Introduction
Regrettably, the emergence of multidrug resistant
(MDR) strains of bacteria during the last 2–3 decades has failed to
ignite intensive research and development efforts towards the
discovery of novel antimicrobial drugs. This is exemplified by the
fact that only 2 new classes of antibiotics have been marketed
since 1970 (1,2). The Gram-negative bacterium
Pseudomonas aeruginosa (PA) is an opportunistic pathogen and
may be regarded as a nosocomial ‘predator’ especially in
immunocompromised patients (3,4).
PA infections may cause life-threatening conditions such as
ventilator-associated and hospital-acquired pneumonias as well as
bloodstream and urinary tract infections in susceptible patients
(5,6). Additionally, PA plagues cystic
fibrosis (CF) patients with at least 50% of the ∼60,000 population
in the US and EU permanently colonized with the bacterium (7,8).
Indeed, PA is a major cause of morbidity and mortality in these
patients (7,9). A remarkable feature of PA lies not
only in its genomic diversity with a plethora of ORFs capable of
degrading antibiotics (10) but
also its capacity to acquire external genomic elements providing
additional resistance mechanisms (11). Ultimately, MDR PA can be a grave
problem, especially in the critical care setting where mortality
rates for certain infections may range from 38 to >70% (12). Thus, designing the appropriate
antimicrobial therapy may be a clinical dilemma due to the paucity
of effective and safe drugs that can combat MDR strains of PA.
An alternative and highly innovative anti-bacterial
therapeutic approach apart from classical antibiotics is to disarm
bacterial pathogens thereby augmenting the host’s innate immune
system to clear the infection (13). The notion of disarmament of
bacterial pathogens by targeting virulence factors has allowed a
resurgence in the importance and potential therapeutic role of
anti-infective monoclonal antibodies (mAbs). Such advances have
been catalyzed by a number of methodological breakthroughs which
have permitted not only the rapid generation of fully human or
humanized mAb candidates but also the capabilities to recombinantly
express mammalian cell-derived mAbs at g/l quantities (14,15). Pre-clinically, a number of
pharmacological studies have demonstrated the effectiveness of
anti-PA virulence mAbs which have justified their clinical
evaluation (16–18). For example, KB001 and panobacumab,
human mAbs targeting PA virulence factors PcrV and 011 LPS
serotype, respectively, have demonstrated positive clinical
findings, albeit in a limited number of hospital-associated
pneumonia and CF cases when added to standard-of-care treatment
(19,20). These findings support passive
immunization approaches targeting virulence factors especially
since from an evolutionary point of view, they may be refractory to
selection pressures due to their pivotal roles in bacterial
infectivity and survival.
In accord with the above, we also believe that
passive immunization with anti-PA virulence mAbs is a viable
therapeutic proposition, representing an unfulfilled medical
requirement. In contrast to the PcrV and 011 LPS virulence factors,
our preferred PA target is the surface-expressed, single polar
flagellum of which flagellin is the primary protein component
(21,22). Flagellar structures are pivotal
for a number of PA’s functions including bacterial motility,
attachment and invasion to susceptible cells as well as being
highly pro-inflammatory via the Toll-like receptor 5 (TLR5)
(21–23). Consequently, a neutralizing mAb
targeting the flagella of PA may intervene at a variety of
critically important steps and curtail the catastrophic sequelae of
events that lead to end-organ infection, dysregulated inflammation
and mortality.
It should be noted that in PA, 2 types of flagellin
have been identified and termed type a and type b which may be
discriminated on the basis of molecular size and reactions with
type-specific polyclonal and mAbs (24,25). In contrast to salmonella
flagellins, PA flagellin types a and b do not exhibit phase
variation since a single strain produces a single type of flagellin
with no switching between types a and b (26). Numerous in vivo studies
have pointed to the protective effects of either polyclonal
antibodies or specific anti-flagellin mAbs following infection with
antibiotic-sensitive PA strains harboring the appropriate,
homologous flagellin type (27–31). Indeed, such antibody preparations
provided equivalent protection to imipenem, a standard-of-care
carbapenem antibiotic (30,31). Since >98% PA strains harbor a
phenotypic type a or b flagellin (24,27), an appropriate mAb therapy governed
by a priori knowledge of the infecting flagellin type may
permit a ‘patient-tailored’ anti-infective therapy and thus a
higher probability of clinical success.
