Introduction
Biomolecules, including proteins, nucleic acids and
sugars, are important components of living organisms and perform a
number of functions. The study of protein assembly, interactions
and subcellular distribution is key in understanding their
functions. Protein-protein interactions are the basis of cellular
activities, such as replication, transcription, translation of
genetic information, intercellular signaling and cellular
regulation. Understanding the spatial patterns of protein
localization and interactions is important in understanding
cellular processes (1). The
spatial distances between proteins are short and their interactions
primarily rely on hydrogen, salt and hydrophobic interaction
forces; therefore, we hypothesized that interacting proteins must
be in close proximity to each other. Traditionally, protein
interactions have been studied by affinity purification, preserving
organelles or interactions in cell lysis and purification
complexes; however, this method often fails to preserve weak or
transient interactions and can disrupt native protein complex
conformations, making it challenging to capture physiologically
relevant interactomes (2). To
overcome this, proximity labeling technology combined with mass
spectrometry (MS) detection is gradually becoming a more suitable
method for the identification of organelle protein components and
interactions (3). The principle of
this technique lies in the fusion of a labeling enzyme to a
targeted protein or subcellular compartment by gene fusion.
Among the currently available proximity labeling
techniques, biotin ligases are the most widely used. Together with
ascorbate peroxidases, they constitute the main class of labeling
enzyme systems (4). Biotin ligases
are engineered biotin ligase proteins that include BioID, BioID2,
BASU, TurboID, miniTurbo, Split-BioID and Split-TurboID (5). The first engineered biotin ligase was
BioID, which is an Escherichia coli BirA (bifunctional
ligase or repressor) mutant with a size of 35 kDa. This enzyme has
a reduced affinity for biotin-adenosine 5'-monophosphate (AMP) and
can react with the primary amine of the proximal protein, leading
to its covalent biotinylation (6).
To address the defective target protein localization due to the
large molecular weight of BioID, a new mutant (BioID2) was
identified from the thermophilic bacterium Aquifex aeolicus
with a size of 27 kDa (7). This
achieves a more targeted localization and requires lower biotin
concentration compared with BioID. However, both BioID and BioID2
exhibit the following limitations: First, the long labeling time
(18-24 h) is not favorable for the study of short transient
interacting proteins. Secondly, biotin can be actively transported
into the cytoplasm of mammalian cells and freely diffuse into the
nucleus; however, it has limited access to the secretory pathway,
resulting in lower labeling efficiency in that compartment
(8). In addition, due to the
prolonged BioID labeling, biotinylation of proteins can affect
their function, resulting in false positive or false negative
results.
TurboID and miniTurbo were generated by targeted
modifications of the BioID biotin ligase (9). Compared with BioID and BioID2,
TurboID and miniTurbo require less labeling time (≤10 min) in
addition to their ability to maintain catalytic activity at lower
temperatures (9). The labeling
principle of TurboID uses adenosine 5'-triphosphate (ATP) and
biotin to generate an intermediate substance, biotin-5'-AMP, which
can covalently label neighboring proteins (10), and can be used to extract or
capture proteins more efficiently. Furthermore, the proximity
labeling technique is now increasingly used in animal cell
experiments, including protein complexes (such as nuclear pore
complexes) (11), tissue
proteosomes (such as mitochondrial matrix and intermembrane space)
(12) and native proteomes
(13,14). It has been shown that this
technique not only allows for the detection of proteins that
transiently interact with specific proteins in living cell
(15), but also reduces the
potential losses caused by traditional methods during purification
and enables the precise identification of real-time (16), dynamic interacting proteins within
living cells. In addition, proximity labeling technology has been
extended to the study of transcriptional complexes and histone
modifications (17), providing an
important avenue for exploring biomolecular interactions.
AMP-activated protein kinase (AMPK) is an
evolutionarily conserved serine/threonine protein kinase with
AMP-dependent properties (18). As
a key molecule for cells to sense external environmental changes
and adjust energy metabolism, AMPK not only regulates energy
metabolism at the cellular level, but also serves an important role
in maintaining the energy homeostasis of the body. AMPK responds to
changes in intracellular adenine nucleotide levels, being activated
by an increase in AMP/ADP relative to ATP. Activation of AMPK
increases the rate of catabolic (ATP-generating) pathways and
decreases the rate of anabolic (ATP-utilising) pathways. In the
normal physiological state, the intracellular AMP level is
generally low; when the ATP level decreases after energy
consumption, the AMP level and the AMP/ATP ratio increase, followed
by the activation of AMPK by its upstream regulator (AMPK kinase)
through phosphorylation (19).
Therefore, the state of the cell can be monitored according to the
concentration of ATP, ADP and AMP. A previous study hasshown that
AMPK, a key regulator of energy metabolism, improves glucose
regulation in type 2 diabetes through its activation by drugs such
as metformin; however, its role in insulin secretion is complex and
remains controversial (20). In
addition, AMPK as exhibits an anti-apoptotic effect in
cardiomyocytes, and its activation not only reduces myocardial
energy consumption but also increases energy production, stabilizes
mitochondrial membrane potential to reduce intracellular reactive
oxygen species and serves a protective role against
ischemia/reperfusion injury and doxorubicin-induced cardiotoxicity
in the myocardium, as demonstrated in mouse models and cultured
cardiomyocytes (21). Therefore,
AMPK may represent a new target for the treatment of diabetes
mellitus and myocardial infarction.
In recent years, AMPK has been shown to serve a role
in a number of physiological and pathological conditions, with
broad potential applications. Song et al (22) found that sleeve gastrectomy may
improve renal injury in mice with hyperuricemic nephropathy by
upregulating ATP binding cassette subfamily G member 2
transcription through modulation of the AMPK/nuclear factor
erythroid 2-related factor 2 pathway. A study by Peng et al
(23) revealed that the
mechanistic axis glutamine hydrolysis/AMPK/succinate-CoA ligase
ADP-forming subunit-β/IL-1β regulates inflammation and obesity
progression in adipose tissue macrophages (ATMs). The study
demonstrated that glutamine hydrolysis in ATMs serves as a key
metabolic flux responsible for providing energy (ATP) and
biosynthetic precursors (for example, glutamate, aspartate and TCA
cycle intermediates) and that this process is associated with AMPK
activity, succinate-induced IL-1β production and the development of
obesity. Furthermore, Safaie et al (24) revealed a complex association
between AMPK activation and tyrosine kinase inhibitor-induced
cardiovascular toxicity, demonstrating that AMPK activation may
mitigate these adverse effects. This provides a foundation for
future therapeutic strategies and suggesting that AMPK activation
could be key in balancing effective cancer therapy with
cardiovascular health. Zhang et al (25) reported that succinate overload
enhances susceptibility to atrial fibrillation and remodeling by
impairing AMPK signaling and mitochondrial function.
Enzyme-catalyzed proximity labeling has emerged as a
new approach for studying the spatial distribution and interaction
characteristics of proteins in living cells. Existing proximity
labeling techniques (for example, using engineered ascorbate
peroxidase and BioID) are limited by long labeling times (>18 h)
(3), high reagent toxicity and
poor permeability. To overcome these limitations of
enzyme-catalyzed proximity labeling techniques, previous studies
have employed (9,10) a novel engineered biotin ligase,
TurboID, which can catalyze the conversion of biotin into
biotin-5'-AMP in vivo. This reactive intermediate enables
the efficient covalent labeling of proximal proteins, a method that
is non-toxic and fast (26).
TurboID can be fused to AMPK using a plasmid cloning technique,
enabling TurboID-biotinylated AMPK to be readily enriched using
streptavidin beads and its binding partners subsequently identified
using MS.
Carbonyl cyanide 3-chlorophenylhydrazone (CCCP) is a
potent mitochondrial oxidative phosphorylation uncoupling agent
that acts on the inner mitochondrial membrane to make it permeable
to H+, causing depolarization of the mitochondrial
membrane potential. This induces autophagy to clear the depolarized
mitochondria and further promotes the mitochondrial pathway of
apoptosis (27). CCCP induces DNA
damage, which in turn induces p53 expression (28). It has been shown that p53 activates
AMPK-dependent inhibitory pathways that attenuate the activity of
mTOR complex 1(23). Inhibition of
mitochondrial depolarization by the proton carrier CCCP facilitates
triggers mitophagy and induces apoptosis and autophagy (29). CCCP-induced loss of mitochondrial
membrane potential and mitochondrial dysfunction not only serve as
triggers for apoptosis but also act as key signals for initiating
mitophagy and broader adaptive stress responses. AMPK, as a central
kinase that senses cellular energy and redox status, serves a key
role in coordinating metabolism and survival, particularly in
responding to mitochondrial stress and initiating mitochondrial
quality control (28). DNAJ heat
shock protein family (Hsp40) member A1 (DNAJA1), as an Hsp70
co-chaperone, is important in maintaining mitochondrial
proteostasis, protein folding and translocation (30). Therefore, the present study
hypothesized that under CCCP-induced acute mitochondrial stress,
AMPK activation may regulate the function of DNAJA1 (or other
chaperones) through phosphorylation or other mechanisms, thereby
influencing mitochondrial protein handling and stress signaling.
This may offer a novel perspective for research in this field.
Therefore, in the present study, CCCP was selected
as an induction factor to treat U251 astrocytes stably
overexpressing the AMPK-TurboID fusion gene through lentiviral
infection, enabling investigation of the AMPK interactome under
energy-depleted conditions. The U251 cell line, derived from human
astrocytoma, is widely used as an in vitro model for
neurobiology research due to its well-characterized astrocytic
properties, high transfection efficiency, and stable proliferation
(31). Subsequently, a TurboID
proximity labeling technique was used to find novel interacting
proteins associated with AMPK, and label-free quantitative protein
profiling was used to obtain a number of interacting proteins.
Furthermore, DNAJA1 was selected for immunoprecipitation (IP) and
immunofluorescence validation. In addition, AMPK and DNAJA1 were
found to be jointly involved in anti-apoptotic cell death.
Therefore, the present study provided a theoretical basis to
explore the biological function of AMPK and the potential
development of new targeted drugs.
