Introduction
Ovarian cancer is the most lethal gynecological
malignancy in the world (1). Its
frequency has increased dramatically in the last decade. Despite
the advent of newer screening tools, the majority of patients have
peritoneal dissemination and distant metastasis at the time of
diagnosis. With disseminated disease, treatment is often
unsuccessful and the overall survival is low. Therefore, stage is
one of the most important factors for determining prognosis
(2). The identification of an
invasion-related molecule associated with the early and rapid
spread of ovarian cancer is the current focus of many
investigators.
First-line chemotherapy with platinum drugs and
taxanes yields a response rate of over 80%, but almost all patients
relapse, and in nearly all patients, recurrent disease is
incurable. Although there are well-established surgical and
chemotherapeutic treatments for primary ovarian cancer, there is
significant opportunity to develop drugs targeting specific
molecular pathways to improve survival in recurrent disease. To do
so requires a thorough understanding of the molecular pathways of
ovarian carcinogenesis.
Several genetic alterations are associated with
ovarian carcinogenesis, the most frequent of which are mutations in
P53, KRAS and BRAF (3,4).
Mutations of either BRAF or KRAS lead to constitutive
activation (phosphorylation) of their downstream target,
mitogen-activated protein kinase (MAPK), also known as
extracellular signal-regulated protein kinase (ERK) (5,6).
These mutations are correlated with overexpression of activated
ERK1/2 in ovarian serous tumors (7). Phosphorylation of ERK1/2 activates
downstream cellular targets (8,9),
including a variety of cellular and nuclear proteins.
The RAS/RAF/MEK/MAPK signaling pathway plays a major
role in various cellular activities including proliferation,
differentiation, apoptosis, angiogenesis and migration (10–15).
Suppression of MAPK activity by inhibitors, dominant-negative MEK1
mutants and anti-sense nucleotides reduces the migratory ability of
certain cell types in response to extracellular growth stimulation
(16–20). Given the important role of MAPK in
cell function, we investigated whether the RAS/RAF/MEK/MAPK pathway
is involved in the metastasis of ovarian cancer. In this study, we
assessed the activation of MAPK in ovarian cancer tissues and
examined the effects of a MEK inhibitor on cell motility and
invasion.
Materials and methods
Tissue samples
Formalin-fixed, paraffin-embedded tissue samples of
88 ovarian cancers, including 45 serous, 10 mucinous, 10 clear-cell
and 23 endometrioid carcinomas, were used in this study. The
samples were obtained from the Department of Obstetrics and
Gynecology of the Shimane University Hospital. Diagnosis was based
on a conventional morphological examination of H&E-stained
sections, and tumors were classified according to the WHO
classification. Tumor staging was performed according to the
International Federation of Gynecology and Obstetrics (FIGO)
classification. The clinicopathological characteristics of the
patients included in this study are summarized in Table I. All patients were primarily
treated with cytoreductive surgery and adjuvant platinum and taxane
chemotherapy (CBDCA AUC5, 175 mg/m2 paclitaxel or 70
mg/m2 docetaxel). All cases received 6–12 courses of
this regimen. The acquisition of tumor tissues was approved by the
Shimane University Institutional Review Board. The paraffin tissue
blocks were organized into tissue microarrays, which were made by
removing 3-mm diameter cores of tumor from each block. Selection of
the area to core was made by a gynecologic oncologist (K.N.) and
pathology technician (K.I.) and was based on a review of the
H&E slides.
 | Table I.Association between p-ERK1/2
expression and clinicopathological factors in patients with ovarian
cancer. |
Table I.
Association between p-ERK1/2
expression and clinicopathological factors in patients with ovarian
cancer.
| Factors | Patients | p-ERK1/2
immunostaining
| P-value |
|---|
| Low | High |
|---|
| FIGO stage | | | | |
| I, II | 44 | 20 | 24 | 0.394 |
| III, IV | 44 | 24 | 20 | |
| Grade | | | | |
| G1 | 17 | 10 | 7 | 0.479 |
| G2, G3 | 71 | 34 | 37 | |
| Histology | | | | |
| Serous | 45 | 21 | 24 | 0.522 |
| Other | 43 | 23 | 20 | |
| Age (years) | | | | |
| <60 | 40 | 22 | 22 | 0.392 |
| ≥60 | 48 | 26 | 22 | |
| Residual tumor | | | | |
| <1 cm | 47 | 22 | 25 | 0.522 |
| ≥1 cm | 41 | 22 | 19 | |
Cell culture and cell lines
ES2 (clear-cell carcinoma) human ovarian cancer cell
lines were obtained from the American Tissue Culture Center
(Rockville, MD, USA). The human ovarian carcinoma cell line KF28
(serous carcinoma) was a kind gift from Dr Yoshihiro Kikuchi (Ohki
Memorial Kikuchi Cancer Clinic for Women, Saitama, Japan) (10). The MPSC1 cell line was established
from a low-grade serous carcinoma and was a kind gift from Dr
Le-Ming Shih (Johns Hopkins Medical Institutions, Baltimore, MD,
USA).