In the present study, we applied a battery of in
vitro and in vivo tests to characterize our fully human,
lead mAb termed LST-007 that targets PA flagellin type b. To our
knowledge, this is the first report demonstrating that an
anti-infective PA mAb administered as a monotherapy, can curb
lethality driven by a clinically relevant MDR PA strain.
Materials and methods
Reagents
All general chemicals were purchased from Sigma
(Rehovot, Israel) and HyLabs (Rehovot, Israel). Imipenem in the
form of the marketed drug Tienam, was acquired from the Department
of Pharmacy, University of Messina. Bacterial PA strains 27853,
25619 and 9721 were purchased from Microbiologics (USA). Ka02, a
hospital-derived MDR PA strain, demonstrated sensitivity only to
amikacin, colistin and partial sensitivity to gentamicin as
determined from Vitek screens. LST-003, a proprietary chimeric mAb
targeting PA flagellin type a was transiently expressed in CHO-K1
cells and used in certain in vitro studies. A human IgG1
isotype control mAb (32) was
used in a number of in vitro and in vivo studies.
Expression and purification of
recombinant PA flagellin type b
The fliC gene (Pa1092 from the PA genome database)
corresponding to flagellin type b of the laboratory strain PAO1,
was used as the template for recombinant expression. This gene
sequence was custom synthesized to permit appropriate codon usage
for optimal expression in E. coli without modifying its
primary amino acid sequence (Geneart, Germany). This cDNA sequence
was engineered with 5′ NdeI and 3′ HindIII
restriction sites enabling direct ligation into a pET28a expression
plasmid cleaved with these respective enzymes. The resultant
recombinant protein harbored an N’-terminal histidine-6 tag
enabling immobilized metal affinity chromatography (IMAC)
purification. The pET28a/ flagellin type b recombinant plasmid was
transformed into BL21 DE3 STAR cells (Invitrogen, Carlsbad, CA,
USA) and a single colony was grown in 10 ml LB containing 50 μg/ml
kanamycin overnight. Thereafter, 0.2 ml of the overnight culture
was taken to inoculate 50 ml pre-warmed LB-kanamycin and at an OD
of 0.3, IPTG was added to a final concentration of 1 mM to induce
expression for 3 h at 37°C. Since the bulk of the recombinant
protein was shown to reside in the insoluble fraction, induced
bacterial pellets were resuspended in 50 mM phosphate buffer (pH
7.4) containing IGEPAL, sonicated and following centrifugation, the
insoluble pellet was taken for IMAC as follows. The pellet was
solubilized in buffer containing 6 M GuHCl and subsequently
purified by IMAC (HiTrap chelating 5 ml column charged with
nickel). Recombinant PA flagellin type b was refolded on the column
using a gradient 6-0 M GuHCl and an elution was performed using an
imidazole gradient (0–500 mM). Peak fractions were pooled, dialyzed
against PBS, quantified using BCA and adjusted with PBS to a stock
concentration of 1 mg/ ml and stored at −20°C. Coomassie gel
staining demonstrated that the Mw of PA flagellin type b was ∼55
kDa with >95% purity. In small-scale expression and purification
studies, recombinant PA flagellin type a was produced in an
identical manner as described for flagellin type b and yielded a
protein of Mw ∼45 kDa with >95% purity.
Expression and purification of
LST-007
LST-007 was isolated from EBV-transformed human B
cells (unpublished data) and its respective VH and
VL cDNA chains were taken for expression using a
commercially available, platform technology. In brief, following
cloning and expansion of transduced LST-007 VH and
LST-007 VL pools, shaker flasks of CHO cells supported
in serum-free media were co-infected and clarified supernatant were
taken for standard purification using a MabSelect medium. Purified
LST-007 was concentrated in a stirred cell, dialyzed vs. phosphate
buffer and quantitated at 280 nm. Bioburden of sterile-filtered
material was 0 cfu/ml with levels of endotoxin <0.04 EU/mg.
Final concentration of LST-007 was quantified at 13.15 mg/ml and
stored as a number of 1-ml aliquots at −20 and −80°C.
