Introduction
Papillary thyroid cancer (PTC) is characterized by
lymphatic spread, and up to 50% of patients with PTCs have positive
lymph node metastases (LNM) at the time of surgery (1). In addition, lymph nodes are the most
common site of thyroid cancer recurrences, and the prevalence of
nodal involvement developing after surgery was reported to be
38.5–58.8%. Therefore, understanding the molecular mechanisms that
control thyroid cancer cell invasion and survival in metastatic
sites is critical for the development of effective therapy.
Cancer cell invasion and the development of
metastases are associated with the induction of epithelial to
mesenchymal transition (EMT) in experimental tumors and advanced
stage human carcinomas (2,3). Epithelial cells undergoing EMT are
characterized by the loss of cell-to-cell contacts and acquisition
of a migratory phenotype. The CDH1 (16q22.1) gene encodes
the transmembrane glycoprotein E-cadherin, which plays an important
role in maintaining cell-cell adhesion in epithelial tissues.
Diminished E-cadherin expression has been considered a hallmark of
EMT (4).
Activation of signaling pathways due to oncogene
mutations contributes to the induction of EMT. Previous studies
have shown that HGF/cMET (5),
TGF-β/Smurf1 (6) and MAPK/ERK
(7) are involved in the regulation
of EMT. Epigenetic mechanisms also control EMT through the
regulation of E-cadherin expression. Diminished expression of
E-cadherin due to its gene promoter hypermethylation has been
demonstrated in various malignancies, including leukemia (8), breast cancer (9) and primary colorectal carcinomas
(10).
The intracellular location of E-cadherin is critical
for its function and is regulated by phosphorylation (11). Activation of tyrosine kinases
results in the loss of cadherin-mediated cell-cell adhesion by
activating E-cadherin endocytosis. The role of PKC signaling has
been demonstrated in the regulation of E-cadherin trafficking,
endocytosis and recycling. It has been shown that cancer cell
treatment with phorbol esters increased the rate of E-cadherin
endocytosis, resulting in decreased levels of membranous E-cadherin
(12). The inflammatory mediator
tumor necrosis factor (TNF)-α induces EMT through PKC dependent
activation of TGF-β (13).
In the thyroid, cDNA microarray analysis and
immunohistochemical studies have demonstrated marked
down-regulation of E-cadherin expression in poorly differentiated
cancer (14,15). Aberrant E-cadherin promoter
methylation was demonstrated in 23–83% of PTCs (16,17).
These studies also suggest that the loss of
E-cadherin expression plays a role in the development of thyroid
cancer metastases. We recently demonstrated that invasive thyroid
cancer cells exhibit a gene expression profile consistent with EMT
(18). However, whether
mesenchymal features of invasive thyroid cancer cells are
maintained in metastatic sites is unknown.
In this study, we examined the methylation status of
the E-cadherin gene promoter in a series of PTCs and corresponding
LNM. We also used thyroid cancer cell lines to establish
relationships between E-cadherin gene expression and cancer cell
motility.
Materials and methods
Thyroid tissue samples
Tumor specimens and clinical data were obtained from
an archival tissue bank and the corresponding computerized
database, which is maintained under IRB approval of the Walter Reed
Army Medical Center. Thyroid tissue samples from 66 patients were
used in this study. All tumors were diagnosed as PTCs with H&E
staining according to the WHO classification criteria. Oncogenic
mutation status was assessed in all primary PTCs as previously
described (18). In 34 cases,
samples from LNM were available for analysis.
DNA extraction and methylation-specific
PCR
Genomic DNA was prepared from tumor tissues and cell
cultures using TRIzol Reagent (Invitrogen, Carlsbad, CA). The
sodium bisulfite reaction was carried out on 1 μg of DNA according
to the protocol specified for the CpGenome™ DNA Modification kit
(Chemicon, Temecula, CA). Methylation-specific PCR (MSP)
amplifications of E-cadherin were performed from 2-μl aliquots in a
25-μl reaction mixture according to the established protocol for
the CpG WIZ™ E-cadherin Amplification kit (Chemicon). The amplified
products were visualized after electrophoresis on a 2%
polyacrylamide gel. The methylated and unmethylated CDH1 exon 1 CpG
islands were 206 and 212 bp, respectively. Genomic DNA from the CpG
WIZ™ E-cadherin Amplification kit was subjected to bisulfite
modification and used as a positive control for methylated
alleles.