Immunohistochemistry
Expression levels of the active (phosphorylated)
form of ERK1/2 and Twist were assessed by immunohistochemistry. The
antibody used in this study was a rabbit polyclonal antibody that
reacted exclusively with phosphorylated ERK1/2 (p-ERK1/2; Cell
Signaling Technology). After antigen retrieval in a sodium citrate
buffer, slides were incubated overnight at 4°C with antibodies to
p-ERK1/2 (Cell Signaling Technology) and Twist (Santa Cruz
Biotechnology, Santa Cruz, CA, USA) at dilutions of 1:1,000 and
1:100, respectively. This was followed by incubation with a
biotinylated linker and streptavidin-horseradish peroxidase (LSAB2
System-HRP; Dako Cytomation, Carpinteria, CA, USA). The signals
were visualized using ABC+ (Dako Cytomation) as the
substrate-chromagen at room temperature for 10 min. Sections were
counterstained with hematoxylin and mounted. The percentage of
positive cells was estimated by randomly counting ∼500 tumor cells
from three different high-power fields (x40) within one specimen. A
positive reaction was defined as discrete localization of the brown
chromagen in the nucleus or cytoplasm. Slides for all samples were
evaluated with a light microscope by two researchers; the
researchers were blind to the clinicopathological factors. The
antibody staining intensity was then analyzed in the cancer cell
nuclei using the HSCORE (21).
This modified HSCORE was calculated as follows: HSCORE =
ΣPi(i), where i is the intensity of
staining (0, undetectable; 1, weakly positive; 2, moderately
positive; 3, intensely positive) and Pi is a
score based on the percentage of stained cells for each intensity,
varying from 0 to 100%.
Western blot analysis
Cell lysates were prepared by dissolving cell
pellets in Laemmli sample buffer (BioRad, Hercules, CA, USA)
supplemented with 5% β-mercaptoethanol (Sigma, St. Louis, MO, USA).
Western blotting was performed on ovarian cancer cell
lines/cultures including ES2, MPSC1 and KF28. Similar amounts of
total protein from each lysate were loaded and separated on 10%
Tris-glycine-SDS polyacrylamide gels (Novex, San Diego, CA, USA)
and electroblotted to Millipore Immobilon-P polyvinylidene
difluoride membranes. The membranes were probed with an active
ERK1/2 antibody (pTEpY, 1:5,000; Cell Signaling Technology)
followed by a peroxidase conjugated anti-mouse or anti-rabbit
immunoglobulin (1:20,000). The same membrane was probed with an
antibody that reacted with total ERK1/2 (1:5,000; Cell Signaling
Technology) for loading controls. Western blots were developed by
chemiluminescence (Pierce, Rockford, IL, USA).
Simulated wound healing assay to assess
cell motility
ES2, MPSC1 and KF28 cells were allowed to grow to
confluence in 24-well plates in the presence of 5 μmol/l
Cl-1040 or 5 μmol/l DMSO (control). A linear wound was
created by scraping the wells with an ART-1000E pipette tip
(Molecular BioProduct, San Diego, CA, USA). The floating cells were
removed by gentle washes in culture medium. The wound was observed
24 h later, and the number of individual cells in the wound was
quantified as an average from multiple fields (at least five) at
magnification x200 for each experiment.
Matrigel invasion assay
The invasion chamber assay was performed according
to the manufacturer’s instructions. Briefly, Matrigel-precoated
transwell chambers with PET membranes containing 8-μm pores
(BD Bioscience, Bedford, MA, USA) were soaked in Dulbecco’s
modified Eagle’s medium and incubated for 60 min at 37°C. ES2,
MPSC1 and KF28 cells were pre-treated with 5 μmol/l CI-1040
or DMSO (as vehicle control) for 30 min. For each well,
5×104 cells in 0.5 ml of culture medium were added to
the upper compartment of the transwell chambers. As a control, an
equal number of uncoated BD Falcon TC companion plates were seeded
with cells in parallel. After 24 h of incubation in the medium
containing CI-1040 or DMSO, all non-invading cells in the upper
compartment were removed using a cotton-tipped swap. Viable cells
were stained with H&E and photographed (magnification x200).
Cells were counted in several fields of triplicate membranes. Data
were expressed as the percentage of cells that invaded through the
Matrigel matrix-coated membrane relative to the cells that migrated
through the control membrane.
Statistical analysis
Overall survival was calculated from the date of
diagnosis to the date of death or last follow-up. There was no
relationship between p-ERK1/2 expression and performance status
distributions. Data were plotted as Kaplan-Meier curves, and the
statistical significance was determined by the log-rank test. Data
were censored when patients were lost to follow-up. Results are
expressed as the mean ± SD. A value of P<0.05 was considered
statistically significant. To generate the P-value, the Student’s
t-test was used. The Pearson correlation coefficient test was used
to examine statistical significance in the immunohistochemical
analysis values.