Binding of LST-007 towards recombinant PA
flagellin type b in ELISA
Wells of Maxisorp ELISA plates (#442404; Nunc Brand
Products) were coated with 250 ng of recombinant PA flagellin type
b and incubated with orbital shaking for 2 h at room temperature.
Thereafter, unbound antigen was removed and 200 μl blocking
solution comprised of PBS-10% fetal bovine serum (FBS; #04-001-1A;
Biological Industries) was added overnight at 4°C. Following
removal of blocking buffer, 50 μl LST-007 in blocking solution was
added over a range of 0–208 pM (0–31.25 ng/ml) and allowed to
incubate for 2 h at room temperature. LST-007 was decanted and the
wells were washed 3 times with 200 μl of PBS containing 0.05%
Tween-20. A secondary antibody consisting of a 1:10,000 dilution of
goat anti-human IgG-Fc-HRP conjugate (#A80-104P; Bethyl) in
blocking solution was added for 1 h at room temperature. Following
identical washing as described above, 50 μl of TMB solution
(#ES001; Millipore) was added after which the colorimetric reaction
was quenched following the addition of 50 μl 10%
H2SO4. The plates were read in an ELISA
reader at OD 450 nm. Blocking buffer with the sole presence of
primary or secondary antibodies was used as blank controls.
Binding of LST-007 towards immobilized,
whole PA bacteria in ELISA
PA strains were grown in 5 ml LB medium at 37°C in
an orbital shaker for 18 h, pelleted and washed twice with PBS.
Following resuspension of the bacterial pellet with a small volume
of PBS, the volume was adjusted to obtain an OD 600 nm of 0.2. To
permit bacterial binding, 50 μl of poly-L-lysine (PLL) of a 1 μg/ml
solution was added to the wells of Maxisorp ELISA plates and
incubated for 30 min at room temperature with orbital shaking.
Following removal of PLL, 50 μl of the PA suspensions were added to
plates and immediately centrifuged at 1,500 rpm for 20 min.
Thereafter, supernatants were carefully removed with a
multi-channel pipettor and adsorbed PA bacteria were irreversibly
fixed to wells by adding 75 μl 0.2% formaldehyde for 15 min at room
temperature. Following removal of formaldehyde and brief drying,
plates were blocked overnight at 4°C with 200 μl PBS-10% FBS. The
blocking solution was removed and 50 μl of 0.5 μg/ml LST-007,
LST-003 or human isotype control mAb was added for 60 min at 37°C.
The ELISA was continued in an identical manner with recombinant PA
flagellin type b as described above.
Isoelectric focusing (IEF) studies
IEF gels (5% T/3% C; 0.25 mm thick) were prepared by
polymerization of acrylamide and N,N’-methylenebisacrylamide on
GelBond® PAG film (124×258 mm), followed by washing,
drying, and reconstitution with 2% Pharmalyte 3–10, 10 mM glutamic
acid, 10 mM lysine, and 32% (v/v) glycerol. IEF was performed on a
horizontal unit, at 15°C, under a nitrogen atmosphere, 10 cm
between electrodes. Gels were prefocused for 500 V-h (500 V, 60
min). Samples were loaded as drops on the gel surface and focused
for 9,500 V-h (500 V, 30 min; 1,000 V, 30 min; 2,000 V, 30 min;
3,500 V, 133 min). Gels were fixed in 20% (w/v) trichloroacetic
acid and stained with Coomassie G-250 using the colloidal
methodology (33).
Surface plasmon resonance (SPR)
studies
SPR was performed on a Biacore 3000 instrument
(Biacore, Uppsala, Sweden). Recombinant PA flagellin type b was
diluted in 100 mM sodium acetate (pH 4.6) to a final concentration
of 50 μg/ml and immobilized on a CM5 Biacore sensor chip. The
antigen was then streamed over the sensor chip for 5 min at a rate
of 10 ml/min. The binding assay was performed by injecting LST-007
at 16 different concentrations ranging from 0.0 to 50 nM at a flow
rate of 20 ml/min at 25°C. These conditions resulted in a linear
relation between the concentration of LST-007 and the maximal
(steady-state) response, indicating the pseudo first-order regime
in relation to the immobilized ligand. The net signal was obtained
by subtracting the blank signal (dextran matrix). The association
phase for LST-007 binding was monitored for 4 min, while the
dissociation phase for 3 min. Responses were monitored as a
function of time by generating the traces (sensorgrams) at 25°C.
