Introduction
Cervical cancer is the second most common
gynecologic malignancy in Chinese women (1). Persistent infection of specific
types of genital human papillomaviruses (HPVs), especially the
oncogenic or high-risk (HR) types, has been generally recognized as
the principle cause of this disease and its precursor, cervical
intraepithelial neoplasia (CIN). HPV-52 is one of 13 anogenital
tract infected HR-HPV types (2).
According to a meta-analysis in 2003 (3), it is the seventh most common
detected HR-HPV type in invasive cervical cancer (ICC) worldwide.
However, in South-East Asia, HPV-52 is one of most common prevalent
HR-HPV types, and a large proportion of cervical cancers is
associated with HPV-52, which is the second or third most common
HPV type in ICC (4,5).
Each HPV type comprises numerous genomic variants,
and an up to 2% nucleotide difference has often been identified.
This difference confers each variant a biologically distinct
characterization and pathogenic risks (6). HPV-52 is phylogenetically related to
HPV-16 (7), whose variants could
be divided into Asian-American, African and European lineages,
whose carcinogenic risks have been extensively studied (8,9).
Similarly, the variants of HPV-52 could be classified as Asian and
European lineages (10), with a
mean sequence difference of 1.8–2.0% (11). However, limited data of HPV-52
variants is available from the mainland of China (12).
The genomic characterization of HPV variants is
necessary for understanding the intrinsic geographical relations
and contribute to research of their pathogenicity. Traditional
studies on the variability of HPV types are very laborious and
costly. First, all target genes need to be sequenced following a
set of polymerase chain reaction (PCR), and then the resulting
sequences need to be compared with the reference sequence to
identify mismatches. The high resolution melting (HRM) analysis
provided scientists a new approach to detect and classify HPV
variant in recent years. This kind of method combines DNA
amplification by PCR and subsequent melting of the amplicons into
one reaction. After data normalization, the sequence variation
could be identified by the shape of melting curves (13). This new approach has been
successfully used to study the variability of HPV type 16 (14).
Based on an epidemiologic screening for HPV
infections in Chaozhou, eastern Guangdong province from 2009–2010,
HPV-52 was found to be the most common HR-HPV type in this area.
More than one third (971/2907) of HR-HPV infectors were singly or
multiply infected with HPV-52. The present study explored the
nucleotide variability and phylogeny of HPV-52, in samples from the
HPV infections screening mentioned above. For this purpose, the E6
and L1 genes were firstly sequenced by the traditional method, then
the HRM analysis was used to group identified variants. The
pathogenicity of the most common variants was compared.
Unexpectedly, several abnormal peaks (double peaks) were found in
direct sequencing results. We speculated these specimens may
contain two or more different HPV-52 variants, and the restriction
enzyme analysis was performed to validate this hypothesis.
Materials and methods
Sample collection
From December 2009 to September 2010, an
epidemiologic screening for HPV infections was organized by the
Chaozhou municipal government, and more than 48,000 females
participated in this screening. Multiplex real-time PCR was firstly
performed to detect 13 types of HR-HPV infection. Then, the HPV
GenoArray test (15,16) was used to identify the specific
HPV types with the same samples. HPV DNA positive women were
advised to receive ThinPrep liquid-based cytology test (LCT), and
histological diagnosis was performed if necessary. In this study, a
woman was eligible to participate in the study if she i)
participated in the HPV infection screening described above; ii)
was singly infected with HPV-52; iii) received LCT and/or
histological diagnosis; and iv) was willing to be a subject in the
present study. The study was carried out with the approval of the
Ethics Committee of Chaozhou Central Hospital, Chaozhou, China, and
patient consent was obtained for the collection of cervical
exfoliated cells.
In accordance with the suggestion of gynaecological
practitioners, 186 HPV-52 infectors received LCT. Among them, 102
women were excluded since they were infected with multiple HPV
types or did not agree to participate in the study. In total, 84
eligible cases of single HPV-52 infection were enrolled into the
study.
DNA extraction and amplification
The cervical exfoliated cells were collected using
plastic cervical swabs, and then immediately stored at 4°C. The DNA
was extracted by the alkaline lysis method using DNA extraction
kits (Hybribio Biotechnology Corp.). Detailed protocols for this
assay have been described previously (15). The quality of extracted DNA was
checked by PCR amplification of the glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) gene (forward, 5′-CAT GAC CAC AGT CCA TGC CAT
CAC T-3′ and reverse primer, 5′-TGA GGT CCA CCA CCC TGT TGC TGT
A-3′).
For complete E6 and L1 gene amplification, two and
three sets of primers (Table I)
were designed respectively based on the sequence of the prototype
TJ49-52 (GenBank ID: GQ472848). The complete 1.8 kb genomes of the
L1 gene were amplified in three or four overlapping fragments
according to the primers used. The amplification protocol was as
follows: initial denaturation for 5 min at 93°C, followed by 35–40
cycles of denaturation for 1 min at 93°C, annealing for 1 min at
the appropriate temperature (Table
I) and elongation for 10 min at 72°C. The amplified DNA
fragments were identified by electrophoresis on 2% agarose gels
stained with ethidium bromide, and submitted for sequencing of both
strands at the Invitrogen Biotechnology Co., Ltd., Guangzhou,
China.
 | Table I.Primers for E6 and L1 genes
amplification. |
Table I.
Primers for E6 and L1 genes
amplification.
| Gene | Target | Primer name | Sense | Antisense | Annealing
temperature (°C) |
|---|
| E6 | 102–548 | 52E6Set1 | 92–113 | 686–713 | 61 |
| | 52E6Set2 | 26–48 | 550–574 | 57 |
| L1 | 5593–7182 | 52L1Set1-A | 5517–5537 | 6024–6042 | 57 |
| | 52L1Set1-B | 5590–6007 | 6515–6536 | 57 |
| | 52L1Set1-C | 6513–6533 | 7091–7108 | 57 |
| | 52L1Set1-D | 6867–6887 | 7290–7311 | 58 |
| | 52L1Set2-A | 5517–5537 | 6024–6042 | 57 |
| | 52L1Set2-B | 5590–6007 | 6515–6536 | 57 |
| | 52L1Set2-C | 6513–6533 | 7091–7108 | 57 |
| | 52L1Set2-D | 6865–6887 | 7416–7438 | 59 |
| | 52L1Set3-A | 5517–5537 | 6024–6042 | 57 |
| | 52L1Set3-B | 5953–5972 | 6645–6664 | 58 |
| | 52L1Set3-C | 6572–6591 | 7205–7225 | 60 |
Sequence analysis and phylogenetic
evaluations
The mismatches were analyzed and determined using
the Blast 2.0 software server (http://www.ncbi.nlm.nih.gov/blast). The HPV-52
prototype TJ49-52 was used as standard for comparison. Each unique
sequence variation was verified by repeated amplification and
opposite strand sequencing. The resulting sequences were aligned
with ClustalX software, the neighbor-joining and unweighted pair
group method with arithmetic average (UPGMA) trees were constructed
by using the MEGA software version 5 (17,18).
