Introduction
Type 2 diabetes (T2D) accounts for >90% of all
diabetes cases and represents a rapidly escalating global health
crisis (1). The 2025 International
Diabetes Federation estimated that ~589 million adults worldwide
are currently living with diabetes, a figure projected to rise to
853 million by 2050(2). Globally,
T2D is now the eighth leading cause of disease burden, responsible
for ~6.7 million mortalities annually and is predicted to become
the second leading cause of mortality by 2050(3). Characterized by insulin resistance
and impaired insulin secretion, T2D is associated with persistent
hyperglycemia and is often accompanied by dyslipidemia (4) and increased oxidative stress
(5). If poorly managed, T2D can
lead to severe micro- and macrovascular complications, including
cardiovascular disease, nephropathy, retinopathy and neuropathy,
markedly reducing quality of life and escalating healthcare costs.
Furthermore, ~70% of individuals with T2D are concurrently
diagnosed with metabolic dysfunction-associated steatotic liver
disease (MASLD). This comorbidity markedly elevates the risk of
advanced liver conditions such as metabolic dysfunction-associated
steatohepatitis (MASH), cirrhosis and hepatocellular carcinoma
(6,7).
Current therapeutic strategies for T2D primarily aim
to mitigate hyperglycemia by enhancing insulin secretion or
improving insulin sensitivity. Standard treatment regimen typically
involves lifestyle modifications, oral antidiabetic agents,
injectable therapies or their combination, with goals extending
beyond glycemic control to address associated metabolic
disturbances such as dyslipidemia and hypertension (8). However, numerous existing
interventions often fall short of comprehensively addressing the
complex network of underlying metabolic derangements driving T2D
and its multifaceted complications.
Mitochondrial dysfunction has emerged as a central
contributor to the pathogenesis of various metabolic diseases.
Previous evidence indicates that inducing mild mitochondrial
uncoupling can alleviate metabolic disorders by promoting energy
expenditure, enhancing mitochondrial respiratory efficiency and
improving insulin sensitivity and hepatic lipid metabolism
(9-11).
These insights have renewed a focus on developing mitochondrial
uncouplers as potential therapeutic agents for T2D, obesity and
MASLD. SR4 is a small-molecule mitochondrial uncoupler previously
identified by our research group for its potent anticancer activity
in vitro and in tumor xenograft models (12-14).
Unlike traditional protonophore uncouplers such as carbonyl
cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), BAM15 and
niclosamide, SR4 belongs to a newly defined class of fatty
acid-activated proton transporters that facilitate the
translocation of fatty acid anions across the mitochondrial inner
membrane, inducing controlled proton leak and mitochondrial
uncoupling (15,16). An earlier study demonstrated that
SR4 improves metabolic parameters in high-fat diet (HFD)-induced
obese mice, including mitigating weight gain, hyperglycemia and
hepatic steatosis (17).
Over the years, numerous mouse models of obesity and
diabetes have been developed, providing valuable insights into the
cellular and molecular mechanisms regulating energy metabolism and
homeostasis. The present study aimed to evaluate the metabolic
effects of SR4 in db/db mice, a well-established genetic
model of obesity and T2D characterized by severe, progressive
hyperglycemia, insulin resistance, dyslipidemia and hepatic
steatosis (18). To elucidate the
mechanistic basis of SR4's action, the present study specifically
focused on the liver, given its central role in energy homeostasis
and nutrient metabolism.
Materials and methods
Chemicals and reagents
SR4 was previously synthesized from a validated
protocol at the Chemical GMP Synthesis Facility, Beckman Research
Institute of the City of Hope (CA, USA) (14). All other chemicals and reagents
were purchased from Sigma-Aldrich (Merck KGaA), unless otherwise
specified.
Animal studies
All animal experiments were conducted in accordance
with a protocol approved by the City of Hope National Medical
Center's Institutional Animal Care and Use Committee (Duarte, USA;
approval no. 12004). Male 9-week-old db/db mice
(BKS.Cg-Dock7m +/+ Leprdb/J) weighing 40-45 g
were obtained from Jackson Laboratory. A total of 32 animals were
divided into two groups: Vehicle control group and SR4-treated
group; eight animals per group were used for the efficacy studies
and another eight animals per group were used for the
metabolic/indirect calorimetry studies. Mice were kept in specific
pathogen-free conditions at 22˚C with a 12/12 h light/dark cycle.
The animals had unrestricted access to water and standard chow diet
(LabDiet® 5K52; Scott Distributing; ScottPharma
Solutions). After 1 week of acclimatization, mice received either
the vehicle control (4% DMSO in corn oil) or SR4 dissolved in
vehicle (10 mg/kg of BW) via oral gavage (14). Dosing was performed three times a
week (Mon, Wed and Fri) in the early morning across a 5-week
period. Food consumption was monitored throughout the present
study. The animals were monitored daily for health and behavior.
Mice were weighed weekly and body composition (lean mass and fat
mass) was measured by quantitative magnetic resonance imaging
(EchoMRI™ 3-in-1; v2.1; Echo Medical Systems). All animals survived
until the end of the present study and did not meet any of the
humane endpoints for early euthanasia, which included failure to
eat or drink for 24 h, inability to make normal posture and
behavior, signs of severe distress and >20% weight loss. At
study completion, mice were euthanized via CO2
inhalation (30-70% total chamber volume/min) followed by cervical
dislocation and observation of cessation of breathing to confirm
mortality. Blood was then collected through cardiac puncture and
the plasma was separated for subsequent analysis. Tissue samples
were collected and either snap-frozen and stored at -80˚C or
preserved in 10% neutral buffer formalin for further biochemical
and immunohistochemical analyses, respectively.
Glucose tolerance tests
To assess glucose tolerance, mice were fasted for 16
h overnight. A 2 g/kg BW D-glucose solution was then administered
intraperitoneally. Tail vein blood (~5-10 µl) was collected
serially at 0, 30, 60, 90 and 120 min post-challenge through the
tail-nick method (19) The blood
collection procedure was approved by the Institutional Animal Care
and Use Committee of City of Hope in accordance with the NIH
‘Guidelines for Survival Blood Collection in Mice and Rats’
(https://oacu.oir.nih.gov/). Blood
glucose concentration was measured with an Accu-Chek Compact Plus
glucometer (Roche Diagnostics Ltd.). The total area under the curve
(AUC) was calculated using the trapezoidal method.
Indirect calorimetry
Indirect calorimetry was performed in a separate
experiment using the PhenoMaster (version 5.9.3; cat. no.
2016-5420; TSE Systems) at the Comprehensive Metabolic Phenotyping
Core at Beckman Research Institute (CA, USA). Mice were treated
with SR4 or vehicle for 5 weeks (n=8/group). The animals were
allowed to acclimate to the cages for 2 days before two cycles of
24 h measurements. Both oxygen consumption (V02) and
carbon dioxide output (VC02) were normalized to lean
mass as determined by EchoMRI™. The respiratory exchange
ratio (RER) was calculated as VC02/V02. With
this, the energy expenditure (EE), standardized for BW, was
calculated using the following formula: EE=(3.185 + 1.232 x RER) x
V02.
Biochemical analysis of blood and
liver samples
Plasma was extracted from blood samples following
centrifugation at 2,000 x g at 4˚C for 10 min. Plasma insulin was
measured using a commercial kit according to the manufacturer's
protocol (Crystal Chem Ultra Sensitive Mouse Insulin Elisa kit;
cat. no. 90080; Crystal Chem, Inc.). Concentrations of plasma
alanine transaminase (ALT), aspartate aminotransferase (AST), total
triglycerides (TG) and cholesterol were quantified using
commercially available kits (ALT Activity Assay Kit, cat. no.
MET-512; AST Assay Kit, cat. no. MET-5127; Triglyceride
Quantification kit, cat. no. STA-39; Total Cholesterol Assay Kit,
cat. no. STA-384) from Cell BioLabs, Inc., according to the
manufacturer's instructions. Levels of glycated hemoglobin (HbA1c)
in the blood were measured using the mouse HbA1c assay kit (cat.
no. 80310; Crystal Chem, Inc) according to the manufacturer's
instructions. Liver TG were measured as previously described
(17). Hepatic nucleotides (ATP
and AMP) were measured using UV-high-performance liquid
chromatography. Briefly, 100 µl of liver homogenates was removed
and mixed with 100 µl of deionized water. Furthermore, 18.5 µl of 1
M potassium hydroxide, 30 µl of 150 mM monopotassium phosphate
(KH2PO4)/150 mM potassium chloride (KCl)
solution (Ph 6.0) and 0.5% acetonitrile were added. The final
mixture was centrifuged at 17,000 x g for 5 min at room
temperature. The supernatant was then collected and transferred to
an auto-injector vial and 10 µl was injected into a Thermo ODS
Hypersil™ C18 column (3 µm; 150x4.6 mm; Thermo Fisher
Scientific, Inc.). Isocratic separation at 25˚C was achieved using
a mobile phase consisting of 150 mM KH2PO4,
150 mM KCl (pH 6.0) and 0.5% acetonitrile. The flow rate was set at
0.8 ml/min and a total run time of 20 min. Nucleotides were
detected by their absorbance at 260 nm using a Shimadzu SPD-40
UV-Vis detector (Shimadzu Scientific Instruments, Inc.) and
compared with the elution position of standards (17).
Liver pathology and histology
At the end of the present study, the liver from each
mouse was harvested and weighed. Liver samples were either
flash-frozen or directly fixed in 10% formalin for downstream
assays. Formalin-fixed liver samples were sectioned (5 µm) and
stained with H&E for 10-15 min at room temperature. Separately,
frozen liver samples from each group were embedded in OCT compound
(Sakura Finetek USA, Inc.), sliced and stained with Oil Red O
(Sigma-Aldrich; Merck KGaA) at room temperature for 15 min. All
stained slides were viewed and images were captured under a bright
field microscope (cat. no. AX70; Olympus Corporation) equipped with
a digital camera.
Analysis of liver oxidative stress and
antioxidant activity
Multiple enzymes associated with oxidative stress
and antioxidant defense, including glutathione (GSH), glutathione
S-transferase (GST) and glutathione peroxidase (GPx), were measured
in liver crude homogenates using established methods as previously
described (20). The level of
malondialdehyde (MDA), a marker of lipid peroxidation, was
quantified using the thiobarbituric acid reactive substances assay
as described previously (21).
Liver mitochondria isolation and
bioenergetic analysis
Fresh mitochondria were isolated from the livers of
mice (n=4-5/group) as described previously (12). Briefly, mice were euthanized by
CO2 inhalation and cervical dislocation as
aforementioned, before livers were immediately removed, minced and
placed in ~10-fold volume of ice-cold mitochondria isolation medium
[250 mM sucrose, 10 mM Tris/hydrogen chloride (HCl), 1 mM EGTA and
1% fatty acid/endotoxin-free BSA; pH 7.4]. Tissues were homogenized
with 25-30 strokes in a Potter-Elvehjem tissue grinder
(Sigma-Aldrich; Merck KGaA). After centrifugation at 800 x g for 10
min at 4˚C, the fats and lipids were carefully aspirated. The
remaining supernatant was then filtered through a sterile 0.40 µm
nylon mesh membrane and centrifuged at 8,000 x g for 10 min at 4˚C.
The supernatant and any white debris were removed. The
mitochondrial pellet was resuspended in ice-cold mitochondrial
assay solution (MAS buffer; 70 mm sucrose, 220 mM mannitol, 10 mM
KH2PO4, 5 mm magnesium chloride, 2 mM HEPES, 1 mM EGTA
and 0.2% fatty acid-free BSA; pH 7.2) and this centrifugation was
repeated. The final pellet was resuspended in a minimal volume of
MAS buffer. Mitochondrial protein concentration was measured using
the DC protein assay kit according to the manufacturer's protocol
(Bio-Rad Laboratories, Inc.) with BSA as a standard. Isolated liver
mitochondria were seeded at 1 µg/well in a Seahorse 96-well plate.
Basal respiration (mitochondrial respiration state 2),
ADP-stimulated respiration (mitochondrial respiration state 3),
non-ADP-stimulated respiration (mitochondrial respiration state 4;
proton leak), FCCP-stimulated respiration (mitochondrial
respiration state 3u; maximal respiration) and spare respiratory
capacity (SRC) were quantified using a Seahorse XF96 flux analyzer
(Seahorse Biosciences, Inc.) as described previously (22).
Protein extraction and western
blotting
Liver tissue samples were homogenized in a lysis
buffer containing 50 mM Tris/HCl (pH 7.4), 150 mM sodium chloride,
1 mM EDTA, 5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 50
mM sodium fluoride, 1% NP-40, 1 mM PMSF and a protease inhibitor
cocktail tablet (Roche Diagnostics Ltd.). Homogenates were then
centrifuged for 15 min at 9,600 x g at 4˚C and supernatants were
collected. Protein concentration was determined using a DC protein
assay kit according to the manufacturer's instructions. For western
blotting, 10 µg of protein per sample were resolved by SDS-PAGE on
4-15% Criterion TGX™ gels (Bio-Rad Laboratories, Inc.)
and then transferred to nitrocellulose membranes. Membranes were
blocked with 5% skimmed milk in Tris-buffered saline containing
0.05% Tween 20 for 1 h at room temperature before overnight
incubation at 4˚C with a 1:1,000 dilution of primary antibodies
against total and phosphorylated AMPK and total and phosphorylated
ACC (cat. nos. 2532, 2537, 8578 and 3661; Cell Signaling
Technology, Inc.). After a series of washes, the membranes were
stained with a 1:3,000 dilution of peroxidase-labeled secondary
antibodies (goat anti-rabbit IgG HRP conjugate; cat. no. 1706515;
Bio-Rad Laboratories, Inc.) at room temperature for 2 h, and an ECL
system (Western Lightning® Chemiluminescence Reagent;
cat. no. NEL104001EA; Revvity, Inc.) was used to visualize the
immunoreactive proteins. Equal loading of proteins was confirmed by
stripping and reprobing the membranes with β-actin antibodies
(1:3,000 dilution; cat. no. 3700; Cell Signaling Technology,
Inc.).