Multi-concentration data were globally fitted using the
BIAevaluation 3.2 software supplied by Biacore to calculate
affinity constant of LST-007.
In vitro PA motility assays
Motility studies were performed as previously
described (34). In brief,
freshly prepared PA colonies were taken for motility experiments as
follows. Soft liquid agar was prepared by autoclaving LB media
containing 1% tryptone, 0.5% NaCl, 0.3% yeast extract and 0.3% soft
agar. This solution was transferred to a water bath at 40°C for 30
min. After 30 min, the solution was aliquoted into working volumes
and mAbs or diluted Cmax sera were added at the desired
concentrations. In some assays, 10 cm plates were used whereby 20
ml of the mAb-containing soft agar was poured. In some assays,
24-well plates were used, with 1 ml of the mAb-containing soft agar
poured/well. Plates were allowed to solidify for ∼30 min at room
temperature within a bacterial culture hood and wells were
inoculated with the desired freshly prepared PA strain by picking a
colony with a sterile toothpick and carefully stabbing a few mm
into the mAb-impregnated agar. Plates were wrapped in parafilm to
prevent dehydration and incubated at 30°C for 12–16 h to maximize
flagellin expression. Wells were photographed and the motility was
measured as the diameter of the halo phenotype that surrounds the
central bacterial growth.
LST-007 pharmacokinetic studies
Female CD-1 mice, age 10–12 weeks (20–25 g) were
used. Animal handling was performed according to the National
Institute of Health (NIH) and the Association for Assessment and
Accreditation of Laboratory Animal Care (AAALAC). During
acclimation (5 days) and following LST-007 dosing, mice were housed
in a specific pathogen-free environment with 3 mice/cage, in
polypropylene cages fitted with solid bottoms and filled with
autoclaved sawdust as bedding material. Animals were provided ad
libitum with a commercial rodent diet and free access to
autoclaved drinking water supplied to each cage. Automatically
controlled environment conditions were set to maintain a
temperature of 22–25°C with a 12 h light/12 h dark cycle and air
changes in the study room.
LST-007 was prepared at a concentration of 1 mg/ml
and injected at 5 ml/kg to achieve a target dose of 5 mg/kg. A
total of 27 mice were taken for simultaneous PK sampling from
bleeds and bronchoalveolar lavage (BAL) fluid, with 3 mice/time
point sacrificed for both samplings. Sampling time points were 5
min (Cmax), 4, 8, 24, 48, 96, 120, 168 and 240 h. Additionally, 3
mice were sampled at 5 min following the injection of saline at 5
ml/kg. In brief, mice were injected intravenously (i.v.) with
LST-007 in the tail vein using a tuberculin syringe and a 30G
needle. At the designated time points, mice were anesthetized using
an intraperitoneal (i.p.) injection of 85 mg/kg xylazine and 5
mg/kg ketamine and bled, ∼500 μl from the orbital sinus. The blood
was collected into 1.5 ml Eppendorf tubes, centrifuged and the
upper sera layer was aliquoted and stored at −80°C until required.
While the mice were still under anesthesia, they were placed on
their backs and the airway was exposed for collection of BAL fluid
by connection of a veinflow to the airway attached to a 26G needle
and 1 ml syringe. A total volume of 700 μl of saline was used to
wash the lungs with return BAL volumes ∼500 μl. Following
centrifugation, the clarified BAL supernatant was removed,
aliquoted and stored at −80°C until assay.
For sampling LST-007, an ELISA was adapted whereby
dilutions of sera (1:500–1:2,000) or BAL fluid (1:100–1:400) were
added to ELISA plates coated with a goat anti-human IgG Fc specific
antibody. Following incubation and blocking (see above), wells were
incubated with a detecting anti-human κ-HRP antibody followed by
colorimetry with TMB. Plates were read at 450 nm after quenching
with H2SO4. Since the BAL fluid compartment
undergoes ‘dilution’ via flushing with saline, recovered BAL
samples were taken for BUN determinations (AML, Herzliya, Israel),
enabling normalization. As an example, BUN levels in BAL were
measured at 1.67 mg/dl as compared to concentrations of 25 mg/dl in
mouse sera. Thus, BAL-derived LST-007 concentrations determined in
ELISA were normalized following a 15-fold multiplication.