RNA extraction and reverse transcription
(RT)-PCR
Total RNA was prepared from tumor tissues and cell
cultures using TRIzol Reagent. Using specific primer pairs as
previously described, RT-PCR was performed using total RNA, and the
products were visualized after electrophoresis on a 2% agarose gel.
cDNA was probed with specific primers for E-cadherin (CDH1 RT² PCR
Primer Set; SuperArray Bioscience Corporation, Frederick, MD). The
conditions used were as follows: 95°C, 15 min; 40 cycles of (95°C,
30 sec; 55°C, 30 sec and 72°C, 30 sec) 72°C, 10 min. This was
confirmed by electrophoresis on a 2% agarose gel.
Immunohistochemical analysis
Sections were deparaffinized, then soaked in alcohol
after microwave treatment in antigen unmasking solution for 10 min.
These were then incubated in 3% hydrogen peroxide for 15 min to
quench endogenous peroxidase activity. Sections were incubated at
4°C overnight with anti-E-cadherin antibodies (sc-21791; Santa Cruz
Biotechnology, Santa Cruz, CA). Immunostaining was performed by use
of the Vectastain Universal Quick kit according to the
manufacturer's instructions. Peroxidase staining was revealed in
3,3-diaminobenzidine. A negative control was applied by omission of
antiserum. The intensity of staining and cellular localization were
evaluated by two independent observers.
For immunofluorescence, species-appropriate
secondary antibodies conjugated with Texas Red (Invitrogen) were
applied for 1 h in the dark at room temperature. Cover slides were
mounted with the Prolong anti-fade kit (Invitrogen) and examined
using fluorescence microscopy.
Cell culture, migration and cell
growth
Human thyroid cancer cell lines (TPC1, FTC133,
FTC236, FTC238 and WRO82-1) were propagated in RPMI-1640 medium
(Invitrogen). Pharmacological inhibitor of PI3K/AKT signaling
(LY-294002), TNF-α and 5-aza-2′-deoxycytidine (5-aza) were from
Sigma Chemical Co. (St. Louis, MO), and MEK1/2 inhibitor (U-0126)
was from Cell Signaling Technology.
Cell migration experiments were performed using a
Boyden chamber or Matrigel-coated membrane (8-μm pore size). The
efficiency of migration was evaluated by membrane staining using a
Diff-Quick staining kit (Dade Behring Inc., Newark, DE). The
migration percentage of total cells was calculated as previously
described (18). All migration
experiments were performed in triplicate.
Cell growth was examined using Vi-CELL™ Cell
Viability Analyzer from Beckman Coulter (Fullerton, CA) according
to the manufacturer's instructions.
Results
Methylation of E-cadherin gene promoter
and E-cadherin expression in human thyroid cancer
We examined the methylation status of the E-cadherin
promoter by using MSP in PTCs and in thyroid tissue adjacent to the
tumor tissue. In PTCs, hypermethylation of the 5′ CpG islands
within the E-cadherin promoter was evident in 26/66 (39.3%) of the
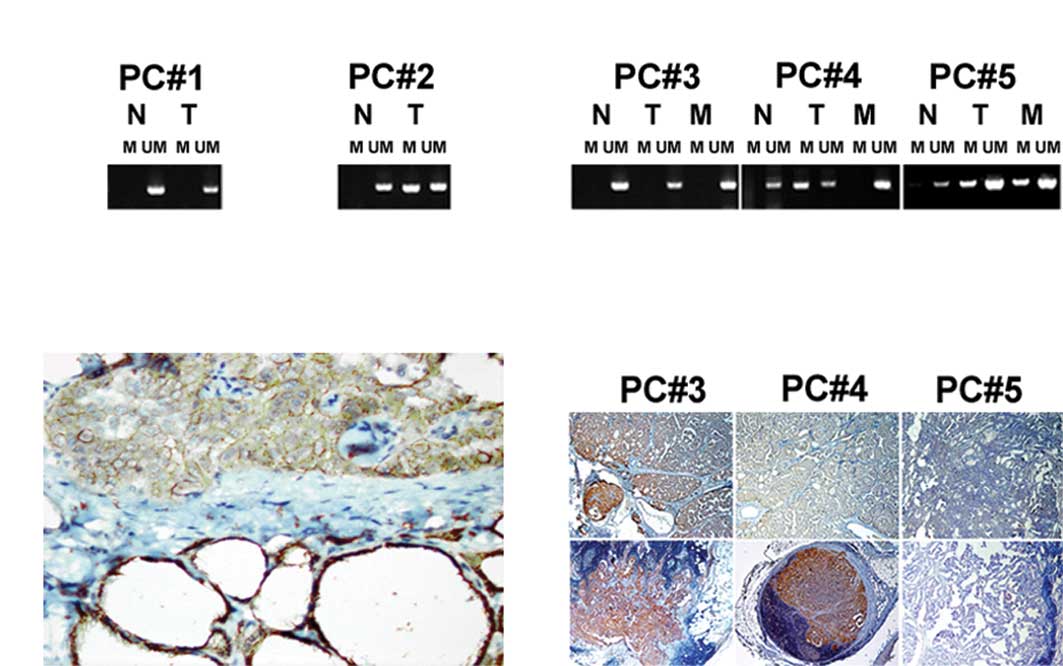
cases (Fig. 1A). In the
corresponding normal thyroid tissues, methylation of E-cadherin was
detected in 1/22 (4.5%) of the analyzed samples. Morphological
evidence of thyroiditis was observed in this case.