Quantitative PCR (qPCR)
Total RNA was isolated from liver samples using the
RNeasy kit (Qiagen, Inc.) according to the manufacturer's
instructions. Complementary DNA (cDNA) was synthesized from the
isolated RNA using the High-Capacity cDNA Reverse Transcription Kit
(Thermo Fisher Scientific, Inc.). qPCR was performed on an ABI-7500
PCR system (Thermo Fisher Scientific, Inc.) using Power
SYBR® Green master mix (Thermo Fisher Scientific, Inc.)
for gene amplification and detection. The list of primer pairs used
in the present study and their sequences are listed in Table I. The following thermocycling
conditions were used for the PCR: Initial denaturation at 50˚C for
2 min; cDNA was denatured at 95˚C for 10 min; and 40 cycles 95˚C
for 15 sec and 60˚C for 60 sec. The relative mRNA levels of all the
target genes were quantified using the comparative
2-ΔΔCq method with β-actin as an internal control
(23).
 | Table IPrimer sequences used for qPCR. |
Table I
Primer sequences used for qPCR.
| Gene | Sequence
(5'-3') |
|---|
| Acaca | F:
CCTCCGTCAGCTCAGATACA |
| | R:
TTTACTAGGTGCAAGCCAGACA |
| Acly | F:
AGGAAGTGCCACCTCCAACAGT |
| | R:
CGCTCATCACAGATGCTGGTCA |
| β-actin | F:
ACCTTCTACAATGAGCTGCG |
| | R:
CTGGATGGCTACGTACATGG |
| Cebpa | F:
GCAAAGCCAAGAAGTCGGTGGA |
| | R:
CCTTCTGTTGCGTCTCCACGTT |
| Fasn | F:
GCGATGAAGAGCATGGTTTAG |
| | R:
GGCTCAAGGGTTCCATGTT |
| Pparg | F:
GCCCTTTGGTGACTTTATGGA |
| | R:
GCAGCAGGTTGTCTTGGATG |
| Scd1 | F:
CTGTACGGGATCATACTGGTTC |
| | R:
GCCGTGCCTTGTAAGTTCTG |
| Srebf1 | F:
ATCCAGGTCAGCTTGTTTGCGATG |
| | R:
TGGACTACTAGTGTTGGCCTGCTT |
Transcriptome analysis of whole
liver
Total RNA was extracted from liver tissue samples
(n=3 control and n=2 SR4 treatment) using the RNeasy Mini Kit
(Qiagen, Inc.), per the manufacturer's protocol. RNA libraries were
prepared using the TruSeq Stranded mRNA Library Prep Kit (Illumina,
Inc.) and sequenced on the HiSeq® 2500 platform
(Illumina, Inc), generating single-end 51 bp reads. Sequence
alignment to the mouse reference genome (GRCm38/mm10) was performed
using TopHat2(version 2.1.1; http://ccb.jhu.edu/software/tophat/index.shtml). Read
quantification and normalization utilized the ‘Empirical Analysis
of Digital Gene Expression in R’ (‘edgeR’; version 4.1) package
(24), based on raw count data.
Differentially expressed genes (DEGs) analysis was conducted using
the ‘DESeq2’ package (25),
applying the Benjamini-Hochberg method to control the false
discovery rate (FDR). Genes with an FDR<0.05 and an absolute
fold change (FC) ≥1.5 were considered DEGs. A volcano plot was
generated using VolcaNoseR (version v1.0.3; https://huygens.science.uva.nl/VolcaNoseR/), with
Manhattan distance used to identify the top 10 DEGs (26).
Functional enrichment analysis of DEGs was performed
using the Database for Annotation, Visualization and Integrated
Discovery platform, version 6.8(27), incorporating Gene Ontology (GO;
https://geneontology.org/) biological processes
and Kyoto Encyclopedia of Genes and Genomes (KEGG; https://www.kegg.jp) pathway annotations. Significant
enrichment was defined by adjusted P<0.05 and gene counts ≥3.
Furthermore, visualization was conducted using SRplot (https://www.bioinformatics.com.cn/en).
To explore protein-protein interactions (PPIs), a PPI network was
constructed using the STRING database (version 12.0; https://string-db.org/) (28), with a minimum confidence score of
0.7. Evidence channels for the interactions included in the network
were derived from experimental data, curated databases,
co-expression, gene fusion, co-occurrence, neighborhood and text
mining. The resulting network was visualized and analyzed in
Cytoscape (version 3.10.3; https://cytoscape.org/) (29). Significant network modules were
identified using the ‘Molecular Complex Detection’ (‘MCODE’)
Cytoscape plugin (30), with the
following parameters: K-core=2, maximum depth=100, node score
cut-off=0.2 and degree cut-off=2. Module functional annotation was
further analyzed using STRING. Finally, hub genes within the PPI
network were identified using the Maximal Clique Centrality (MCC)
algorithm in the ‘cytoHubba’ plugin (31).
Statistical analysis
Statistical analyses were performed using GraphPad
Prism (version 10.3.1; GraphPad; Dotmatics). Data are presented as
mean ± SEM. BW changes were compared between groups using repeated
measure two-way ANOVA coupled with post hoc Tukey's test.
Comparison between two groups was analyzed by Welch's t-test to
adjust for unequal sizes and variances between samples. P<0.05
was considered to indicate a statistically significant
difference.
Results
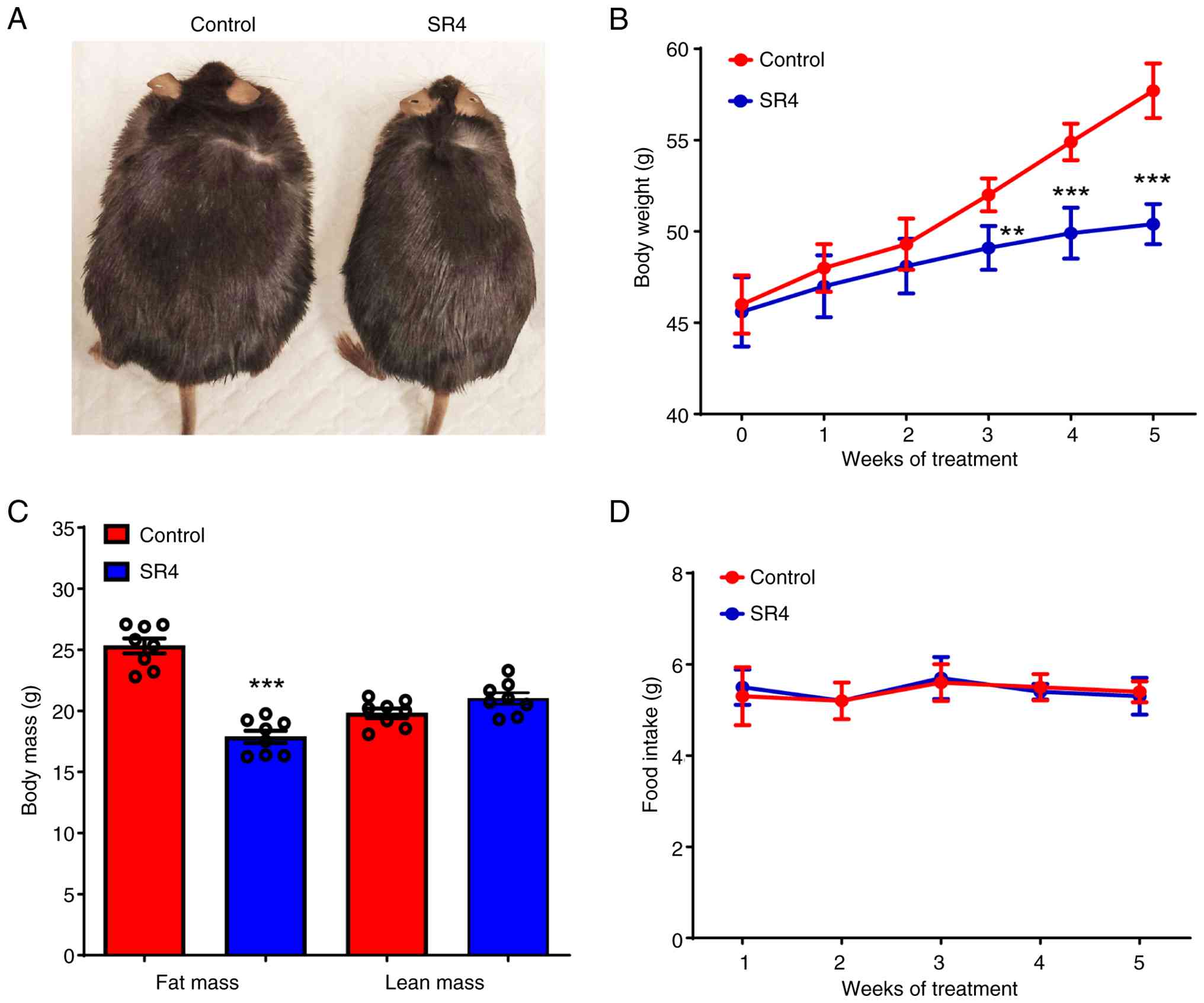
SR4 reduces BW and body fat mass
without altering food intake in db/db mice
To investigate the effects of SR4 in diabetic mice,
either vehicle control (4% DMSO in corn oil) or SR4 was
administered through oral gavage to male db/db mice three
times a week for 5 weeks. SR4-treated mice showed significant
weight loss after only 3 weeks of treatment and at the end of the
present study these animals exhibited ~13% overall weight loss
compared with vehicle control (50.4±0.4 vs. 57.7±0.5 g,
respectively; Fig. 1A and B). In addition, SR4-treated animals
displayed a greater reduction in fat mass without any marked change
in lean mass (Fig. 1C). Notably,
the decrease in BW and improvement in body mass composition were
not associated with less food consumption as both SR4 treatment and
control demonstrated similar daily food intake (Fig. 1D).
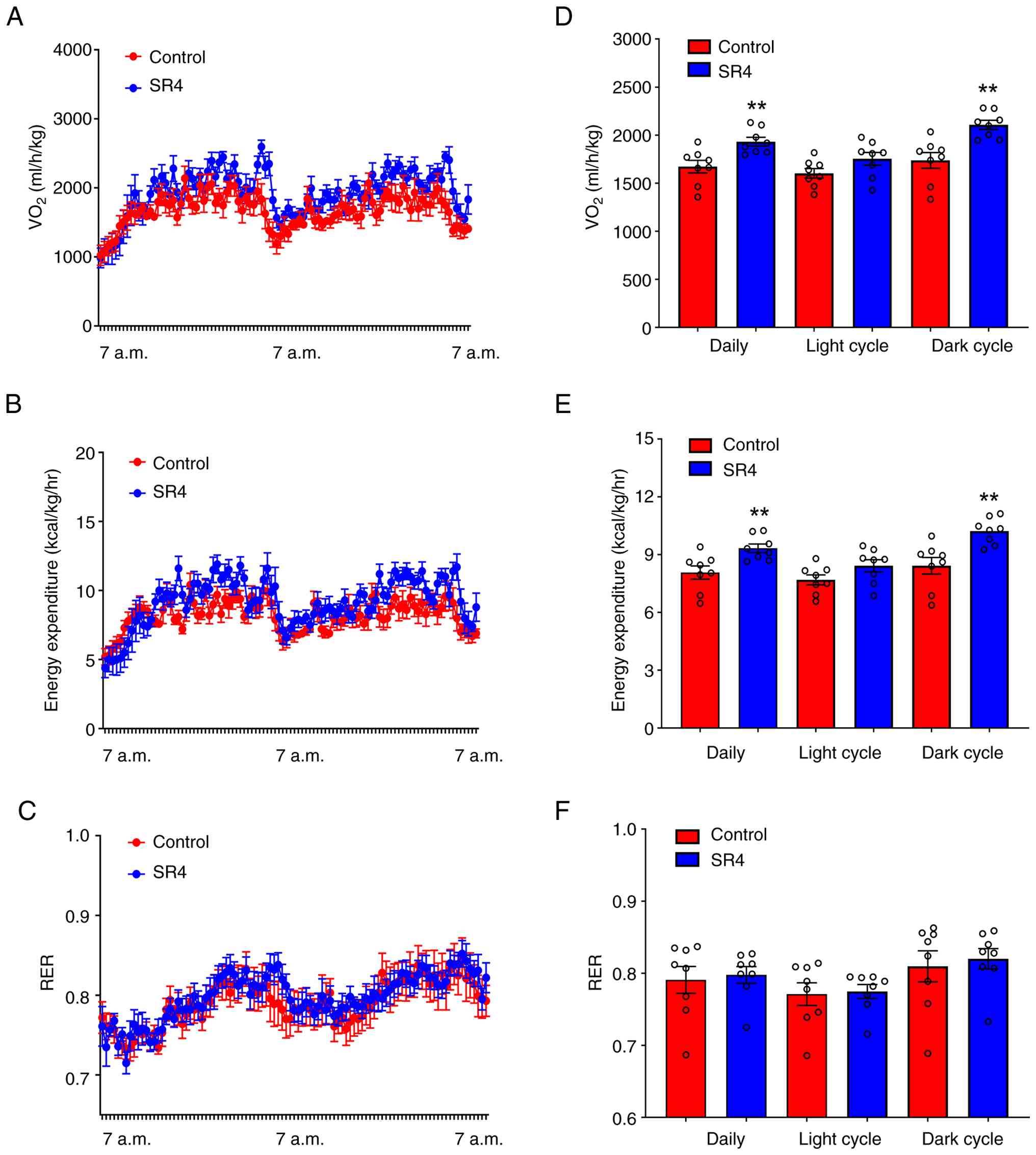
SR4 increases oxygen consumption and
energy expenditure in db/db mice
To determine if the reductions in weight and
improved body composition by SR4 was associated with energy
expenditure, indirect calorimetry experiments were performed over a
3-day period (with one day acclimation). Both oxygen consumption
and total daily expenditure, but not RER, were significantly
increased in animals treated with SR4 compared with the control
(Fig. 2A-C). Notably, the apparent
increase in oxygen consumption and energy expenditure were only
detected in the dark phase and not in the light phase (Fig. 2D and E). No significant change was observed in
the daily RER in both dark and light cycles between SR4 and control
animals (Fig. 2F).