Mouse model of pneumonia
Adult C57 mice (25 g; Harlan Nossan, Milan, Italy)
were housed in a controlled environment and provided with a
standard rodent diet and water. Animal care was in compliance with
the Italian regulations for protection of animals used for
experimental and other scientific purposes (DM 116192) as well as
the EEC regulations (OJ of ECL 358/1 12/18/1986). The mice were
housed in cages with filter tops in specific pathogen-free
conditions. They were briefly anesthetized with inhaled Sevorane
(Abbot Laboratories) in an oxygenated chamber and placed in a
supine position with their heads elevated ∼30°. Initially, a
limited number of mice were dedicated to the animal surgery and
intratracheal (i.t.) procedure by validating 100% animal survival
and normal behavior throughout the 7 days post instillation of 50
μl lactated Ringer’s solution into the left lung. Thereafter, a
LD80 pneumonia model was established at 3 days
post-infection, elicited by the i.t. administration of the MDR PA
strain Ka02 at 106 cfu in 50 μl lactated Ringer’s
solution. Using this LD80, the biological activity of
LST-007 or control treatments (i.v. 6 ml/kg) on animal survival
were investigated in the following 5 experimental groups: group 1
(n=20): i.t. Ka02 followed by i.v. formulation buffer (FB) at +60
min and +25 h after infection; group 2 (n=20): i.t. Ka02 followed
by i.v. LST-007 (20 mg/kg) at +60 min and FB at +25 h; termed
‘LST-007x1’; group 3 (n=20): i.t. Ka02 followed by i.v. LST-007 (20
mg/kg) at +60 min and + 25 h (20 mg/kg); termed ‘LST-007x2’; group
4 (n=20): i.t. Ka02 followed by freshly prepared, i.p. imipenem (25
mg/kg; 0.25 ml of a 2.5 mg/ml solution) given b.i.d. for 3 days
post-infection at time points of +60 min, +5, +24, +29, +48 and +53
h; group 5 (n=15): i.t. Ka02 followed by i.v. human isotype mAb (20
mg/kg) at +60 min and + 25 h (20 mg/kg). For groups 2, 3 and 5, mAb
stock concentrations of 3.33 mg/ml were provided from which 0.15 ml
were injected per mouse (i.e. 6 ml/kg).
Statistics were run in Stata version 7 (Stata Corp.,
College Station, TX, USA). The differences between the antibody or
imipenem treated groups vs. FB-treated groups were analyzed.
Dichotomous outcomes were compared using Fisher’s exact test and
continuous variables by the Student’s t-test. All statistical tests
were two-tailed. Differences were considered to be statistically
significant with a P-value ≤0.05.
Results
In vitro characterization of
mammalian-expressed LST-007
Recombinant LST-007 was expressed in CHO cells and
purified to homogeneity. The upper panel of Fig. 1 shows the Coomassie gel staining
of an SDS-PAGE loaded with clarified media containing secreted
LST-007 (lane 2), a washed fraction (lane 3) and 4 μg of the final
eluted LST-007 product (lane 4) depicting the heavy and light
chains. Lane 1 is a molecular weight (Mw) marker of bands 14, 19,
28, 39, 51, 64, 97 and 191 kDa. LST-007, prepared as a
sterile-filtered solution at 13.15 mg/ml, was verified to be free
of bacterial burden with LPS <0.04 EU/mg. In ELISA studies
employing recombinantly expressed flagellin type b as the
immobilized antigen, LST-007 demonstrated a concentration-dependent
increase in OD which peaked at 208 pM (=31.25 ng/ml LST-007). No
binding was observed whatsoever with PA flagellin type a (Fig. 1). In IEF studies, when loaded at
various locations on the IEF gel at different amounts, LST-007 was
demonstrated to be a very basic protein with a pI of 9.3 (Fig. 1, lower panel). When added
maximally at 25 μg (lane 9), two very light additional bands were
observed next to the main band at pH 9.2. While it is possible that
the heavy pH 9.3 band consists of several unresolved bands, for the
most part, the isoelectric focusing data at pH 9.2 demonstrates a
high level of LST-007 electrostatic homogeneity. The basic pI of
LST-007 is similar to the pIs of a number of marketed mAbs from
theoretical and experimental measurements (35).