Clinicopathological data of the patients as a
function of E-cadherin promoter methylation status are presented in
Table I. Histological examination
of thyroid tumors revealed that the E-cadherin gene promoter was
more frequently hypermethylated in PTCs with a typical papillary
pattern of growth compared to the follicular variant of PTCs.
Methylation of the E-cadherin promoter was associated with
pathological features suggesting an aggressive thyroid cancer such
as extrathyroidal invasion and metastases.
 | Table I.Clinical features and E-cadherin
methylation in papillary thyroid carcinomas. |
Table I.
Clinical features and E-cadherin
methylation in papillary thyroid carcinomas.
| Clinical
features | E-cadherin promoter
methylation status
| P-value |
|---|
| Hypermethylated (26
cases) | Unmethylated (40
cases) |
|---|
| Age (year) | 43.1±17.1 | 35.3±12.2 | 0.2949 |
| Tumor size (mm) | 33±0.6 | 22±0.8 | 0.4902 |
| Gender
(male:female) | 6:20 | 16:24 | 0.1881 |
| Invasion
(no:yes) | 4:22 | 18:22 | 0.0164 |
| Multifocal growth
(no:yes) | 12:14 | 26:14 | 0.2021 |
| Metastases
(no:yes) | 1:25 | 16:24 | 0.0011 |
Hypermethylation of the E-cadherin promoter was
detected in 8/30 (26.6%), 1/8 (12.5%) and 0/5 (0%) of tumors with
BRAF, RET and RAS mutations, respectively. In PTCs without
mutations, E-cadherin hypermethylation was detected in 8/28 (28.5%)
cases. The correlation between E-cadherin promoter methylation and
thyroid oncogene mutations was not statistically significant.
E-cadherin expression in the PTCs and normal thyroid
tissues was examined by immunohistochemistry. Normal thyroid
tissues showed intense membranous staining while the stromal cells
did not express E-cadherin (Fig.
1B). Decreased intensity of E-cadherin immunostaining was found
in all PTCs with hypermethylated E-cadherin gene promoter, and in
16/40 (40%) of PTCs with unmethylated E-cadherin. Loss of
membranous E-cadherin expression was associated with lymphocyte
infiltration of tumor stroma and the presence of metastases. No
significant correlation was found between thyroid oncogenic
mutation status and the level of E-cadherin protein expression.
Methylation of the E-cadherin promoter in
the invasive front of thyroid cancer and in lymph node
metastases
We hypothesized that thyroid cancer cells located in
the invasive front of thyroid cancer acquire increased motility due
to hypermethylation of the E-cadherin promoter and decreased
E-cadherin expression. Therefore, we examined samples dissected
from the invasive front and central areas of the same tumor in 10
widely invasive PTCs. In 7 cases, the methylation patterns of the
E-cadherin promoter and patterns of E-cadherin immunostaining were
the same in the central and invasive areas. In 3 cases, there was a
switch in E-cadherin promoter methylation status from unmethylated
in the central area to hypermethylated in the invasive area of the
tumor.