Restriction enzyme analysis
Abnormal peaks (double peaks) were found in seven
sequence traces, and their corresponding PCR products were named
Ab1 to Ab7, respectively. In order to eliminate samples
cross-contamination, the DNA of these specimens was re-extracted,
amplified and sequenced again.
For this rare phenomenon, we speculated that the PCR
products contained two kinds of amplicons. In order to validate
this hypothesis, restriction enzymes TaqI, XbaI,
BspEI, RsaI, MspI and MlyI (all
purchased from Fermentas) were selected according to the
neighboring sequence of abnormal peaks. The selection criteria were
i) either one hypothetical amplicon or another was cleavable; and
ii) there was only one recognition site of restriction enzyme on
the selected amplicon. The PCR products were digested with specific
enzymes according to the recommended protocols for digestion of PCR
products directly after amplification. The 50 μl digestion mixture
included 15 μl PCR reaction mixture, 29 μl nuclease-free water, 3
μl 10X buffer and 3 μl (10 U/μl) restriction enzyme. The detailed
protocols for digestion are listed in Table II. The resulting fragments were
run on 2% agarose gels stained with ethidium bromide, and the
uncleavaged fragments were purified and submitted for
sequencing.
| Table II.Details for restriction enzyme
analysis. |
Table II.
Details for restriction enzyme
analysis.
| PCR product | Primers | Amplicon
length | Restriction
enzyme | Recognition
site | Digestion | Inactivation | Fragment
length |
|---|
| Ab1 | 52E6Set2 | 549 | RsaI | 5′-GT↓AC-3′ | 37°C for 24 h | 80°C for 20
min | 198, 351 |
| Ab2-5a | 52L1Set1-A | 526 | TaqI | 5′-T↓CGA-3′ | 65°C for 3 h | EDTA
solutionb | 252, 274 |
| | | XbaI | 5′-T↓CTAGA-3′ | 37°C for 24 h | 65°C for 20
min | 254, 272 |
| Ab6 | 52L1Set1-A | 526 | BspEI | 5′-T↓CCGGA-3′ | 55°C for 3 h | 80°C for 20
min | 108, 418 |
| Ab7 | 52L1Set3-B | 712 | MspI | 5′-C↓CGG-3′ | 37°C for 24 h | 80°C for 20
min | 518, 194 |
| | | MlyI |
3′-CTCAG(n)5↓-5′ | 37°C for 8 h | 65°C for 20
min | 532, 180 |
HRM analysis
All of the specimens which could be successfully
amplified with PCR, were subjected to HRM analysis. The primers
(forward, 5′-TGT ATT ATG TGC CTA CGC TTT T-3′ and reverse, 5′-GGC
GTT TGA CAA ATT ATA CAT C-3′) used for HRM analysis were designed
based on the mismatches identified on the E6 gene. PCR reactions
were performed on the LightCycler® 480 II. The
instrument (Roche Diagnostics) was equipped with the software
LightCycler® 480 Gene Scanning Software Version 1.5
(Roche Diagnostics). Approximately 50 ng of DNA was amplified in a
total volume of 20 μl containing 0.2 μM of each primer, 1X PCR
buffer, 0.2 mM of each dNTP, 0.3 unit TaqDNA polymerase (Dongsheng
Biotech Co., Ltd. Guangzhou, China) and 1 μl LC Green plus (Idaho
Technology). The reaction conditions were 95°C for 3 min, followed
by 35 cycles at 98°C for 20 sec, 60°C for 20 sec, and 72°C for 20
sec. The melting program included three steps: denaturation at 95°C
for 1 min, renaturation at 40°C for 1 min and then melting in a
continuous fluorescent reading from 60–95°C at 20
acquisitions/°C.
Liquid-based cytology test (LCT) and
pathological diagnosis
LCT was performed in the Chaozhou Central Hospital.
The detailed protocols were previously described (15). The results were evaluated using
the Bethesda system (19). The
evaluation system included: i) negative (A0), ii) atypical squamous
cells (ASC), iii) low-grade squamous intraepithelial lesion (LSIL),
iv) high-grade squamous intraepithelial lesion (HSIL), and v)
squamous cell carcinoma (SCC). The LSIL, HSIL and SCC cases further
received biopsies and the samples were processed and diagnosed in
the Department of Pathology, Chaozhou Central Hospital.
Statistical analysis
For pathogenic risks assessment, binary and
multinomial logistic regression analysis was used. Firstly, the
pathogenic risks were compared (binary logistic regression) between
two main lineages variants according to the phylogenetic trees
constructed. Then, the pathogenic risks of the four most common
variants were compared (multinomial logistic regression). All data
were analyzed using SPSS software version 16. P-values were
two-sided, and statistical significance was accepted if the P-value
was ≤0.05.
Results and Discussion
Phylogenetic and geographical relatedness
of HPV52 variants
Of the total 84 specimens, five specimens were
excluded since they were negative for GAPDH amplification, and the
complete E6 and L1 genes were amplified and sequenced successfully
in 79 cases. Among them, six cases were not suitable for genetic
variability evaluation because they were infected with two kinds of
HPV-52 variants. Thus, the HPV-52 genetic variability was evaluated
in 73 HPV-52 singly infected females (median age 44.5 years, range
35.5–59.4 years).
At the nucleotide level, a total of 21 HPV-52
variants were identified when E6 and L1 sequences were combined and
the prototype TJ49-52 was used as the standard for comparison. The
sequences of these 21 variants have been submitted to the GenBank.
The GenBank IDs are JN874437-JN874457 for the E6 gene and
JN874416-JN874436 for the L1 gene (Table III). The maximum nucleotide
diversity was 1.1% (22/2037) observed between isolates CZ52A375 and
CZ52E429.
| Table III.The GenBank IDs for the isolates
reported in this study. |
Table III.