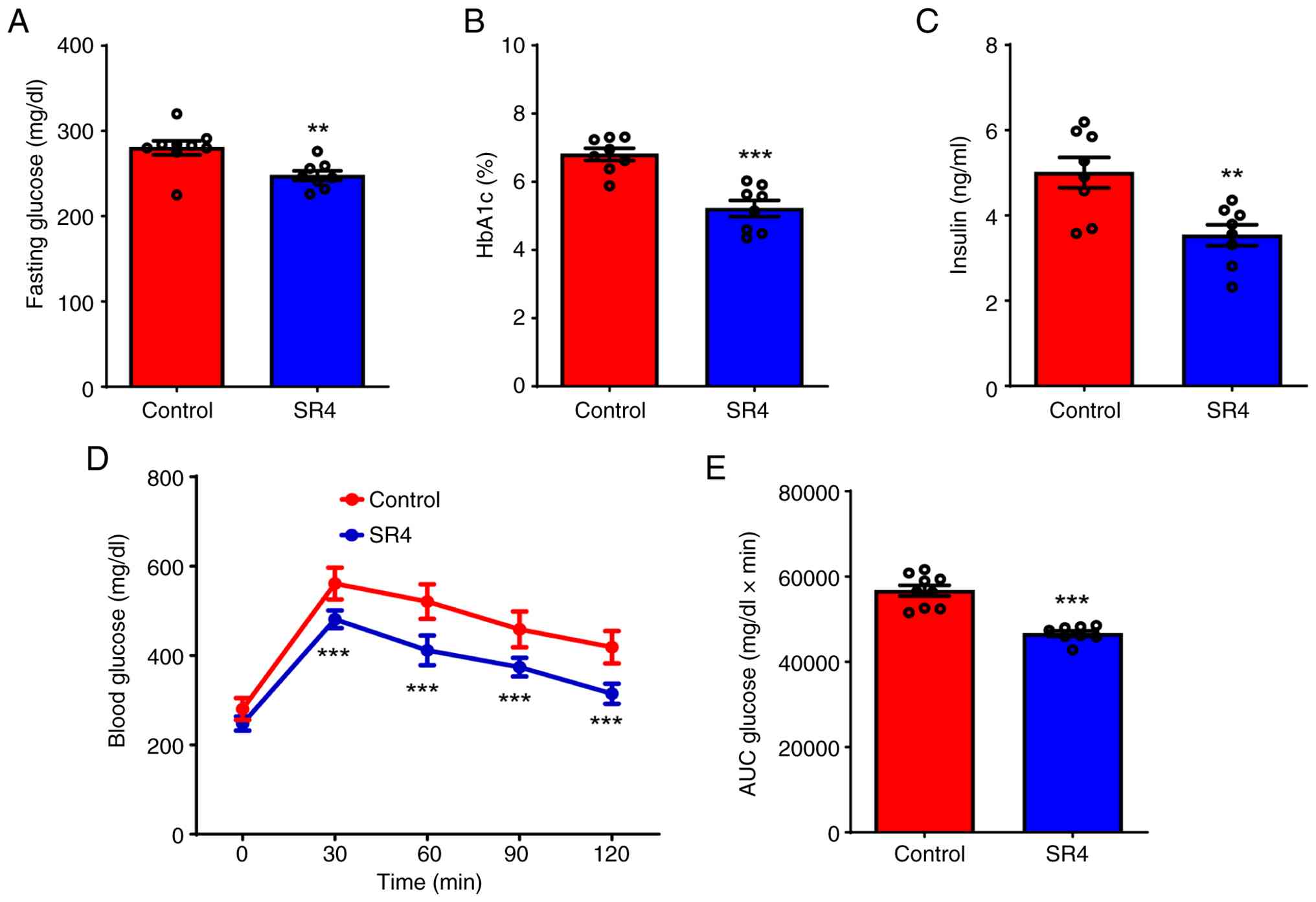
SR4 improves glycemic control and
insulin sensitivity in db/db mice
Db/db mice are characterized by a
non-functional leptin receptor, leading to obesity and insulin
resistance and ultimately causing hyperglycemia. This hyperglycemia
develops progressively, with initial insulin resistance followed by
a decrease in insulin secretion, resulting in sustained high blood
glucose levels (32). As obesity
is one of the key risk factors for insulin resistance in
db/db mice, the effects of SR4 on glucose and insulin
sensitivity were investigated. SR4 treatment significantly reduced
fasting blood glucose after 5 weeks of treatment, with mean plasma
glucose of 280.2±0.2 mg/dl in the control group vs. 245±0.4 mg/dl
in the SR4-treated group (Fig.
3A). Similarly, HbA1c levels were also significantly decreased
by SR4 at the end of treatment (Fig.
3B). Additionally, SR4 reduced the plasma insulin levels
significantly (Fig. 3C). To
further investigate the effects of SR4 on hyperglycemia,
intraperitoneal glucose tolerance tests were performed after a 16 h
fast. Glucose tolerance was quantified as the AUC integrated from
0-120 min. Control db/db mice exhibited impaired glucose
tolerance and SR4 treatment significantly reduced the peak glucose
levels after the glucose load (Fig.
3D). Consequently, glucose AUC was significantly suppressed in
SR4-treated mice compared with control (Fig. 3E). Together, these results
indicated that SR4 could improve glycemic control and insulin
sensitivity in db/db mice.
SR4 prevents dyslipidemia, hepatic
steatosis and liver injury in db/db mice
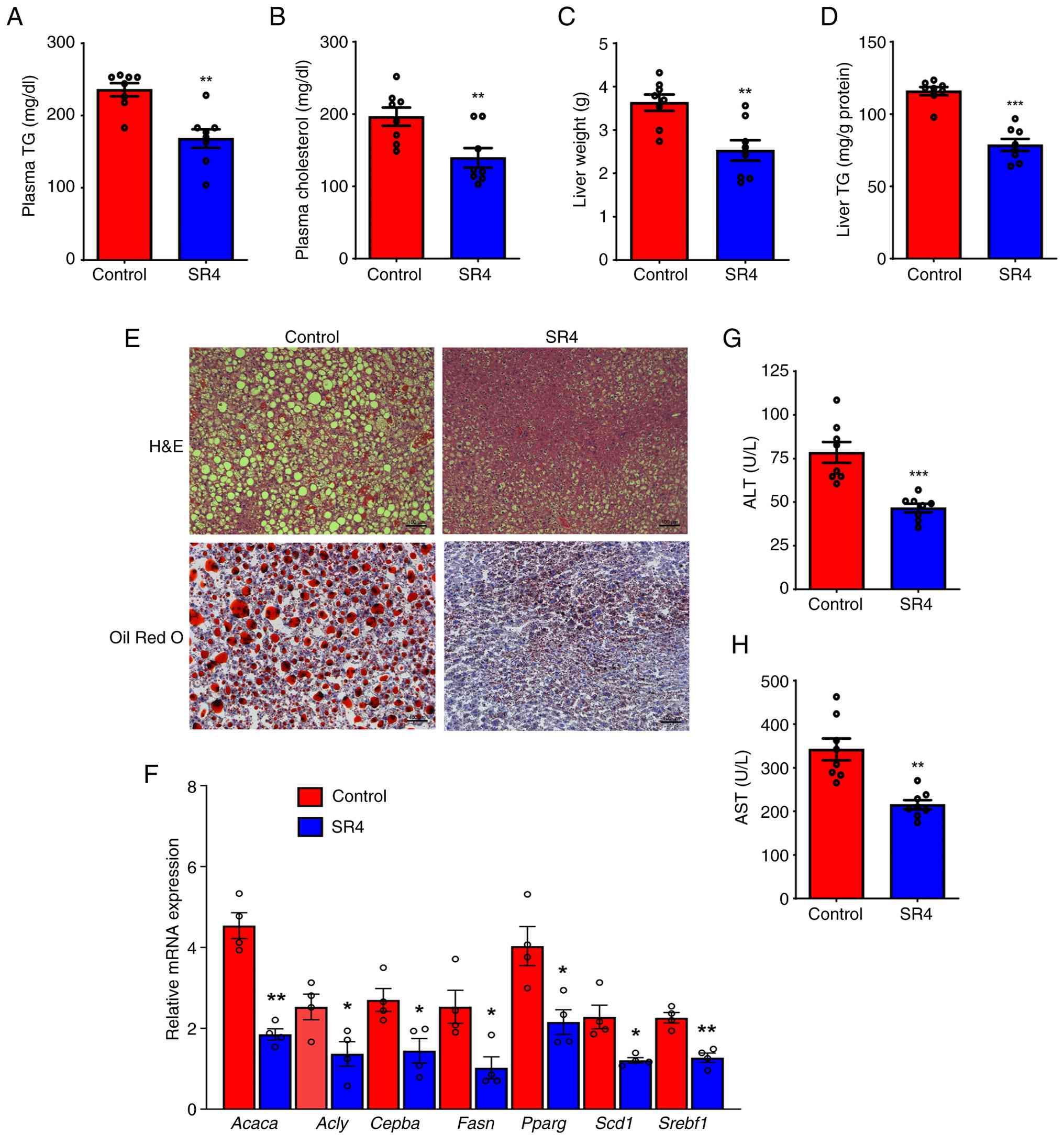
Plasma TG levels of SR4-treated mice were lower by
28% compared with that of the vehicle control group (168.1±12.8 vs.
235.8±9.0 mg/dl; Fig. 4A). SR4
also significantly decreased total cholesterol by 29% compared with
control (139.6±18.2 vs. 196.5±14.3 mg/dl; Fig. 4B). As T2D is associated with the
development of fatty liver in the db/db mice (33), the liver pathology was next
examined upon SR4 treatment. Compared with the control group, SR4
treated mice had 30% lower liver wet weight (3.6±0.2 vs. 2.5±0.2 g;
Fig. 4C). Biochemical analyses
also revealed that SR4 significantly decreased hepatic TG contents
by 32% compared with control animals (78.6±4.1 vs. 116.0±2.8 mg/g
protein; Fig. 4D).
 | Figure 4SR4 ameliorates dyslipidemia and
hepatic steatosis in db/db mice. Plasma (A) TG and (B)
cholesterol levels, (C) liver weight and liver TG content (D) of
control and SR4-treated mice at the end of the present study. (E)
Representative images of liver sections stained with H&E and
Oil Red O. Magnification, x160, scale bar 100 µm. (F) Quantitative
PCR analysis showing the relative mRNA expression levels of lipid
metabolism-associated genes Acaca, Acly,
Cebpa, Fasn, Pparg, Scd1 and
Srepbf1 in liver of mice with and without SR4 treatment.
Plasma levels of liver injury enzymes (G) ALT and (H) AST. All
quantitative data are presented as the mean ± SEM, with n=8 animals
per group for panels A-D and G-H and n=4 animals per group for
panel F. Statistical significance is indicated as follows:
*P<0.05, **P<0.01 and
***P<0.001 for SR4 vs. control. ALT, alanine
transaminase; AST, aspartate aminotransferase; Acaca,
acetyl-coenzyme A carboxylase; Acly, ATP citrate lyase;
Cebpa, CCAAT/enhancer-binding protein a; Fasn, fatty
acid synthase; Pparg, peroxisome proliferator-activated
receptor g; Scd1, stearoyl-coenzyme A desaturase 1;
Srebf1, sterol regulatory element binding protein-1c; TG,
triglycerides. |
Liver tissues were further analyzed to observe
changes in lipid accumulation. H&E staining of liver sections
revealed that db/db control animals exhibited numerous large
vacuoles filled with excess lipids, which is characteristic of the
hepatic steatosis that develops in these mice (Fig. 4E, upper panel). In addition, Oil
Red O staining demonstrated an accumulation of numerous larger fat
droplets in these mice (Fig. 4E,
lower panel). In the SR4-treated group, hepatocellular vacuolation
was minimally observed and a notable improvement in lipid
accumulation with a marked reduction in the number and size of
lipid droplets was detected in the liver of these animals. To
determine the molecular mechanisms involved in the inhibitory
effect of SR4 on hepatic steatosis, the expression of numerous
known hepatic genes was measured genes using qPCR. SR4
significantly suppressed the expression of a number of genes
involved in fatty acid and cholesterol synthesis, including
acetyl-CoA carboxylase (ACC; Acaca), ATP citrate lyase
(Acly), CCAAT/enhancer-binding protein α (Cebpa),
fatty acid synthase (Fasn), PPARγ (Pparg), sterol
regulatory element binding protein-1c (Srebf1) and
stearoyl-coenzyme A desaturase 1 (Scd1; Fig. 4F). Excessive lipid accumulation in
the liver is often associated with liver damage, including elevated
levels of liver enzymes ALT and AST. SR4 treatment significantly
decreased the plasma levels of these enzymes by 40 and 37%,
respectively, compared with the control group (Fig. 4G and H). These results indicated that SR4
improved the overall pathology of the liver and attenuated the
obesity-induced hepatic steatosis and liver injury in db/db
mice.