We also compared the methylation status of the
E-cadherin promoter between the primary PTC and LNM in 34 cases,
for which both primary tumors and corresponding metastases were
available for analysis. In 20 of the 34 examined cases (60%),
methylation of the E-cadherin promoter was identical in the primary
tumor and in the corresponding LNM (Fig. 1C). The E-cadherin promoter was
unmethylated in both the primary tumor and in the corresponding LNM
in 10 cases. In 10 cases, the E-cadherin promoter was
hypermethylated in both the primary tumor and in the corresponding
LNM. In 14/34 (40%) cases, the methylation status of E-cadherin was
different in the primary PTC and in the corresponding LNM. In 12
cases (35%), the E-cadherin gene promoter was hypermethylated in
the primary PTC but unmethylated in LNM. Immunostaining showed an
increased level of E-cadherin expression in these LNM (Fig. 1D). In two cases (5%), a
hypermethylated E-cadherin gene promoter was detected only in
metastases, and not in the primary tumor.
E-cadherin expression and migratory
ability of thyroid cancer cell lines
To determine the role of E-cadherin in the
regulation of thyroid cancer cell motility, we examined the
methylation status of the E-cadherin promoter in thyroid cancer
cell lines with different phenotypes. TPC-1, FTC133, FTC236 and
FTC238 cells showed spindle, mesenchymal-like morphology, but WRO
cells demonstrated round, epithelial-like characteristics. In
mesenchymal-like thyroid cancer cells, the E-cadherin promoter was
hypermethylated, and E-cadherin expression was not detected at the
mRNA and protein levels. In the epithelial-like thyroid cancer cell
lines, unmethylated copies of the E-cadherin promoter were
detected, and E-cadherin expression was evident at the mRNA and
protein levels. Immunostaining demonstrated membranous patterns of
E-cadherin expression in epithelial-like WRO cells (Fig. 2A).
E-cadherin-negative thyroid cancer cells with
mesenchymal-like morphology efficiently migrated in the Boyden
chamber assay. These cells were also able to invade through the
Matrigel-coated membrane. By contrast, epithelial-like
E-cadherin-positive WRO cells did not migrate (Fig. 2B).
To determine whether E-cadherin methylation status
and the migratory ability of thyroid cancer cells is affected by
treatment with demethylating agents, we incubated thyroid cancer
cells with 5-aza-2′-deoxycytidine. Thyroid cancer cell treatment
with 5-aza (2.5 and 5 μM for up to 72 h) did not affect the
methylation status of the E-cadherin gene promoter or E-cadherin
expression, and did not inhibit cell migration. In mesenchymal
like-cells, 5-aza treatment was associated with inhibition of cell
growth. By contrast, 5-aza treatment had no significant effect on
growth in the epithelial-like thyroid cancer cells.
We previously demonstrated that PI3K/AKT and RAS/ERK
signaling plays a role in the regulation of thyroid cancer cell
migration. It has also been shown that the activation of PKC
signaling by PMA induces morphological changes in WRO cells.
To determine whether these signaling control thyroid
cancer cell motility through the regulation of E-cadherin, we
examined also E-cadherin promoter methylation status and E-cadherin
expression after cell exposure to PI3K/AKT inhibitor (LY-294002),
RAS/ERK inhibitor (U-0126) and after treatment with PMA.
In mesenchymal-like thyroid cancer cells treatment
with LY-294002 (20 μM) and U-0126 (20 μM) was associated with the
inhibition of migration. However, these migratory inhibitory
effects were not associated with changes in E-cadherin promoter
methylation status, E-cadherin expression and conversion to an
epithelial phenotype. PMA treatment had no significant effect on
mesenchymal-like thyroid cancer cell migration, but dramatically
altered the morphology of epithelial-like cells. PMA treatment (100
nM for 24 h) was associated with loss of cell-to-cell contacts and
acquisition of mesenchymal like morphological features in WRO
cells. Immunostaining showed that chronic exposure to PMA was
associated with the loss of membranous E-cadherin staining in WRO
cells (Fig. 2C) and the induction
of migration (Fig. 2D).
PMA-inducible re-localization of E-cadherin from the cell membrane
to the cytoplasm was not associated with changes in E-cadherin
promoter methylation status or in the level of E-cadherin
expression.
Since decreased membranous E-cadherin expression in
human PTCs was associated with lymphocyte infiltration, we
hypothesized that these effects may be mediated by the cytokine
TNF-α. Chronic TNF-α treatment (100 nM for 24 h) resulted in the
loss of membranous E-cadherin and induction of cancer cell
migration (Fig. 2D). Similarly to
PMA, TNF-inducible re-localization of E-cadherin from the cell
membrane to the cytoplasm was not associated with changes in
E-cadherin promoter methylation status or in the level of
E-cadherin expression.