Materials and methods
Materials
Monoclonal antibodies against Flag (cat. no.
ab205606) and DNAJA1 (cat. no. ab192904), were purchased from
Abcam. AMPKα2 (cat. no. A7339), BAX (cat. no. A12009), Bcl-2 (cat.
no. A0208) and α-tubulin (cat. no. AC012) antibodies were purchased
from ABclonal Biotech Co., Ltd. The streptavidin-peroxidase
antibody (cat. no. SA00001-0) was obtained from Proteintech Group,
Inc. CCCP (cat. no. C6700) was purchased from Beijing Solarbio
Science & Technology Co., Ltd. PVDF membranes were obtained
from MilliporeSigma. Dynabeads™ MyOne™
streptavidin C1 (cat. no. 65001), biotin (cat. no. B20656) and
liposomal transfection reagent (Lipofectamine® 2000;
cat. no. 11668500) were obtained from Thermo Fisher Scientific,
Inc. RIPA buffer (cat. no. G2002-100ML), DAPI (cat. no.
G1012-100ML) and anti-fluorescence quenching sealing agent (cat.
no. G1401-25ML) were purchased from Wuhan Servicebio Technology
Co., Ltd. The anti-DYKDDDDK (Flag) affinity gel (cat. no.
20585ES03), lentivirus concentration solution (cat. no. 41101ES50)
and protein silver staining kit (cat. no. 36244ES25) were purchased
from Shanghai Yeasen Biotechnology Co., Ltd. A DNA purification kit
(cat. no. DP204) and a high purity plasmid extraction kit (cat. no.
DP104) were obtained from Tiangen Biotech Co., Ltd. Lastly,
inorganic salts were purchased from Sinopharm Chemical Reagent Co.,
Ltd.
Cell lines and culture
293T (human embryonic kidney epithelial) and U251
(malignant astrocyte) cell lines were obtained from American Type
Culture Collection. U251 and 293T cells were cultured in DMEM
(Gibco; Thermo Fisher Scientific, Inc.) containing 10% FBS (Gibco;
Thermo Fisher Scientific, Inc.) and 0.5% penicillin-streptomycin.
All cells were cultured in a CO2 incubator at 37˚C with
5% CO2.
Construction of AMPK-TurboID
overexpression vector
TurboID cDNA was synthesized and integrated into a
plenti-cytomegalovirus (CMV)-EGFP (Beijing Tsingke Biotech Co.,
Ltd.) plasmid replacing the EGFP fragment (plenti-CMV-TurboID).
This was conducted by Beijing Tsingke Biotech Co., Ltd. The AMPK
gene was amplified using human cDNA as a template and the following
primers: AMPK forward,
5'-atagaagacaccgactctagaATGGCTGAGAAGCAGAAGCAC-3'; AMPK reverse,
5'-catacgcgtgatatccccgggACGGGCTAAAGTAGTAATCAG-3' (the lowercase
letters indicate restriction enzyme sites and protective bases, and
the uppercase letters indicate the gene-specific sequences that
anneal to the template). The thermocycling conditions were as
follows: Initial denaturation at 94˚C for 5 min; followed by 30
cycles of denaturation at 94˚C for 30 sec, annealing at 56˚C for 30
sec and extension at 72˚C for 2 min; with a final extension at 72˚C
for 8 min. The plasmid plenti-CMV-TurboID was double digested using
XbaI and SmaI and the AMPK fragment was purified
using a DNA purification kit and combined with the linearized
plenti-CMV-TurboID plasmid. Positive clones were screened following
transformation into E. coli TOP10 cells and selection on
ampicillin (100 µg/ml)-containing LB agar plates. Plasmids were
extracted using a high purity plasmid extraction kit, and then the
extracted plasmids were processed through enzymatic digestion and
sent to Beijing Qingke Biotechnology Co., Ltd. for sequencing of
the insert. The selected plasmid was named AMPK-TurboID and was
tagged with Flag.
Lentiviral packaging of AMPK-TurboID
and plenti-CMV-EGFP plasmids
Firstly, 293T cells were cultured in a 10-cm dish
until their cell density reached 80-90% for transfection. At the
beginning of transfection, the plasmids were diluted with
serum-free high-glucose medium. A total of two 1.5 ml Eppendorf
(EP) tubes were prepared, one EP tube with 15 µg of psPAX2
(packaging plasmid), 6 µg of pMD2.G (envelope plasmid) and 10 µg of
AMPK-TurboID (transfer plasmid) and the other EP tube with 15 µg of
psPAX2, 6 µg of pMD2.G and 10 µg of plenti-CMV-EGFP (transfer
plasmid). Subsequently, an appropriate amount of liposomal
transfection reagent (~60 µl per 10-cm dish) was diluted with
serum-free medium and incubated at room temperature for 5 min.
Next, the diluted DNA plasmid and the diluted liposomal nucleic
acid transfection reagent were gently mixed and incubated at room
temperature for 30 min. Subsequently, the incubated DNA liposome
complexes were evenly dropped into 293T cell culture dishes that
had been replaced with pre-warmed complete high-glucose DMEM
(containing 10% FBS) without antibiotics DMEM and incubated
overnight at 37˚C in a cell culture incubator with 5%
CO2. The next day, the medium was replaced with fresh
medium and incubation continued for 48-72 h when the cell
supernatant was collected. The collected medium supernatant was
filtered by aspiration with a 5-ml syringe onto a 0.45-mm membrane
filter, after which the filtrate was mixed with lentivirus
concentration reagent in a 4:1 ratio and incubated overnight at
4˚C. After incubation, the supernatant was removed by
centrifugation (4˚C; 6,580 x g; 40-60 min) and fresh medium was
added to the sediment and dispensed into a number of 1.5 ml EP
tubes.
Construction and validation of AMPK
overexpression cell lines
U251 cells were grown in 10-cm cell culture dishes
and when cell confluence reached 80-90%, an appropriate number of
cells (2x105) were seeded in 24-well cell culture
plates. When the cell density reached 70-80%, lentivirus
concentrate carrying either the AMPK-TurboID plasmid or the
plenti-CMV-EGFP plasmid was dropwise added to different wells of
the plate, and the cells were infected (multiplicity of infection
of 10) for 6-8 h at 37˚C and replaced with fresh medium. The
fluorescence intensity of plenti-CMV-EGFP plasmid-infected cell
culture plates was visualized using an inverted fluorescence
microscope. after 48 h. Successful infection was demonstrated when
stronger fluorescence appeared. Resistance screening was performed
with medium containing puromycin (1.5 µg/ml) and cell status was
observed after 24-48 h. Maintenance was performed in 0.5 µg/ml
puromycin. Viable cells were cultured into 12-well plates and
sequentially passaged to the number of cells that met the
requirements of subsequent experiments. The time interval from
transduction to subsequent experiments was ~2 weeks. During this
period, the cell status was observed daily and medium was renewed
once every 2 days. A number of cells were used to obtain cell
lysate for SDS-PAGE gel electrophoresis and determine whether
stable transfection was successful. Cells stably transfected with
the plenti-CMV-EGFP plasmid were named EGFP-U251 cells and cells
stably transfected with the AMPK-TurboID plasmid were named
AMPK-U251 cells.
Screening of AMPK biotin labeling
time
AMPK-U251 cells were grown into 6-well culture
plates and experiments were performed when their density reached
~80%. Cell medium was replaced with complete medium containing
biotin (500 µmol/l), magnesium chloride (MgCl2; 1
µmol/l) and ATP (200 µmol/l) and incubated at 37˚C, and seven
labeling time gradients were set, namely 0 and 10 min, 1, 3, 6, 12
and 24 h. Cells were collected, cell lysis was performed and cell
lysates were subjected to western blotting using a
streptavidin-peroxidase antibody.
Western blotting
To confirm successful stable transfection,
whole-cell lysates were prepared from the generated EGFP-U251 and
AMPK-U251 cells, as well as control U251 cells. Cells were washed
twice with ice-cold PBS and lysed in RIPA lysis buffer (cat. no.
P0013B; Beyotime Biotechnology) supplemented with a protease
inhibitor cocktail on ice for 30 min. The lysates were clarified by
centrifugation at 12,000 x g for 20 min at 4˚C. Protein
concentrations were determined using an Enhanced BCA Protein Assay
Kit (cat. no. P0009; Beyotime Biotechnology). Equal amounts of
protein (10 µg per lane) were denatured in 5X SDS-PAGE loading
buffer at 95˚C for 10 min. The samples were then separated by
SDS-PAGE on a 10% polyacrylamide gel at 120 V for 1.5 h. Following
electrophoresis, the proteins were transferred onto a PVDF membrane
(cat. no. IPVH00010; MilliporeSigma) using a wet transfer system at
250 mA for 150 min at 4˚C. The membrane was blocked with Quick
Block™ Blocking Buffer (Beyotime Biotechnology) for
15-30 min at room temperature. Subsequently, the membrane was
incubated overnight at 4˚C with primary antibodies diluted in
blocking buffer. The following primary antibodies were used: Rabbit
anti-AMPK polyclonal antibody (cat. no. 10929-2-AP; Proteintech
Group, Inc.) and rabbit anti-FLAG polyclonal antibody (cat. no.
80801-2-RR; Proteintech Group, Inc.), both diluted at 1:1,000.
GAPDH was used as a loading control. After washing three times with
TBST (10 min each), the membrane was incubated with the
HRP-conjugated goat anti-rabbit IgG secondary antibody (cat. no.
SA00001-2; diluted at 1:5,000; Proteintech Group, Inc.) for 1 h at
room temperature. Protein bands were visualized using an enhanced
chemiluminescence detection reagent (cat. no. BL523A; Biosharp Life
Sciences). The signals were captured using a chemiluminescence
imaging system (ChemiDoc™ MP Imaging System; Bio-Rad
Laboratories, Inc.). Densitometric analysis of the bands was
performed using ImageJ software (Version 1.53, National Institutes
of Health, Bethesda, MD, USA).
Effect of adding ATP on biotin
labeling
AMPK-U251 cells were cultured into 6-well culture
plates and left to reach a density of ~70-80% for subsequent
experiments. The original culture medium was discarded and replaced
with biotin-labeling buffer containing five varying concentrations
(0, 50, 100, 200 and 500 µmol/l; 37˚C) after two washes with PBS,
in order to collect sufficient labeled protein and prevent
oversaturation of labeling efficiency. The biotin labeling buffer
was composed of complete medium containing biotin (500 µmol/l),
MgCl2 (1 µmol/l) and ATP (200 µmol/l) and biotin
labeling buffer without ATP had only the ATP component removed.
Cells were collected, cell lysis was performed and lysates were
subjected to western blotting using a streptavidin-peroxidase
antibody.