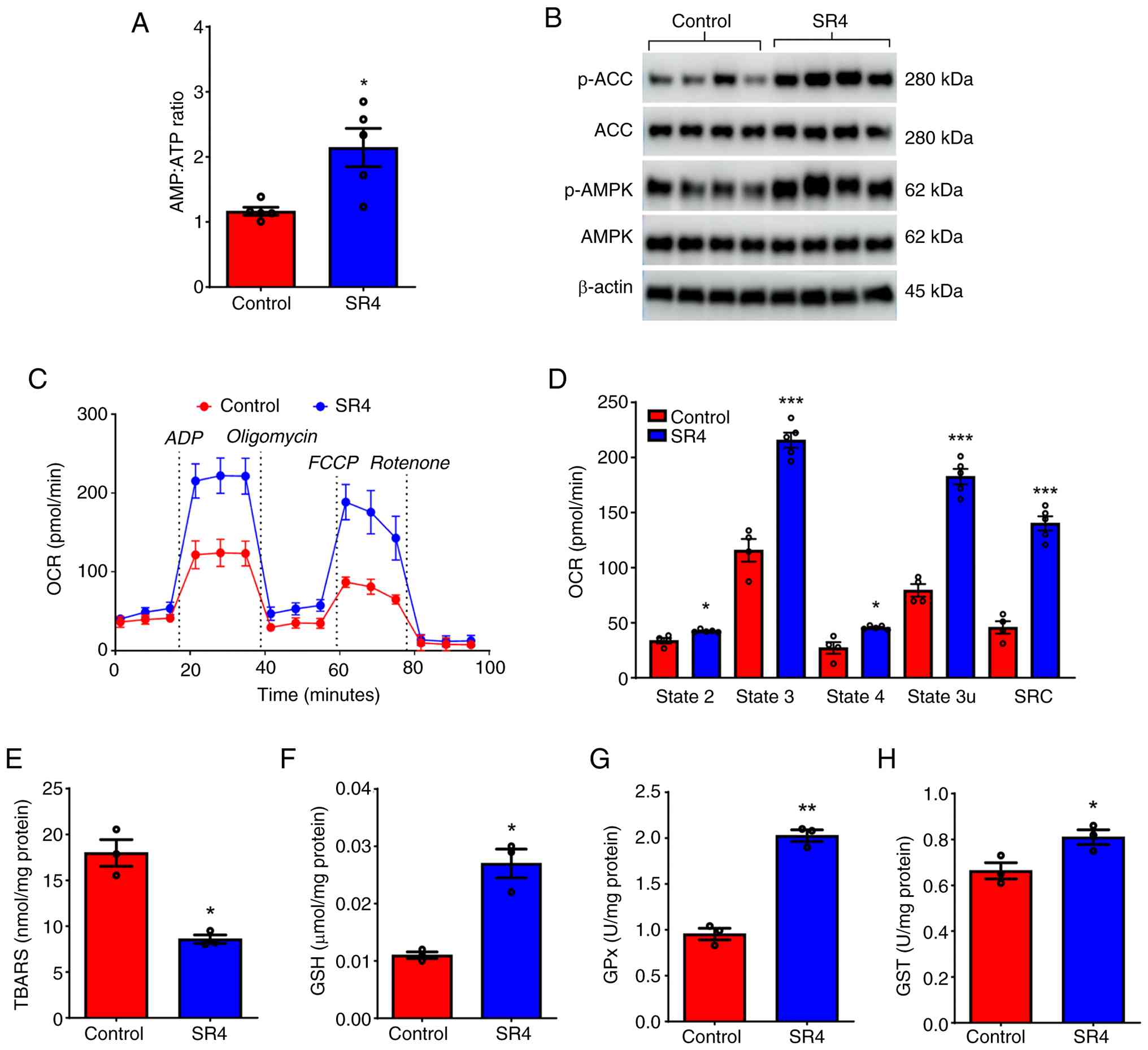
SR4 activates hepatic AMPK, increases
mitochondria respiration and prevents oxidative stress in db/db
mice
Mitochondrial uncouplers such as SR4 are known to
activate AMPK by disrupting mitochondrial function and reducing ATP
production, which can lead to an increase in the AMP:ATP ratio, a
key signal for AMPK activation (12). Chronic treatment with SR4 in
db/db mice led to a significant increase in the hepatic AMP:ATP
ratio (Fig. 5A) as well as an
increase in phosphorylation of AMPK and its downstream target ACC
(Fig. 5B).
 | Figure 5SR4 activates hepatic AMPK, increases
liver mitochondrial respiration and alleviates oxidative stress in
db/db mice. (A) Liver AMP:ATP ratio in control and
SR4-treated animals (n=5). (B) Representative western blotting
analysis of protein lysates from liver (n = 3). Lysates were
subjected to SDS-PAGE and immuno-blotted with antibodies specific
to phosphorylated and total AMPK, phosphorylated and total ACC or
β-actin. (C) Seahorse analysis of mitochondrial function and
bioenergetics of freshly isolated liver mitochondria (n=4-5 per
group) after 5 weeks of SR4 treatment. Real time OCR and key
respiration parameters such as (D) basal respiration (state 2),
ADP-linked respiration (state 3), maximal respiration (state 3u),
proton leak (state 4) and spare respiratory capacity are shown. (E)
Hepatic levels of MDA, measured as TBARS and antioxidants (F) GSH,
(G) GPx and (H) GST in liver lysates of control and SR4-treated
mice (n=3). Statistical significance is indicated as follows:
*P<0.05, **P<0.01 and
***P<0.001 for SR4 vs. control. p-, phosphorylated.
ACC, acetyl-CoA carboxylase; OCR, oxygen consumption rate; MDA,
malondialdehyde; FCCP, carbonyl
cyanide-p-trifluoromethoxyphenylhydrazone; GSH, glutathione; GST,
glutathione S-transferase; GPx, glutathione peroxidase; TBARS,
thiobarbituric acid reactive substances. |
An additional known effect of mitochondria
uncouplers is to increase oxygen consumption and mitochondria
respiration (9,12,15).
Therefore, the mechanisms by which SR4 treatment can affect liver
mitochondria function and bioenergetics using the Seahorse flux
analyzer were examined (Fig. 5C).
Mitochondria freshly isolated from the livers of mice treated with
SR4 showed an increase in basal respiration (state 2) of 19.0%
compared with control. Similarly, ADP-linked respiration (state 3)
increased to ~80% while maximal respiration (state 3u) further
increased to 120%, demonstrating increased levels of oxygen
consumption linked to ATP production (Fig. 5C). This increase in oxygen
consumption rate (OCR) further coincides with a 40% increase in
proton leak across the inner mitochondrial membrane. Furthermore,
SR4-treatment significantly increased the SRC, the capability to
increase energy production when faced with higher demands and often
used as a parameter in assessing mitochondria function, by ~195%
(Fig. 5D). A previous study showed
that impaired mitochondrial function in db/db mice can lead
to oxidative stress (5). Next, the
effects of SR4 on liver oxidative stress and GSH-linked antioxidant
system were examined. Hepatic TBAR levels of mice treated with SR4
were lower by 53% compared with those of the control group
(8.6±0.45 nmol MDA/mg protein vs. 17.9±1.45 nmol MDA/mg protein;
Fig. 5E). Concomitantly, the
activity levels of antioxidant enzymes GSH, GST and GPx were
significantly higher in the liver of SR4-treated mice compared with
the control (Fig. 5F-H). Together,
these data suggest that SR4 uncoupling activity could activate
hepatic AMPK, improve mitochondria function and alleviate oxidative
stress.
SR4 alters the expression of metabolic
genes associated with key pathways regulating lipid metabolism,
energy homeostasis and oxidative stress
To further investigate the effects of SR4 treatment
on liver function and metabolic signaling pathways, untargeted
whole-transcriptome sequencing of liver tissues isolated from
db/db mice treated with or without SR4 was performed. This
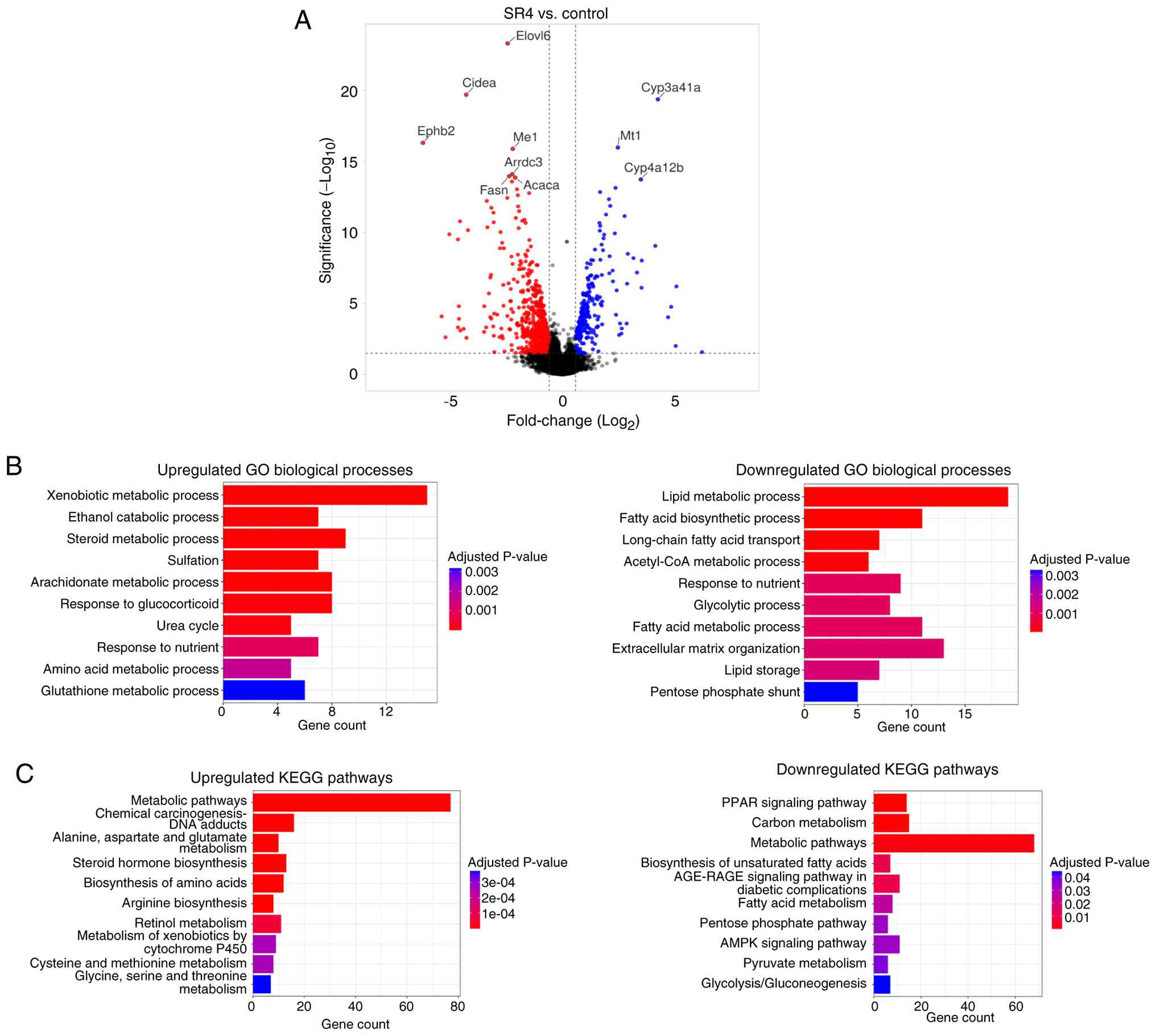
analysis identified 642 DEGs, with 217 being upregulated and 425
downregulated by SR4 treatment. Fig.
6A presents a volcano plot highlighting these DEGs based on
significance criteria (adjusted P<0.05 and absolute FC≥1.5). The
top 10 most strongly regulated genes included the elongation of
very long chain fatty acids protein 6 (Elovl6), ephrin
type-B receptor 2, cell death-inducing DFFA-like effector A, malic
enzyme 1, arrestin domain-containing 3, fatty acid synthase
(Fasn), ACC a (Acaca), cytochrome P450 3A41A,
cytochrome P450 4A12B and metallothionein 1. The functional
relevance of these DEGs to metabolic liver diseases, including
diabetes, obesity and hepatic steatosis, is summarized in Table II (34-53).
Overall, the genes most highly modulated by SR4 were primarily
associated with fatty acid synthesis, lipid accumulation and liver
steatosis, inflammatory responses and fibrotic processes, which
suggested that SR4 may exert protective effects through the
coordinated regulation of hepatic metabolic and stress-response
pathways.
| Table IITop 10 transcripts regulated by SR4
treatment in the liver of db/db mice and their functions and
association with liver metabolic diseases, diabetes and
obesity. |
Table II
Top 10 transcripts regulated by SR4
treatment in the liver of db/db mice and their functions and
association with liver metabolic diseases, diabetes and
obesity.
| Gene | Effect of SR4 | Gene function and
association | (Refs.) |
|---|
| Elovl6 | Downregulated | Elovl6
encodes fatty acid elongase 6, an enzyme primarily involved in the
elongation of C12-C16 saturated and monounsaturated fatty acids to
C18 species. Hepatic Elovl6 expression is highly upregulated
in genetically obese mouse models, such as ob/ob and
db/db mice. Notably, Elovl6 deficiency has been shown
to improve glycemic control in db/db mice. In humans,
Elovl6 has been implicated in the development and
progression of MASH by contributing to increased hepatic oxidative
stress, inflammation and fibrosis. | (34,35) |
| Ephb2 | Downregulated | The Ephb2
gene encodes EphB2, a member of the Eph receptor family of
transmembrane receptor tyrosine kinases that serves a key role in
cell-cell signaling, often through interaction with ephrin-B
ligands. EphB2 has been implicated in promoting inflammation and
fibrosis in MASH. Specifically, increased expression of
Ephb2 in liver tissue is associated with exacerbated
fibrosis, and studies in preclinical models have shown that
Ephb2 deficiency attenuates liver fibrosis and
inflammation. | (36,37) |
| Cidea | Downregulated | The Cidea
gene encodes the protein CIDEA, which serves a notable role in
various metabolic processes, particularly in the regulation of
lipid droplet formation and energy homeostasis. CIDEA functions as
a sensor for dietary saturated fatty acids and is a known
transcriptional target of sterol regulatory element-binding protein
1. Consistent with its role in lipid metabolism, Cidea
expression is markedly elevated in the livers of obese mice and
humans. The hepatic overexpression of Cidea increases lipid
accumulation and promotes the formation of larger lipid droplets.