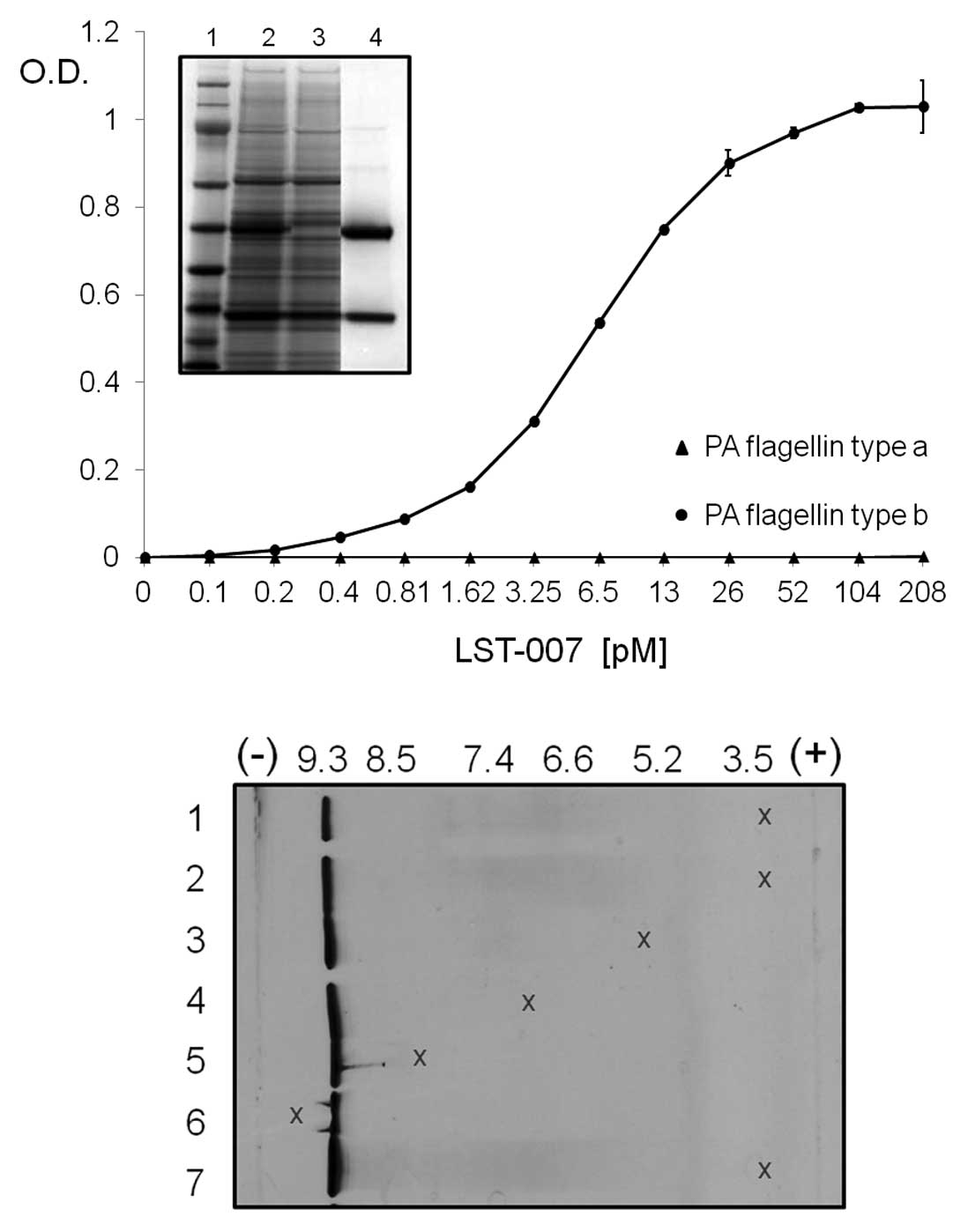 | Figure 1.In vitro properties of
recombinant LST-007 expressed and purified from CHO cells. Upper
panel depicts purification profile of LST-007 as shown by Coomassie
gel staining of clarified CHO cell supernatant (lane 2), wash
fraction (lane 3) and eluted LST-007 product (lane 4), under
reducing conditions. Lane 1 is a Mw (kDa) marker of sizes 191, 97,
64, 51, 39, 28, 19 and 14. ELISA curve demonstrates
concentration-dependent binding of LST-007 from at least 3
independent experiments towards E. coli-expressed, purified
PA flagellin type b (circles) with no reactivity towards flagellin
type a (triangles). Lower panel demonstrate IEF properties of
LST-007. Samples of 2 mg/ml LST-007 were loaded as 5 μl drops to
the gel surface at designated positions (denoted by x) at 1 cm
(lane 2), 3 cm (lane 3), 5 cm (lane 4), 7 cm (lane 5) and 9 cm
(lane 6) from the anode. Additionally, LST-007 was loaded at 1 cm
from the anode at a concentration of 1 mg/ml (lane 1) and 5 mg/ml
(lane 7). The results show that LST-007 is a very basic protein of
pI 9.3. |