The GenBank IDs for the isolates
reported in this study.
| Isolates | GenBank ID for E6
gene | GenBank ID for L1
gene | Lineage |
|---|
| CZ52A105 | JN874437 | JN874416 | Asian |
| CZ52A207 | JN874438 | JN874417 | Asian |
| CZ52A255 | JN874439 | JN874418 | Asian |
| CZ52A620 | JN874441 | JN874420 | Asian |
| CZ52A1404 | JN874442 | JN874421 | Asian |
| CZ52A1230 | JN874443 | JN874422 | Asian |
| CZ52A375 | JN874444 | JN874423 | Asian |
| CZ52A1569 | JN874446 | JN874425 | Asian |
| CZ52A264 | JN874447 | JN874426 | Asian |
| CZ52A247 | JN874448 | JN874427 | Asian |
| CZ52A277 | JN874449 | JN874428 | Asian |
| CZ52A336 | JN874450 | JN874429 | Asian |
| CZ52A1023 | JN874451 | JN874430 | Asian |
| CZ52A228 | JN874453 | JN874432 | Asian |
| CZ52A168 | JN874454 | JN874433 | Asian |
| CZ52D416 | JN874455 | JN874434 | Asian |
| CZ52D417 | JN874456 | JN874435 | Asian |
| CZ52A921 | JN874457 | JN874436 | Asian |
| CZ52E928 | JN874440 | JN874419 | European |
| CZ52E136 | JN874445 | JN874424 | European |
| CZ52E429 | JN874452 | JN874431 | European |
There were 10 and 15 kinds of variability found on
E6 and L1 gene, respectively. Across the E6 gene 2.5% nucleotide
sites (11/447) and 1.3% encoded amino acids (2/149) were variable
(Table IV), while 1.9% nucleotide
sites (31/1590) and 1.1% encoded amino acids (6/530) were variable
across the L1 gene (Table V).
| Table IV.Mismatches on the E6 gene. |
Table IV.
Mismatches on the E6 gene.
|
Nucleotide
position
|
|---|
| Isolate | 90 | 136 | 168 | 228 | 249 | 255 | 277 | 278 | 336 | 366 | 429 |
|---|
| CZ52A105 | T | C | C | T | T | G | A | G | T | C | A |
| CZ52A1569 | C | C | C | T | T | G | A | G | T | C | A |
| CZ52A336 | T | C | C | T | T | G | A | G | C | C | A |
| CZ52A228 | T | C | C | C | T | G | A | G | T | C | A |
| CZ52A255 | T | C | C | T | T | A | A | G | T | C | A |
| CZ52A168 | T | C | T | T | T | G | A | G | T | C | A |
| CZ52A277 | T | C | C | T | T | G | C | G | T | Aa | A |
| CZ52E928 | T | C | C | T | G | G | A | Ab | T | C | A |
| CZ52E429 | T | C | C | T | T | G | A | Ab | T | C | G |
| CZ52E136 | T | T | C | T | T | G | A | Ab | T | C | G |
| Table V.Mismatches on the L1 gene. |
Table V.
Mismatches on the L1 gene.
|
Nucleotide
position
|
|---|
| Isolate | 14 | 207 | 247 | 264 | 345 | 375 | 408 | 444 | 519 | 546 | 620 | 621 | 654 | 879 | 921 | 975 | 999 | 1023 | 1134 | 1137 | 1192 | 1200 | 1230 | 1246 | 1260 | 1353 | 1377 | 1404 | 1488 | 1548 | 1569 |
|---|
| CZ52A105 | T | G | A | A | A | A | C | G | G | A | A | T | A | C | A | T | A | G | G | G | G | C | G | G | T | A | A | A | A | G | G |
| CZ52A207 | T | A | A | A | A | A | C | G | G | A | A | T | A | C | A | T | A | G | G | G | G | C | G | G | T | A | A | A | A | G | G |
| CZ52A620 | T | G | A | A | A | A | C | G | G | A | Ca | T | A | C | A | T | A | G | G | G | G | C | G | G | T | A | A | A | A | G | G |
| CZ52A1404 | T | G | A | A | A | A | C | G | G | A | A | T | A | C | A | T | A | G | G | G | G | C | G | G | T | A | A | Cb | A | G | G |
| CZ52A1230 | T | G | A | A | A | A | C | G | G | A | A | T | A | C | A | T | A | G | G | G | G | C | A | G | T | A | A | A | A | G | G |
| CZ52A1569 | T | G | A | A | A | A | C | G | G | A | A | T | A | C | A | T | A | G | G | G | G | C | G | G | T | A | A | A | A | G | A |
| CZ52A264 | T | G | A | G | A | A | C | G | G | A | A | T | A | C | A | T | A | G | G | G | G | C | G | G | T | A | A | A | A | G | G |
| CZ52A1023 | T | G | A | A | A | A | C | G | G | A | A | T | A | C | A | T | A | A | G | G | G | C | G | G | T | A | A | A | A | G | G |
| CZ52A277 | T | G | A | A | A | A | C | G | G | A | A | T | A | C | A | T | A | G | G | G | G | C | G | Ac | T | A | G | A | A | G | G |