Conversely, Cidea knockout in mice reduces these lipid
abnormalities, highlighting its key role in regulating hepatic
lipid metabolism. | (38,39) |
| Me1 | Downregulated | The Me1 gene
encodes malic enzyme 1, a cytosolic enzyme that catalyzes the
oxidative decarboxylation of malate to pyruvate, concomitantly
generating NADPH. This enzyme serves a key role in linking
glycolysis and the citric acid cycle, and the NADPH produced is
essential for various anabolic pathways, including de novo
fatty acid and cholesterol biosynthesis. Consistent with its role
in lipogenesis, studies in mice have demonstrated that Me1
knockdown can notably reduce adiposity and hepatic steatosis in
obese models. | (40,41) |
| Arrdc3 | Downregulated | The Arrdc3
gene encodes ARRD3, a member of the arrestin family of proteins
that regulate various cellular processes, including protein
trafficking and G-protein-coupled receptor signaling. ARRD3 has
been shown to directly interact with SCD1, a key enzyme in
monoun-aturated fatty acid synthesis. This interaction stabilizes
SCD1 protein levels, thereby promoting increased lipid production
and accumulation. In mice, liver-specific deletion of Arrdc3
markedly increases insulin sensitivity, while whole-body
Arrdc3 knockout mice exhibit resistance to the metabolic
complications of obesity, including reduced adiposity and hepatic
steatosis. | (42,43) |
| Fasn | Downregulated | The Fasn
gene encodes fatty acid synthase, a key enzyme in de novo
lipogenesis that catalyzes the synthesis of long-chain fatty acids
from ACC and malonyl-CoA. Consistent with its central role in lipid
synthesis, hepatic expression of Fasn is increased in both
mouse and human livers affected by steatosis. Furthermore,
pharmacological inhibition of Fasn has demonstrated efficacy
in ameliorating steatosis in mouse models and in human patients
with MASLD. | (44,45) |
| Acaca | Downregulated | The Acaca
gene encodes ACC1, a crucial enzyme in fatty acid biosynthesis.
ACC1 catalyzes the rate-limiting step in the conversion of ACC to
malonyl-CoA, which serves as a key precursor for long-chain fatty
acid synthesis. Consistent with its notable role in lipo-genesis,
hepatic overexpression of Acaca is associated with increased
fat accumulation and can contribute to MASLD. Importantly,
pharma-cological inhibition of ACC1 has been shown to reduce
hepatic steatosis and improve insulin sensitivity in both mice and
humans. | (46,47) |
|
Cyp3a41a | Upregulated | The Cyp3a41a
gene encodes the cytochrome P450 enzyme 3A41. CYP3A enzymes,
including Cyp3a41a, are known for their broad substrate
specificity, metabolizing a vast array of endogenous compounds such
as steroid hormones, bile acids and cholesterol, thereby
contributing to their regulation and systemic homeostasis. The
related isoform CYP3A4 is reduced in animals and individuals with
MASLD and MASH. | (48,49) |
|
Cyp4a12b | Upregulated | In mice,
Cyp4a12b encodes a cytochrome P450 ω-hydroxylase involved in
the metabolism of fatty acids and their oxygenated derivatives
(oxylipins). It catalyzes the ω-hydroxylation of arachidonic acid,
forming 20-hydroxyeicosatetraenoic acid. Cyp4a12b expression
is reduced during the development of liver fibrosis and
inflammation. | (50,51) |
| Mt1 | Upregulated | Hepatic expression
of Mt1, a gene encoding metallothionein 1, is markedly
downregulated in liver tissues of patients with MASH and in
high-fat diet-induced mouse models. This hepatic downregulation of
Mt1 is associated with increased TG and total cholesterol
levels, exacerbated lipid accumulation, and elevated liver fibrosis
and inflammation | (52,53) |
To evaluate the biological significance of the DEGs,
GO and KEGG pathway enrichment analyses were performed. GO
biological process analysis indicated that the 217 upregulated DEGs
were significantly enriched in pathways related to ‘Xenobiotic
metabolic process’, ‘Steroid metabolic process’, ‘Amino acid
metabolic process’, ‘Urea cycle’ and ‘Glutathione metabolic
process’ (Fig. 6B). By contrast,
the 425 downregulated DEGs were primarily associated with ‘Lipid
metabolic process’, ‘Fatty acid biosynthetic process’, ‘Lipid
storage’, ‘Glycolytic process’ and the ‘Pentose phosphate shunt’.
KEGG pathway analysis revealed that upregulated DEGs were enriched
in ‘Metabolic pathways’, ‘Alanine, aspartate and glutamate
metabolism’, ‘Chemical carcinogenesis-DNA adducts’, ‘Steroid
hormone biosynthesis’, ‘Biosynthesis of amino acids’, ‘Retinol
metabolism’ and ‘Metabolism of xenobiotics by cytochrome P450’
(Fig. 6C). Downregulated DEGs were
significantly associated with the ‘PPAR signaling pathway’, ‘Carbon
metabolism’, ‘Fatty acid metabolism’, ‘AMPK signaling pathway’,
‘Glycolysis/Gluconeogenesis’ and the ‘Pentose phosphate pathway’.
These results indicated that the transcriptional changes induced by
SR4 treatment involve key pathways regulating nutrient metabolism,
energy homeostasis and oxidative stress, processes that are
involved in the pathogenesis of T2D and progression to MASLD.
To assess the functional relationships among the
identified DEGs, a PPI network was constructed using the STRING
database with a high-confidence interaction score threshold of 0.7.
The resulting network, derived from the 642 DEGs, comprised 328
nodes (representing 328 of the DEGs) and 764 edges, forming its
largest connected component (Fig.
7A). This number of observed edges was significantly greater
than expected for a random network of similar size
(P≤1x10-16), indicating that the encoded proteins are
more interconnected than would occur by chance and are likely
involved in biologically related processes. Among the 328 genes in
the network, 118 were upregulated and 210 were downregulated.
Network clustering analysis using the ‘MCODE’ plugin in Cytoscape
identified ≥20 subnetwork modules. The top five modules were
functionally enriched in distinct metabolic processes. Cluster 1
was associated with steroid hormone biosynthesis and retinol
metabolism, cluster 2 with the pentose phosphate pathway (PPP) and
glycolysis, cluster 3 with fatty acid synthesis and pyruvate
metabolism, cluster 4 with lipid droplet metabolism and cluster 5
with cellular amino acid catabolic pathways (Fig. 7B-F).
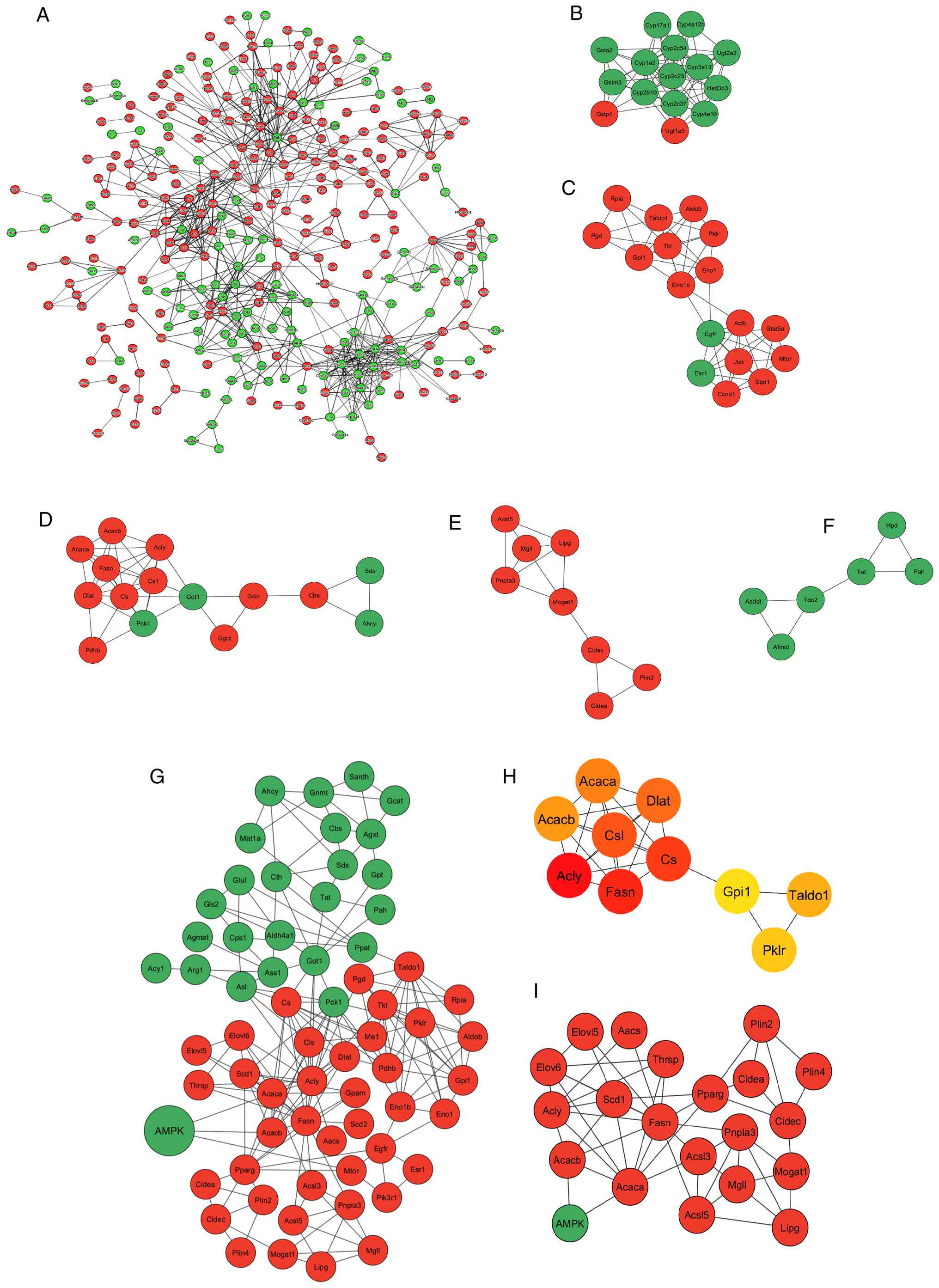 | Figure 7PPI networks and subnetworks
(modules) of DEGs in SR4-treated db/db mice livers. (A) PPI
network of all 642 DEGs. For clarity, disconnected nodes are not
shown. Circles represent genes (nodes) and lines between them
represent interactions (edges). The top 5 most significant modules
from the PPI network of DEGs, identified by MCODE screening. These
modules are associated with (B) steroid hormone biosynthesis, (C)
pentose phosphate pathway and glycolysis, (D) fatty acid synthesis
and pyruvate metabolism, (E) lipid droplet metabolism and (F)
metabolism of amino acids and derivatives. (G) PPI subnetwork for
genes associated with AMPK and nutrient and energy metabolic
pathways, as identified by GO and KEGG enrichment analyses. (H) Top
10 hub genes identified in the subnetwork in (G) using the ‘Maximal
Clique Centrality’ plugin of CytoHubba. (I) Putative
AMPK/PPARγ-associated regulatory axis for fatty acid and TG
synthesis, TG production, lipid droplet formation and storage. In
panels (A-G) and (I), green and red circles represent upregulated
and downregulated genes, respectively. In panel (H), red circles
indicate hub genes with high-ranking scores, while yellow
rectangles represent hub genes with lower-ranking scores. PPARγ,
peroxisome proliferator-activated receptor γ; PPI, protein-protein
interaction; TG, triglycerides; DEGs, differentially expressed
genes. |
Due to the central role of AMPK in regulating
nutrient and energy metabolism, along with its increased protein
phosphorylation and pathway enrichment in the present study, a
focused PPI subnetwork was constructed to explore functionally
connected gene modules. This subnetwork encompassed DEGs implicated
in AMPK signaling, amino acid metabolism, lipid metabolism and
glucose metabolism, including key components of the PPP and
pyruvate metabolism, as identified through GO and KEGG enrichment
analyses. The resulting network comprised 65 nodes and 191 edges,
underscoring a high degree of interconnectivity among genes
involved in core metabolic processes (Fig. 7G). Notably, multiple interactions
within the subnetwork converged on key AMPK regulatory nodes,
including Acaca and Acacb (encoding ACC 1 and 2,
respectively), Pparg (PPARγ) and mammalian target of
rapamycin (mTor), highlighting the intricate integration of
lipid biosynthesis with energy-sensing pathways. To pinpoint
central regulatory elements within the subnetwork, hub gene
analysis was performed using the MCC algorithm within the
‘cytoHubba’ plugin. This analysis identified a number of prominent
hub genes, with Fasn and Acly exhibiting the highest
MCC scores (Fig. 7H).
Additionally, a putative AMPK/PPARγ-associated regulatory network
was identified, linking fatty acid synthesis, TG production, lipid
droplet formation and storage (Fig.
7I). Collectively, these findings suggested that genes involved
in lipid metabolism serve key roles in mediating the SR4-induced
metabolic reprogramming in the liver.
Discussion
Mitochondrial uncouplers have emerged as a promising
class of therapeutics for metabolic disorders such as obesity, T2D,
MASLD and certain cancer types. These compounds act by dissipating
the proton gradient across the mitochondrial inner membrane. By
uncoupling oxidative phosphorylation from ATP synthesis, they
increase energy expenditure and improve metabolic parameters,
making them promising candidates for treating diseases
characterized by nutrient excess and metabolic dysfunction
(9-11,17).
