Introduction
Over the past decade, suicide gene therapies have
been widely studied in a broad array of human cancer types. This
strategy involves the transfer of suicide genes into cancer cells,
and followed by harmless prodrug treatment. A common approach used
is the herpes simplex virus thymidine kinase/ganciclovir
(HSV-TK/GCV) system, and its clinical trials are in the phase III
stage. According to these findings, multisubstrate
deoxyribonucleoside kinase of Drosophila melanogaster
(Dm-dNK) has received noticeable attention as another
potential therapeutic agent for cancer treatment. Similar to herpes
simplex virus deoxyribonucleoside kinase, Dm-dNK first
catalyzes the nucleoside analog into its monophosphate form, which
is considered the rate-limiting step throughout the activation.
This monophosphate form is further catalyzed into other
phosphorylated forms by normal cellular kinases, resulting in the
arrest of DNA replication and induced cell apoptosis. Dm-dNK
as a suicide gene also displays increased sensitivity to several
nucleoside analogs. Bertoli et al (1) reported that pyrimidine nucleoside
analogs (E)-5-(2-bromovinyl)-2′-deoxyuridine (BVDU) and
1-b-D-arabinofuranosylthymine (araT) are valuable for enhancing the
efficacy of Dm-dNK suicide gene therapy. Knecht et al
(2)demonstrated that gemcitabine
2′,2′-difluoro-deoxycytidine (dFdC), an anti-cancer drug, is an
efficient substrate for Dm-dNK, which can efficiently
phosphorylate dFdC, and the 2.2 Å resolution structure of
Dm-dNK is important. These data reveal that the residues
Tyr70 and Arg105 take into consideration the positioning of dFdC,
suggesting its significant implications for therapeutic efforts due
to the broad substrate acceptance of Dm-dNK. Characterized
by its high catalytic property and broad substrate acceptance,
Dm-dNK has therapeutic potential and is used in the
treatment of several cancer types (3–5).
Despite these advantages, one issue is the limited
efficiency, suggesting that the discovery of new approaches is
urgently required. A previous study has demonstrated an approach to
selectively kill c-Myc-expressing lung cancer cells by fusing the
c-Myc gene promoter with TK gene (6). In particular, in order to increase
the potency of efficient killing in a wide range of human cancers,
human telomerase reverse transcriptase (hTERT) serves as an ideal
biomarker, and hTERT promoter-directed suicide gene therapy can be
employed as a modality for a targeted suicide gene therapy for
hTERT-positive tumors (7,8). In the present study, we investigated
the selective tumor cell killing of hTERT promoter in the treatment
of human cancers. We selected the hTERT promoter to drive
Dm-dNK gene expression and thus generated a tumor-selective
viral vector through dFdC administration. Recently, adenoviral
vector-mediated Dm-dNK expression has been established and
is considered auspicious (9).
However, its clinical use is still limited due to the short-term
expression and vector-induced toxicity. With respect to the
application of efficient and safe gene delivery vehicles,
lentiviral vectors have become potentially useful vehicles since
they provide stable and long-term expression of the transgene by
incorporation into the target cell DNA. Therefore, the feasibility
of hTERT promoter-induced Dm-dNK gene expression via
lentiviral vector is expected. In our present study, we first
developed a Lenti-hTERT-dNK/dFdC system and tested its effect on
the targeted therapeutic approach both in vitro and in
vivo. Our results demonstrated that this system could be a
valuable tumor targeting strategy for tumor control.
Materials and methods
Cells and culture
Human breast cancer cell line Bcap37 and human
gastric cancer cell line SGC7901 were both obtained from the Cancer
Institution of China Medical University (Shenyang, China). Normal
human fetal lung fibroblast cell line WI-38 was purchased from ATCC
(American Type Cell Culture, Manassas, VA, USA). Bcap37 was
cultured in Roswell Park Memorial Institute (RPMI)-1640 medium.
SGC7901 and WI-38 were cultured in high glucose Dulbecco’s modified
Eagle’s medium (DMEM). Cells were maintained in a medium
supplemented with 10% heat inactivated fetal bovine serum (FBS)
(Gibco-BRL, Germany), 1% L-glutamine, 100 U/ml penicillin and 100
U/ml streptomycin at 37°C in a humidified incubator supplied with
5% CO2.
Construction of the lentiviral
vectors
The Dm-dNK coding sequence was amplified from
plasmid PLXSN-dNK using a polymerase chain reaction (PCR) technique
with the following primers: upstream, 5′-CCG GAA TTC (EcoRI)
ACC ATG GCG CAG GCA-3′ and downstream, 5′-CGC GGA TCC (BamH)
TCA TTA TCT GGC GAC-3′. The synthetic DNA sequence was released
with endonucleases EcoRI and BamH (New England
Biolabs, Beverly, MA, USA). Dm-dNK-3Flag was amplified by
PCR and ligated into plasmid PGC-FU (GeneChem, Shanghai, China)
consisting of a 5′-long terminal repeat (LTR), cytomegalovirus
(CMV) promoter, multiple clone site, a green fluorescent protein
(GFP) sequence and a 3′-LTR. Moreover, Dm-dNK-3Flag was also
ligated into plasmid PGC-FU-hTERT (GeneChem) consisting of a
5′-LTR, hTERT promoter, multiple clone site, GFP sequence and a
3′-LTR. In order to generate the recombinant plasmid PGC-FU-dNK or
PGC-FU-hTERT-dNK, GFP was removed with endonucleases AgeI
and EcoRI (New England Biolabs Ltd., UK). Subsequently, the
plasmids together with two packaging plasmids PHelper1.0 (gag, pol
and rev, component) and plasmid PHelper2.0 (VSVG, component) were
packed and co-transfected into a human embryonic kidney cell line
(HEK293T) using Lipofectamine™ 2000 reagent (Invitrogen, USA)
according to the manufacturer’s instructions. Lentivirus-containing
medium was filtered from the post-transfection supernatant and used
for transductions. Concentrated viruses of Lenti-GFP,
Lenti-hTERT-dNK and Lenti-CMV-dNK were obtained, respectively, and
then stored at −80°C. The titer of lentiviral vectors was
determined by dilution. Lenti-GFP was used to determine the
infectivity between cell lines. In order to increase the infection
efficiency, all lentivirus-infected cells were cultured in the
medium containing Polybrene (6 μg/ml) (Sigma, USA).