To determine whether constitutive TNF stimulation is
required for the maintenance of the mesenchymal phenotype in
thyroid cancer cells we performed time course experiments. Analysis
of thyroid cancer cells showed that the TNF-inducible loss of
membranous E-cadherin expression was transient. The switch from
medium containing TNF to TNF-free medium resulted in the
re-establishment of epithelial like morphology and loss of
migratory ability in WRO cells.
Discussion
The acquisition of a migratory phenotype is an
essential property of invading and metastasizing cancer cells. Cell
migration is commonly understood as the movement of individual
cells that undergo polarized extension-contraction cycles. Reduced
expression of E-cadherin is regarded as one of the main molecular
events involved in individual cancer cell invasion.
In the present study, we demonstrated that PTCs
presenting with metastases at the time of surgery more frequently
display E-cadherin promoter methylation compared to PTCs without
metastases. These findings are consistent with previously reported
data showing a high frequency of aberrant E-cadherin promoter
methylation in poorly differentiated metastatic thyroid cancers
(19,20). Our data suggest that thyroid cancer
cells with the highest density of methylation and the most
diminished E-cadherin protein expression may be responsible for
dissociation from the primary tumor and dissemination to lymph
nodes.
Comparative analysis of E-cadherin in primary tumors
and corresponding LNM revealed dynamic changes in E-cadherin
methylation status and E-cadherin expression during metastatic
progression. In a significant proportion of cases, E-cadherin
expression was increased in lymph node metastases compared to
primary tumors, and this phenomenon was related to the loss of
epigenetic inhibition. These results suggest that the mesenchymal
phenotype of invading cancer cells may be reversed in metastatic
sites, and thyroid cancer cells may undergo a mesenchymal to
epithelial transition in lymph node metastases.
The possible role of mesenchymal to epithelial
transition in metastatic sites is supported by previously reported
data showing that epigenetic inactivation of E-cadherin is unstable
and may be influenced by micro-environmental factors during breast
cancer metastatic progression (21). There is evidence suggesting that
E-cadherin plays a role in cancer cell survival within the
lymphatic and metastatic sites. Strong expression of E-cadherin was
observed in intra-lymphatic emboli in patients with breast lobular
carcinomas (22). The
re-expression of E-cadherin was demonstrated in lymph node
metastasis from E-cadherin-negative colorectal carcinomas (23).
We found that, in certain cases, PTCs presenting
with metastases were characterized by E-cadherin expression in both
the primary and metastatic lesions. These findings suggest that
thyroid cancer metastases may arise from tumor cells that have
never lost E-cadherin expression. Previous studies underlined the
role of E-cadherin in the regulation of a specific type of cellular
translocation referred to as collective cell migration (24,25).
In contrast to single-cell migration, during collective cell
migration cancer cells maintain cell-cell contact and migrate as a
group of cells forming a protruding sheet or nest. In the course of
tumor progression, tumor clusters may enter lymphatic vessels and
can be isolated from peripheral blood (26). It is possible that thyroid cancer
cells may use this mechanism and invade lymphatic vessels as a
group of interconnected cancer cells expressing a high level of
cell adhesion molecules.
To explore the possible role of E-cadherin in the
regulation of thyroid cancer cell migration, we performed in
vitro studies. Consistent with human data, our in vitro
results demonstrated a direct link between the
methylation-associated loss of E-cadherin expression and thyroid
cancer cell motility. Thyroid cancer cells with the highest density
of methylation and the most diminished E-cadherin protein
expression were characterized by a high migratory ability.
We also explored the relationships between the
activation of thyroid oncogene-inducible signaling and epigenetic
mechanisms in the regulation of thyroid cancer cell motility. Our
results indicate that PI3K/AKT and RAS/ERK signaling contributes to
thyroid cancer cell motility independently of E-cadherin
methylation, E-cadherin expression and localization. By contrast,
the effects of TNF-inducible signaling on cell migration involve
changes in intracellular E-cadherin localization.
To determine whether the pharmacological targeting
of methylation induces E-cadherin expression and converts
mesenchymal-like cells to an epithelial phenotype, we examined the
effects of 5-aza on thyroid cancer cells. Though treatment with
5-aza did not induce E-cadherin expression, it was associated with
the inhibition of growth in mesenchymal-like thyroid cancer cells.