TurboID-based neighboring biotin
labeling technology
This method was performed with reference to that
previously reported by Branon et al (9). The wild-type U251 cell line and
AMPK-U251 cell line were grown in T75 cell culture flasks and the
experiment was set up in three groups: i) Blank group, two T75
culture flasks for the wild-type U251 cell line; ii) experimental
group I, two T75 culture flasks for the AMPK-U251 cell line; and
iii) experimental group II, two T75 culture flasks for the
AMPK-U251 cell line treated with 15 µmol/l CCCP. All three groups
of cells were cultured using DMEM medium containing biotin (500
µmol/l) and MgCl2 (1 µmol/l) at 37˚C, and the cells were collected
after 6 h for sample preparation.
Affinity purification of
biotin-labeled proteins
Cells in the T75 cell culture flasks were collected
by centrifugation (11,200 x g for 15 min at 4˚C) into 1.5-ml
centrifuge tubes, resuspended by adding 1 ml of RIPA buffer
(containing PMSF), and placed on ice for ~20 min for lysis. In
parallel, the Dynabeads were centrifuged at 11,200 x g for 15 min
at 4˚C. The supernatant was transferred to a 1.5-ml centrifuge
tube. Subsequently, the streptavidin-coated magnetic beads were
washed with 1 ml RIPA buffer for ~2 min at 37˚C, the supernatant
was clarified by adsorption on a magnetic stand, RIPA buffer was
discarded and washing was repeated 5 times at 37˚C. After the last
wash, the beads were divided into three equal parts (200 µl per
aliquot), and after precipitation on a magnetic stand the RIPA
buffer was discarded. The protein lysate (1 ml) was added to the
beads separately, placed at 4˚C and shaken slowly overnight using a
360˚ shaker. The next day, the supernatant was discarded and the
samples were washed once with 1 ml potassium chloride, three times
with Buffer 1 [3M NaCl 33.5 ml, 1M Tris-HCl (pH 7.4) 0.5 and 16 ml
Ultrapure Water] configured in advance, once with 1 ml sodium
carbonate buffer and then 2 min (maximum 3 min) with 1 ml 10% SDS,
after which 1 ml RIPA buffer was added and soaked continuously for
1 min at 50˚C in a metal bath.
Subsequently, one-third of each sample was removed
and processed for silver staining, while the rest of the samples
were washed twice with RIPA buffer and PBS, then stored at -20˚C.
Cells were counted and lysed at equal cell densities. Protein
samples (10 µg per lane) were separated by 10% SDS-PAGE. Subsequent
experiments were performed using a silver staining kit (fixative,
sensitizing, silver staining, development and termination
solutions) following the manufacturer's protocol. the gel was
removed and placed in 100 ml fixative on a 60-70 rpm shaker for 20
min. The fixative was discarded and placed in 100 ml 30% ethanol on
a slow shaker for 10 min. The fixative was discarded and 100 ml 30%
ethanol was added with the samples shaken slowly for 10 min. The
ethanol was then discarded, 200 ml deionized water was added and
shaken slowly for 10 min. The water was replaced with 100 ml 1X
sensitizing solution and shaken for 4-5 min. The sensitizing
solution was replaced with 200 ml deionized water and shaken for 1
min, the process was repeated and the water discarded. A total of
100 ml 1X silver staining solution was added and shaken for 30-40
min. Then, 100 ml deionized water was added, shaken for 30 sec,
repeated once and then the water was discarded. Subsequently, 100
ml color development solution was added and shaken until bands
appeared (3-10 min). The color development solution was replaced
with 100 ml termination solution and shaken for 10 min. The
termination solution was discarded, 100 ml deionized water was
added and shaken for 30 sec, repeated once and the water discarded.
Images were captured with a camera. All incubations and washes were
performed at room temperature.
Liquid chromatography-tandem MS
analysis
The remaining two-thirds of the protein samples were
sent to Spectrum Zhonghe (Wuhan) Life Science Technology Co., Ltd.,
for label-free quantitative MS (32). The LC-MS/MS analysis was performed
using an Orbitrap Exploris 480 mass spectrometer coupled with an
EASY-nLC 1200 liquid chromatography system (both Thermo Fisher
Scientific, Inc.). Ionization was carried out in positive
electrospray ionization (ESI+) mode with a spray voltage of 2,000
V. The mass spectrometer was operated in data-dependent acquisition
mode, acquiring full MS scans over a mass range of 350-1,250 m/z at
a resolution of 15,000, followed by higher-energy collisional
dissociation (HCD) fragmentation with a normalized collision energy
of 28. The ion transfer tube temperature was set to 320˚C, and the
nebulizer (sheath gas) pressure and auxiliary gas flow rate were
both set to 3 arbitrary units (these values are not directly
provided in psi or l/min, as they are controlled by the instrument
software). No multiple reaction monitoring transitions were
assessed in this study, as the analysis was performed using
label-free quantification. Enrichment analysis was performed on the
data obtained. The online analysis tool Sangerbox (version 3.0;
http://vip.sangerbox.com/home.html)
was used for Gene Ontology (GO) and Kyoto Encyclopedia of Genes and
Genomes (KEGG) analysis. Significantly changed proteins were
identified using a threshold of FDR <0.05 and |log2FC|>1. A
total of 1,045 interacting proteins in the AMPK overexpression
group were selected for analysis.
Validation of protein interactions
between DNAJA1 and AMPK
Protein interactions were initially verified using a
co-IP assay. Wild-type U251 cells and AMPK-U251 cells were
collected into EP tubes and labeled as A1 and B1, respectively. To
tubes A1 and B1, 1 ml of IP lysis buffer and 10 µl of PMSF were
added. The cells were then gently pipetted to disperse and placed
on a rotator at 4˚C for 30 min for lysis. The lysed cells were
centrifuged in a refrigerated centrifuge at 4˚C and 11,200 x g for
10 min, and protein concentrations were determined using a BCA
assay. Two additional EP tubes were labeled as A2 and B2, and 50 µl
of Anti-DYKDDDDK magnetic beads were added to each tube.
Subsequently, 1 ml of IP lysis buffer was added to tubes A2 and B2,
and the tubes were placed on a rotator at 4˚C for 10 min to wash
away impurities; this washing step was repeated twice. After
discarding the IP lysis buffer from tubes A2 and B2, 900 µl of the
protein lysate from tube A1 was transferred to tube A2, and the
same procedure was performed for tube B1 using lysate from tube B1.
Tubes A2 and B2 containing the lysate-bead mixtures were placed on
a rotator at 4˚C and incubated overnight. The next day, tubes A2
and B2 were removed, the original solution was discarded, and 1 ml
of PBS was added. The tubes were placed on a rotator at 4˚C for 10
min to wash; this washing step was repeated twice. After discarding
the PBS from tubes A2 and B2, an appropriate amount of 1X loading
buffer was added. An appropriate amount of loading buffer was also
added to tubes A1 and B1. All four EP tubes were placed in a thermo
mixer at 100˚C for 10 min to denature the proteins. Finally,
SDS-PAGE gel electrophoresis was performed.
In a second step, AMPK-TurboID cells
(5x104 cells/cm2) were seeded in a 3.5-cm
cell confocal dish, the medium was discarded when the density
reached 80%, PBS added to wash the cells for 3 min and washing was
repeated twice. Subsequently, 4% paraformaldehyde was added (37˚C)
for 15 min to fix the cells in the dish and then discarded. PBS was
added for 3 min and discarded, and washing was repeated twice.
Next, 0.5% Triton X-100 was added for 20 min at room temperature
and then PBS was added for 3 min and repeated twice. PBS was
absorbed from the dish with absorbent paper and 200 µl of normal
goat serum (cat. no. AR0009; Wuhan Boster Biological Technology,
Ltd.) was added dropwise and incubated at room temperature for 30
min for blocking. The solution was removed with absorbent paper,
and a primary antibody solution (2.5 µl DNAJA1 primary antibody;
1:200 dilution; 1 µl Flag; 1:500 dilution primary antibody and 500
µl primary antibody dilution) was added and incubated at 4˚C
overnight. The cells were washed for 3 min with PBS twice and PBS
was removed with absorbent paper. The secondary antibodies,
Cy3-conjugated goat anti-mouse IgG (1:250 dilution; cat. no. A0516;
Beyotime Biotechnology) and FITC-conjugated goat anti-mouse IgG
(1:250 dilution; cat. no. A0568; Beyotime Biotechnology), were
diluted in 250 µl of PBS and added to the cells. After incubation
at room temperature for 1 h in the dark, the cells were incubated
with DAPI (1 µg/ml; 37˚C) for 5 min and washed three times with PBS
for 5 min. After absorbing the PBS with blotting paper, the slides
were sealed with anti-fluorescent quenching agent and cells were
observed under a confocal Olympus FV3000 microscope (Olympus
Corporation).
Statistical analysis
Data analysis and graphing were conducted with
GraphPad Prism (version 10.1.2; Dotmatics). Each group of data as
the mean ± SD from three experimental replicates. Comparisons among
groups were performed using one-way ANOVA followed by Tukey's
honestly significant difference test. P<0.05 was considered to
indicate a statistically significant difference.