Results and Discussion
Relationship between p-ERK1/2 expression
and clinicopathological factors
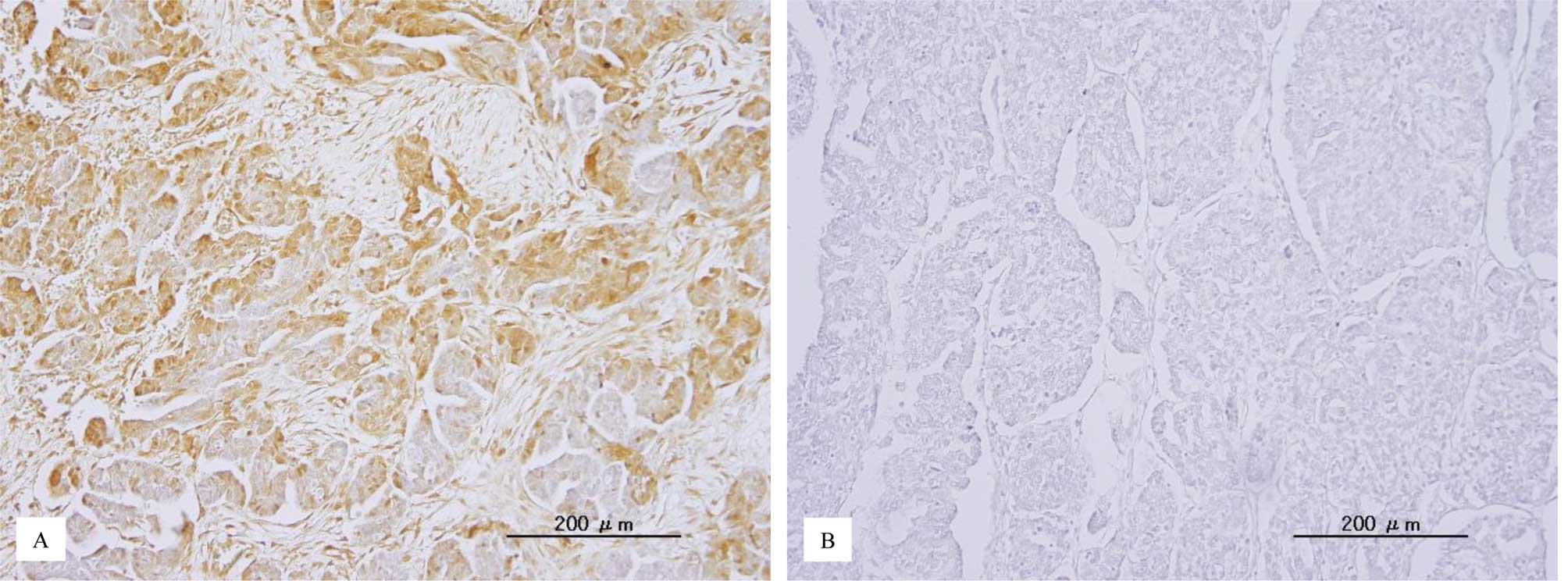
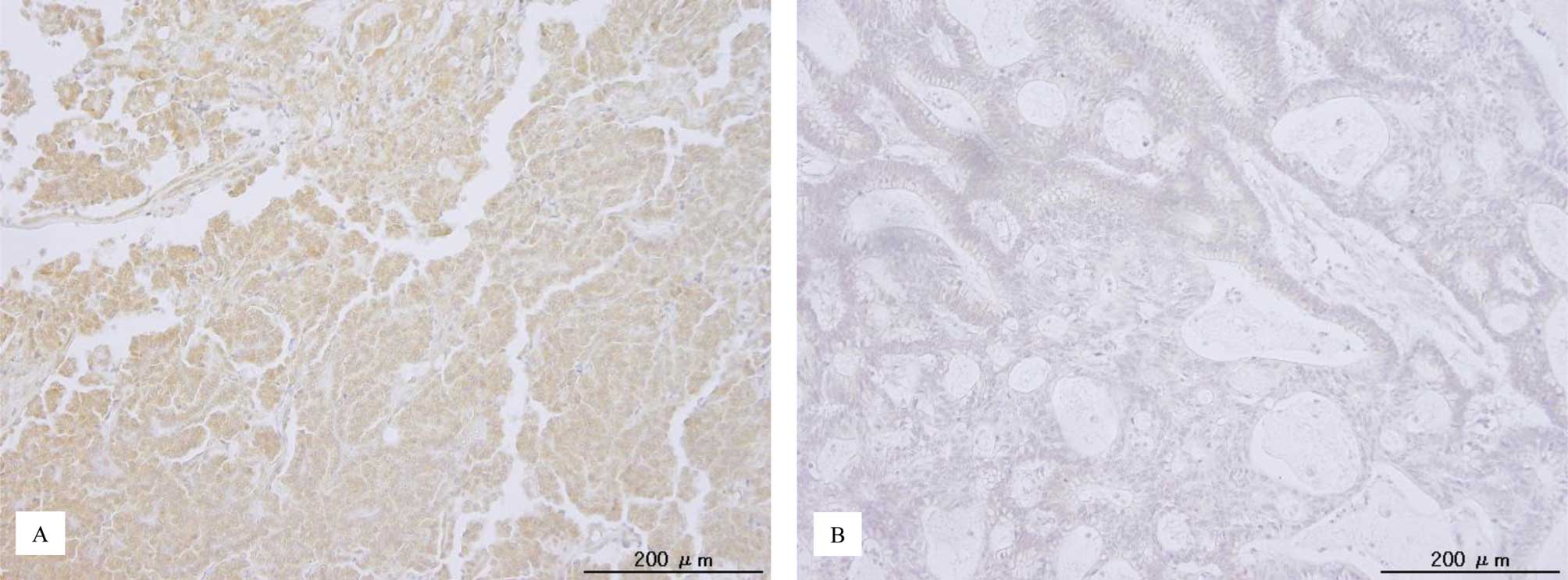
p-ERK1/2 immunoreactivity was detected in both the
nuclei and cytoplasm of the tumor cells (Fig. 1). This is consistent with a
previous report (22). The median
HSCORE of nuclear p-ERK1/2 and the range were 18 and 0–288,
respectively. Patients were stratified into one of two groups
depending on the median p-ERK1/2 immunohistochemical HSCORE. The
relationships between p-ERK1/2 expression and clinicopathological
factors are shown in Table I.