Numerous uncouplers have demonstrated both metabolic efficacy and
favorable safety profiles in preclinical models and are now
advancing through clinical development. Niclosamide, originally
developed as an antihelmintic agent, exhibits mitochondrial
uncoupling properties and is currently under clinical evaluation
for cancer therapy (trial no. NCT02519582 and NCT05188170) with
additional metabolic benefits reported in a preclinical study
(54). Other uncouplers such as
TLC-6740, under investigation for obesity and T2D (trial no.
NCT05822544) and HU6, which is in phase II trials for MASH (trial
no. NCT04874233 and NCT05979779), highlight the increasing clinical
momentum for this therapeutic strategy.
SR4 represents a newer class of mitochondrial
uncouplers known as synthetic anion fatty acid transporters
(15,16). These compounds facilitate the
transport of fatty acid anions across the mitochondrial inner
membrane, where the anions act as proton shuttles to induce
controlled mitochondrial uncoupling. Unlike classical protonophores
such as FCCP and 2,4-dinitrophenol which cause rapid and complete
collapse of the mitochondrial membrane potential, leading to full
depolarization, severe off-target effects including plasma membrane
disruption and dose-dependent cytotoxicity (55), fatty acid anion transporters such
as SR4 may provide greater mitochondrial specificity and safety
through gradual, physiologically controlled depolarization. This
mechanism of action renders SR4 particularly promising for
long-term treatment of chronic metabolic conditions such as
obesity, T2D and MASH, where sustained and safe increase in energy
expenditure and metabolism is therapeutically advantageous.
Previous studies have shown that a BW loss of 5-10%
is sufficient to markedly improve insulin sensitivity, lipid
profiles and hepatic steatosis, thereby lowering the risk for T2D
and MASLD (56,57). In the present study, SR4 treatment
led to a significant 13% reduction in BW in db/db mice
across 5 weeks. This weight loss occurred without any decrease in
food intake, indicating that SR4 primarily enhances metabolism and
energy expenditure rather than suppressing appetite. Notably, this
weight reduction was associated with a favorable shift in body
composition as SR4 specifically reduced fat mass while preserving
lean mass. This outcome is clinically desirable as numerous
existing weight loss drugs such as glucagon-like peptide-1 receptor
(GLP-1R) agonists and dual GLP-1R/glucose-dependent insulinotropic
polypeptide receptor agonists, can cause a loss of both fat and
lean tissue mass (58). Preserving
lean mass during weight loss reduction is key for maintaining
muscle strength and healthy metabolism and decreasing risk of
physical injury (59).
In the present study, SR4 treatment improved
hyperglycemia and dyslipidemia and exerted potent anti-steatotic
effects in db/db mice, as evidenced by reduced liver weight,
lower TG content and fewer hepatic lipid droplets. Mechanistically,
SR4 increased the hepatic AMP:ATP ratio, leading to activation of
AMPK, a central regulator of energy homeostasis that promotes
catabolic pathways while inhibiting anabolic processes such as
de novo lipogenesis (60).
Transcriptomic and qPCR analyses revealed that SR4 suppressed
several key lipogenic genes and transcription factors, including
Acaca, Acly, Fasn, Scd1, Cebpa,
Srebf1 and Pparg. with additional enrichment analyses
confirming downregulation of fatty acid biosynthesis and lipid
storage pathways. PPI networks identified gene clusters downstream
of AMPK and PPARγ signaling, implicating a coordinated regulatory
axis controlling fatty acid synthesis, TG accumulation and lipid
droplet formation. These results are consistent with previous
observations in HFD-fed obese mice (17) and align with previous studies
linking AMPK-dependent pathways involving Acaca,
Fasn, Scd1, Elovl6 and Mogat1 as key
mediators of de novo lipid synthesis (61). SR4 has also been shown to prevent
hepatic steatosis, fibrosis and liver injury in TERF2 interacting
protein-deficient MASH mice through AMPK activation (62). Thus, SR4 appears to improve hepatic
metabolic homeostasis by activating AMPK and reprogramming lipid
metabolism.
Both qPCR and transcriptomic analyses also revealed
that SR4 downregulated hepatic PPARγ expression. While PPARγ is
predominantly expressed in adipose tissue, the ectopic induction of
PPARγ is observed in the steatotic liver. Genetic deletion or
pharmacological inhibition of hepatic PPARγ reduces steatosis and
inflammation (63,64). Furthermore, PPARγ activity is
negatively regulated by AMPK and mTOR, with AMPK phosphorylation
suppressing its transcriptional activity and mTOR inhibition
attenuating lipogenesis (65,66).
Consistent with this, SR4 activated AMPK and inhibited mTOR,
providing a plausible mechanism for reduced PPARγ expression and
subsequent improvement in hepatic lipid metabolism.
Mitochondrial respiratory dysfunction is a hallmark
of metabolic diseases. A decrease in mitochondrial respiration has
been observed in obese T2D mice (67). Using Seahorse extracellular flux
analyses, the present study found that SR4 significantly enhanced
mitochondrial respiration, increasing OCR across multiple
respiratory states including basal (state 2), ADP-stimulated (state
3), uncoupled maximal respiration (state 3u), proton leak and SRC.
This improved respiratory capacity suggests that SR4 helps to
improve the use of metabolic substrates for energy by the liver,
contributing to increased whole-body energy expenditure. Similar
effects have been observed with the mitochondrial uncoupler BAM15,
which enhances mitochondrial oxidative capacity partly by
peroxisome proliferator-activated receptor γ coactivator-1 α
activation (68). Whether SR4
promotes mitochondrial biogenesis through similar pathways remains
to be investigated.
SR4 also modulated oxidative stress, a key
contributor to liver injury in metabolic disease. The present study
found that SR4 treatment significantly reduced hepatic MDA levels,
indicating attenuation of lipid peroxidation. This effect was
accompanied by increased activity of GSH-linked antioxidant
enzymes, including GPx and GST. These enzymes serve key roles in
detoxifying reactive oxygen species as GPx reduces hydrogen
peroxide and lipid hydroperoxides using reduced GSH, while GSTs
catalyze the conjugation of GSH to electrophilic substrates,
facilitating their excretion. Transcriptomic analysis further
supported this observation, revealing significant upregulation of
genes involved in GSH metabolism, such as GSH S-transferase α 2,
GSH S-transferase κ 1, GSH S-transferase mu 7 and GSH S-transferase
θ 3. This dual action, enhancing mitochondrial respiratory function
while bolstering antioxidant defenses, could be an important
mechanism by which SR4 prevents liver injury in db/db
mice.
Beyond lipid metabolism, transcriptomic analysis
also revealed that SR4 induces a broader metabolic reprogramming in
the liver of db/db mice. SR4 upregulated genes involved in
‘Amino acid metabolic process’, while downregulating those
associated with ‘Glycolysis/Gluconeogenesis’, ‘Pyruvate metabolism’
and the ‘PPP’. This shift resembles the adaptive metabolic response
observed during caloric restriction or fasting, where anabolic
pathways are suppressed and catabolic processes are enhanced to
maintain energy homeostasis and preserve mitochondrial function
under nutrient-limited conditions. A key feature of this adaptive
response is the enhancement of glutamine metabolism. Through
glutaminolysis, glutamine contributes to anaplerosis by
replenishing TCA cycle intermediates, thereby supporting
mitochondrial ATP production when glycolytic flux is reduced
(69). Glutamine also serves as a
precursor for GSH synthesis, thereby enhancing cellular antioxidant
defenses (70). These functions
are especially important under conditions of energetic stress,
where metabolic flexibility is vital for cellular survival and
function. Additionally, during amino acid catabolism, the removal
and transfer of the amino group through deamination and
transamination provides carbon skeletons that can be utilized in
the TCA cycle for energy generation, while the α-amino nitrogen
atoms are converted into ammonia, which is then detoxified and
converted to urea through the urea cycle for safe excretion
(71). Notably, SR4 increased the
expression of urea cycle genes, reflecting greater amino acid
turnover and nitrogen disposal.
The downregulation of the PPP in liver of
SR4-treated mice further underscores a redirection of glucose flux
away from anabolic biosynthesis. As a major source of NADPH, the
PPP primarily supports reductive biosynthesis and maintains redox
balance through the regeneration of reduced GSH (72). Suppression of this pathway may
therefore indicate a systemic reduction in overall biosynthetic
activity, aligning with the energy-conserving, catabolic state
induced by SR4. Additionally, inhibition of pyruvate metabolism may
restrict the production of ACC, a central metabolic intermediate
required for both TCA cycle activity and de novo
lipogenesis. Limiting ACC availability could contribute to the
observed suppression of lipogenic gene expression and the reduction
in hepatic TG content seen with SR4 treatment. Collectively, these
findings suggest that SR4 promotes a hepatic metabolic program
reminiscent of nutrient deprivation, characterized by metabolic
flexibility, enhanced oxidative metabolism and inhibition of
lipogenesis, which may underlie its therapeutic benefits in
obesity, insulin resistance and MASLD.
Despite these promising findings, the present study
has some limitations. First, only male db/db mice were used,
which may not capture sex-dependent metabolic responses, as female
mice often exhibit different disease progression and therapeutic
sensitivity (73). Future studies
should include both sexes to enhance translational relevance.
Second, the absence of comparisons with standard anti-diabetic or
anti-steatosis drugs limits direct clinical interpretation.
Notably, a recent head-to-head study of female db/db mice
compared the mitochondria uncoupler BAM15 with semaglutide,
rosiglitazone and niclosamide and reported that BAM15 exhibited
greater weight loss reduction and improvements in dyslipidemia and
steatosis (74). Third, the
present investigation focused solely on hepatic outcomes, leaving
the systemic impact of SR4 on other metabolically active tissues
such as skeletal muscles and adipose tissues to be determined.
Finally, the absence of pharmacokinetic and bioavailability studies
limits the understanding of SR4 in vivo. Our preliminary
study has shown that SR4 preferentially accumulates in adipose
tissues and liver, consistent with its high lipophilic properties.
Future work will need to address these gaps to fully elucidate the
therapeutic potential and mechanism of action of SR4.
In conclusion, the present study provided evidence
that SR4 is an effective orally administered agent that ameliorates
multiple hallmark features of metabolic dysfunction in T2D,
including hyperglycemia, insulin resistance, dyslipidemia, hepatic
steatosis and liver injury. SR4 promoted BW loss and improved body
composition by reducing fat mass while preserving lean mass,
without suppressing appetite, which highlighted its potential as a
safe and sustainable therapeutic strategy. Mechanistically, these
effects appeared to be mediated through mitochondrial uncoupling,
AMPK activation, modulation of mTOR and PPARγ signaling, improved
mitochondrial bioenergetics, antioxidant defense and global
metabolic reprogramming (Fig. 8).
Together, these findings support further preclinical and clinical
development of SR4 and related fatty acid anion transporters as a
novel therapeutic class for treating metabolic diseases.
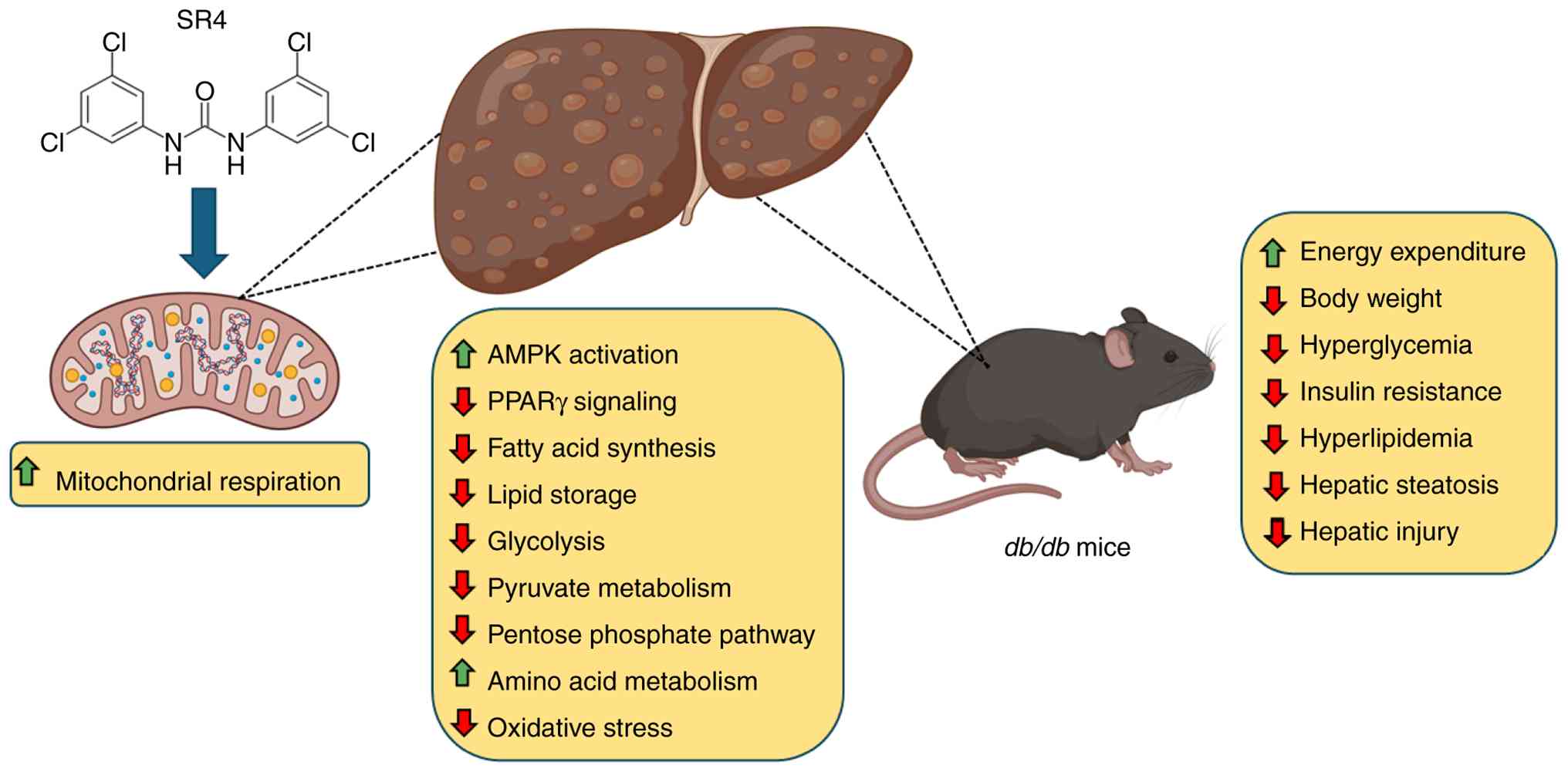 | Figure 8Summary of the metabolic effects of
SR4 on the liver of db/db mice. SR4 functions as a
mitochondrial uncoupler by facilitating fatty acid-activated proton
transport across the inner mitochondrial membrane. As a bisaryl
urea-based anion transporter, SR4 enhances the flip-flop movement
of deprotonated fatty acids across the inner mitochondrial
membrane, promoting proton leak. This uncoupling disrupts the
mitochondrial proton gradient, leading to reduced ATP synthesis.