Results
Construction of overexpression
plasmids and TurboID tagging flow chart
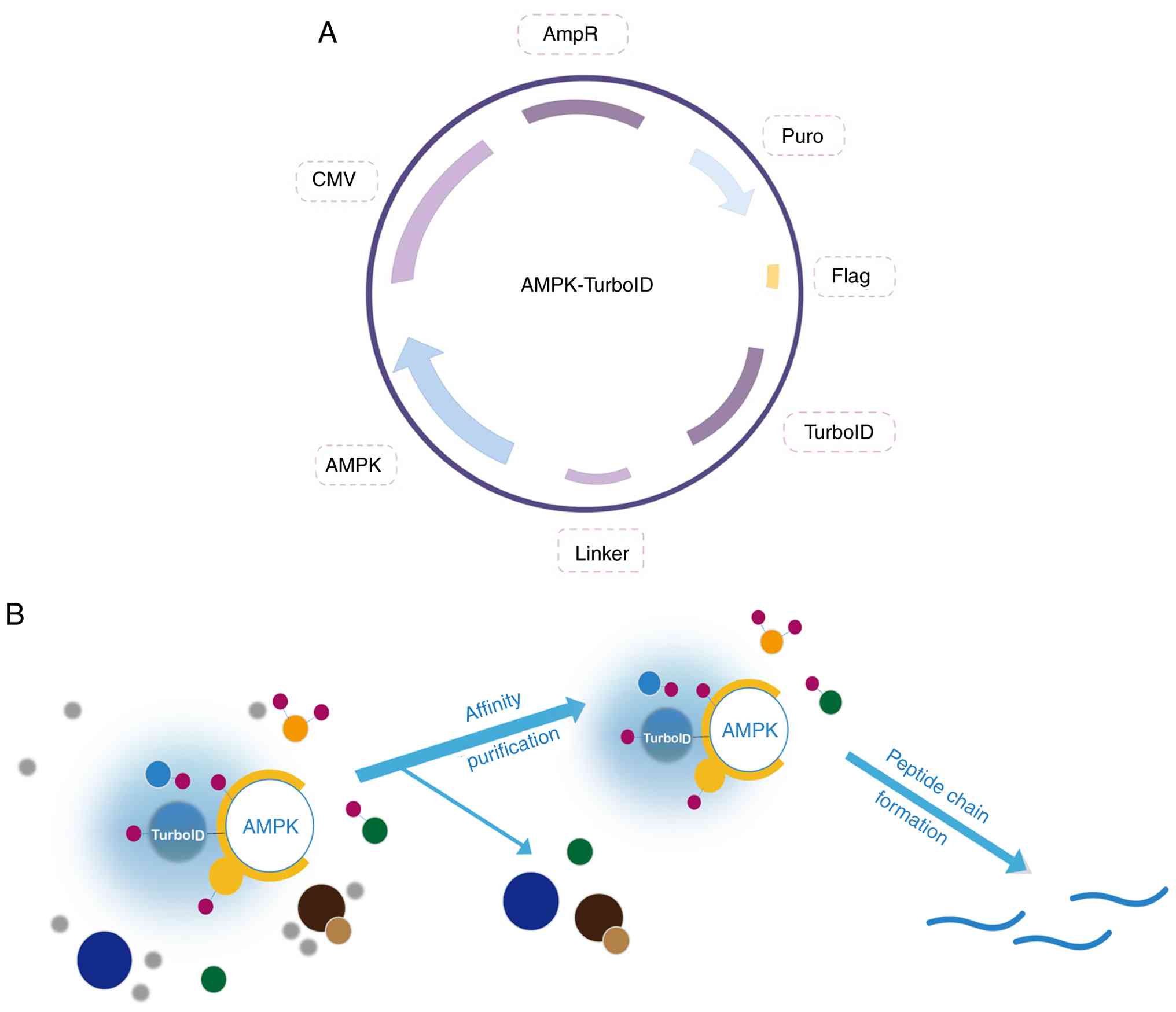
Fig. 1A shows the
composition of the AMPK-TurboID overexpression plasmid, including
the target gene AMPK, the biotin labeling enzyme TurboID and the
tag protein Flag, as well as two different resistance screening
markers, ampicillin and puromycin, whereby ampicillin was used for
the screening of positive clones in Esherichia coli and
puromycin was used for the screening of stably transfected cells.
Fig. 1B illustrates the biotin
labeling experiment. First, AMPK-U251 cells that were successfully
and stably transfected were placed in a biotin environment with a
suitable concentration, so that free biotin could fully enter the
cells. Adjacent proteins were covalently labeled with biotin by
TurboID. After the protein affinity purification experiment, other
proteins that were not labeled with biotin were excluded and the
proteins that had been labeled with biotin were enriched. Lastly,
the labeled proteins were analyzed by label-free quantitative
MS.
Construction of AMPK-TurboID
overexpression stable cell line
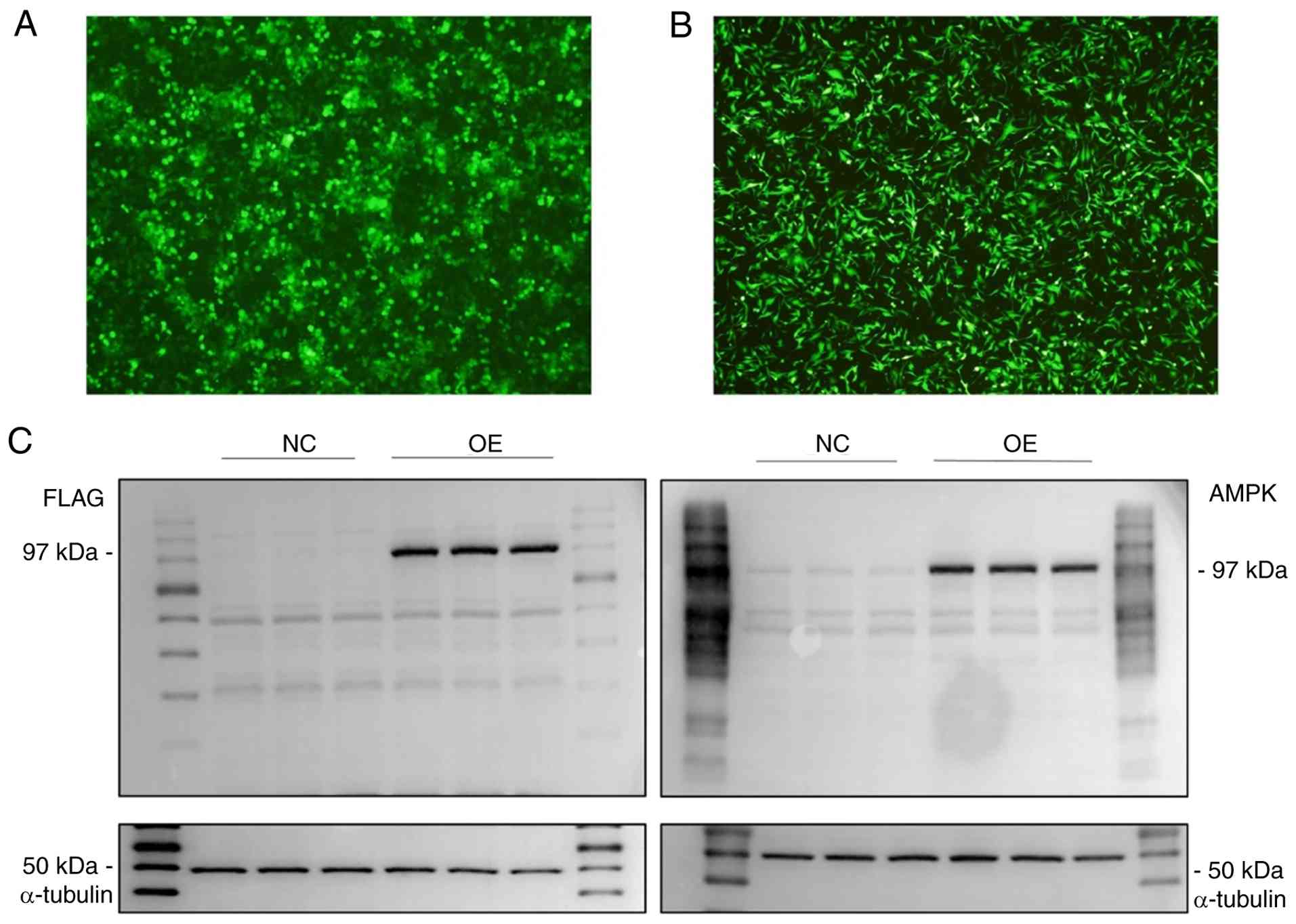
As shown in Fig.
2A, a large amount of fluorescence was observed under the
microscope in plenti-CMV-EGFP-transfected 293T cells after 48 h,
determining that 293T cells had been successfully transfected and
the viral fluid supernatant could be collected to infect cells.
Subsequently, as demonstrated in Fig.
2B, U251 cells infected with the lentivirus carrying the
plenti-CMV-EGFP construct for 48 h exhibited a large amount of
fluorescence, implying that the concurrent AMPK-TurboID gene was
also effectively infected into U251 cells with high
probability.
Validation of AMPK-TurboID
overexpression in stably transduced cell lines
Comparative analysis of stably transduced
AMPK-TurboID and TurboID-U251 cell lines was conducted through
western blotting. Western blotting was performed with both an AMPK
antibody and a Flag antibody (Fig.
2C). The results revealed that the AMPK-TurboID fusion protein
showed a uniform and clear band at a molecular weight of ~97 kDa
(molecular weight of AMPK, 62.3 kDa; molecular weight of TurboID,
35 kDa; Flag size was neglected), while the corresponding protein
region in the TurboID-U251 cell line was blank, indicating that the
stable cell line was successfully constructed.
Biotin labeling and silver
staining
The results of the biotin labeling assay showed that
interacting proteins of AMPK appeared after 10 min and more
markedly after 1 h of labeling (Fig.
3A). To prevent oversaturation of biotin labeling, 6 h was used
as the biotin labeling time of AMPK-U251 cells in the subsequent
experiment. The results showed that there was some improvement in
biotin labeling efficiency with the addition of ATP, however the
effect was limited (groups 4-6; Fig.
3B). Considering the possible effect of the addition of ATP on
the activity of AMPK, ATP was not added to the subsequent
biotin-labeling buffers in the present study.