RT-PCR
Bcap37, SGC-7901 and WI-38 cells were seeded in
6-well plates at a density of 105 cells/well for 24 h,
respectively. Then Dm-dNK at a multiplicity of infection
(MOI) of 10 was added to the cell cultures. Polybrene (6 μg/ml) was
added to all cultures. Total-RNA was extracted from the cultured
cells using TRIzol reagent (Sigma) 3 days after infection. In order
to minimize the genomic DNA contamination, purified RNA was treated
with DNase I. cDNA was synthesized from 1 ng of RNA using the
RT-PCR kit (Takara Bio, Inc., Japan) following the manufacturer’s
protocol. Human glyceraldehyde-3-phosphate dehydyrogenase (GAPDH)
was used as the housekeeping gene. Amplification was performed
using PCR with the following primers: Dm-dNK, upstream,
5′-CCG GAA TTC ACC ATG GAG GCA-3′ and downstream, 5′-CGC GGA TCC
TCA TTA TCT GGC GAC-3′; and GAPDH, upstream, 5′-ACC ACA GTC CAT GCC
ATC AC-3′ and downstream, 5′-TCC ACC ACC CTG TTG CTG TTG CTG TA-3′.
Briefly, following a pre-heating step at 94°C for 4 min,
Dm-dNK was amplified with 35 cycles at a melting temperature
of 94°C for 1 min, an annealing temperature of 60°C (55°C for
GAPDH) for 1 min, and an extension temperature of 72°C for 1.5 min.
The expected length of Dm-dNK amplicon was 779 bp. Relative
expression of Dm-dNK was normalized to GAPDH (452 bp). A
total of 10 μl of each amplicon was separated by electrophoresis on
a 2% agarose gel, and the efficiency of cDNA synthesis was
determined by PCR with GAPDH-specific primers and visualized by
SYBR-Green staining.
Western blotting
Cells were harvested from 6-well plates after
transfection as previously described. For protein detection, the
cells were lyzed with lysis buffer (50 mmol/l HEPES at pH 7.4, 250
mmol/l NaCl, 1 mmol/l NaF, 1 mmol/l EDTA, 1% Triton X-100, 1 mmol/l
DTT) containing protease inhibitors. The cell lysates were then
normalized for protein content using the BCA protein assay kit
(KeyGEN, Shanghai, China). Equal amount of protein containing
sample buffer was separated on SDS-polyacrylamide gel by
electrophoresis and then transferred to polyvinylidene difluoride
(PVDF) membranes (Millipore, Bedford, MA, USA). The membranes were
blocked in Tris-buffered saline (TBS) (10 mM Tris-HCl at pH 8.0,
0.05% Tween-20, 150 mM NaCl) containing 1% bovine serum albumin
(BSA) at room temperature for 2 h and then probed with anti-Flag
(1:1,000) (Abcam, Cambridge, CA, USA) or anti-β-actin (1:500),
which was followed by a secondary horseradish peroxidase-conjugated
antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA).
After several washes, the corresponding blots were developed with a
chemiluminescence reagent (Thermo Fisher Scientific, Inc.,
Rockford, IL, USA). β-actin was used as a control for equal protein
loading.
Enzyme assays
Cell protein extracts were prepared as described
(10) from Bcap37, SGC7901 and
WI-38 cells at 72 h after lentivirus infection. The activity of
purified recombinant enzymes was determined in a 35-ml reaction
mixture containing 50 mM Tris-HCl at pH 7.6, 5 mM MgCL2,
2 mM dithiothreitol, 15 mM NaF, 100 mM KCL, 5 mM ATP, 0.5 mg/ml BSA
and 0.6 mg protein extract. Briefly, 2.5 mM
[methyl-3H]dThd (Moravek Biochemicals, Inc., Brea, CA,
USA) was mixed with an equivalent amount of unlabeled substrate.
Sample aliquots were spotted on Whatman DE-81 filter paper disks
after incubation of 10, 20 and 30 min at 37°C, respectively. The
filters were dried for 1 h and then washed three times in 5 mM
ammonium formate. The filter-bound nucleoside monophosphates were
eluted with 0.5 M KCl, and the radioactivity was measured by
scintillation counting.
Cell viability analysis
Exponentially growing cells were seeded into 96-well
plates (Corning Inc., USA). After overnight culture, cells were
infected with Lenti-GFP, LentihTERT-dNK, Lenti-CMV-dNK and
untreated at the MOI rate of 10. After 3 days, the infecting medium
was replaced with fresh medium containing 10% FBS and dFdC at
various concentrations from 0.001 to 10 μM. Then the plates were
incubated at 37°C for 72 h in the humidified incubator supplied
with 5% CO2. Subsequently, 20 μl of tetrazolium salt
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT)
(Promega, USA) (5 mg/ml) was added to each well, and the plate was
then incubated for 4 h. The cells were lysed in 200 μl of
dimethylsulphoxide (DMSO) and mixed thoroughly. Color reaction was
performed by determining the absorbance at 570 nm, and the number
of viable cells was measured. Each experiment was performed in
triplicate and repeated three times.
Cell proliferation and flow cytometric
apoptosis assays
All cells transfected with Lenti-GFP,
Lenti-hTERT-dNK and Lenti-CMV-dNK or untreated cells were seeded in
24-well plates with a pre-coat of dFdC (1 μM). During the
incubation, cells were trypsinized and suspended in a serum-free
medium. Cell counting was carried out under a microscope using a
hemocytometer. The cell numbers were obtained from an average of
three experiments. To further evaluate the apoptosis extent and
clarify the mechanism of this suicide gene therapy, an Annexin
V-FITC/propidium iodide (PI) double staining kit (GenMed
BioScience, China) was used to detect the apoptosis ratio. Briefly,
Bcap37, SGC7901 and WI-38 cells were cultured in 6-well plates and
incubated with 1 μM dFdC for 72 h after the infection with
Lenti-hTERT-dNK, Lenti-CMV-dNK, Lenti-GFP and untreated
respectively for 72 h. The cells were then harvested by
trypsinization, washed twice with cold PBS and centrifuged at room
temperature for 10 min. After the supernatant was discarded, the
cell pellet was resuspended with 200 μl of 1X binding buffer with
addition of 5 μl Annexin V-FITC (20 μg/ml). Following a 15-min
incubation in the dark, 200 μl binding buffer and 10 μl of PI (50
μg/ml) were added to the cell suspension. The percentage of cells
undergoing apoptosis was then determined by a FACScan flow
cytometer [equipped with CellQuest and ModFITLT for Mac V1.01
software (Becton-Dickinson, San Jose, CA, USA)].