There was no significant correlation between p-ERK1/2 expression
and the tested clinicopathological factors (Table I).
Effect of p-ERK1/2 on the prognosis of
ovarian carcinomas
Next, we examined the prognostic effect of p-ERK1/2
expression. Kaplan-Meier estimates of overall survival are plotted
in Fig. 1. There was no
significant relationship between p-ERK1/2 expression and overall
survival in patients with ovarian carcinoma (P=0.426). A univariate
analysis demonstrated that FIGO stage III, IV (P=0.0068; log-rank
test), patient age ≥60 years (P=0.0078; log-rank test), residual
tumor ≥1 cm (P<0.0001; log-rank test) correlated with shorter
overall survival.
Effects of ERK1/2 inactivation by CI-1040
lead to decreased motility and invasive capabilities in vitro
Previously, we reported the p-ERK1/2 expression in
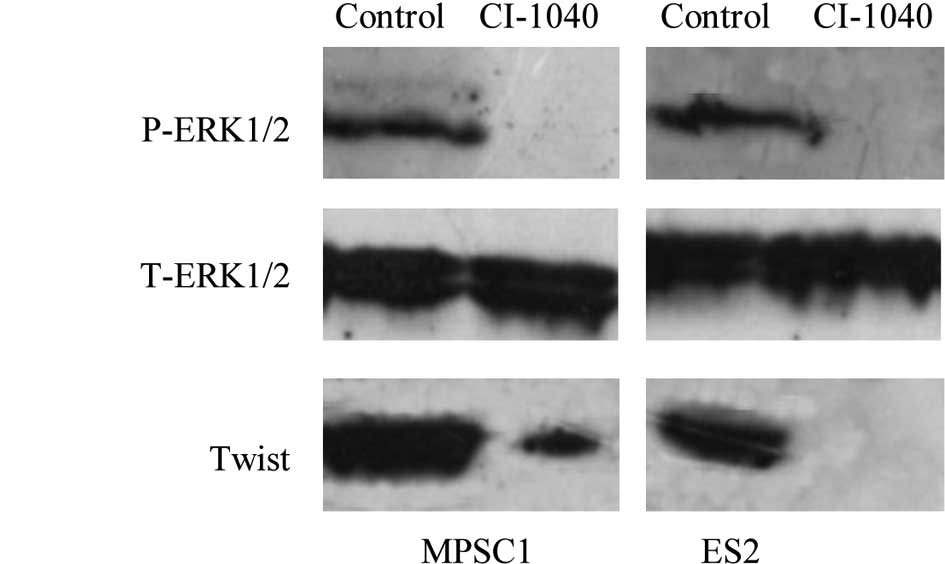
various ovarian cancer cell lines (23). Western blot analysis showed a
dose-dependent effect on the expression of active ERK1/2 in MPSC1
and ES2 cells, and active ERK1/2 was not detectable 6 h after
treatment of the cells with CI-1040 at a concentration of 5
μM (Fig. 2A). To understand
the phenotypic characteristics of inhibition by p-ERK1/2, we
analyzed the motility and invasive abilities of MPSC1 and ES2 cells
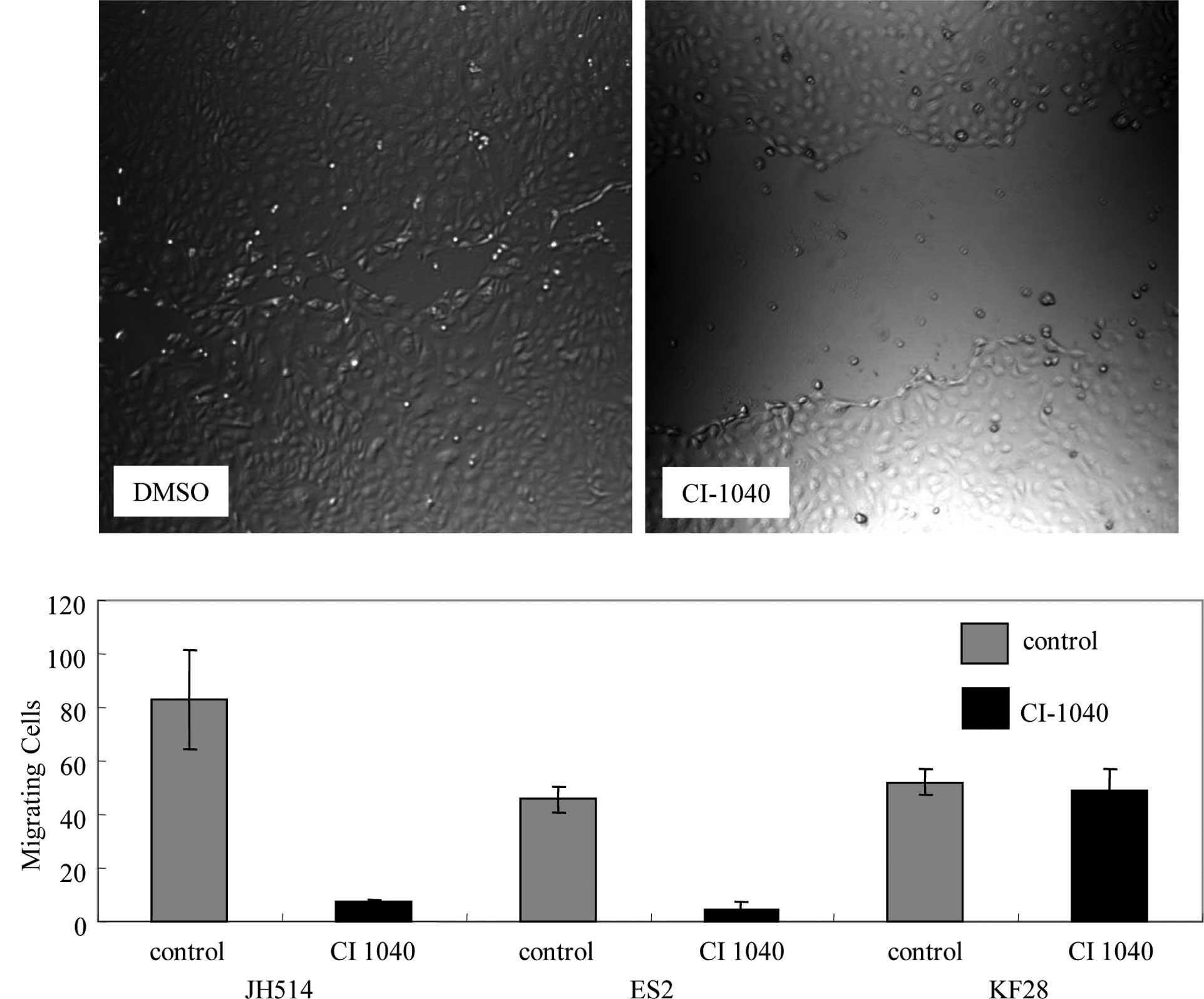
which expressed p-ERK1/2. Cell motility was investigated with a
wound-healing assay. CI-1040-treated MPSC1 and ES2 cells showed 90%
reduced cell motility compared to the DMSO control (P<0.01)
(Fig. 3A and B). Following the
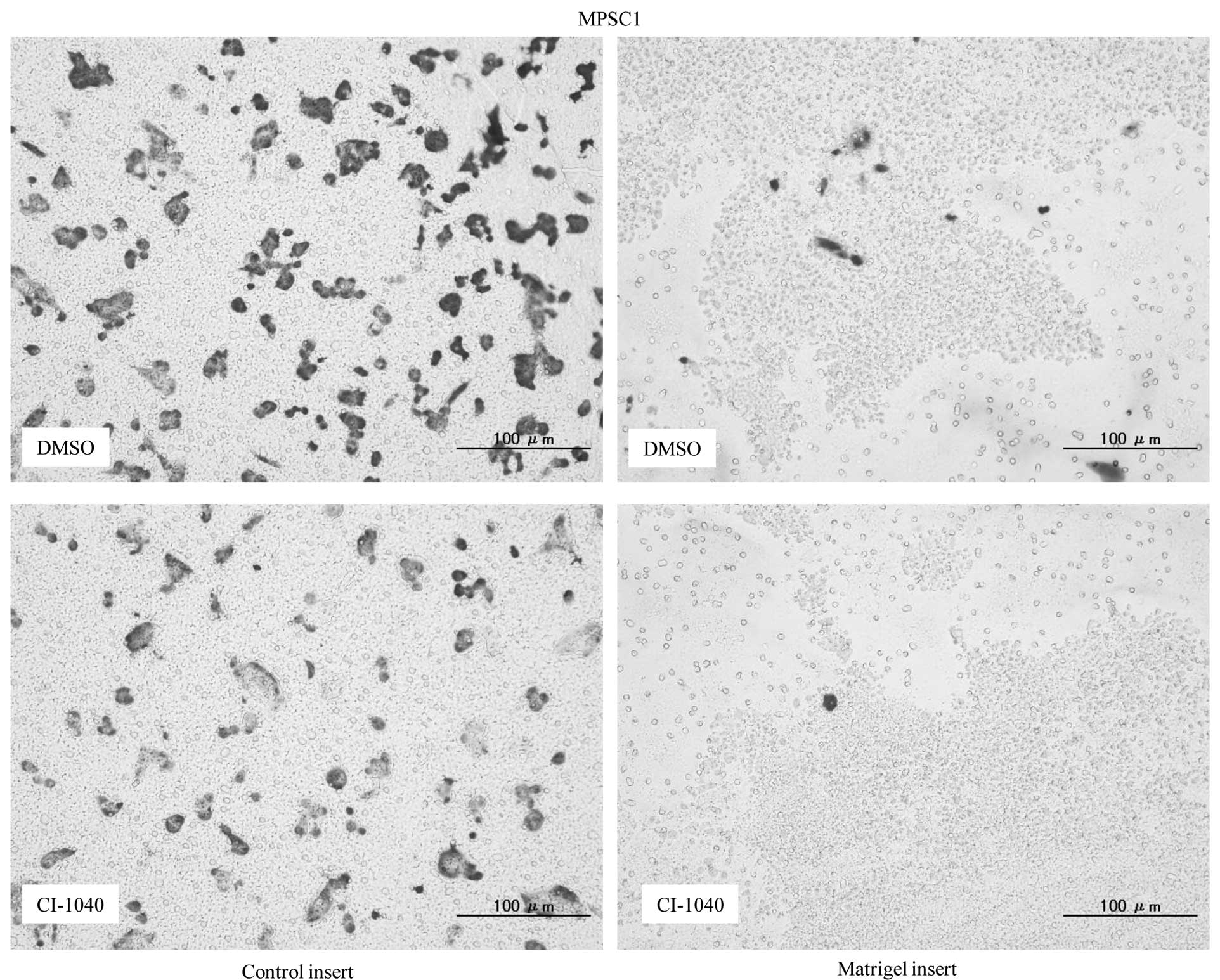
Matrigel invasion assay, an 85 and 79% decrease in cell invasion
was observed in CI-1040-treated MPSC1 and ES2 cells in comparison