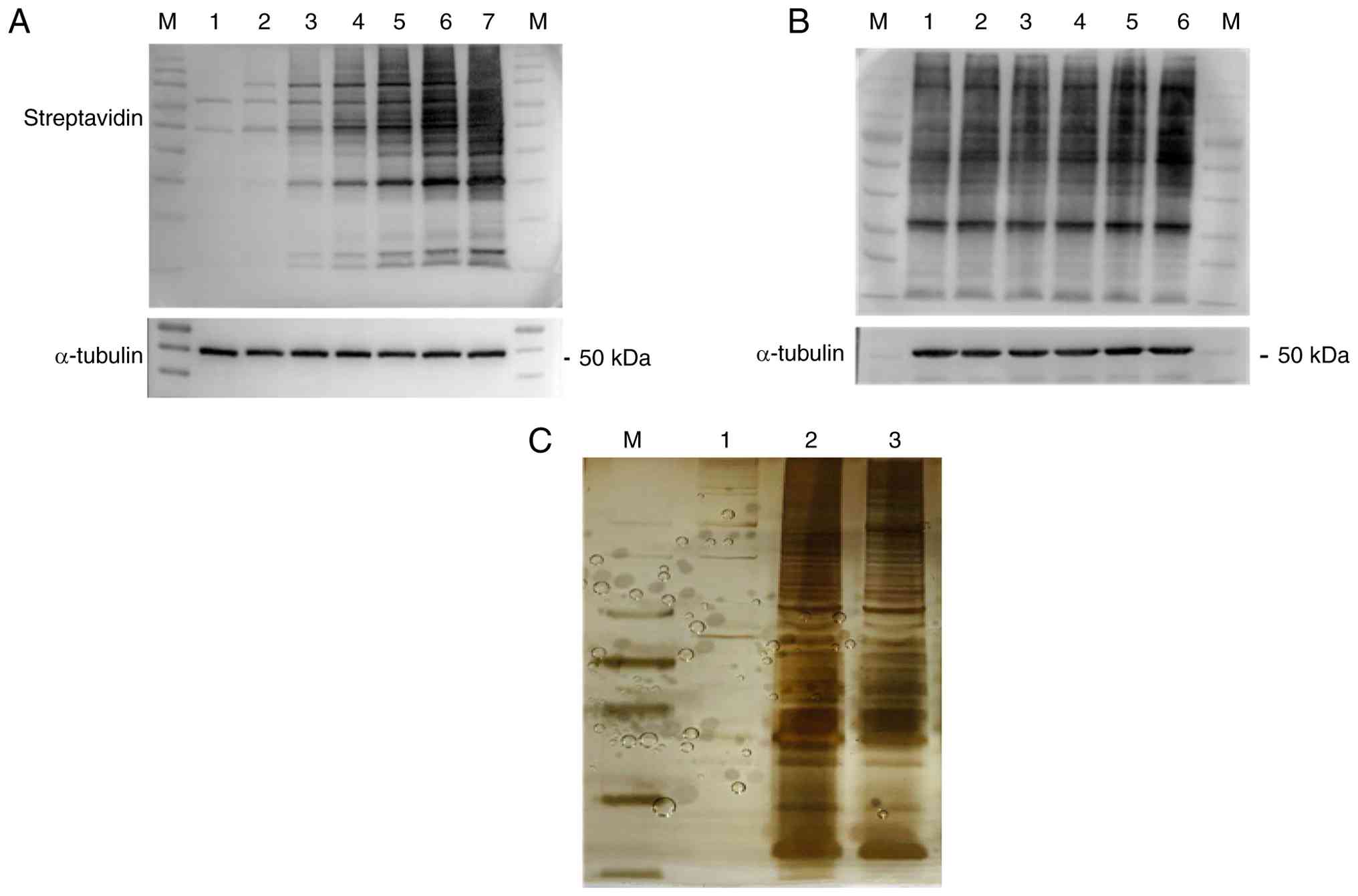 | Figure 3Biotin labeling and silver staining.
(A) Biotin labeling time screening. 1, 0 min; 2, 10 min; 3, 1 h; 4,
3 h; 5, 6 h; 6, 12 h; and 7, 24 h. (B) Effect of adding ATP on
biotin labeling. 1, 2, 3 indicate repeats without ATP added; 4, 5,
6 indicate repeat groups with ATP added. (C) Identification by
silver staining. 1, U251; 2, AMPK-U251; 3, AMPK-U251 cells treated
with carbonyl cyanide 3-chlorophenylhydrazone. M, molecular weight
standard; AMPK, AMP-activated protein kinase. |
Subsequently, the U251 and AMPK-U251 cell lines
underwent affinity purification and silver staining. As shown in
Fig. 3C, U251 as a negative
control pulled only a limited number of proteins. Since U251 cells
do not express TurboID, they theoretically should not enrich
biotin-labeled proteins; therefore, the few protein bands observed
likely represent non-specific binding or experimental background
noise. Therefore these data were excluded. The remaining two sets
of data indicated that AMPK overexpressing cell lines pulled a
higher number of biotin-interacting proteins.
MS data analysis
Analysis of the MS data revealed a total of 1,808
non-redundant interacting proteins identified by proximity labeling
across all groups after removing duplicates, with 329 proteins
identified in the U251 blank group and 1,045 proteins identified in
the AMPK overexpression group and 1,705 proteins in the
CCCP-treated group. Furthermore, after excluding 129 proteins that
were also identified in the blank group, 916 interacting proteins
remained (Table SI). In addition,
1,705 proteins were identified in the CCCP-treated group and after
excluding 296 proteins that were also identified in the blank
group, 1,409 interacting proteins remained (Table SII).
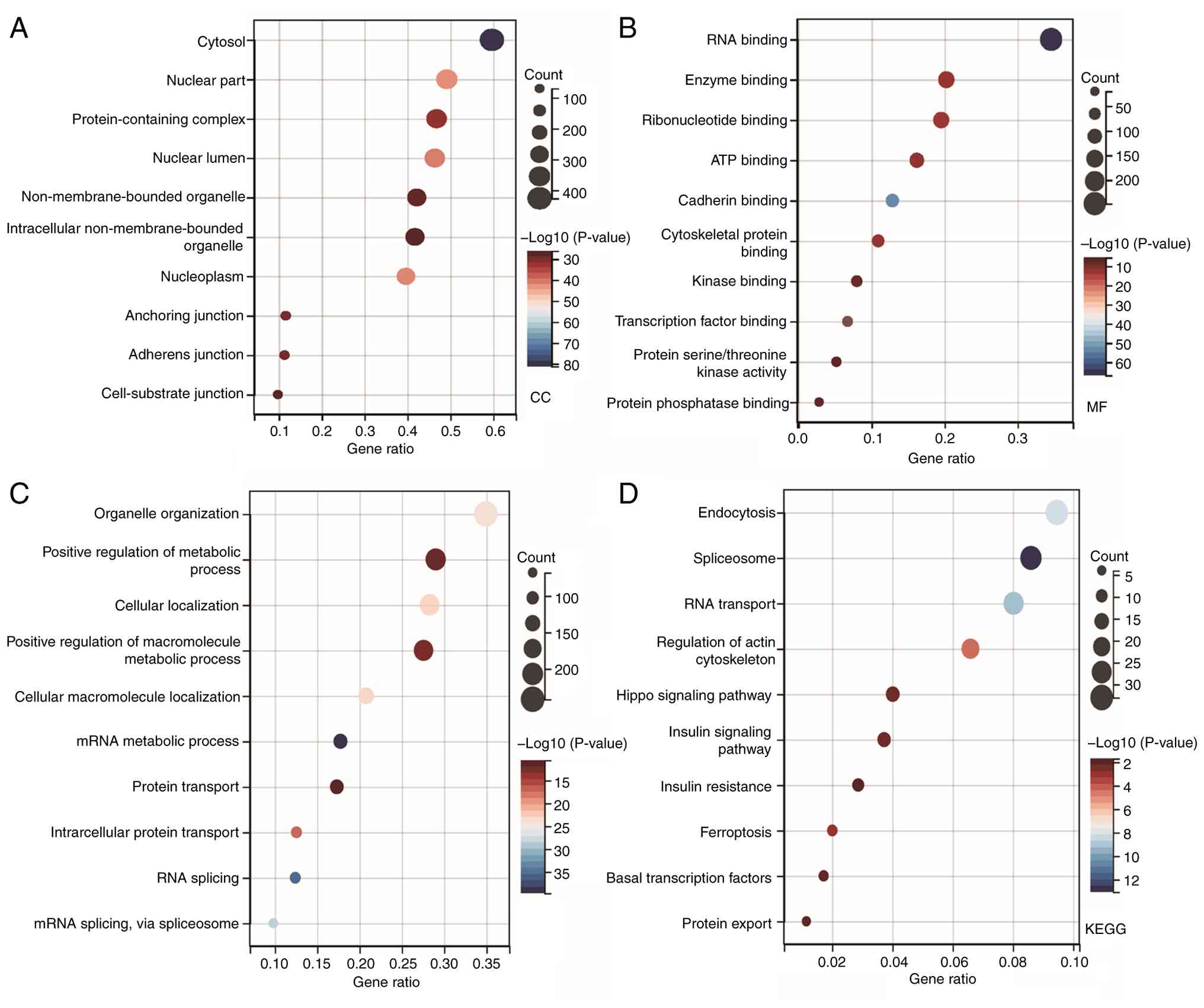
The online analysis tool Sangerbox 3.0 was used for
GO and KEGG analysis. A total of 1,045 interacting proteins in the
AMPK overexpression group were selected for analysis. In the
analysis of cellular components, the terms ‘cytosol’, ‘nuclear
part’, ‘protein-containing complex’, ‘nuclear lumen’ and
‘non-membrane-bounded organelle’ were enriched (Fig. 4A). In the analysis of molecular
functions, the search revealed enrichment of ‘RNA binding’, ‘enzyme
binding’, ‘ribonucleotide binding’ and ‘ATP binding’ (Fig. 4B). The enrichment of ‘ATP binding’
reflects AMPK's function as a cellular energy sensor, while ‘RNA
binding’ suggests a potential role in post-transcriptional
regulation. In addition, a number of significantly enriched terms
were identified in the analysis of biological processes, including
‘organelle organization’, ‘positive regulation of metabolic
process’, ‘cellular localization’, ‘positive regulation of
macromolecule metabolic process’ and ‘cellular macromolecule
localization’ (Fig. 4C). These
terms directly reflect AMPK's central role in coordinating
metabolism and organelle dynamics. In the KEGG analysis, pathways
that were found to be enriched included ‘endocytosis’,
‘spliceosome’, ‘RNA transport’, ‘regulation of actin cytoskeleton’,
‘Hippo signaling pathway’, ‘insulin signaling pathway’, ‘insulin
resistance’ and ‘ferroptosis’ (Fig.
4D).
DNAJA1 interacts with AMPK
MS data revealed that DNAJA1 is an interaction
partner of AMPK. DNAJA1, a key protein in the protection against
apoptosis, acts as a co-chaperone protein for heat shock protein
family A member 1B and negatively regulates the transport of Bax
from the cytoplasm to the mitochondria under conditions of cellular
stress (33). Co-IP assays
revealed that DNAJA1 interacts with AMPK. As shown in Fig. 5A, AMPK may specifically precipitate
DNAJA1.