Antitumor effect of Lenti-hTERT-dNK/dFdC
system in vivo
To detect the antitumor effectiveness of
Lenti-hTERT-dNK/dFdC system in vivo, 24 female BALB/C nude
mice, 6- to 7-weeks old were purchased from the Experimental Animal
Center, Chinese Academy of Sciences (Shanghai, China). All
experiments followed the Guide for the Care and Use of Laboratory
Animals (National Research Council, 1996). A total of
1.0×107 Bcap37 cells were suspended in 100 μl PBS and
then inoculated in the flanks of nude mice. Once the tumor reached
a volume of ∼100 mm3, mice were randomly assigned into 4
groups (6 per group) as follows: i) dFdC with PBS; ii) dFdC with
Lenti-GFP; iii) dFdC with Lenti-hTERT-dNK and iv) dFdC with
Lenti-CMV-dNK. The animals in the lentivirus treatment groups
received a series of intratumoral injections at a dose of
109 plaque forming units (pfu) three times with a 2-day
interval. Subsequently, 5 mg/kg dFdC was daily administered into
the peritoneal cavity over 7 consecutive days. Xenograft tumor
burdens were inspected every 5 days and recorded with a caliper for
up to 30 days. Tumor volume was calculated according to the
formula: (1/2 × length × width2).
Statistical analysis
All data are expressed as mean ± SD and analyzed
using the statistical software SPSS version 10.1 (SPSS, Inc.,
Chicago, IL, USA). P<0.05 was considered statistically
significant.
Results
Dm-dNK expression
In order to determine the infection efficiency, we
used lentivector plasmids PGC-FU, PGC-FU-dNK-3Flag and
PGC-FU-hTERT-dNK-3Flag. To construct plasmid PGC-FU-dNK-3Flag, the
GFP expression cassette in plasmid PGC-FU was replaced by a
dNK-3Flag expression cassette. We also constructed a plasmid
PGC-FU-hTERT-dNK-3Flag, in which the Dm-dNK gene was under
the control of the hTERT promoter after the CMV promoter was
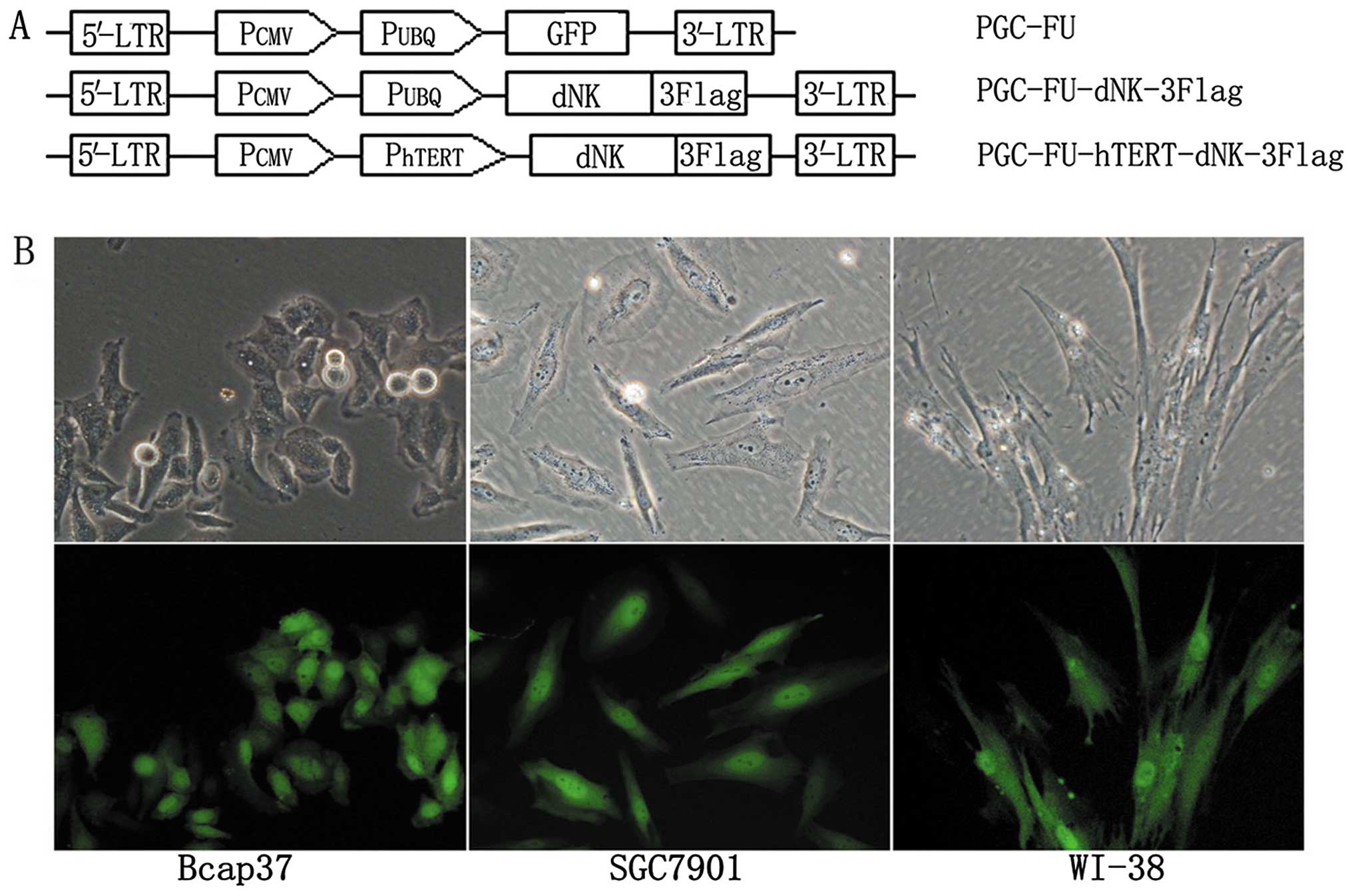
removed from the plasmid PGC-FU-dNK-3Flag (Fig. 1A). The infectivity of the cell
lines was determined by GFP expression in the Lenti-GFP. Fig. 1B shows that >90% of the cancer