to the DMSO control (P<0.05) (Fig.
4A). These results were not observed in p-ERK1/2-negative KF28
cells following CI-1040 treatment (Fig. 4B).
ERK1/2 inactivation by CI-1040 leads to
decreased motility and invasion via down-regulation of Twist
Tumor progression and invasion are complex
biological processes that involve the remodeling of stromal tissue
by invading cells. Twist appears to play a key role in these
processes. Twist expression activates dormant developmental
pathways in invading tumor cells (24). Twist has recently been associated
with metastasis in ovarian (25),
liver (26) and breast cancer
(27). Exogenous expression of
Twist promotes colony formation in anchorage-independent assays
(28). Our results demonstrated
that MEK inhibition by CI-1040 leads to suppression of invasion and
migration. Therefore, suppression of invasion-related molecules
such as Twist may be one mechanism by which p-ERK1/2 suppresses
metastasis. In this study, ERK1/2 inactivation by CI-1040 decreased
the expression of Twist with a subsequent significant reduction in
cell motility and invasiveness (Fig.
2B). Next, we analyzed p-ERK1/2 and Twist expression by
immunostaining in the ovarian carcinoma samples. The Twist
immunohistochemical HSCORE significantly correlated with the p-ERK
immunohistochemical HSCORE (r=0.370, P=0.003; Fig. 5). Expression of Twist (Twist
immunohistochemical HSCORE >0) was observed in 24% (21/88) of
the analyzed tumors. Consideration of these in vitro
findings in the context of our in vivo results suggests that
Twist may be an important downstream participant in MAPK signaling.
In the present model, p-ERK1/2 expression was positively correlated