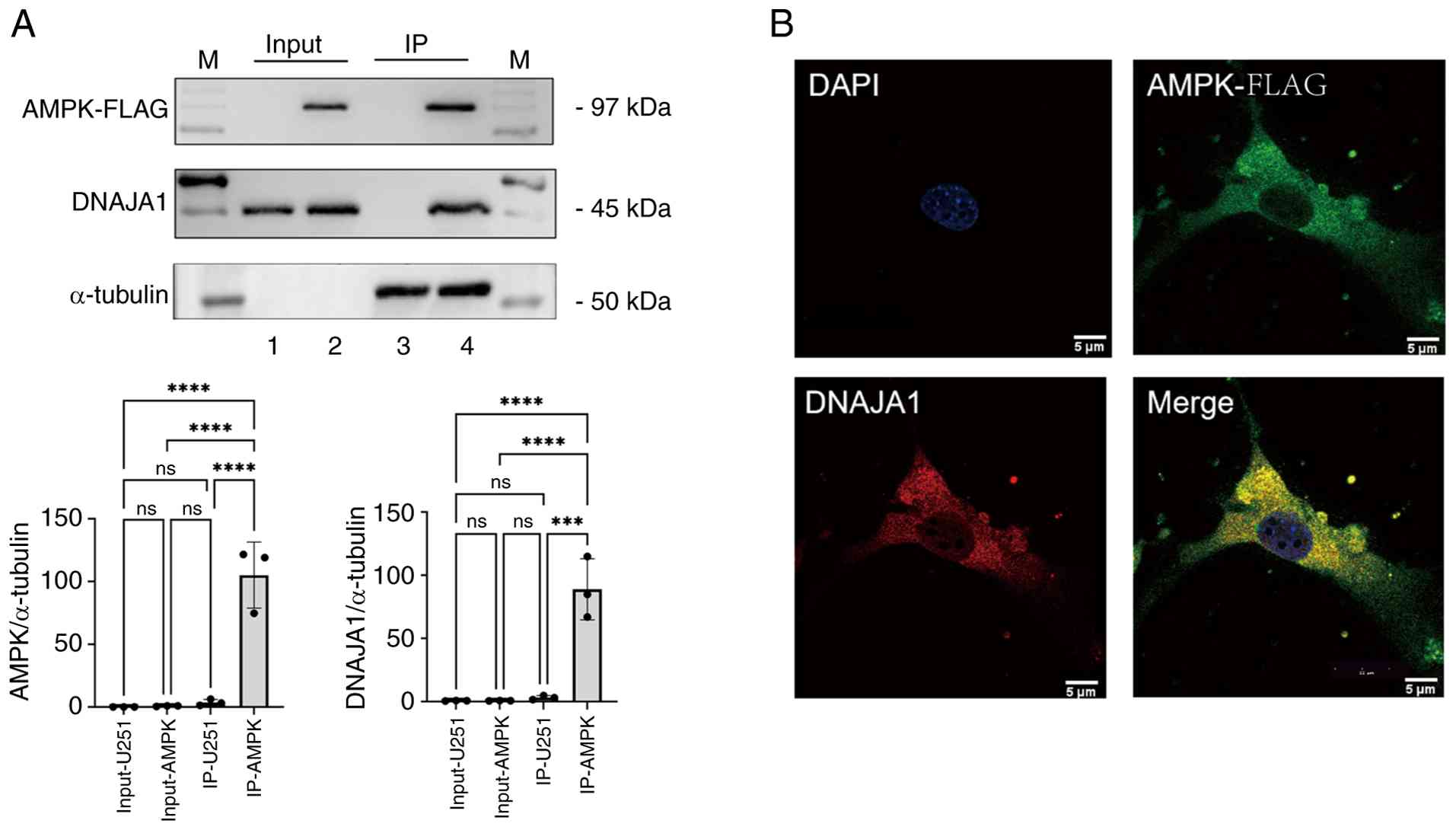 | Figure 5Validation of the interaction between
AMPK and DNAJA1. (A) Co-IP was used to verify the interaction
between AMPK and DNAJA1. 1, Input of U251 cell line; 2, input of
AMPK overexpressing cell line; 3, co-IP eluate of U251 cell line
after Flag-magnetic beads affinity adsorption; and 4, co-IP eluate
of AMPK overexpressing cell line after Flag-magnetic beads affinity
adsorption. (B) Immunofluorescence validation of the interaction
between AMPK and DNAJA1. ***P<0.001,
****P<0.0001; ns, not significant. AMPK,
AMP-activated protein kinase; DNAJA1, DNAJ heat shock protein
family (Hsp40) member A1; IP, immunoprecipitation; ns, not
significant; M, molecular weight standard. |
Immunofluorescence detection of the localization of
AMPK and DNAJA1 in U251 cells further provided a basis for the
interaction between AMPK and DNAJA1. Fig. 5B shows that both AMPK and DNAJA1
were localized in the cytoplasm, providing a spatial and temporal
basis to further determine the interactions between AMPK and
DNAJA1.
AMPK interacts with DNAJA1 to
participate in anti-apoptosis
It has previously been established (33) that DNAJA1 is involved in the
negative regulation of Bax and thus participates in the
anti-apoptotic process. The present study demonstrated the
existence of an interaction between AMPK and DNAJA1 and therefore
hypothesized that AMPK may interact with DNAJA1 to participate in
the anti-apoptotic pathway of cells. As shown in Fig. 6, the expression levels of DNAJA1
were showed a slight non-significant increase and the expression
level of Bax was decreased in the AMPK overexpressing cell line
compared with those in the U251 cell line, while Bcl2 expression
showed no significant change, indicating that the apoptotic process
may be attenuated in AMPK overexpressing cells. CCCP is a
mitochondrial uncoupler that disrupts mitochondrial membrane
potential and depletes cellular ATP levels, thereby inhibiting AMPK
activity (34). The CCCP-treated
AMPK-TurboID overexpressing cell line exhibited significantly
weakened DNAJA1 and Bcl2 levels but enhanced Bax levels compared
with those in the AMPK overexpressing cells without CCCP
treatment.
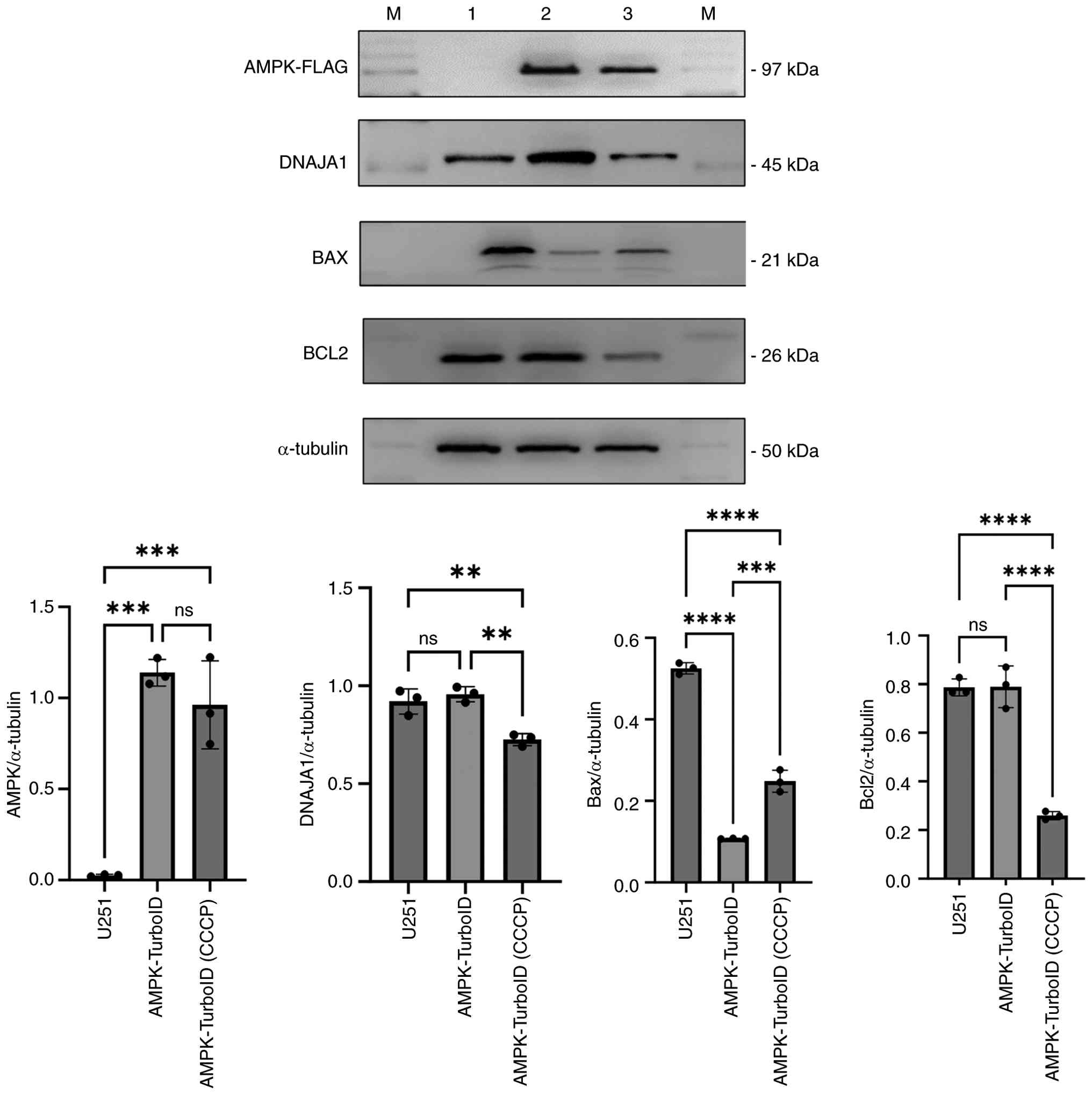 | Figure 6Western blotting validation of the
interaction between AMPK and DNAJA1 in the anti-apoptosis pathway.
1, U251; 2, AMPK-U251; 3, AMPK-U251 cells treated with CCCP; AMPK,
AMP-activated protein kinase; DNAJA1, DNAJ heat shock protein
family (Hsp40) member A1; CCCP, carbonyl cyanide
3-chlorophenylhydrazone; ns, not significant; M, molecular weight
standard. **P<0.01, ***P<0.001,
****P<0.0001; ns, not significant. |
Discussion
Proteins are not only the material basis of life
activities but also important components of cells, and are one of
the primary regulators of physiological functions and metabolism in
the human body. Proteins act in two main forms, either alone or in
specific complexes with other proteins or chaperone molecules
(35,36). The study of protein interactions is
an important aspect of protein functional study (37). Based on the concept that
interacting proteins must be in proximity to each other, proximity
labeling techniques have gradually become an emerging research
topic. Among them, TurboID stands out among the proximity labeling
techniques due to its advantages of high catalytic activity, fast
labeling, no in vivo toxicity and easy operation. The
principle of action involves catalyzing the biotinylation of the
fused protein and target proteins in a proximity-dependent manner
(38). In order to consistently
and stably express the target gene and study protein interactions,
a stably transfected cell line was constructed in the present
study. Stable overexpression of genes can overcome the disadvantage
of relatively limited and uncontrollable transient overexpression
(39) and increase the
reproducibility of experimental results.