| CZ52A921 | T | A | A | A | A | A | C | G | G | A | A | T | A | C | G | T | A | G | G | G | G | C | G | G | T | A | A | A | A | G | G |
| CZ52A247 | T | G | Gd | A | A | A | C | A | G | A | A | T | A | C | G | A | A | G | G | G | G | C | G | G | T | A | A | A | A | G | G |
| CZ52A375 | T | G | A | G | A | G | C | G | G | A | Ca | C | A | C | A | T | G | G | G | G | Ae | C | G | G | T | A | A | A | A | G | G |
| CZ52D416 | T | G | A | A | A | A | C | G | A | G | A | T | A | T | A | T | A | G | G | G | G | C | G | G | T | A | A | A | A | G | G |
| CZ52E928 | T | A | A | A | A | A | T | G | G | G | A | T | G | C | A | T | A | G | G | T | G | T | A | G | C | C | A | A | G | G | G |
| CZ52E429 | Cf | A | A | A | G | A | T | G | A | G | A | T | A | T | A | T | A | G | A | T | G | T | A | G | C | A | A | A | G | A | G |
Great similarity was found between the phylogenetic
trees constructed based on the E6 or L1 gene. Partial and complete
HPV-52 sequence reports in different regions of the world formed
evolutionary trees with two main branches driven by variants with
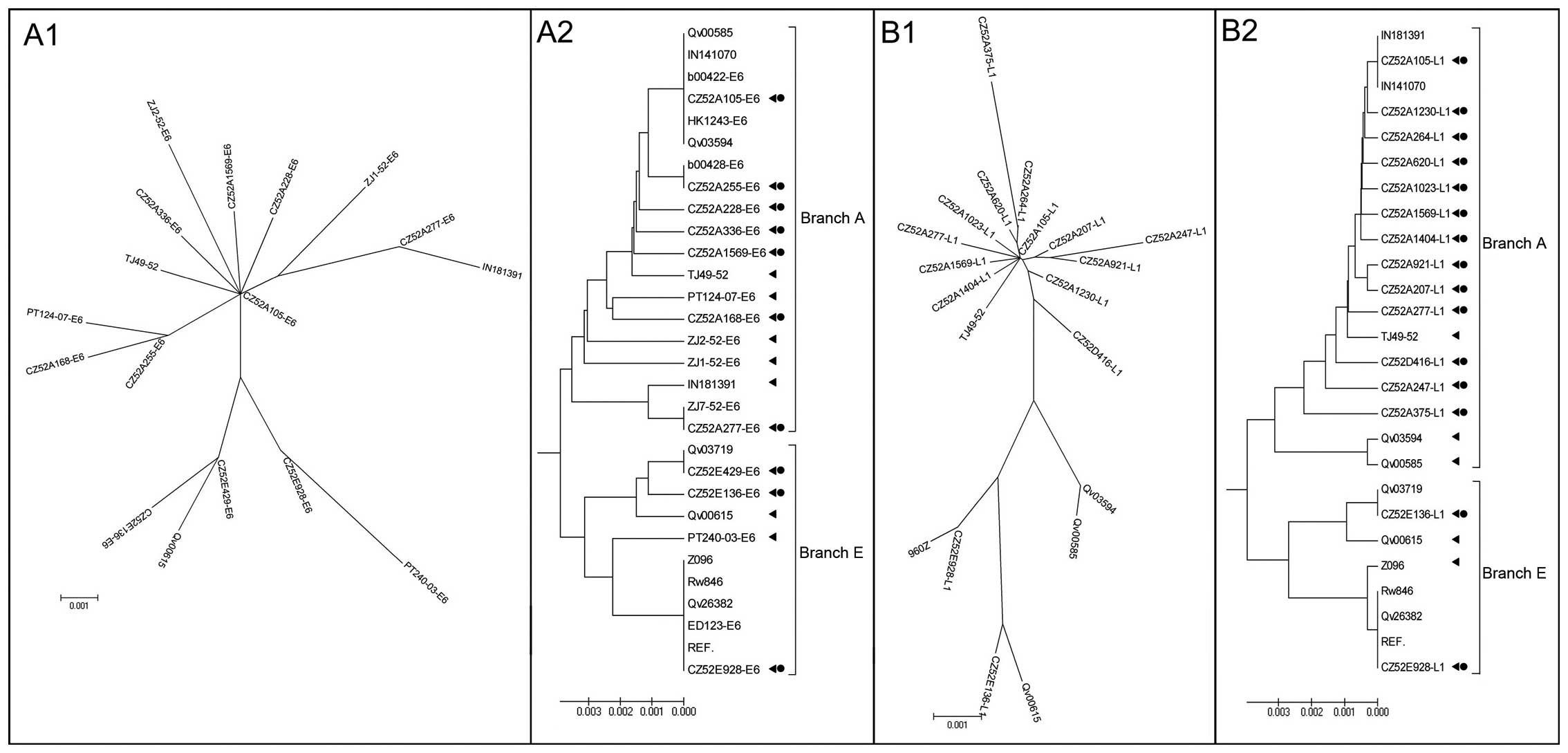
high prevalence in Asia (Fig. 1,
branch A) or Europe (Fig. 1,
branch E), which was coincident with the previous study (10). The details of the sequences used
for phylogenetic tree construction are presented in Table VI.
| Table VI.Details of the sequences used for
phylogenetic tree construction. |
Table VI.
Details of the sequences used for
phylogenetic tree construction.
| Isolate | Sequence type | GenBank ID | City/Country | Lineage |
|---|
| b00422 | E6 genes, complete
cds | EU924143 | Guangzhou,
China | Asian |
| b00428 | E6 genes, complete
cds | EU924144 | Guangzhou,
China | Asian |
| HK1243 | E6 genes, complete
cds | DQ057293 | Hongkong,
China | Asian |
| ZJ1-52 | E6 genes, complete
cds | HM004116 | Hangzhou,
China | Asian |
| ZJ2-52 | E6 genes, complete
cds | HM004115 | Hangzhou,
China | Asian |
| ZJ7-52 | E6 genes, complete
cds | HM004117 | Hangzhou,
China | Asian |
| PT124-07 | E6 genes, complete
cds | FJ002423 | Portugal | Asian |
| TJ49-52 | Complete
genome | GQ472848 | Tianjin, China | Asian |
| IN141070 | Complete
genome | HQ537743 | Chiang Mai,
Thailand | Asian |
| IN181391 | Complete
genome | HQ537742 | Chiang Mai,
Thailand | Asian |
| Qv00585 | Complete
genome | HQ537741 | Guanacaste, Costa
Rica | Asian |
| Qv03594 | Complete
genome | HQ537740 | Guanacaste, Costa
Rica | Asian |
| ED123 | E6 genes, complete
cds | DQ057290 | Scotland | European |
| PT240-03 | E6 genes, complete
cds | EF566934 | Portugal | European |
| REF. | Complete
genome | X74481 | Germany | European |
| Rw846 | Complete
genome | HQ537735 | Rwandan | European |
| Z096 | Complete
genome | HQ537736 | Zambia | European |
| Qv03719 | Complete
genome | HQ537745 | Guanacaste, Costa
Rica | European |
| Qv26382 | Complete
genome | HQ537732 | Guanacaste, Costa
Rica | European |
| Qv00615 | Complete
genome | HQ537746 | Guanacaste, Costa
Rica | European |
The isolate CZ52A105 represented the most common
variant on branch A with 45.2% (33/73) of the cases infected with
this kind of variant (Table VII).
Following was isolate CZ52A255 (8/73) and CZ52A207 (8/73). There
was one synonymous mutation compared with CZ52A105 on E6 for the
former and on L1 for the latter. The fourth most common variant
were isolates CZ52A228 (2/73) and CZ52A264 (2/73). There was one
synonymous mutation compared with CZ52A105 on E6 for the former,
and one synonymous mutation on E6 and L1 respectively for the
latter. The sequences of other variants located on branch A were
all unique.
| Table VII.LCT results for the most frequent
variants. |
Table VII.