with the Twist expression level in the regulation of metastasis.
Further studies are required to confirm whether Twist is a
downstream target of ERK signaling.
The development of a highly invasive ovarian cancer
phenotype requires coordinated up- or down-regulation of many
signaling pathways. Identification of the active MAPK-regulated
molecules in ovarian cancer is important to gain further insight
into the role of the MAPK signaling pathway in the metastasis of
ovarian cancer. In the present study, we observed a profound
reduction in cell motility, as evidenced by wound healing and
invasion assays in CI-1040-treated MPSC1 and ES2 cells with
positive p-ERK1/2. The lack of effect of the MEK inhibitor on
motility and invasion in KF28 ovarian cancer cells was expected
given that that they do not express p-ERK1/2.
While we measured Twist expression as an effect of
MAPK, other downstream effectors may also contribute to cell
motility and invasion in ovarian cancer. Several studies have shown
that MAPK activation leads to extracellular matrix-dependent cell
spreading and migration (13,17,19).
The mechanisms for this likely involve integrin activation
(29), integrin-dependent adhesion
or cytoskeletal organization, and phosphorylation of myosin light
chain kinase, calpain or focal adhesion kinase (17–19).
These activities directly or indirectly enhance cell motility. In
addition to promoting cell motility and migration, constitutive
activation of MAPK enhances matrix metalloproteinase activation, a
key event during cellular invasion (30). The present study is consistent with
previous reports showing that MAPK pathway inhibitors are potent in
suppressing cell migration in tumor cells (17,31,32).