cells (Bcap37 and SGC7901) and normal cells (WI-38) were transduced
with Lenti-GFP at an MOI of 10. Then we analyzed the expression of
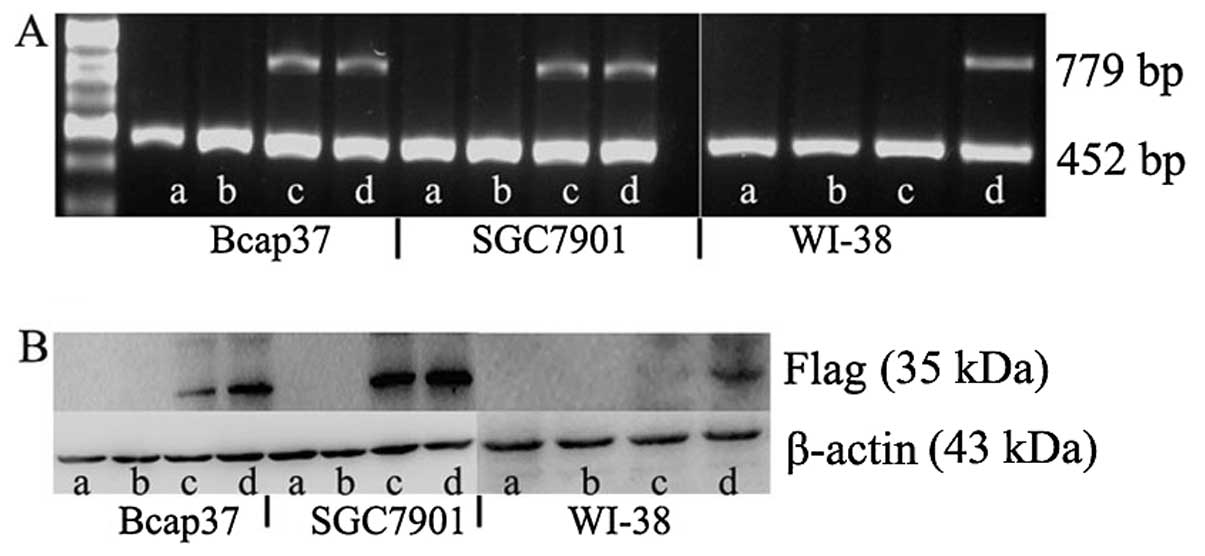
Dm-dNK at the mRNA and protein levels by RT-PCR and western
blotting, respectively. The null lentivirus and untreated control
induced cells did not exhibit Dm-dNK expression at both the
mRNA and protein levels. The Dm-dNK expression at the mRNA
level was not significantly different in the cancer cells (Bcap37
and SGC7901) infected with Lenti-hTERT-dNK and Lenti-CMV-dNK
(Fig. 2A). The Dm-dNK
expression at the protein level was slightly decreased in the
Bcap37 cells infected with Lenti-hTERT-dNK compared with that of
Lenti-CMV-dNK-infected cells, whereas it was nearly identical at
the protein level in the SGC7901 cells infected with
Lenti-hTERT-dNK and Lenti-CMV-dNK (Fig. 2B). In contrast, we also observed
that the Dm-dNK expression at the mRNA and protein levels
was low or completely absent in WI-38 cells.
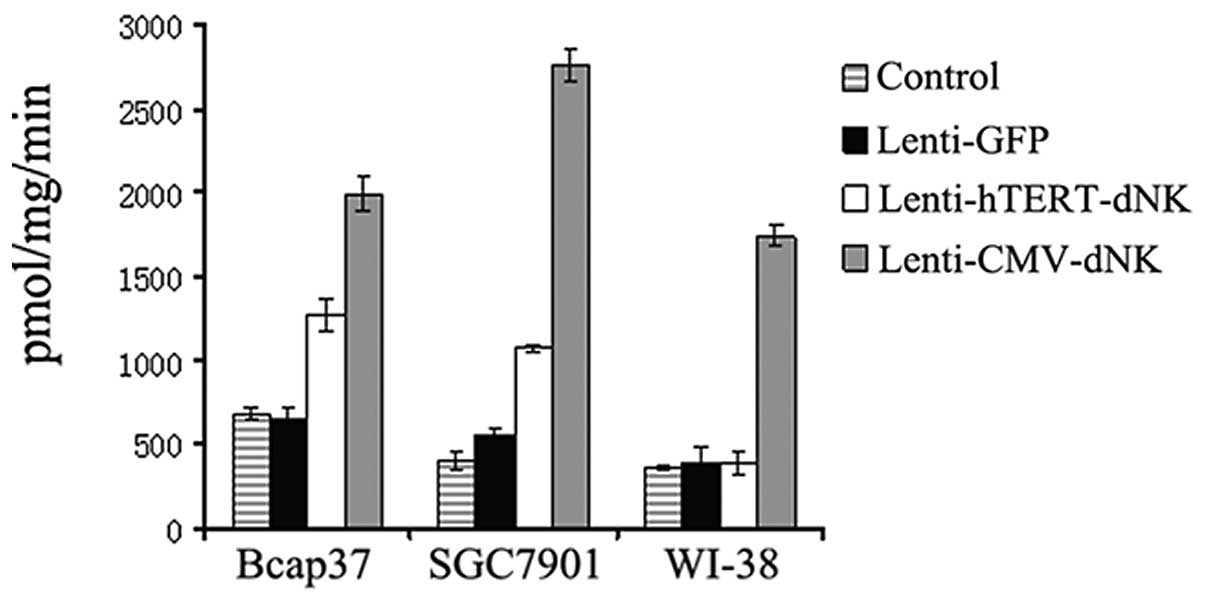
To further evaluate the essential role of nucleoside
kinase in the infected cells, we determined the phosphorylation of
dThd in the cell protein extracts. As expected, the enzymatic
activity of nucleoside kinase was strong in the Bcap37 and SGC7901
cells infected with Lenti-hTERT-dNK, which was ∼2-fold higher than
that in the cells infected with the lentiviral vector alone.
Although the Dm-dNK activity of the hTERT promoter in the
Bcap37 and SGC7901 cells was consistently lower than that of the
CMV promoter, the incidence of activity with the hTERT promoter was
similar to that with the CMV promoter. In contrast, the
Dm-dNK activity in WI-38 cells infected with Lenti-hTERT-dNK
was not different from that of its mock cells (Fig. 3). These results were consistent
with its expression levels in the previous study, establishing a
connection between the enzymatic activity of Dm-dNK and its
expression at the mRNA and protein levels in these experimental
cell lines.