LCT results for the most frequent
variants.
| | LCT results, n
| |
|---|
| Isolate
(lineagea) | Cases, n | ASC | LSIL | HSIL and SCC | LCT+
(%) | OR (95% CI)b |
|---|
| CZ52A105 (A) | 33 | 10 | 3 | 1 | 14 (42.4) | Reference |
| CZ52A207 (A) | 8 | 4 | 0 | 0 | 4 (50.2) | 1.36
(0.29–6.38) |
| CZ52A255 (A) | 8 | 5 | 0 | 0 | 5 (62.5) | 2.26
(0.46–11.08) |
| CZ52E429 (E) | 4 | 3 | 0 | 0 | 3 (75.0) | 4.07
(0.38–43.39) |
| Total Asian
variants | 66 | 23 | 8 | 2 | 33 (50.0) | Reference |
| Total European
variants | 7 | 4 | 0 | 0 | 4 (57.1) | 1.33
(0.28–6.43) |
The isolate CZ52A105 located at the bifurcation of
branch A, 83.6% (61/73) samples were identical with CZ52A105 at
amino acid level. Moreover, its E6 and L1 sequences were completely
matched with isolate IN141070 (11,20) reported in Chiang Mai (N 18.56, E
72.49). The E6 sequence was also identical with isolates b00422 and
b00433 reported in Guangzhou (N 23.70, E 113.15) and isolate HK1243
and HK2571 (17) reported in
HongKong (N 22.65, E 113.86). The latitude of Chaozhou (N 23.40, E
116.38) and of these 3 cities all ranged from N 18 to N 24. Taken
all above mentioned into consideration, we suggest that the isolate
CZ52A105 represents the most common and ancient HPV-52 variant in
South-East Asia.
Nearly one tenth (7/73) of the cases were
phylogenetically related to European lineages, and three variants
were identified. The isolate CZ52E429 (4 cases) represented the
most frequent variant, followed by isolate CZ52E928 (2 cases).
Compared with CZ52E429, one non-synonymous mutant was observed on
L1, two and seven synonymous mutants were identified on E6 and L1
respectively. The other isolate located on branch E was CZ52E136 (1
case), there was only one synonymous mutant found on E6 if the
sequence of isolate CZ52E429 was used as a reference. The isolate
CZ52E928 located at the bifurcation of branch E, it may represent
the most widely spread variant. The same E6 and L1 sequences were
also found in Europe (Germany) (21), Africa (Rwanda) and North America
(Costa Rica) (11,22,23). We speculate that this variant may
be derived from the worldwide migration of the European host.
Variants pathogenicity assessment and HRM
analysis application
Despite phylogenetic relatedness, a
geographic-related pathogenicity difference has been found in
HPV-16 and HPV-18 variants. The carcinogenic risk for
Asian-American or African HPV16 variants is greater than that of
European variants (8), and
non-European HPV-18 variants are more common in cancer tissues than
European variants (9).
A preliminary pathogenicity comparison between
HPV-52 variants was performed in this study. The LCT results of the
most frequent variants are shown in Table VII. A total of 73 specimens were
firstly divided into 2 groups, one was made up of the Asian lineage
related specimens (66 cases), and the other was composed of
European lineage related specimens (7 cases). The pathogenic risk
was assessed by binary logistic regression analysis. The median age
of the two groups was 45.4 and 42.2-year-old, no significant
difference was found between the pathogenic risk of these two