Blockade of the MAPK pathway by treatment with MEK inhibitors
correlates well with the inhibition of invasion of tumor cells
derived from rhabdomyosarcoma, fibrosarcoma, bladder carcinoma,
colon carcinoma, prostate carcinoma and breast carcinoma cell lines
(33). These results suggest that
the inactivation of the MAPK pathway may have an anti-metastatic
effect on tumor cells.
Our results provide compelling evidence that the
biological effects of the ERK signaling pathway depend on the
p-ERK1/2 status. We found that ovarian carcinomas with p-ERK1/2
expression were more sensitive to cell migration and invasion
inhibition by the MEK inhibitor, CI-1040. This observation suggests
that ovarian carcinomas with p-ERK1/2 expression are more highly
dependent on the activation of the MEK-ERK pathway for cell
migration and invasion than those without p-ERK1/2 expression.
Thus, inactivation of ERK1/2 results in marked inhibition of cell
migration and invasion of ovarian carcinomas with p-ERK1/2
expression in comparison to only a modest effect on
p-ERK1/2-negative tumors. The above observations lend strong
support to the view of ‘kinase addiction’, in which dependence on a
particular kinase pathway confers susceptibility to a kinase
inhibitor (34,35).
In light of our in vivo and in vitro
findings, we propose that recurrent ovarian cancer patients with
p-ERK1/2 expression should be considered for MEK inhibitor
(CI-1040) therapy. Additionally, patients with primary tumors that
express p-ERK1/2 may also benefit from MEK inhibitor therapy in
combination with conventional platinum and taxane chemotherapy. The
use of a MEK inhibitor at the time of initial chemotherapy may
decrease the invasive potential of any residual tumor cells,
thereby extending the disease-free interval.
Thus far, the MEK inhibitor CI-1040 has fared poorly
in clinical trials for breast, colon and lung cancer (36). It is possible that this may be due
to non-selective use of the inhibitor. It is possible that more
favorable outcomes may result when patients are stratified based on
p-ERK1/2 expression status. We recommend that stratification is
used in further clinical trials of MEK inhibitors in ovarian cancer
patients.
In summary, we demonstrated that the phenotypic
changes in cell migration and invasion in ovarian carcinomas in
response to MEK inhibition depend on p-ERK1/2 status. The findings
of this study provide new insight into the biological roles of MAPK
signaling in ovarian carcinomas. Additionally, our observations
have an important therapeutic implication for patients with ovarian
cancers expressing p-ERK1/2. Therefore, detection of p-ERK1/2 in
ovarian cancers may identify patients who will benefit from CI-1040
therapy.
Acknowledgements
This study was supported by grants
from the Ministry of Education, Culture, Sports, Science and
Technology of Japan and the Sagawa Cancer Research Foundation.
References
|
1.
|
Wingo PA, Tong T and Bolden S: Cancer
statistics, 1995. CA Cancer J Clin. 45:8–30. 1995. View Article : Google Scholar
|
|
2.
|
Faleiro-Rodrigues C, Macedo-Pinto I,
Pereira D and Lopes CS: Prognostic value of E-cadherin
immunoexpression in patients with primary ovarian carcinomas. Ann
Oncol. 15:1535–1542. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Singer G, Oldt R III, Cohen Y, et al:
Mutations in BRAF and KRAS characterize the development of
low-grade ovarian serous carcinoma. J Natl Cancer Inst. 95:484–486.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Nakayama K, Nakayama N, Kurman RJ, et al:
Sequence mutations and amplification of PIK3CA and AKT2 genes in
purified ovarian serous neoplasms. Cancer Biol Ther. 5:779–785.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Wan PT, Garnett MJ, Roe S, et al:
Mechanism of activation of the RAF-ERK signaling pathway by
oncogenic mutations of B-RAF. Cell. 116:855–867. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Olson JM and Hallahan AR: p38 MAP kinase:
a convergence point in cancer therapy. Trends Mol Med. 10:125–129.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Hsu CY, Bristow R, Cha MS, et al:
Characterization of active mitogen-activated protein kinase in
ovarian serous carcinomas. Clin Cancer Res. 10:6432–6436. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Peyssonnaux C and Eychene A: The
Raf/MEK/ERK pathway: new concepts of activation. Biol Cell.
93:53–62. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Allen LF, Sebolt-Leopold J and Meyer MB:
CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK
(MAPKK). Semin Oncol. 30:105–116. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Yamamoto K, Kikuchi Y, Kudoh K and Nagata
I: Modulation of cisplatin sensitivity by taxol in
cisplatin-sensitive and -resistant human ovarian carcinoma cell
lines. J Cancer Res Clin Oncol. 126:168–172. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Holmstrom TH, Tran SE, Johnson VL, et al:
Inhibition of mitogen-activated kinase signaling sensitizes HeLa
cells to Fas receptor-mediated apoptosis. Mol Cell Biol.
19:5991–6002. 1999.PubMed/NCBI
|
|
12.
|
Cowley GP and Smith ME: Modulation of
E-cadherin expression and morphological phenotype in the
intravascular component of adenocarcinomas. Int J Cancer.