Cell cytotoxicity, proliferation and
induction of apoptosis
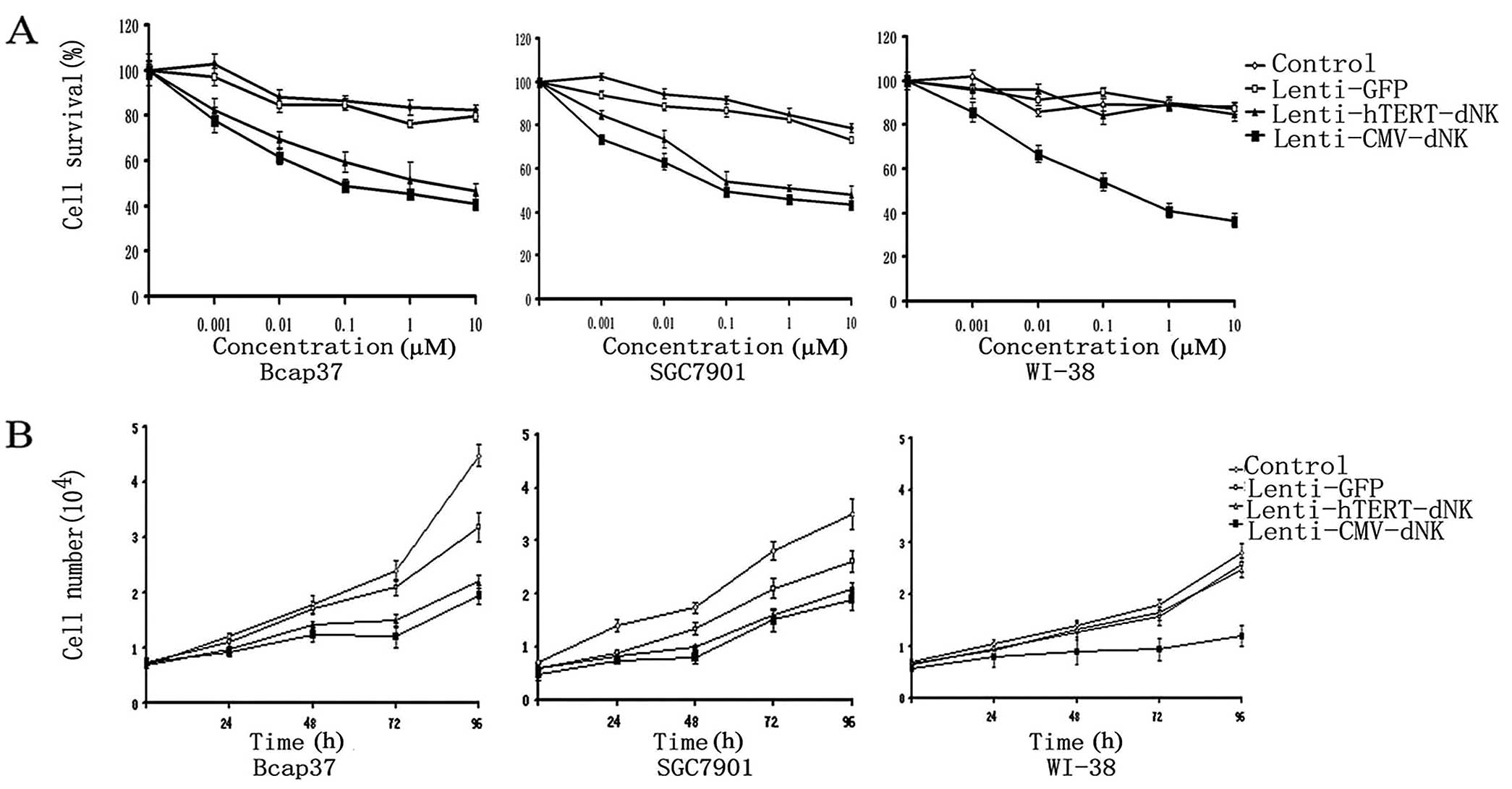
In order to evaluate the effects of Lenti-hTERT-dNK
on the prodrug dFdC in the infected cells, we infected Bcap37,
SGC7901 and WI-38 cells with the lentiviral vector and then treated
them with increasing concentrations of dFdC (0–10 μM) for 5 days.
Subsequently, the cell viability was determined using the MTT
cytotoxicity assay. Cell viability was decreased in cancer cells
(Bcap37 and SGC7901) in the presence of dFdC, demonstrating the
cytotoxicity of the Lenti-hTERT-dNK/dFdC system (Fig. 4A). In contrast, slight
cytotoxicity was observed in the WI-38 cells in the presence of
dFdC. As the control, we observed significant cytotoxicity in the
cells infected with Lenti-CMV-dNK/dFdC. The cell survival rate of
Bcap37 and SGC7901 cells was ∼80% with 0.001 μM of dFdC, ∼60% with
0.01 and 0.1 μM of dFdC, and ∼40% with 10 μM of dFdC. However, data
showed that the normal fibroblast cells (WI-38) infected with the
Lenti-hTERT-dNK/dFdC system were resistant to dFdC. Its survival
rate remained the same as before (∼90% of cells survived even with
10 μM of dFdC). These results illustrated that cancer cells
responded to dFdC in a dose-dependent manner from 0.001 to 10 μM in
the Lenti-hTERT-dNK/dFdC system, whereas the normal fibroblast
cells were completely unaffected within that dose range. These data
suggest that the Lenti-hTERT-dNK/dFdC system is able to selectively
kill hTERT-positive cells in vitro.
Compared with dFdC-induced cytotoxicity in cancer
cells infected with Lenti-hTERT-dNK and Lenti-CMV-dNK, the
cytotoxicity of the Lenti-hTERT-dNK/dFdC system was weaker than
that of the Lenti-CMV-dNK/dFdC system. This also occurred at low
concentration of dFdC (0.01 μM, P<0.05) in both Bcap37 and
SGC7901 cells. Moreover, it was noteworthy that the cytotoxicity of
the Lenti-hTERT-dNK/dFdC system was not as weaker as we expected.
For example, when the dFdC concentration was 1 μM, the difference
was not statistically significant (P>0.05) between the two
cancer cell lines.
Conversely, the cell proliferation was apparently
inhibited after the treatment with 1 μM of dFdC in the cancer cells
infected with Lenti-hTERT-dNK or Lenti-CMV-dNK (Fig. 4B). During the proliferation of
WI-38 cells, we did not observe significant inhibition of cell
growth in the Lenti-hTERT-dNK/dFdC system (up to 4 days).