The resulting decline in cellular ATP levels activates AMPK, a key
regulator of energy homeostasis. In the liver, AMPK activation
suppresses lipogenic pathways, including PPARγ signaling, while
promoting catabolic processes that support mitochondrial function
and energy utilization. This metabolic reprogramming enhances
mitochondrial respiration and oxidative capacity, reduces oxidative
stress and improves metabolic flexibility. Collectively, these
effects contribute to broad metabolic improvements in db/db
mice, including increased energy expenditure and metabolic
activity, reduced nody weight, improved glycemic control, enhanced
insulin sensitivity, correction of dyslipidemia and attenuation of
hepatic steatosis and injury. Figure partly created in BioRender.
PPARγ, peroxisome proliferator-activated receptor γ. |
Acknowledgements
The authors wish to thank Dr Patrick Fueger
(Comprehensive Metabolic Phenotyping Core Facility) for assistance
in the indirect calorimetry studies, Dr Lu Yang and Dr Xiwei Wu
(Integrative Genomics and Bioinformatics Core) and Dr Yate-Ching
Yuan (Division of Research Informatics) for the help in RNA
sequencing data analyses and database submission, and Mrs. Leslie
Smith-Powell for the nucleotide measurements (Analytical
Pharmacology Core).
Funding
Funding: The present study was partly supported by the Samuel
Rahbar Diabetes and Drug Discovery Endowment, Arthur Riggs Diabetes
& Metabolism Research Institute and innovation grant from the
Wanek Family Project for Type 1 Diabetes at City of Hope.
Availability of data and materials
The data generated in the present study may be found
in the National Center for Biotechnology Information Gene
Expression Omnibus database under accession number GSE308701 or at
the following URL: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE308701.
Authors' contributions
JLF conceptualized, designed and conducted the
experiments, collected and analyzed data and wrote the manuscript.
SS conducted experiments, collected data and analyzed results and
reviewed the manuscript. JS performed experiments, collected data
and reviewed the manuscript. All authors confirm the authenticity
of all the raw data. All authors read and approved the final
version of the manuscript.
Ethics approval and consent to
participate
All animal experiments performed in the present
study were approved by the Institutional Animal Care and Use
Committee of the City of Hope National Medical Center (approval no.
12004; Duarte, USA).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Use of artificial intelligence tools
During the preparation of this work, artificial
intelligence tools were used to improve the readability and
language of the manuscript or to generate images, and subsequently,
the authors revised and edited the content produced by the
artificial intelligence tools as necessary, taking full
responsibility for the ultimate content of the present
manuscript.
References
|
1
|
American Diabetes Association Professional
Practice Committee. Classification and diagnosis of diabetes: 2.
Diagnosis and Classification of Diabetes: Standards of Care in
Diabetes-2024. Diabetes Care. 47 (Suppl 1):S20–S42. 2024.PubMed/NCBI View Article : Google Scholar
|
|
2
|
International Diabetes Federation: IDF
Diabetes Atlas, 11th edition. Brussels, Belgium, IDF, 2025.
|
|
3
|
Khan MAB, et al: Global burden of diabetes
and its projections to 2050: A systematic review. Lancet Diabetes
Endocrinol, 2023.
|
|
4
|
Mooradian AD: Dyslipidemia in type 2
diabetes mellitus. Nat Clin Pract Endocrinol Metab. 5:150–159.
2009.PubMed/NCBI View Article : Google Scholar
|
|
5
|
Caturano A, D'Angelo M, Mormone A, Russo
V, Mollica MP, Salvatore T, Galiero R, Rinaldi L, Vetrano E,
Marfella R, et al: Oxidative stress in type 2 diabetes: Impacts
from pathogenesis to lifestyle modifications. Curr Issues Mol Biol.
45:6651–6666. 2023.PubMed/NCBI View Article : Google Scholar
|
|
6
|
Younossi ZM, Golabi P, Price JK, Owrangi
S, Gundu-Rao N, Satchi R and Paik JM: The global epidemiology of
nonalcoholic fatty liver disease and nonalcoholic steatohepatitis
among patients with type 2 diabetes. Clin Gastroenterol Hepatol.
22:1999–2010.e8. 2024.PubMed/NCBI View Article : Google Scholar
|
|
7
|
Younossi ZM, Kalligeros M and Henry L:
Epidemiology of metabolic dysfunction-associated steatotic liver
disease. Clin Mol Hepatol. 31 (Suppl):S32–S50. 2025.PubMed/NCBI View Article : Google Scholar
|
|
8
|
Davies MJ, Aroda VR, Collins BS, Gabbay
RA, Green J, Maruthur NM, Rosas SE, Del Prato S, Mathieu C,
Mingrone G, et al: Management of hyperglycemia in type 2 diabetes,
2022. A consensus report by the American diabetes association (ADA)
and the european association for the study of diabetes (EASD).
Diabetes Care. 45:2753–2786. 2022.PubMed/NCBI View Article : Google Scholar
|
|
9
|
Goedeke L and Shulman GI: Therapeutic
potential of mitochondrial uncouplers for the treatment of
metabolic associated fatty liver disease and NASH. Mol Metab.
46(101178)2021.PubMed/NCBI View Article : Google Scholar
|
|
10
|
Alexopoulos SJ, Chen SY, Brandon AE,
Salamoun JM, Byrne FL, Garcia CJ, Beretta M, Olzomer EM, Shah DP,
Philp AM, et al: Mitochondrial uncoupler BAM15 reverses
diet-induced obesity and insulin resistance in mice. Nat Commun.
11(2397)2020.PubMed/NCBI View Article : Google Scholar
|
|
11
|
Kanemoto N, Okamoto T, Tanabe K, Shimada
T, Minoshima H, Hidoh Y, Aoyama M, Ban T, Kobayashi Y, Ando H, et
al: Antidiabetic and cardiovascular beneficial effects of a
liver-localized mitochondrial uncoupler. Nat Commun.
10(2172)2019.PubMed/NCBI View Article : Google Scholar
|
|
12
|
Figarola JL, Singhal J, Tompkins JD,
Rogers GW, Warden C, Horne D, Riggs AD, Awasthi S and Singhal SS:
SR4 Uncouples mitochondrial oxidative phosphorylation, modulates
AMP-dependent kinase (AMPK)-mammalian target of rapamycin (mTOR)
signaling, and inhibits proliferation of HepG2 hepatocarcinoma
cells. J Biol Chem. 290:30321–30341. 2015.PubMed/NCBI View Article : Google Scholar
|
|
13
|
Singhal SS, Figarola J, Singhal J,
Nagaprashantha L, Berz D, Rahbar S and Awasthi S: Novel compound
1,3-bis (3,5-dichlorophenyl) urea inhibits lung cancer progression.
Biochem Pharmacol. 86:1664–1672. 2013.PubMed/NCBI View Article : Google Scholar
|
|
14
|
Figarola JL, Singhal J, Singhal S, Kusari
J and Riggs A: Bioenergetic modulation with the mitochondria
uncouplers SR4 and niclosamide prevents proliferation and growth of
treatment-naïve and vemurafenib-resistant melanomas. Oncotarget.
9:36945–36965. 2018.PubMed/NCBI View Article : Google Scholar
|
|
15
|
Rawling T, MacDermott-Opeskin H, Roseblade
A, Pazderka C, Clarke C, Bourget K, Wu X, Lewis W, Noble B, Gale
PA, et al: Aryl urea substituted fatty acids: A new class of
protonophoric mitochondrial uncoupler that utilises a synthetic
anion transporter. Chem Sci. 11:12677–12685. 2020.PubMed/NCBI View Article : Google Scholar
|
|
16
|
York E, McNaughton DA, Roseblade A,
Cranfield CG, Gale PA and Rawling T: Structure-activity
relationship and mechanistic studies of bisaryl urea anticancer
agents indicate mitochondrial uncoupling by a fatty Acid-Activated
Mechanism. ACS Chem Biol. 17:2065–2073. 2022.PubMed/NCBI View Article : Google Scholar
|
|
17
|
Figarola JL, Singhal P, Rahbar S, Gugiu
BG, Awasthi S and Singhal SS: COH-SR4 reduces body weight, improves
glycemic control and prevents hepatic steatosis in high fat
diet-induced obese mice. PLoS One. 8(e83801)2013.PubMed/NCBI View Article : Google Scholar
|
|
18
|
Lau JK, Zhang X and Yu J: Animal models of
non-alcoholic fatty liver disease: Current perspectives and recent
advances. J Pathol. 241:36–44. 2017.PubMed/NCBI View Article : Google Scholar
|
|
19
|
Benedé-Ubieto R, Estévez-Vázquez O,
Ramadori P, Cubero FJ and Nevzorova YA: Guidelines and
considerations for metabolic tolerance tests in mice. Diabetes
Metab Syndr Obes. 13:439–450. 2020.PubMed/NCBI View Article : Google Scholar
|
|
20
|
Noeman SA, Hamooda HE and Baalash AA:
Biochemical study of oxidative stress markers in the liver, kidney
and heart of high fat diet induced obesity in rats. Diabetol Metab
Syndr. 3(17)2011.PubMed/NCBI View Article : Google Scholar
|
|
21
|
Fraga CG, Leibovitz BE and Tappel AL:
Lipid peroxidation measured as thiobarbituric acid-reactive
substances in tissue slices: Characterization and comparison with
homogenates and microsomes. Free Radic Biol Med. 4:155–1561.
1988.PubMed/NCBI View Article : Google Scholar
|
|
22
|
Iuso A, Repp B, Biagosch C, Terrile C and
Prokisch H: Assessing mitochondrial bioenergetics in isolated
mitochondria from various mouse tissues using Seahorse XF96
analyzer. Methods Mol Biol. 1567:217–230. 2017.PubMed/NCBI View Article : Google Scholar
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-delta delta C(T)) method. Methods. 25:402–408.
2001.PubMed/NCBI View Article : Google Scholar
|
|
24
|
Robinson MD, McCarthy DJ and Smyth GK:
edgeR: A Bioconductor package for differential expression analysis
of digital gene expression data. Bioinformatics. 26:139–140.
2010.PubMed/NCBI View Article : Google Scholar
|
|
25
|
Love MI, Huber W and Anders S: Moderated
estimation of fold change and dispersion for RNA-seq data with
DESeq2. Genome Biol. 15(550)2014.PubMed/NCBI View Article : Google Scholar
|
|
26
|
Goedhart J and Luijsterburg MS: VolcaNoseR
is a web app for creating, exploring, labeling and sharing volcano
plots. Sci Rep. 10(20560)2020.PubMed/NCBI View Article : Google Scholar
|
|
27
|
Huang DW, Sherman BT, Lempicki RA and
Lemoine S: DAVID: A web server for functional enrichment analysis
and functional annotation of gene lists (2021 update). Nucleic
Acids Res. 50:W216–W221. 2022.PubMed/NCBI View Article : Google Scholar
|
|
28
|
Szklarczyk D, Nastou K, Koutrouli M,
Kirsch R, Mehryary F, Hachilif R, Hu D, Peluso ME, Huang Q, Fang T,
et al: The STRING database in 2025: Protein networks with
directionality of regulation. Nucleic Acids Res. 53:D730–D737.
2025.PubMed/NCBI View Article : Google Scholar
|
|
29
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003.PubMed/NCBI View Article : Google Scholar
|
|
30
|
Bader GD and Hogue CW: An automated method
for finding molecular complexes in large protein interaction
networks. BMC Bioinformatics. 4(2)2003.PubMed/NCBI View Article : Google Scholar
|
|
31
|
Chin CH, Chen SH, Wu HH, Ho CW, Ko MT and
Lin CY: cytoHubba: Identifying hub objects and sub-networks from
complex interactome. BMC Syst Biol. 8 (Suppl 4)(S11)2014.PubMed/NCBI View Article : Google Scholar
|
|
32
|
Burke SJ, Batdorf HM, Burk DH, Noland RC,
Eder AE, Boulos MS, Karlstad MD and Collier JJ: Db/db mice exhibit
features of human type 2 diabetes that are not present in
weight-matched C57BL/6J mice fed a Western diet. J Diabetes Res.
2017(8503754)2017.PubMed/NCBI View Article : Google Scholar
|
|
33
|
Sahai A, Malladi P, Pan X, Paul R,
Melin-Aldana H, Green RM and Whitington PF: Obese and diabetic
db/db mice develop marked liver fibrosis in a model of nonalcoholic
steatohepatitis: Role of short-form leptin receptors and
osteopontin. Am J Physiol Gastrointest Liver Physiol.