With regard to further advancements in MS and
proteomics, Song et al (40) identified four potential protein
biomarkers in nasal swabs for the diagnosis of coronavirus
disease-19 by combining matrix-assisted laser desorption/ionization
time-of-flight MS analysis with top-down proteomics techniques.
Xiao et al (41), developed
an innovative research framework by integrating targeted proteomics
and metabolomics technologies, enabling the screening of potential
biomarkers for the early detection of hepatocellular carcinoma. The
parallel-flow projection and transfer learning across omics data
method proposed by Hu et al (42) integrates microfluidics with deep
learning, enabling deep and high-resolution spatial localization of
thousands of proteins across intact tissue sections. Since relying
solely on mRNA measurements risks missing important biological
information, Furtwängler et al (43) utilized recent advances in
single-cell proteomics by MS to generate an in vivo
differentiation hierarchy dataset of >2,500 human
CD34+ hematopoietic stem and progenitor cells. Direct
protein-level analysis is essential for comprehensive biological
insights. In the present study, a similar MS-based proteomics
approach, combined with TurboID proximity labeling, was employed to
screen for AMPK-interacting proteins in U251 cells.
The AMPK signaling pathway is also an important
research avenue. AMPK trimeric serine/threonine protein kinase is
composed of a catalytic subunit (α) and two regulatory subunits (β
and γ) (44), which form an enzyme
regulating the general switch of cellular metabolism and
maintaining the balance of nutrient supply and energy demand in
vivo (45). Studying the
interaction between AMPK and other proteins is an important step in
understanding its function. It has been shown that AMPK is
associated with mitochondrial fission factor (MFF), which can
promote AMPK phosphorylation and activate mitochondrial fission. In
2022, Peng et al (46)
demonstrated that high osmotic pressure-induced energy stress can
activate the AMPK/MFF pathway, which subsequently regulates
mitochondrial fission and mitophagy, contributing to the
development of dry eye. Receptor-interacting protein kinase 1
(RIPK1) is a key factor in mediating cell inflammation and cell
death. Zhang et al (47)
found that AMPK may inhibit RIPK1 activation through
phosphorylation at Ser415, suppressing energy stress-induced cell
death. These findings provide important insights regarding the
development of potential therapeutic drugs aimed at preventing
ischemia-induced cell death and tissue damage. Penfluridol can
activate the AMPK/FOXO3a/BIM signaling pathway and inhibit
glycolysis-induced apoptosis, thereby inhibiting tumor formation.
Zheng et al (48) found
that penfluridol markedly inhibited the growth of patient-derived
xenograft (PDX) tumors and the therapeutic effect of penfluridol
was associated with the expression of phosphofructokinase, liver
type (PFKL). PDX was subcutaneously inoculated with esophageal
squamous cell carcinoma in mice, and it was found that penfluridol
targeted PFKL-inhibited glycolysis and inhibited the occurrence of
esophageal cancer in an AMPK/FOXO3a/BIM-dependent manner. In
addition, AMPK also participates in numerous physiological
processes, such as cell proliferation, metabolism (49), autophagy (50) and apoptosis (51).
Despite U251 cells being astrocytes with a metabolic
environment different from that of liver, muscle, heart or
pancreatic cells, they may be still intricately associated with
systemic diseases, such as diabetes. Han et al (52) found that the an abnormally high
level of D-ribose was detected in the urine of type 2 diabetic
patients, accelerated the formation of advanced glycation
end-products (AGEs) in U251 and U87MG cells as well as in mouse
brains, and induced the upregulation of the receptor for AGEs
(RAGE). The results indicated that D-ribose-derived AGEs induced
spatial cognitive impairment in mice, which was associated with the
activation of RAGE-dependent inflammatory responses mediated by
astrocytes. The present study focused on the screening of
AMPK-interacting proteins. The human U251 astrocytoma cell line was
used for cell model establishment, which exhibits a high efficiency
of lentiviral infection (53,54),
and resulted in the successful establishment of a stable cell line
expressing the AMPK-TurboID fusion protein. This provided a solid
foundation for subsequent proximity labeling and proteomic
analyses. Subsequently, biotin labeling experiments were conducted
and a number of proteins interacting with AMPK were found. Finally,
AMPK-interacting proteins were screened by label-free MS, with GO
and KEGG enrichment analysis performed on the AMPK overexpression
group.
Apoptosis is a fundamental process. AMPK, a key
cellular energy sensor, and DNAJA1, a co-chaperone protein, have
both been implicated in the regulation of apoptosis, although their
potential interplay in this process remains unclear. DNAJA1 can
negatively regulate the pro-apoptotic protein Bax by inhibiting its
translocation from the cytoplasm to the mitochondria during
cellular stress, thus participating in the anti-apoptotic process
(33). AMPK activates NF-κB to
increase the expression of the anti-apoptotic protein Bcl2, thereby
promoting cell survival and inhibiting apoptosis (41). The present study demonstrated the
existence of an interaction between AMPK and DNAJA1 and therefore
proposed the hypothesis that AMPK interacts with DNAJA1 to
participate in the anti-apoptotic pathway of cells. The expression
levels of Bax and Bcl2, two apoptosis markers, were compared in
U251 cells, AMPK-TurboID cells and CCCP drug-treated AMPK-TurboID
cells. The results showed that compared with the U251 cell line,
the AMPK overexpressing cell line exhibited decreased BAX
expression, indicating an attenuation of the apoptotic process. By
contrast, CCCP-treated AMPK-TurboID cells exhibited significantly
weaker expression of DNAJA1 and Bcl2 and enhanced expression of Bax
compared with AMPK overexpressing cells without CCCP treatment,
supporting the notion that AMPK can interact with DNAJA1 to inhibit
apoptosis.
Lastly, despite the present study having
successfully constructed an AMPK overexpression cell line,
determined its interacting proteins and showed that AMPK may
inhibit apoptosis, the application of the findings in treating
clinical diseases where AMPK may be implicated in, such as diabetes
and myocardial infarction, require further investigation.
Supplementary Material
Reciprocal proteins pulled from the
AMP-activated protein kinase overexpression group.
Reciprocal proteins pulled from the
carbonyl cyanide 3-chlorophenylhydrazone processing group.
Acknowledgements
Not applicable.
Funding
Funding: The present study was supported by The Scientific
Innovation Team of Hubei University of Science and Technology
(grant no. 2023T11), The Hubei Provincial Natural Science
Foundation and Xianning Innovation and Development Project (grant
no. 2025AFD403) and The Hubei Institute of Science and Technology
Horizontal Research Project (grant nos. 2022HX135 and
2023HX188).
Availability of data and materials
The proteomics data generated in the present study
may be found in the iProX database under accession number PXD044248
or at the following URL: (https://www.iprox.cn/page/project.html?id=IPX0006806000).
The other data generated in the present study may be requested from
the corresponding author.
Authors' contributions
XL and WL conceived and designed the study. ML, JG,
YX and QW performed the experiments and acquired the data. JG, YX
and QW contributed to data analysis and interpretation. JG, YX and
QW prepared the figures. ML, JG and YX contributed to methodology
development. QW and SG validated the experimental results. XL, ML
and WL drafted the manuscript. SG, JG and YX reviewed and edited
the manuscript. WL and SG supervised the project and acquired
funding. All authors have read and approved the final manuscript.
XL and WL confirm the authenticity of all the raw data.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Huber LA, Pfaller K and Vietor I:
Organelle proteomics: Implications for subcellular fractionation in
proteomics. Circ Res. 92:962–968. 2003.PubMed/NCBI View Article : Google Scholar
|
|
2
|
Wang X and Huang L: Identifying dynamic
interactors of protein complexes by quantitative mass spectrometry.
Mol Cell Proteomics. 7:46–57. 2008.PubMed/NCBI View Article : Google Scholar
|
|
3
|
Roux KJ, Kim DI, Raida M and Burke B: A
promiscuous biotin ligase fusion protein identifies proximal and
interacting proteins in mammalian cells. J Cell Biol. 196:801–810.
2012.PubMed/NCBI View Article : Google Scholar
|
|
4
|
Bosch JA, Chen CL and Perrimon N:
Proximity-dependent labeling methods for proteomic profiling in
living cells: An update. Wiley Interdiscip Rev Dev Biol.
10(e392)2021.PubMed/NCBI View
Article : Google Scholar
|
|
5
|
Qin W, Cho KF, Cavanagh PE and Ting AY:
Deciphering molecular interactions by proximity labeling. Nat
Methods. 18:133–143. 2021.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Samavarchi-Tehrani P, Samson R and Gingras
AC: Proximity dependent biotinylation: Key enzymes and adaptation
to proteomics approaches. Mol Cell Proteomics. 19:757–773.
2020.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Kim DI, Jensen SC, Noble KA, Kc B, Roux
KH, Motamedchaboki K and Roux KJ: An improved smaller biotin ligase
for BioID proximity labeling. Mol Biol Cell. 27:1188–1196.
2016.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Zempleni J: Uptake, localization, and
noncarboxylase roles of biotin. Annu Rev Nutr. 25:175–196.
2005.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Branon TC, Bosch JA, Sanchez AD, Udeshi
ND, Svinkina T, Carr SA, Feldman JL, Perrimon N and Ting AY:
Efficient proximity labeling in living cells and organisms with
TurboID. Nat Biotechnol. 36:880–887. 2018.PubMed/NCBI View
Article : Google Scholar
|
|
10
|
Cho KF, Branon TC, Udeshi ND, Myers SA,
Carr SA and Ting AY: Proximity labeling in mammalian cells with
TurboID and split-TurboID. Nat Protoc. 15:3971–3999.