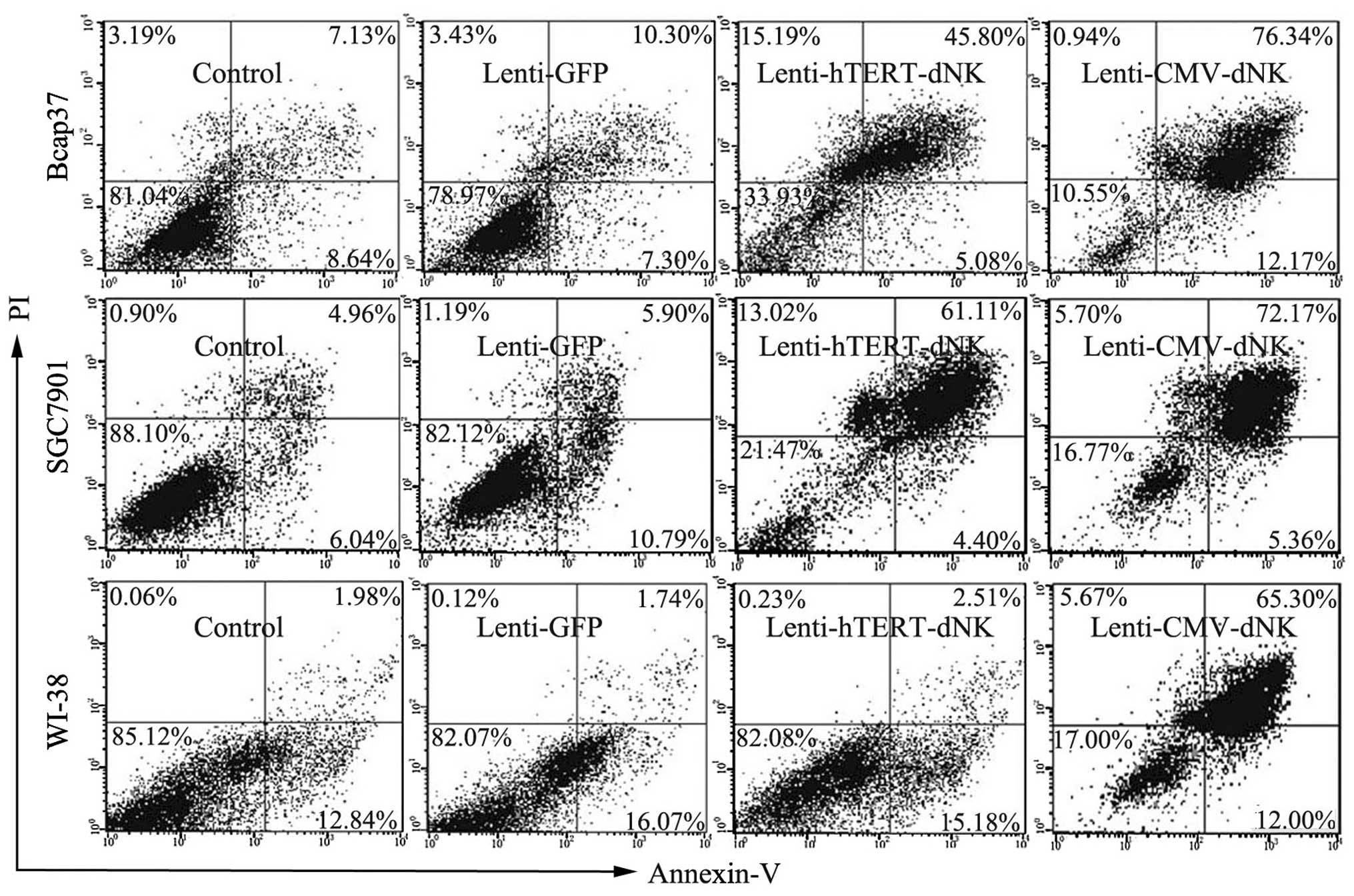
In our present study, we performed quantitative flow
cytometry in order to determine the percentage of apoptotic cells.
Cells infected with the Lenti-CMV-dNK/dFdC system showed the
highest percentage of apoptosis, ranging from 77.3 (WI-38) to
88.51% (Bcap37). In the Lenti-hTERT-dNK/dFdC system, Bcap37 and
SGC7901 cells both showed a higher percentage of apoptosis (50.88
and 65.11%, respectively) (Fig.
5). Conversely, the percentage of apoptosis was only 17.69% in
the human embryonic lung fibroblast WI-38 cells, indicating that
Dm-dNK had minimum contribution to the cytotoxic effects on
normal cells. In addition, the WI-38 cells infected with the
Lenti-hTERT-dNK/dFdC system showed nearly an identical apoptosis
ratio compared with the control cells not expressing Dm-dNK
(17.69 and 14.82%, respectively). These findings were consistent
with the previous observation (Fig.
4). Taken together, these data demonstrated that the
Lenti-hTERT-dNK/dFdC system efficiently induced the apoptosis of
cancer cells.
Antitumor efficacy of
Lenti-hTERT-dNK/dFdC in vivo
Since our in vitro data clearly indicated
that Lenti-hTERT-dNK/dFdC had specific cytopathic effects on tumor
cells, we further examined the therapeutic potential on nude mice
in order to identify whether the Lenti-hTERT-dNK/dFdC system was
able to specifically inhibit tumor growth in vivo.
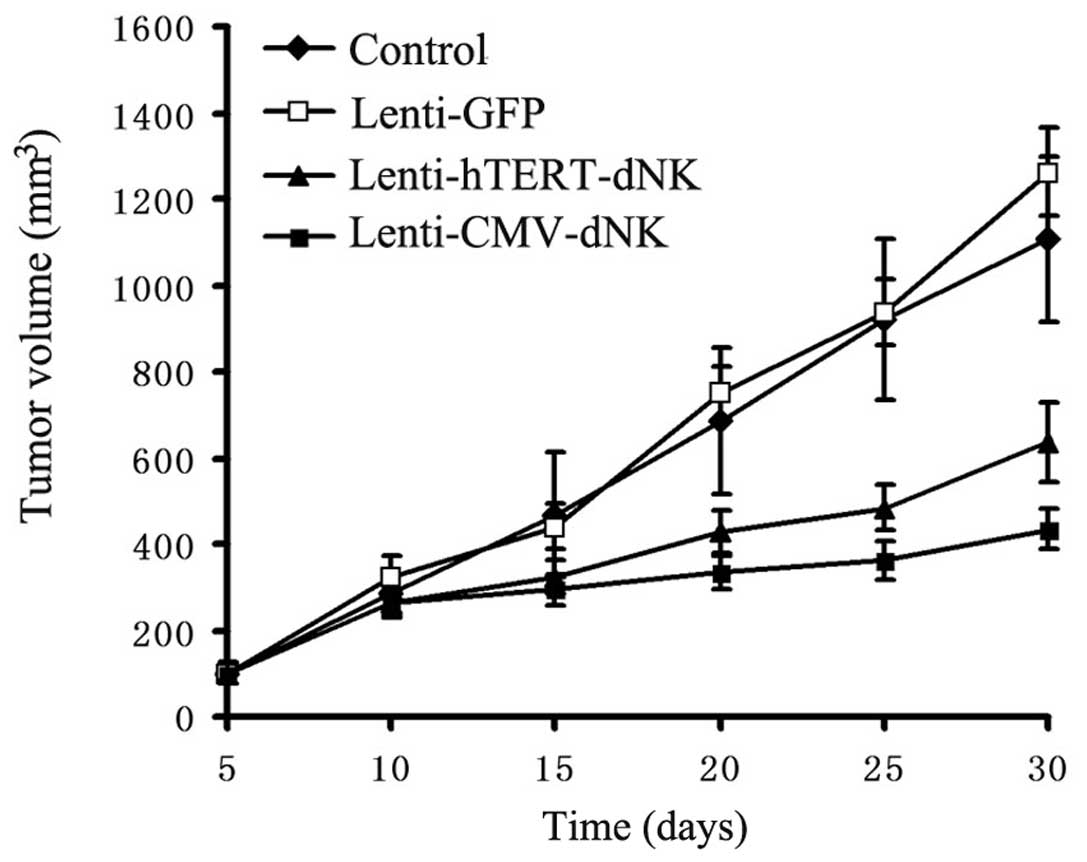
Either Lenti-hTERT-dNK or Lenti-CMV-dNK combined
with dFdC treatment resulted in a significant inhibition of tumor
growth in vivo compared with PBS or Lenti-GFP combined with
dFdC treatment (Fig. 6). At Day
30, the mean volume of tumors obtained by PBS or Lenti-GFP
injection combined with dFdC (1,107.64±193.10 and 1,262.25±100.84
mm3, respectively) was larger than that of
Lenti-hTERT-dNK combined with dFdC (636.96±92.39 mm3)
(P<0.05). Moreover, Lenti-hTERT-dNK combined with dFdC did not
show an improved therapeutic benefit in terms of tumor volume on
Day 30 compared with Lenti-CMV-dNK combined with dFdC (435.54±46.58
mm3) (P<0.05).