Clinical trials in humans have shown the anti-neoplastic activity
of 5-aza (Decitabine) in patients with leukemia and myelodysplastic
syndrome, and a clinical trial evaluating Decitabine in the
treatment of patients with metastatic thyroid cancer is ongoing.
Our data showed that E-cadherin-positive and -negative thyroid
cancer cells are characterized by different responses to treatment
with 5-aza, and suggested that a demethylating agent may have a
variable effect against cancer cells located in primary tumors and
in metastatic lesions.
In summary, in the present study we demonstrated
that E-cadherin expression in thyroid cancer cells is regulated by
epigenetic mechanisms, and dynamic changes in E-cadherin
methylation status may occur during metastatic progression.
Epigenetic mechanisms and activation of TNF-inducible signaling
independently contribute to thyroid cancer cell invasiveness
through regulation of E-cadherin expression and intracellular
E-cadherin localization. These mechanisms may play a role in the
induction of epithelial to mesenchymal transition in the primary
tumor, as well as in the conversion from the mesenchymal to the
epithelial phenotype in lymph node metastases.
Acknowledgements
This study was funded by the
Department of Pediatrics, Uniformed Services University of the
Health Sciences (USUHS). We thank Ildy Katona, Chair, Department of
Pediatrics, for the support of this project.
References
|
1.
|
Puxeddu E and Filetti S: The 2009 American
Thyroid Association Guidelines for Management of Thyroid Nodules
and Differentiated Thyroid Cancer: progress on the road from
consensus- to evidence-based practice. Thyroid. 19:1145–1147. 2009.
View Article : Google Scholar
|
|
2.
|
Shook D and Keller R: Mechanisms,
mechanics and function of epithelial-mesenchymal transitions in
early development. Mech Dev. 120:1351–1383. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Thiery JP: Epithelial-mesenchymal
transitions in development and pathologies. Curr Opin Cell Biol.
15:740–746. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Takeichi M: Cadherin cell adhesion
receptors as a morphogenetic regulator. Science. 251:1451–1455.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Matteucci E, Ridolfi E and Desiderio MA:
Hepatocyte growth factor differently influences Met-E-cadherin
phosphorylation and downstream signaling pathway in two models of
breast cells. Cell Mol Life Sci. 63:2016–2026. 2006. View Article : Google Scholar
|
|
6.
|
Wang H, Radjendirane V, Wary KK and
Chakrabarty S: Transforming growth factor beta regulates cell-cell
adhesion through extracellular matrix remodeling and activation of
focal adhesion kinase in human colon carcinoma Moser cells.
Oncogene. 23:5558–5561. 2004. View Article : Google Scholar
|
|
7.
|
Honma N, Genda T, Matsuda Y, Yamagiwa S,
Takamura M, Ichida T and Aoyagi Y: MEK/ERK signaling is a critical
mediator for integrin-induced cell scattering in highly metastatic
hepatocellular carcinoma cells. Lab Invest. 86:687–696. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Corn PG, Smith BD, Ruckdeschel ES, Douglas
D, Baylin SB and Herman JG: E-cadherin expression is silenced by 5′
CpG island methylation in acute leukemia. Clin Cancer Res.
6:4243–4248. 2000.
|
|
9.
|
Gagnon J, Shaker S, Primeau M, Hurtubise A
and Momparler RL: Interaction of 5-aza-2′-deoxycytidine and
depsipeptide on anti-neoplastic activity and activation of
14-3-3sigma, E-cadherin and tissue inhibitor of metalloproteinase 3
expression in human breast carcinoma cells. Anticancer Drugs.
14:193–202. 2003.
|
|
10.
|
Lind GE, Thorstensen L, Lovig T, Meling
GI, Hamelin R, Rognum TO, Esteller M and Lothe RA: A CpG island
hypermethylation profile of primary colorectal carcinomas and colon
cancer cell lines. Mol Cancer. 3:282004. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Jaggi M, Rao PS, Smith DJ, Wheelock MJ,
Johnson KR, Hemstreet GP and Balaji KC: E-cadherin phosphorylation
by protein kinase D1/protein kinase C{mu} is associated with
altered cellular aggregation and motility in prostate cancer.
Cancer Res. 65:483–492. 2005.PubMed/NCBI
|
|
12.
|
Le TL, Joseph SR, Yap AS and Stow JL:
Protein kinase C regulates endocytosis and recycling of E-cadherin.
