trends Biochem Sci. 21:83–86.
1996.PubMed/NCBI
|
|
19
|
Ellgaard L and Helenius A: Quality control
in the endoplasmic reticulum. Nat Rev Mol Cell Biol. 4:181–191.
2003.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Mei Y, Thompson MD, Cohen RA and Tong X:
Endoplasmic reticulum stress and related pathological processes. J
Pharmacol Biomed Anal. 1(1000107)2013.PubMed/NCBI
|
|
21
|
Schroder M and Kaufman RJ: The mammalian
unfolded protein response. Annu Rev Biochem. 74:739–789.
2005.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007.PubMed/NCBI View
Article : Google Scholar
|
|
23
|
Bhatti JS, Bhatti GK and Reddy PH:
Mitochondrial dysfunction and oxidative stress in metabolic
disorders-A step towards mitochondria based therapeutic strategies.
Biochim Biophys Acta Mol Basis Dis. 1863:1066–1077. 2017.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Mittal M, Siddiqui MR, Tran K, Reddy SP
and Malik AB: Reactive oxygen species in inflammation and tissue
injury. Antioxid Redox Signal. 20:1126–1167. 2014.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Hansen PR, Rieneck K and Bendtzen K:
Spironolactone inhibits production of proinflammatory cytokines by
human mononuclear cells. Immunol Lett. 91:87–91. 2004.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Liu W, Gong W, He M, Liu Y, Yang Y, Wang
M, Wu M, Guo S, Yu Y, Wang X, et al: Spironolactone protects
against diabetic cardiomyopathy in streptozotocin-induced diabetic
rats. J Diabetes Res. 2018(9232065)2018.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Mayyas F, Alzoubi KH and Bonyan R: The
role of spironolactone on myocardial oxidative stress in rat model
of streptozotocin-induced diabetes. Cardiovasc Ther.
35(e12242)2017.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Wang CC, Lee AS, Liu SH, Chang KC, Shen MY
and Chang CT: Spironolactone ameliorates endothelial dysfunction
through inhibition of the AGE/RAGE axis in a chronic renal failure
rat model. BMC Nephrol. 20(351)2019.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Do MH, Hur J, Choi J, Kim M, Kim MJ, Kim Y
and Ha SK: Eucommia ulmoides ameliorates glucotoxicity by
suppressing advanced glycation end-products in diabetic mice
kidney. Nutrients. 10(265)2018.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Chhokar VS, Sun Y, Bhattacharya SK, Ahokas
RA, Myers LK, Xing Z, Smith RA, Gerling IC and Weber KT: Loss of
bone minerals and strength in rats with aldosteronism. Am J Physiol
Heart Circ Physiol. 287:H2023–H2026. 2004.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Carbone LD, Cross JD, Raza SH, Bush AJ,
Sepanski RJ, Dhawan S, Khan BQ, Gupta M, Ahmad K, Khouzam RN, et
al: Fracture risk in men with congestive heart failure risk
reduction with spironolactone. J Am Coll Cardiol. 52:135–138.
2008.PubMed/NCBI View Article : Google Scholar
|