Discussion
Gene delivery with a viral vector is a promising
approach for the treatment of human cancer. Studies on suicide gene
therapy combining herpes simplex virus thymidine kinase gene
transfer and ganciclovir treatment have been widely investigated in
human breast, gastric, colon, prostate and bladder cancer cells and
hepatocarcinoma cells (11–16). The feasibility of suicide gene
strategy has been demonstrated. However, subsequent studies have
also shown that the use of the gene strategy is limited due to its
poor delivery efficiency and targeted delivery problems. Therefore,
an improved therapeutic strategy is required.
In our present study, we focused on the enzyme
Dm-dNK. In contrast to HSV-1 TK and other enzymes, it has a
broad substrate specificity and a high catalytic rate when
transferred in human cells. A previous study has shown that
retroviral transduction with encoding Dm-dNK has
cytotoxicity to several nucleoside analogs when expressed in human
cells (17). However, low
efficiency of the transgene and some vector-induced toxicity are
still obstacles. Therefore, it is essential to design the long-term
gene expression with stable integration of the transgene. Recently,
lentiviral vectors have generated novel perspectives for gene
therapy, and they have become a tool for gene transfer in a broad
field (18). Due to their high
delivery efficiency and long-term expression of the transgene,
lentiviral vectors appear to be promising candidates and can be
used in clinics to cure acquired disorders. To the best of our
knowledge, our study was the first to express Dm-dNK using a
lentiviral vector. It has been reported that Dm-dNK
expression together with dFdC treatment exerts a negative effect on
cell survival via adenovirus. However, its effect via lentivirus
remains unclear. Therefore, we developed a lentiviral vector
expressing Dm-dNK and further investigated its suitability
for medical therapy.
Another important consideration is the target of the
suicide gene. It is widely accepted that hTERT is expressed in a
vast majority of tumors, and its expression levels are associated
with the malignancy of different types of cancer cells. The absence
of hTERT results in tumor cell senescence and hence shortens the
life-span of cells. In order to obtain a tumor-specific gene
delivery system, we developed a lentiviral vector expressing
Dm-dNK under the control of the hTERT promoter instead of
the CMV promoter, whereas a CMV promoter cassette was designed as a
control. We performed functional studies aimed at targeted killing
of cancer cells by Dm-dNK transfer, and our data confirmed
that the constitutive version can be used to induce apoptosis in
cancer cells. Our study successfully demonstrated that
Dm-dNK expression at the mRNA and protein levels under the
control of the hTERT promoter was apparent in cancer cells, whereas
its expression was low or absent in the normal cells. As far as the
gene expression regulated by the CMV promoter was concerned, the
expression of Dm-dNK was obvious in all cells.
It is widely accepted that cancer is promoted by an
accumulation of multiple genetic and epigenetic alterations. In
cancer gene therapy, it has been recognized as a crucial step to
target the expression of suicide genes in a wide range of human
malignant cancer cells while avoiding severe side effects in normal
cells. The activity of the hTERT promoter is significantly higher
in most telomerase-positive cells than that in telomerase-negative
cells (19,20). It is feasible to drive the
expression of many therapeutic genes by the hTERT promoter, such as
Bax, caspase-8 and FADD (21). In
our present study, the hTERT promoter also showed the potential for
targeted killing of cancer cells. However, the activity of the
hTERT promoter in most cancer cells is suboptimal and usually
weaker than that of commonly used promoters, such as the CMV
promoter, SV40 promoter and RSV-LTR. Therefore, the mechanisms by
which the promoter performs so differentially among cells remains
unclear. Further investigation is required to clarify the
mechanisms before this strategy is used as a routine clinical
practice.
Previous reports have indicated that a limited
number of key single amino acid residues highlight the importance
of substrate specificity (22).
In 2011, Zhu et al (23)
discovered that the catalytic rates are relatively increased by the
alteration at the sites of 244E, 245S, 251S and 252R of the last 10
amino acids. Based on these studies, we further investigated its
important structural features and structure-function
relationships.
Recently, a wide variety of other gene therapies
have been investigated in many malignancies, such as
antiangiogenic, tumor-suppressor, cytokine-based and
oxidative-based gene therapy (24). In addition, cell cycles regulate
growth and differentiation. The regulation of the different cell
cycles is another hallmark of cancer control (25). Suicide gene therapies can be
combined with these gene therapies, and the combination strategy
may provide new potentials to deal with malignancies in future
clinical application.
In summary, our study demonstrated that the
recombinant suicide gene lentiviral Dm-dNK driven by
cancer-associated hTERT promoter combined with dFdC for cancer gene
therapy was an effective and promising treatment to use. It is
conceivable that wide clinical applications may eventually be
achieved through the development of this suicide gene strategy.
Acknowledgements
This study was supported by grants
from the National Natural Science Foundation of China (nos.