60:325–329. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Huang C, Jacobson K and Schaller MD: MAP
kinases and cell migration. J Cell Sci. 117:4619–4628. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Mansour SJ, Matten WT, Hermann AS, et al:
Transformation of mammalian cells by constitutively active MAP
kinase kinase. Science. 265:966–970. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Mansour SJ, Resing KA, Candi JM, et al:
Mitogen-activated protein (MAP) kinase phosphorylation of MAP
kinase kinase: determination of phosphorylation sites by mass
spectrometry and site-directed mutagenesis. J Biochem. 116:304–314.
1994.
|
|
16.
|
Anand-Apte B, Zetter BR, Viswanathan A, et
al: Platelet-derived growth factor and fibronectin-stimulated
migration are differentially regulated by the Rac and extracellular
signal-regulated kinase pathways. J Biol Chem. 272:30688–30692.
1997. View Article : Google Scholar
|
|
17.
|
Klemke RL, Cai S, Giannini AL, et al:
Regulation of cell motility by mitogen-activated protein kinase. J
Cell Biol. 137:481–492. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Webb DJ, Donais K, Whitmore LA, et al:
FAK-Src signalling through paxillin, ERK and MLCK regulates
adhesion disassembly. Nat Cell Biol. 6:154–161. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Cheresh DA, Leng J and Klemke RL:
Regulation of cell contraction and membrane ruffling by distinct
signals in migratory cells. J Cell Biol. 146:1107–1116. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Lai CF, Chaudhary L, Fausto A, et al: Erk
is essential for growth, differentiation, integrin expression, and
cell function in human osteoblastic cells. J Biol Chem.
276:14443–14450. 2001.PubMed/NCBI
|
|
21.
|
Mao TL, Seidman JD, Kurman RJ and Shih IM:
Cyclin E and p16 immunoreactivity in epithelioid trophoblastic
tumor – an aid in differential diagnosis. Am J Surg Pathol.
30:1105–1110. 2006.PubMed/NCBI
|
|
22.
|
Mizumoto Y, Kyo S, Mori N, et al:
Activation of ERK1/2 occurs independently of KRAS or BRAF status in
endometrial cancer and is associated with favorable prognosis.
Cancer Sci. 98:652–658. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Nakayama N, Nakayama K, Yeasmin S, et al:
KRAS or BRAF mutation status is a useful predictor of sensitivity
to MEK inhibition in ovarian cancer. Br J Cancer. 99:2020–2028.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Kang Y and Massague J:
Epithelial-mesenchymal transitions: twist in development and
metastasis. Cell. 118:277–279. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Terauchi M, Kajiyama H, Yamashita M, et
al: Possible involvement of TWIST in enhanced peritoneal metastasis
of epithelial ovarian carcinoma. Clin Exp Metastasis. 24:329–339.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Niu RF, Zhang L, Xi GM, et al:
Up-regulation of Twist induces angiogenesis and correlates with
metastasis in hepatocellular carcinoma. J Exp Clin Cancer Res.
26:385–394. 2007.PubMed/NCBI
|
|
27.
|
Mironchik Y, Winnard PT, Vesuna F, et al:
Twist overexpression induces in vivo angiogenesis and correlates
with chromosomal instability in breast cancer. Cancer Res.
65:10801–10809. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Maestro R, Dei Tos AP, Hamamori Y, et al:
Twist is a potential oncogene that inhibits apoptosis. Genes Dev.
13:2207–2217. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Chou FL, Hill JM, Hsieh JC, et al: PEA-15
binding to ERK1/2 MAPKs is required for its modulation of integrin
activation. J Biol Chem. 278:52587–52597. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Kurata H, Thant AA, Matsuo S, et al:
Constitutive activation of MAP kinase kinase (MEK1) is critical and
sufficient for the activation of MMP-2. Exp Cell Res. 254:180–188.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Webb DJ, Nguyen DH and Gonias SL:
Extracellular signal-regulated kinase functions in the urokinase
receptor-dependent pathway by which neutralization of low density
lipoprotein receptor-related protein promotes fibrosarcoma cell
migration and Matrigel invasion. J Cell Sci. 113:123–134. 2000.
|
|
32.
|
Takino T, Miyamori H, Watanabe Y, et al:
Membrane type 1 matrix metalloproteinase regulates
collagen-dependent mitogen-activated protein/extracellular
signal-related kinase activation and cell migration. Cancer Res.
64:1044–1049. 2004. View Article : Google Scholar
|
|
33.
|
Tanigawara Y, Kita T, Hirono M, et al:
Identification of N-acetyltransferase 2 and CYP2C19 genotypes for
hair, buccal cell swabs, or fingernails compared with blood. Ther
Drug Monit. 23:341–346. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Sebolt-Leopold JS, Dudley DT, Herrera R,
et al: Blockade of the MAP kinase pathway suppresses growth of
colon tumors in vivo. Nat Med. 5:810–816. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Arteaga CL and Baselga J: Tyrosine kinase
inhibitors: Why does the current process of clinical development
not apply to them? Cancer Cell. 5:525–531. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Rinehart J, Adjei AA, Lorusso PM, et al:
Multicenter phase II study of the oral MEK inhibitor, CI-1040, in
patients with advanced non-small-cell lung, breast, colon and
pancreatic cancer. J Clin Oncol. 22:4456–4462. 2004. View Article : Google Scholar : PubMed/NCBI
|