lineages variants (P=0.72). Then, the pathogenic risks of the four
most common variants were evaluated. There was no statistically
significant difference between the groups (P=0.51). However, there
were some limitations for this conclusion, because there was a
great unbalance between the case numbers of the variants. In order
to validate this conclusion, it is necessary to perform this
experiment in a larger sample.
The HRM analysis provided us a more economic and
rapid method to deal with large scale screening HPV-52 variants. As
a preliminary attempt, the E6 gene was used to group identified
HPV-52 variants by HRM analysis. The choice of E6 gene for the
analysis of the variability by HRM was based on the fact that it
was the most common gene examined by sequencing worldwide, and
because it has an optimal variable region (6,14).
A shorter amplicon was usually required for HRM
analysis because as amplicon size increase, it becomes increasingly
difficult to identify mismatches (13). For this reason, the amplicon
(nucleotide 187–326, 140 bp) used for HRM analysis was only one
third of the complete E6 gene. This amplicon involved five
mismatches found on the E6 gene in our study, and these mismatches
led to six kinds of variants. One variant involved two mismatches,
the other five contained only one mismatch. It is worth pointing
out that the consensus mismatch between the Asian and European
lineages was located at nucleotide 278 (G>A) (Table IV), and it was also located in the
amplicon.
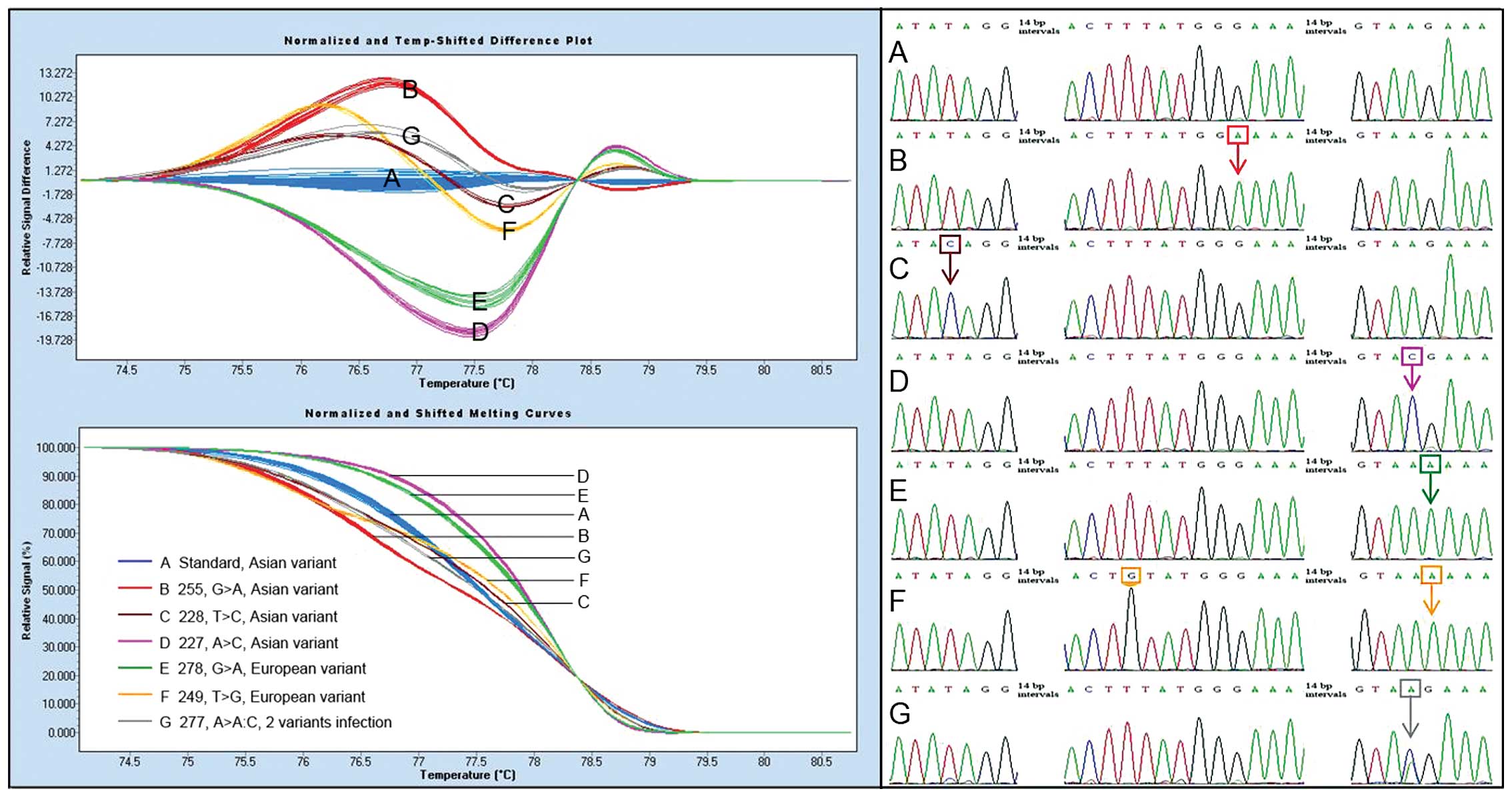
After all the data were normalized, a total of 79
specimens (including six multiple variants infection cases) were
divided into six groups (Fig. 2),
and all of six variants on the amplicon could be divided into
distinct groups. Four groups (Fig.
2A–D) were composed of Asian variants, and the other two
(Fig. 2E and F) were made of
European variants. In this sense, Asian and European variants could
be distinguished by HRM analysis with a complete accuracy.
Therefore, the application of HRM would facilitate the HPV-52
variants pathogenic comparison between Asian and European lineages.
In addition, one case with two HPV-52 variants infection (Ab1)
could also be identified by the shape of the resulting melting
curve (Fig. 2G).
Multiple variant infections
In this study, some double peaks were found in 7
direct sequencing results, and 6 specimens were involved in these
events. The Ab6 and Ab7 were consecutive fragments derived from one
specimen, and the other 5 came from different specimens.
Interestingly, the same double peaks were found in four of them
(Ab2-Ab5) at the same nucleotide position.
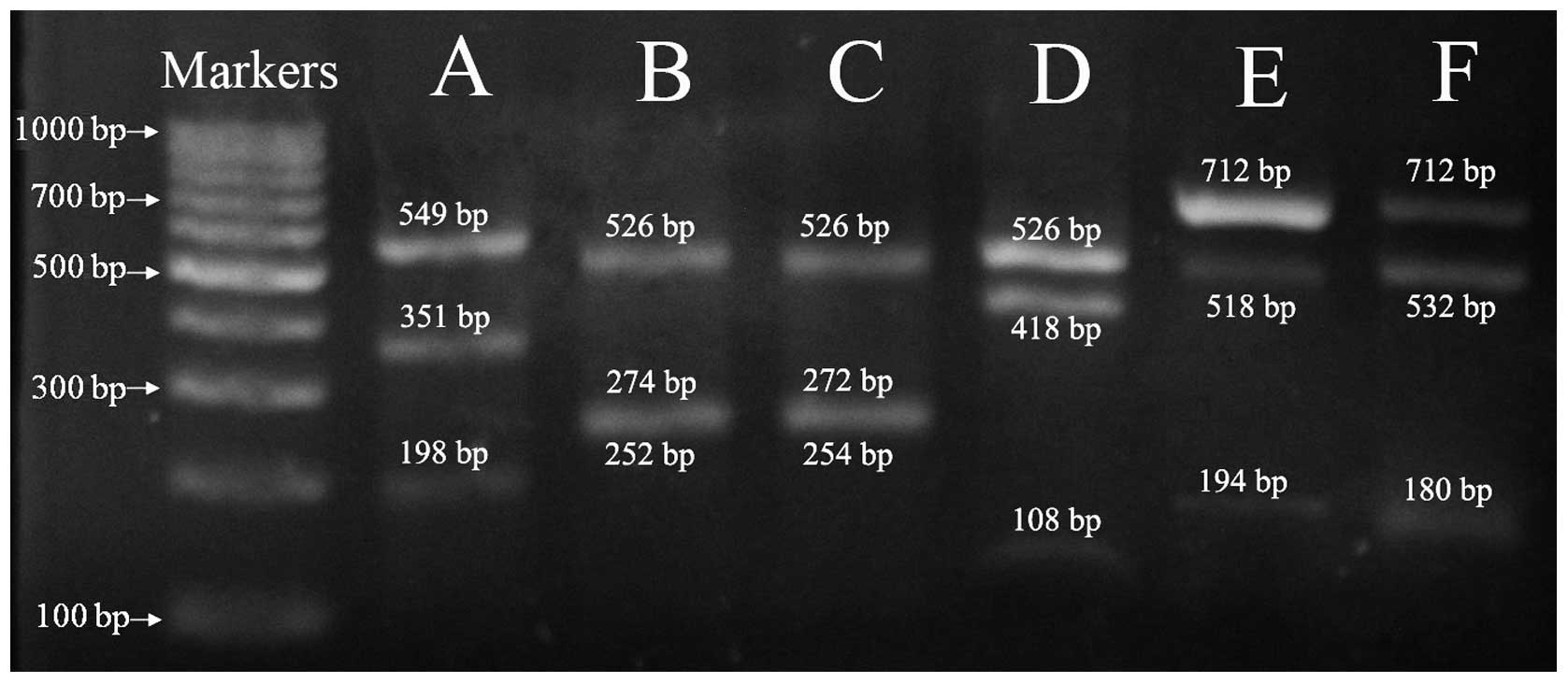
After digestion with specific restriction enzymes,
three electrophoresis bands could be found, including two new
generated bands and one uncleavable band (Fig. 3), which was purified and submitted
for sequencing. The double peaks disappeared in the sequence trace
of the uncleavable fragments, which were replaced by a single base
at the corresponding positions (Fig.