81071900 and 81172199), the Scientific Research Foundation for
Returned Scholars, the Ministry of Education of China (2008) and
the Hi-Tech Research Development Program of China (863 program,
2006AA02Z493).
References
|
1.
|
A BertoliM FrancoJ BalzariniM JohanssonA
KarlssonAltered deoxyribonucleotide pools in T-lymphoblastoid cells
expressing the multisubstrate nucleoside kinase of Drosophila
melanogasterFEBS
J27239183928200510.1111/j.1742-4658.2005.04808.x16045762
|
|
2.
|
W KnechtNE MikkelsenAR
ClausenDrosophila melanogaster deoxyribonucleoside kinase
activates gemcitabineBiochem Biophys Res
Commun382430433200910.1016/j.bbrc.2009.03.04119285960
|
|
3.
|
S MaL ZhaoZ ZhuThe multisubstrate
deoxyribonucleoside kinase of Drosophila melanogaster as a
therapeutic suicide gene of breast cancer cellsJ Gene
Med133053112011
|
|
4.
|
Z ZhuL MaoL ZhaoSynergistic therapeutic
effect in gastric cancer cells produced by oncolytic adenovirus
encoding Drosophila melanogaster deoxyribonucleoside
kinaseCancer Biol
Ther11874882201110.4161/cbt.11.10.1518221383545
|
|
5.
|
M ItoY SudaH HarashimaH KamiyaCytotoxic
effect of Drosophila deoxynucleoside kinase gene on
replicating plasmid in HeLa cellsBiol Pharm Bull33122312272010
|
|
6.
|
K NishinoT OsakiT
KumagaiAdenovirus-mediated gene therapy specific for small cell
lung cancer cells using a Myc-Max binding motifInt J
Cancer91851856200110.1002/1097-0215(200002)9999:9999%3C::AID-IJC1120%3E3.0.CO;2-111275991
|
|
7.
|
ST YuC LiMH LuNoninvasive and real-time
monitoring of the therapeutic response of tumors in vivo with an
optimized hTERT
promoterCancer11818841893201210.1002/cncr.2647622009660
|
|
8.
|
B YuY ZhangY ZhanCo-expression of herpes
simplex virus thymidine kinase and Escherichia coli
nitroreductase by an hTERT-driven adenovirus vector in breast
cancer cells results in additive antitumor effectsOncol
Rep26255264201121573500
|
|
9.
|
S MaW QuL MaoAntitumor effects of
oncolytic adenovirus armed with Drosophila melanogaster
deoxyribonucleoside kinase in colorectal cancerOncol
Rep2714431450201222294034
|
|
10.
|
MP SandriniAR ClausenSL OnFM AarestrupB
Munch-PetersenJ PiskurNucleoside analogues are activated by
bacterial deoxyribonucleoside kinases in a species-specific mannerJ
Antimicrob Chemother60510520200710.1093/jac/dkm24017615154
|
|
11.
|
LM AndersonS SwaminathanI ZackonAK
TajuddinB ThimmapayaSA WeitzmanAdenovirus-mediated tissue-targeted
expression of the HSVtk gene for the treatment of breast cancerGene
Ther6854864199910.1038/sj.gt.330090910505111
|
|
12.
|
Q TangD ZhangM WanL JinExperimental study
of the RV-HSV-TK/GCV suicide gene therapy system in gastric
cancerCancer Biother
Radiopharm22755761200710.1089/cbr.2007.34618158766
|
|
13.
|
JG PanX ZhouR LuoRF HanThe
adeno-associated virus-mediated HSV-TK/GCV suicide system: a
potential strategy for the treatment of bladder carcinomaMed
OncolOct202011(Epub ahead of print)
|
|
14.
|
FQ ZhengY XuRJ YangCombination effect of
oncolytic adenovirus therapy and herpes simplex virus thymidine
kinase/ganciclovir in hepatic carcinoma animal modelsActa Pharmacol
Sin30617627200910.1038/aps.2009.33
|
|
15.
|
M AhnSJ LeeX LiEnhanced combined
tumor-specific oncolysis and suicide gene therapy for prostate
cancer using M6 promoterCancer Gene
Ther167382200910.1038/cgt.2008.5918772902
|
|
16.
|
JF ZhangF WeiHP WangPotent antitumor
activity of telomerase-dependent and HSV-TK armed oncolytic
adenovirus for non-small cell lung cancer in vitro and in vivoJ Exp
Clin Cancer Res2952201010.1186/1756-9966-29-5220487549
|
|
17.
|
X ZhengM JohanssonA KarlssonRetroviral
transduction of cancer cell lines with the gene encoding
Drosophila melanogaster multisubstrate deoxyribonucleoside
kinaseJ Biol
Chem2753912539129200010.1074/jbc.M00621220010993893
|
|
18.
|
D TronoLentiviral vectors: turning a
deadly foe into a therapeutic agentGene
Ther72023200010.1038/sj.gt.330110510680011
|
|
19.
|
M TakakuraS KyoT KanayaCloning of human
telomerase catalytic subunit (hTERT) gene promoter and
identification of proximal core promoter sequences essential for
transcriptional activation in immortalized and cancer cellsCancer
Res595515571999
|
|
20.
|
YS CongJ WenS BacchettiThe human
telomerase catalytic subunit hTERT: organization of the gene and
characterization of the promoterHum Mol
Genet8137142199910.1093/hmg/8.1.1379887342
|
|
21.
|
J GuB FangTelomerase promoter-driven
cancer gene therapyCancer Biol Ther2Suppl 1S64S70200314508082
|
|
22.
|
W KnechtMP SandriniK JohanssonH EklundB
Munch-PetersenJ PiskurA few amino acid substitutions can convert
deoxyribonucleoside kinase specificity from pyrimidines to
purinesEMBO J2118731880200210.1093/emboj/21.7.187311927571
|
|
23.
|
Z ZhuS MaL ZhaoAdenovirus-mediated
Drosophila melanogaster deoxyribonucleoside kinase mutants
combined with gemcitabine harbor a safe cancer treatment profileInt
J Oncol387457532011
|
|
24.
|
C ZhangQT WangH LiuZZ ZhangWL
HuangAdvancement and prospects of tumor gene therapyChin J
Cancer30182188201110.5732/cjc.010.1007421352695
|
|
25.
|
D Abate-DagaL Garcia-RodriguezL SumoyC
FillatCell cycle control pathways act as conditioning factors for
TK/GCV sensitivity in pancreatic cancer cellsBiochim Biophys
Acta180311751185201010.1016/j.bbamcr.2010.06.00920599444
|