2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Tourasse NJ and Li WH: Selective
constraints, amino acid composition, and the rate of protein
evolution. Mol Biol Evol. 17:656–664. 2000.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Martell JD, Deerinck TJ, Sancak Y, Poulos
TL, Mootha VK, Sosinsky GE, Ellisman MH and Ting AY: Engineered
ascorbate peroxidase as a genetically encoded reporter for electron
microscopy. Nat Biotechnol. 30:1143–1148. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
13
|
Mick DU, Rodrigues RB, Leib RD, Adams CM,
Chien AS, Gygi SP and Nachury MV: Proteomics of primary cilia by
proximity labeling. Dev Cell. 35:497–512. 2015.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Cho IT, Adelmant G, Lim Y, Marto JA, Cho G
and Golden JA: Ascorbate peroxidase proximity labeling coupled with
biochemical fractionation identifies promoters of endoplasmic
reticulum-mitochondrial contacts. J Biol Chem. 292:16382–16392.
2017.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Huang Y, Zhai G, Li Y, Han Y, Chen C, Lu C
and Zhang K: Deciphering the interactome of histone marks in living
cells via genetic code expansion combined with proximity labeling.
Anal Chem. 94:10705–10714. 2022.PubMed/NCBI View Article : Google Scholar
|
|
16
|
Kim HB and Kim KE: Precision proteomics
with TurboID: Mapping the suborganelle landscape. Korean J Physiol
Pharmacol. 28:495–501. 2024.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Yheskel M, Sidoli S and Secombe J:
Proximity labeling reveals a new in vivo network of interactors for
the histone demethylase KDM5. Epigenetics Chromatin.
16(8)2023.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Yan Y, Zhou XE, Xu HE and Melcher K:
Structure and physiological regulation of AMPK. Int J Mol Sci.
19(3534)2018.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Hardie DG, Ross FA and Hawley SA: AMPK: A
nutrient and energy sensor that maintains energy homeostasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012.PubMed/NCBI View
Article : Google Scholar
|
|
20
|
Fu A, Eberhard CE and Screaton RA: Role of
AMPK in pancreatic beta cell function. Mol Cell Endocrinol.
366:127–134. 2013.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Wang S, Song P and Zou MH: AMP-activated
protein kinase, stress responses and cardiovascular diseases. Clin
Sci (Lond). 122:555–573. 2012.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Song K, Kong X, Zhang Z, Xian Y, He M,
Zhang Y, Liao X, Huang Z, Kang A, Xiao D and Ren Y: Sleeve
gastrectomy ameliorates renal injury in obesity-combined
hyperuricemic nephropathy mice by modulating the AMPK/Nrf2/ABCG2
pathway. Sci Rep. 14(22834)2024.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Peng C, Jiang H, Jing L, Yang W, Guan X,
Wang H, Yu S, Cao Y, Wang M, Ma H, et al: Macrophage SUCLA2 coupled
glutaminolysis manipulates obesity through AMPK. Nat Commun.
16(1738)2025.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Safaie N, Idari G, Ghasemi D, Hajiabbasi
M, Alivirdiloo V, Masoumi S, Zavvar M, Majidi Z and Faridvand Y:
AMPK activation; a potential strategy to mitigate TKI-induced
cardiovascular toxicity. Arch Physiol Biochem. 131:329–341.
2025.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Zhang Y, Gong H, Jin L, Liu P, Fan J, Qin
X and Zheng Q: Succinate predisposes mice to atrial fibrillation by
impairing mitochondrial function via SUCNR1/AMPK axis. Redox Biol.
81(103576)2025.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Zhang Y, Li Y, Yang X, Wen Z, Nagalakshmi
U and Dinesh-Kumar SP: TurboID-based proximity labeling for in
planta identification of protein-protein interaction networks. J
Vis Exp. (10.3791/60728)2020.PubMed/NCBI View
Article : Google Scholar
|
|
27
|
de Graaf AO, van den Heuvel LP, Dijkman
HB, de Abreu RA, Birkenkamp KU, de Witte T, van der Reijden BA,
Smeitink JA and Jansen JH: Bcl-2 prevents loss of mitochondria in
CCCP-induced apoptosis. Exp Cell Res. 299:533–540. 2004.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Kane MS, Paris A, Codron P, Cassereau J,
Procaccio V, Lenaers G, Reynier P and Chevrollier A: Current
mechanistic insights into the CCCP-induced cell survival response.
Biochem Pharmacol. 148:100–110. 2018.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Han YH, Kim SH, Kim SZ and Park WH:
Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) as an
O2(*-) generator induces apoptosis via the depletion of
intracellular GSH contents in Calu-6 cells. Lung Cancer.
63:201–209. 2009.PubMed/NCBI View Article : Google Scholar
|
|
30
|
He Y and Wang Z: The roles of HSP40/DNAJ
protein family in neurodegenerative diseases. Zhejiang Da Xue Xue
Bao Yi Xue Ban. 51:640–646. 2022.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Roscoe JA, Gorin FA and Tait RC: Human
astrocytoma U251 RNA and genomic brain glycogen phosphorylase
sequences. Brain Res Mol Brain Res. 10:273–275. 1991.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Su Y, Guo Y, Guo J, Zeng T, Wang T and Liu
W: Study of FOXO1-interacting proteins using TurboID-based
proximity labeling technology. BMC Genomics. 24(146)2023.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Gotoh T, Terada K, Oyadomari S and Mori M:
hsp70-DnaJ chaperone pair prevents nitric oxide- and CHOP-induced
apoptosis by inhibiting translocation of Bax to mitochondria. Cell
Death Differ. 11:390–402. 2004.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Barner HB, Labovitz AJ and Fiore AC:
Prosthetic valves for the small aortic root. J Card Surg. 9 (2
Suppl):S154–S157. 1994.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Mitchem MM, Shrader C, Abedi E and Truman
AW: Novel insights into the post-translational modifications of
Ydj1/DNAJA1 co-chaperones. Cell Stress Chaperones. 29:1–9.
2024.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Liu G, Wong L and Chua HN: Complex
discovery from weighted PPI networks. Bioinformatics. 25:1891–1897.
2009.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Li XL, Foo CS and Ng SK: Discovering
protein complexes in dense reliable neighborhoods of protein
interaction networks. Comput Syst Bioinformatics Conf. 6:157–168.
2007.PubMed/NCBI
|
|
38
|
Zhang Y, Song G, Lal NK, Nagalakshmi U, Li
Y, Zheng W, Huang PJ, Branon TC, Ting AY, Walley JW and
Dinesh-Kumar SP: TurboID-based proximity labeling reveals that UBR7
is a regulator of N NLR immune receptor-mediated immunity. Nat
Commun. 10(3252)2019.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Hua F, Mu R, Liu J, Xue J, Wang Z, Lin H,
Yang H, Chen X and Hu Z: TRB3 interacts with SMAD3 promoting tumor
cell migration and invasion. J Cell Sci. 124:3235–3246.
2011.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Song R, Li D, Hao X, Lyu Q, Ma Q, Chen X
and Qiao L: MALDI-TOF MS analysis of nasal swabs for the
characterization of patients infected with SARS-CoV-2 Omicron.
VIEW. 5(20240015)2024.
|
|
41
|
Xiao J, Liu H, Yao J, Yang S, Shen F, Bu
K, Wang Z, Liu F, Xia N, Yuan Q, et al: The characterization of
serum proteomics and metabolomics across the cancer trajectory in
chronic hepatitis B-related liver diseases. VIEW.
5(20240031)2024.
|
|
42
|
Hu B, He R, Pang K, Wang G, Wang N, Zhu W,
Sui X, Teng H, Liu T, Zhu J, et al: High-resolution spatially
resolved proteomics of complex tissues based on microfluidics and
transfer learning. Cell. 188:734–748.e22. 2025.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Furtwängler B, Üresin N, Richter S,
Schuster MB, Barmpouri D, Holze H, Wenzel A, Grønbæk K,
Theilgaard-Mönch K, Theis FJ, et al: Mapping early human blood cell
differentiation using single-cell proteomics and transcriptomics.
Science. 390(eadr8785)2025.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Herzig S and Shaw RJ: AMPK: Guardian of
metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol.
19:121–135. 2018.PubMed/NCBI View Article : Google Scholar
|
|
45
|
Viollet B and Andreelli F: AMP-activated
protein kinase and metabolic control. Handb Exp Pharmacol. 303–330.
2011.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Peng: Erratum in: AMPK/MFF activation:
role in mitochondrial fission and mitophagy in dry eye. Invest
Ophthalmol Vis Sci. 63(24)2022.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Zhang T, Xu D, Trefts E, Lv M, Inuzuka H,
Song G, Liu M, Lu J, Liu J, Chu C, et al: Metabolic orchestration
of cell death by AMPK-mediated phosphorylation of RIPK1. Science.
380:1372–1380. 2023.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Zheng C, Yu X, Liang Y, Zhu Y, He Y, Liao
L, Wang D, Yang Y, Yin X, Li A, et al: Targeting PFKL with
penfluridol inhibits glycolysis and suppresses esophageal cancer
tumorigenesis in an AMPK/FOXO3a/BIM-dependent manner. Acta Pharm
Sin B. 12:1271–1287. 2022.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Xu J, Ji J and Yan XH: Cross-talk between
AMPK and mTOR in regulating energy balance. Crit Rev Food Sci Nutr.
52:373–381. 2012.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Gwinn DM, Shackelford DB, Egan DF,
Mihaylova MM, Mery A, Vasquez DS, Turk BE and Shaw RJ: AMPK
phosphorylation of raptor mediates a metabolic checkpoint. Mol
Cell. 30:214–226. 2008.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Zhang MH, Fang XS, Guo JY and Jin Z:
Effects of AMPK on apoptosis and energy metabolism of gastric
smooth muscle cells in rats with diabetic gastroparesis. Cell
Biochem Biophys. 77:165–177. 2019.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Han C, Lu Y, Wei Y, Wu B, Liu Y and He R:
D-ribosylation induces cognitive impairment through RAGE-dependent
astrocytic inflammation. Cell Death Dis. 5(e1117)2014.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Liang D, Song Y, Fan G, Ji D, Zhang T, Nie
E, Liu X, Liang J, Yu R and Gao S: Effects of long form of CAPON
overexpression on glioma cell proliferation are dependent on
AKT/mTOR/P53 signaling. Int J Med Sci. 16:614–622. 2019.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Li GH, Wei H, Chen ZT, Lv SQ, Yin CL and
Wang DL: STAT3 silencing with lentivirus inhibits growth and
induces apoptosis and differentiation of U251 cells. J Neurooncol.
91:165–174. 2009.PubMed/NCBI View Article : Google Scholar
|