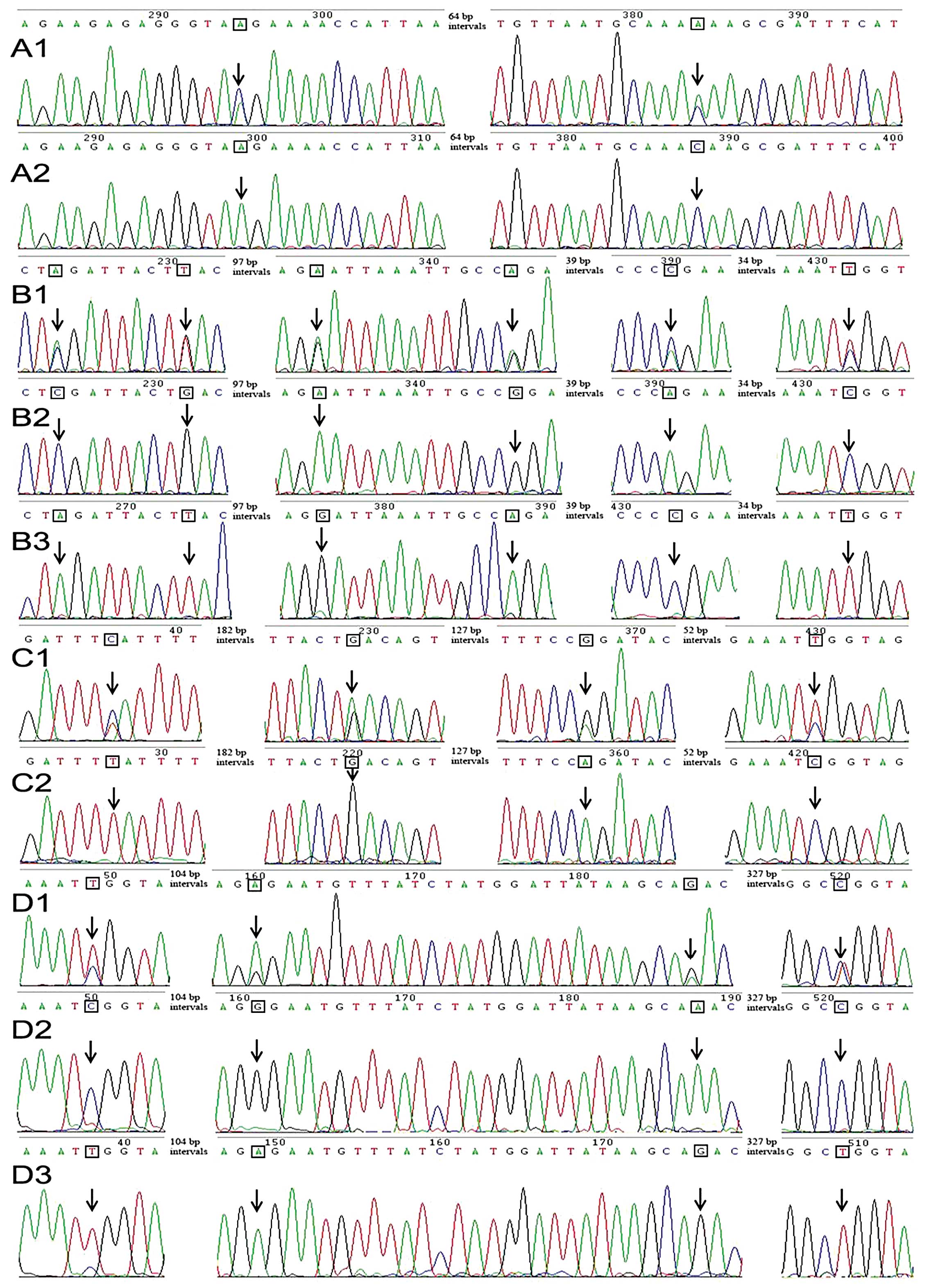
3). Using Ab2 (Fig. 3C) as an
example, six double peaks were observed in its direct sequencing
results (Fig. 4B2). They were
composed of peaks, C:A, G:T, A:G, G:A, A:C and C:T in turn. After
digestion with restriction enzyme XbaI, these double peaks
were replaced with single base C, G, A, G, A and C respectively in
the sequence trace of the uncleavable fragment. On the contrary, if
this PCR product was digested with restriction enzyme TaqI,
these double peaks were replaced with single base A, T, G, A, C and
T, respectively. Based on this result, we believed that the most
possible reason for this event was that the specimens contained two
HPV-52 variants, in other words, these women infected two kinds of
HPV-52 variants. We believe that this phenomenon may be similar to
the multiple type infections. Once a woman is singly infected with
HPV-52, she has an almost equal opportunity of further infection
with other HPV types or infection with HPV-52 again, the former
events leading to multiple infections, and the latter possibly
leading to two kinds of HPV-52 variants infection.
To our knowledge, this rare phenomenon has not so
far been reported worldwide. However, since it was observed in 7.6%
(6/79) specimens in this study, why was it not observed in the
previous studies? We think that there are three possible reasons.
First, this rare phenomenon is preferentially observed in an area
of high HPV-52 prevalence. Second, the primers used for
amplification must have been suitable for two kinds of variants.
Last but not least, the viral loads of two different variants
should be nearly equal in the same sample. However, the lower one
might be ignored in the sequencing trace if there was a great
difference between the viral loads of the two variants. As
described above, HPV-52 was the most common HPV type in eastern
Guangdong, and its high prevalence may increase the opportunity of
multiple variants infection.
Acknowledgements
This study was supported by grants by
the Medical Research Funds of Guangdong Province (A2011760,
B2008179). Reagent discounts and reagent gifts were from the
Hybribio Biotechnology Limited Corp. We offer special recognition
for the excellent work of the study staff in sample collection. We
sincerely thank SY Zheng and JB Liu (Department of Pathology,
Chaozhou Central Hospital) for technical assistance with
pathology.