287:G1035–G1043. 2004.PubMed/NCBI View Article : Google Scholar
|
|
34
|
Zhao H, Matsuzaka T, Nakano Y, Motomura K,
Tang N, Yokoo T, Okajima Y, Han SI, Takeuchi Y, Aita Y, et al:
Elovl6 deficiency improves glycemic control in diabetic db/db mice
by expanding β-Cell mass and increasing insulin secretory capacity.
Diabetes. 66:1833–1846. 2017.PubMed/NCBI View Article : Google Scholar
|
|
35
|
Matsuzaka T, Atsumi A, Matsumori R, Nie T,
Shinozaki H, Suzuki-Kemuriyama N, Kuba M, Nakagawa Y, Ishii K,
Shimada M, et al: Elovl6 promotes nonalcoholic steatohepatitis.
Hepatology. 56:2199–2208. 2012.PubMed/NCBI View Article : Google Scholar
|
|
36
|
Xiao Y, Batmanov K, Hu W, Zhu K, Tom AY,
Guan D, Jiang C, Cheng L, McCright SJ, Yang EC, et al: Hepatocytes
demarcated by EphB2 contribute to the progression of nonalcoholic
steatohepatitis. Sci Transl Med. 15(eadc9653)2023.PubMed/NCBI View Article : Google Scholar
|
|
37
|
Mimche PN, Lee CM, Mimche SM, Thapa M,
Grakoui A, Henkemeyer M and Lamb TJ: EphB2 receptor tyrosine kinase
promotes hepatic fibrogenesis in mice via activation of hepatic
stellate cells. Sci Rep. 8(2532)2018.PubMed/NCBI View Article : Google Scholar
|
|
38
|
Zhou L, Xu L, Ye J, Li D, Wang W, Li X, Wu
L, Wang H, Guan F and Li P: Cidea promotes hepatic steatosis by
sensing dietary fatty acids. Hepatology. 56:95–107. 2012.PubMed/NCBI View Article : Google Scholar
|
|
39
|
Sans A, Bonnafous S, Rousseau D, Patouraux
S, Canivet CM, Leclere PS, Tran-Van-Nhieu J, Luci C, Bailly-Maitre
B, Xu X, et al: The differential expression of Cide family members
is associated with NAFLD progression from steatosis to
steatohepatitis. Sci Rep. 9(7501)2019.PubMed/NCBI View Article : Google Scholar
|
|
40
|
Simmen FA, Pabona JMP, Al-Dwairi A,
Alhallak I, Montales MTE and Simmen RCM: Malic enzyme 1 (ME1)
promotes adiposity and hepatic steatosis and induces circulating
insulin and leptin in obese female mice. Int J Mol Sci.
24(6613)2023.PubMed/NCBI View Article : Google Scholar
|
|
41
|
Al-Dwairi A, Pabona JM, Simmen RC and
Simmen FA: Cytosolic malic enzyme 1 (ME1) mediates high fat
diet-induced adiposity, endocrine profile, and gastrointestinal
tract proliferation-associated biomarkers in male mice. PLoS One.
7(e46716)2012.PubMed/NCBI View Article : Google Scholar
|
|
42
|
Batista TM, Dagdeviren S, Carroll SH, Cai
W, Melnik VY, Noh HL, Saengnipanthkul S, Kim JK, Kahn CR and Lee
RT: Arrestin domain-containing 3 (Arrdc3) modulates insulin action
and glucose metabolism in liver. Proc Natl Acad Sci USA.
117:6733–6740. 2020.PubMed/NCBI View Article : Google Scholar
|
|
43
|
Patwari P, Emilsson V, Schadt EE, Chutkow
WA, Lee S, Marsili A, Zhang Y, Dobrin R, Cohen DE, Larsen PR, et
al: The arrestin domain-containing 3 protein regulates body mass
and energy expenditure. Cell Metab. 14:671–683. 2011.PubMed/NCBI View Article : Google Scholar
|
|
44
|
Dorn C, Riener MO, Kirovski G, Saugspier
M, Steib K, Weiss TS, Gäbele E, Kristiansen G, Hartmann A and
Hellerbrand C: Expression of fatty acid synthase in nonalcoholic
fatty liver disease. Int J Clin Exp Pathol. 3:505–514.
2010.PubMed/NCBI
|
|
45
|
O'Farrell M, Duke G, Crowley R, Buckley D,
Martins EB, Bhattacharya D, Friedman SL and Kemble G: FASN
inhibition targets multiple drivers of NASH by reducing steatosis,
inflammation and fibrosis in preclinical models. Sci Rep.
12(15661)2022.PubMed/NCBI View Article : Google Scholar
|
|
46
|
Mateo-Marín MA and Alves-Bezerra M:
Targeting acetyl-CoA carboxylases for the treatment of MASLD. J
Lipid Res. 65(100676)2024.PubMed/NCBI View Article : Google Scholar
|
|
47
|
Tamura YO, Sugama J, Iwasaki S, Sasaki M,
Yasuno H, Aoyama K, Watanabe M, Erion DM and Yashiro H: Selective
Acetyl-CoA carboxylase 1 inhibitor improves hepatic steatosis and
hepatic fibrosis in a preclinical nonalcoholic steatohepatitis
model. J Pharmacol Exp Ther. 379:280–289. 2021.PubMed/NCBI View Article : Google Scholar
|
|
48
|
Jamwal R, de la Monte SM, Ogasawara K,
Adusumalli S, Barlock BB and Akhlaghi F: Nonalcoholic fatty liver
disease and diabetes are associated with decreased CYP3A4 protein
expression and activity in human liver. Mol Pharm. 15:2621–2632.
2018.PubMed/NCBI View Article : Google Scholar
|
|
49
|
Woolsey SJ, Mansell SE, Kim RB, Tirona RG
and Beaton MD: CYP3A activity and expression in nonalcoholic fatty
liver disease. Drug Metab Dispos. 43:1484–1490. 2015.PubMed/NCBI View Article : Google Scholar
|
|
50
|
Ping DB, Sun X, Peng Y and Liu CH:
Cyp4a12-mediated retinol metabolism in stellate cells is the
antihepatic fibrosis mechanism of the Chinese medicine Fuzheng
Huayu recipe. Chin Med. 18(51)2023.PubMed/NCBI View Article : Google Scholar
|
|
51
|
Theken KN, Deng Y, Kannon MA, Miller TM,
Poloyac SM and Lee CR: Activation of the acute inflammatory
response alters cytochrome P450 expression and eicosanoid
metabolism. Drug Metab Dispos. 39:22–29. 2011.PubMed/NCBI View Article : Google Scholar
|
|
52
|
Li X, Zhong S, Sun Y, Huang X, Li Y, Wang
L, Wu Y, Yang M, Yuan HX, Liu J and Zang S: Integration analysis
identifies the role of metallothionein in the progression from
hepatic steatosis to steatohepatitis. Front Endocrinol (Lausanne).
13(951093)2022.PubMed/NCBI View Article : Google Scholar
|
|
53
|
Guan C, Zou X, Shi W, Gao J, Yang C, Ge Y,
Xu Z, Bi S and Zhong X: Metallothionein 1B attenuates inflammation
and hepatic steatosis in MASH by inhibiting the AKT/PI3K pathway. J
Lipid Res. 66(100701)2025.PubMed/NCBI View Article : Google Scholar
|
|
54
|
Laila UE, Zhao ZL, Xu DY, Liu H and Xu ZX:
Pharmacological advances and therapeutic applications of
niclosamide in cancer and other diseases. Eur J Med Chem.
290(117527)2025.PubMed/NCBI View Article : Google Scholar
|
|
55
|
Park KS, Jo I, Pak K, Bae SW, Rhim H, Suh
SH, Park J, Zhu H, So I and Kim KW: FCCP depolarizes plasma
membrane potential by activating proton and Na+ currents in bovine
aortic endothelial cells. Pflugers Arch. 443:344–352.
2002.PubMed/NCBI View Article : Google Scholar
|
|
56
|
Vilar-Gomez E, Martinez-Perez Y,
Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B,
Gonzalez-Fabian L, Friedman SL, Diago M and Romero-Gomez M: Weight
loss through lifestyle modification significantly reduces features
of nonalcoholic steatohepatitis. Gastroenterology. 2:367–378.
2015.PubMed/NCBI View Article : Google Scholar
|
|
57
|
Wilding JP: The importance of weight
management in type 2 diabetes mellitus. Int J Clin Pract.
68:682–691. 2014.PubMed/NCBI View Article : Google Scholar
|
|
58
|
Neeland IJ, Linge J and Birkenfeld AL:
Changes in lean body mass with glucagon-like peptide-1-based
therapies and mitigation strategies. Diabetes Obes Metab. 26 (Suppl
4):S16–S27. 2024.PubMed/NCBI View Article : Google Scholar
|
|
59
|
Cava E, Yeat NC and Mittendorfer B:
Preserving healthy muscle during weight loss. Adv Nutr. 8:511–519.
2017.PubMed/NCBI View Article : Google Scholar
|
|
60
|
Hardie DG, Ross FA and Hawley SA: AMPK: A
nutrient and energy sensor that maintains energy homeostasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012.PubMed/NCBI View Article : Google Scholar
|
|
61
|
Tai T, Shao YY, Zheng YQ, Jiang LP, Han
HR, Yin N, Li HD, Ji JZ, Mi QY, Yang L, et al: Clopidogrel
ameliorates High-fat diet-induced hepatic steatosis in mice through
activation of the AMPK signaling pathway and beyond. Front
Pharmacol. 15(1496639)2024.PubMed/NCBI View Article : Google Scholar
|
|
62
|
Wang Y, Liu S, Ni M, Chen Y, Chen R, Wang
J, Jiang W, Zhou T, Fan S, Chang J, et al: Terf2ip deficiency
accelerates non-alcoholic steatohepatitis through regulating
lipophagy and fatty acid oxidation via Sirt1/AMPK pathway. Free
Radic Biol Med. 220:78–91. 2024.PubMed/NCBI View Article : Google Scholar
|
|
63
|
Matsusue K, Haluzik M, Lambert G, Yim SH,
Gavrilova O, Ward JM, Brewer B Jr, Reitman ML and Gonzalez FJ:
Liver-specific disruption of PPARgamma in leptin-deficient mice
improves fatty liver but aggravates diabetic phenotypes. J Clin
Invest. 111:737–747. 2003.PubMed/NCBI View Article : Google Scholar
|
|
64
|
Baumann A, Burger K, Brandt A, Staltner R,
Jung F, Rajcic D, Lorenzo Pisarello MJ and Bergheim I: GW9662, a
peroxisome proliferator-activated receptor gamma antagonist,
attenuates the development of non-alcoholic fatty liver disease.
Metabolism. 133(155233)2022.PubMed/NCBI View Article : Google Scholar
|
|
65
|
Sozio MS, Lu C, Zeng Y, Liangpunsakul S
and Crabb DW: Activated AMPK inhibits PPARα and PPARγ
transcriptional activity in hepatoma cells. Am J Physiol
Gastrointest Liver Physiol. 301:G739–G747. 2011.PubMed/NCBI View Article : Google Scholar
|
|
66
|
Blanchard PG, Festuccia WT, Houde VP,
St-Pierre P, Brûlé S, Turcotte V, Côté M, Bellmann K, Marette A and
Deshaies Y: Major involvement of mTOR in the PPARγ-induced
stimulation of adipose tissue lipid uptake and fat accretion. J
Lipid Res. 53:1117–1125. 2012.PubMed/NCBI View Article : Google Scholar
|
|
67
|
Hunter CA, Kartal F, Koc ZC, Murphy T, Kim
JH, Denvir J and Koc EC: Mitochondrial oxidative phosphorylation is
impaired in TALLYHO mice, a new obesity and type 2 diabetes animal
model. Int J Biochem Cell Biol. 116(105616)2019.PubMed/NCBI View Article : Google Scholar
|
|
68
|
Xiong G, Zhang K, Ma Y, Song Y, Zhang W,
Qi T, Qiu H, Shi J, Kan C, Zhang J and Sun X: BAM15 as a
mitochondrial uncoupler: A promising therapeutic agent for diverse
diseases. Front Endocrinol (Lausanne). 14(1252141)2023.PubMed/NCBI View Article : Google Scholar
|
|
69
|
Yoo HC, Yu YC, Sung Y and Han JM:
Glutamine reliance in cell metabolism. Exp Mol Med. 52:1496–1516.
2020.PubMed/NCBI View Article : Google Scholar
|
|
70
|
Amores-Sánchez MI and Medina MA:
Glutamine, as a precursor of glutathione, and oxidative stress. Mol
Genet Metab. 67:100–105. 1999.PubMed/NCBI View Article : Google Scholar
|
|
71
|
Torres N, Tobón-Cornejo S,
Velazquez-Villegas LA, Noriega LG, Alemán-Escondrillas G and Tovar
AR: Amino acid catabolism: An overlooked area of metabolism.
Nutrients. 15(3378)2023.PubMed/NCBI View Article : Google Scholar
|
|
72
|
TeSlaa T, Ralser M, Fan J and Rabinowitz
JD: The pentose phosphate pathway in health and disease. Nat Metab.
5:1275–1289. 2023.PubMed/NCBI View Article : Google Scholar
|
|
73
|
Dungubat E, Kusano H, Mori I, Tawara H,
Sutoh M, Ohkura N, Takanashi M, Kuroda M, Harada N, Udo E, et al:
Age-dependent sex difference of non-alcoholic fatty liver disease
in TSOD and db/db mice. PLoS One. 17(e0278580)2022.PubMed/NCBI View Article : Google Scholar
|
|
74
|
Chen SY, Beretta M, Olzomer EM,
Alexopoulos SJ, Shah DP, Byrne FL, Salamoun JM, Garcia CJ, Smith
GC, Larance M, et al: Head-to-head comparison of BAM15,
semaglutide, rosiglitazone, NEN, and calorie restriction on
metabolic physiology in female db/db mice. Biochim Biophys Acta Mol
Basis Dis. 1870(166908)2024.PubMed/NCBI View Article : Google Scholar
|