Am J Physiol Cell Physiol. 283:C489–C499. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Takahashi E, Nagano O, Ishimoto T, Yae T,
Suzuki Y, Shinoda T, Nakamura S, Niwa S, Ikeda S, Koga H, Tanihara
H and Saya H: TNF-{alpha} regulates TGF-{beta}-dependent
epithelial-mesenchymal transition by promoting
hyaluronan-CD44-Moesin interaction. J Biol Chem. 285:4060–4073.
2010.
|
|
14.
|
Fluge O, Bruland O, Akslen LA, Lillehaug
JR and Varhaug JE: Gene expression in poorly differentiated
papillary thyroid carcinomas. Thyroid. 16:161–175. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Choi YL, Kim MK, Suh JW, Han J, Kim JH,
Yang JH and Nam SJ: Immunoexpression of HBME-1, high molecular
weight cytokeratin, cytokeratin 19, thyroid transcription factor-1,
and E-cadherin in thyroid carcinomas. J Korean Med Sci. 20:853–859.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
16.
|
Graff JR, Greenberg VE, Herman JG, Westra
WH, Boghaert ER, Ain KB, Saji M, Zeiger MA, Zimmer SG and Baylin
SB: Distinct patterns of E-cadherin CpG island methylation in
papillary, follicular, Hurthle's cell, and poorly differentiated
human thyroid carcinoma. Cancer Res. 58:2063–2066. 1998.PubMed/NCBI
|
|
17.
|
Hoque MO, Rosenbaum E, Westra WH, Xing M,
Ladenson P, Zeiger MA, Sidransky D and Umbricht CB: Quantitative
assessment of promoter methylation profiles in thyroid neoplasms. J
Clin Endocrinol Metab. 90:4011–4018. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Vasko V, Espinosa AV, Scouten W, He H,
Auer H, Liyanarachchi S, Larin A, Savchenko V, Francis GL, de la
Chapelle A, Saji M and Ringel MD: Gene expression and functional
evidence of epithelial-to-mesenchymal transition in papillary
thyroid carcinoma invasion. Proc Natl Acad Sci USA. 104:2803–2808.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Brecelj E, Frkovic Grazio S, Auersperg M
and Bracko M: Prognostic value of E-cadherin expression in thyroid
follicular carcinoma. Eur J Surg Oncol. 31:544–548. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Rocha AS, Soares P, Fonseca E,
Cameselle-Teijeiro J, Oliveira MC and Sobrinho-Simoes M: E-cadherin
loss rather than beta-catenin alterations is a common feature of
poorly differentiated thyroid carcinomas. Histopathology.
42:580–587. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Graff JR, Gabrielson E, Fujii H, Baylin SB
and Herman JG: Methylation patterns of the E-cadherin 5′ CpG island
are unstable and reflect the dynamic, heterogeneous loss of
E-cadherin expression during metastatic progression. J Biol Chem.
275:2727–2732. 2000.
|
|
22.
|
Gupta A, Deshpande CG and Badve S: Role of
E-cadherins in development of lymphatic tumor emboli. Cancer.
97:2341–2347. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Batistatou A, Charalabopoulos AK, Scopa
CD, Nakanishi Y, Kappas A, Hirohashi S, Agnantis NJ and
Charalabopoulos K: Expression patterns of dysadherin and E-cadherin
in lymph node metastases of colorectal carcinoma. Virchows Arch.
448:763–767. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Friedl P, Noble PB, Walton PA, Laird DW,
Chauvin PJ, Tabah RJ, Black M and Zanker KS: Migration of
coordinated cell clusters in mesenchymal and epithelial cancer
explants in vitro. Cancer Res. 55:4557–4560. 1995.PubMed/NCBI
|
|
25.
|
Nabeshima K, Moriyama T, Asada Y, Komada
N, Inoue T, Kataoka H, Sumiyoshi A and Koono M: Ultrastructural
study of TPA-induced cell motility: human well-differentiated
rectal adenocarcinoma cells move as coherent sheets via localized
modulation of cell-cell adhesion. Clin Exp Metastasis. 13:499–508.
1995. View Article : Google Scholar
|
|
26.
|
Brandt B, Junker R, Griwatz C, Heidl S,
Brinkmann O, Semjonow A, Assmann G and Zanker KS: Isolation of
prostate-derived single cells and cell clusters from human
peripheral blood. Cancer Res. 56:4556–4561. 1996.PubMed/NCBI
|