References
|
1.
|
HC ChenM SchiffmanCY LinMH PanSL YouLC
ChuangCY HsiehKL LiawAW HsingCJ ChenCBCSP-HPV Study
GroupPersistence of type-specific human papillomavirus infection
and increased long-term risk of cervical cancerJ Natl Cancer
Inst10313871396201110.1093/jnci/djr28321900119
|
|
2.
|
N MuñozFX BoschS de SanjoséR HerreroX
CastellsaguéKV ShahPJ SnijdersCJ MeijerInternational Agency for
Research on Cancer Multicenter Cervical Cancer Study
GroupEpidemiologic classification of human papillomavirus types
associated with cervical cancerN Engl J Med3485185272003
|
|
3.
|
GM CliffordJS SmithM PlummerN MuñozS
FranceschiHuman papillomavirus types in invasive cervical cancer
worldwide: a meta-analysisBr J
Cancer886373200310.1038/sj.bjc.660068812556961
|
|
4.
|
CM HoTY ChienSH HuangBH LeeSF
ChangIntegrated human papillomavirus types 52 and 58 are
infrequently found in cervical cancer, and high viral loads predict
risk of cervical cancerGynecol
Oncol1025460200610.1016/j.ygyno.2005.11.03516386784
|
|
5.
|
K TakeharaT TodaT NishimuraJ SakaneY
KawakamiT MizunoeM NishiwakiK TaniyamaHuman papillomavirus types 52
and 58 are prevalent in uterine cervical squamous lesions from
Japanese womenPatholog Res Int2011246936201121660229
|
|
6.
|
HU BernardIE Calleja-MaciasST DunnGenome
variation of human papillomavirus types: phylogenetic and medical
implicationsInt J
Cancer11810711076200610.1002/ijc.2165516331617
|
|
7.
|
M SchiffmanR HerreroR DesalleA HildesheimS
WacholderAC RodriguezMC BrattiME ShermanJ MoralesD GuillenThe
carcinogenicity of human papillomavirus types reflects viral
evolutionVirology3377684200510.1016/j.virol.2005.04.00215914222
|
|
8.
|
A HildesheimM SchiffmanC BromleyS
WacholderR HerreroA RodriguezMC BrattiME ShermanU ScarpidisQQ
LinHuman papillomavirus type 16 variants and risk of cervical
cancerJ Natl Cancer
Inst93315318200110.1093/jnci/93.4.31511181779
|
|
9.
|
L SicheroS FerreiraH TrottierE
Duarte-FrancoA FerenczyEL FrancoLL VillaHigh grade cervical lesions
are caused preferentially by non-European variants of HPVs 16 and
18Int J Cancer12017631768200710.1002/ijc.2248117230525
|
|
10.
|
T RaiolPS WyantRM de AmorimDM CerqueiraNG
MilaneziM Brígido MdeL SicheroCR MartinsGenetic variability and
phylogeny of the high-risk HPV-31, -33, -35, -52, and -58 in
central BrazilJ Med Virol81685692200910.1002/jmv.2143219235839
|
|
11.
|
Z ChenM SchiffmanR HerreroR DesalleK
AnastosM SegondyVV SahasrabuddhePE GravittAW HsingRD BurkEvolution
and taxonomic classification of human papillomavirus 16
(HPV16)-related variant genomes: HPV31, HPV33, HPV35, HPV52, HPV58
and HPV67PLoS
One6e20183201110.1371/journal.pone.002018321673791
|
|
12.
|
XL WuCT ZhangXK ZhuYC WangDetection of HPV
types and neutralizing antibodies in women with genital warts in
Tianjin City, ChinaVirol
Sin25817201010.1007/s12250-010-3078-420960279
|
|
13.
|
CT WittwerGH ReedCN GundryJG VandersteenRJ
PryorHigh-resolution genotyping by amplicon melting analysis using
LCGreenClin Chem49853860200310.1373/49.6.85312765979
|
|
14.
|
I SabolM CretnikI HadzisejdićA Si-MohamedM
MatovinaB GrahovacS LevanatM GrceA new approach for the evaluation
of the human papillomavirus type 16 variability with high
resolution melting analysisJ Virol
Methods162142147200910.1016/j.jviromet.2009.07.02919664661
|
|
15.
|
M LinLY YangLJ LiJR WuYP PengZY LuoGenital
human papillomavirus screening by gene chip in Chinese women of
Guangdong provinceAust NZ J Obstet
Gynaecol48189194200810.1111/j.1479-828X.2008.00844.x18366494
|
|
16.
|
SS LiuRC LeungKK ChanAN CheungHY
NganEvaluation of a newly developed GenoArray human papillomavirus
(HPV) genotyping assay and comparison with the Roche Linear Array
HPV genotyping assayJ Clin
Microbiol48758764201010.1128/JCM.00989-0920042614
|
|
17.
|
IE Calleja-MaciasLL VillaJC PradoM
KalantariB AllanAL WilliamsonLP ChungRJ CollinsRE ZunaST
DunnWorldwide genomic diversity of the high-risk human
papillomavirus types 31, 35, 52, and 58, four close relatives of
human papillomavirus type 16J
Virol791363013640200510.1128/JVI.79.21.13630-13640.200516227283
|
|
18.
|
K TamuraD PetersonN PetersonG StecherM
NeiS KumarMEGA5: molecular evolutionary genetics analysis using
maximum likelihood, evolutionary distance, and maximum parsimony
methodsMol Biol Evol2827312739201110.1093/molbev/msr121
|
|
19.
|
D SolomonD DaveyR KurmanA MoriartyD
O’ConnorM PreyS RaabM ShermanD WilburT Wright JrN YoungForum Group
Members; Bethesda 2001 Workshop: The 2001 Bethesda System:
terminology for reporting results of cervical
cytologyJAMA28721142119200211966386
|
|
20.
|
K WongworapatR KeawvichitB SirirojnS
DokutaC RuangyuttikarnS SriplienchanA SontiratKT KlaPE GravittDD
CelentanoDetection of human papillomavirus from self-collected
vaginal samples of women in Chiang Mai, ThailandSex Transm
Dis35172173200810.1097/OLQ.0b013e318158af6518216725
|
|
21.
|
H DeliusB HofmannPrimer-directed
sequencing of human papillomavirus typesCurr Top Microbiol
Immunol186133119948205838
|
|
22.
|
R HerreroPE CastleM SchiffmanMC BrattiA
HildesheimJ MoralesM AlfaroME ShermanS WacholderS ChenEpidemiologic
profile of type-specific human papillomavirus infection and
cervical neoplasia in Guanacaste, Costa RicaJ Infect
Dis19117961807200510.1086/42885015871111
|
|
23.
|
DK SinghK AnastosDR HooverRD BurkQ ShiL
NgendahayoE MutimuraA CajigasV BigirimaniX CaiHuman papillomavirus
infection and cervical cytology in HIV-infected and HIV-uninfected
Rwandan womenJ Infect Dis19918511861200910.1086/59912319435429
|