The specificity of the above ELISA data was
re-enforced in surface-plasmon resonance (SPR) studies. Here,
LST-007 (0.5–20 nM) was streamed over immobilized PA flagellin type
b bound to a sensor chip and concentration-dependent increases in
the signal were observed as demonstrated in the sensorgrams
(Fig. 2). Analysis of these data
confirmed the high affinity of LST-007 towards recombinant PA
flagellin type b which was calculated at 0.74 nM.
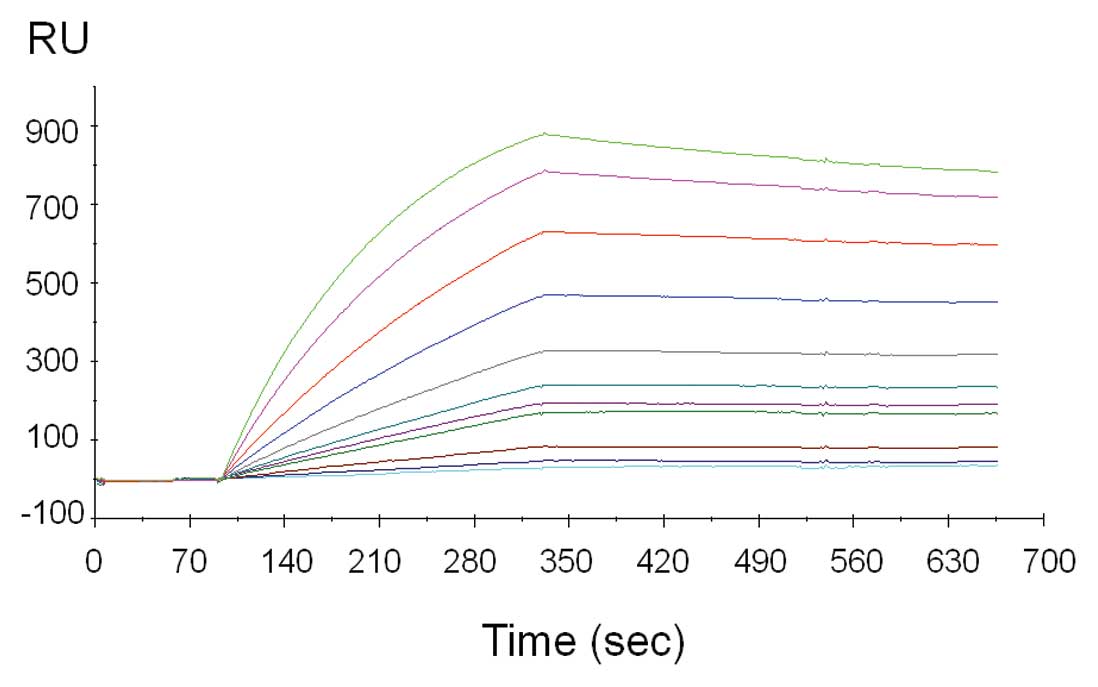 | Figure 2.Concentration-dependent, sensorgram
traces of LST-007 binding towards purified, E.coli-expressed
PA flagellin type b by surface plasmon resonance (SPR) using
Biacore 3000. Binding of purified LST-007 towards its purified
antigen on coated CM5 biosensor chips. Sensorgram traces depict
binding of LST-007 at 0.5, 0.75, 1, 2.5, 3, 4, 5, 7.5, 10, 15 and
20 nM with proportional increases in resonance units (RU). The
affinity of LST-007 affinity towards recombinant PA flagellin type
b was calculated at 7.4×10−10 M. |
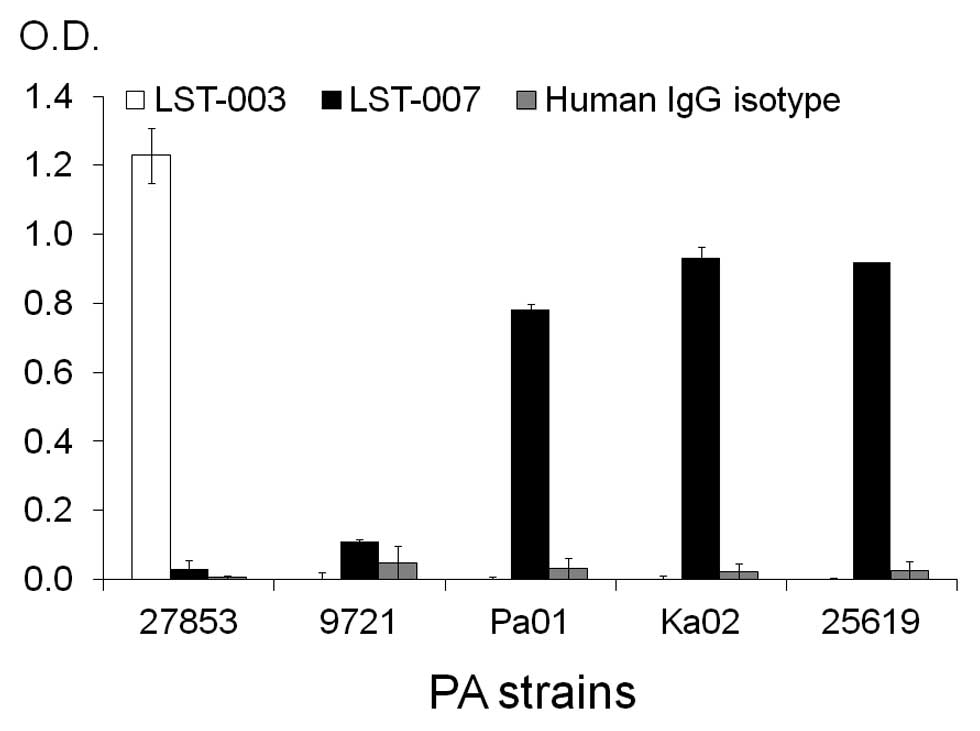
To further scrutinize the specificity of LST-007
binding and assess its capability to bind the ‘naturally-occurring’
flagellin type b, ELISA studies were established whereby the PA
target was in the form of immobilized whole PA bacteria tethered to
ELISA plates via poly-L-lysine (PLL). In these binding studies
(Fig. 3), PA strains 27853 (type
a flagellin), Pa01 (type b flagellin) and 2 additional flagellin
type b containing PA strains Ka02 and 25619 (as determined by PCR
analysis, data not shown) were immobilized on plates. Additionally,
a non-motile PA strain (9721) and thus potentially devoid of a
flagellum was also incorporated in the screen as a negative
control. LST-007 (0.5 μg/ml) specifically bound strains PAO1, Ka02
and 25619 but was devoid of reactivity towards PA strains 27853 and
9721. Conversely, LST-003 solely reacted with strain 27853. To
confirm the specificity of the ELISA, 0.5 μg/ml of a human isotype
control mAb failed to react with any of the PA strains (Fig. 3). These data therefore confirm
that LST-007 is a highly specific mAb and capable of binding
flagellin type b in its natural form as part of intact PA
bacteria.
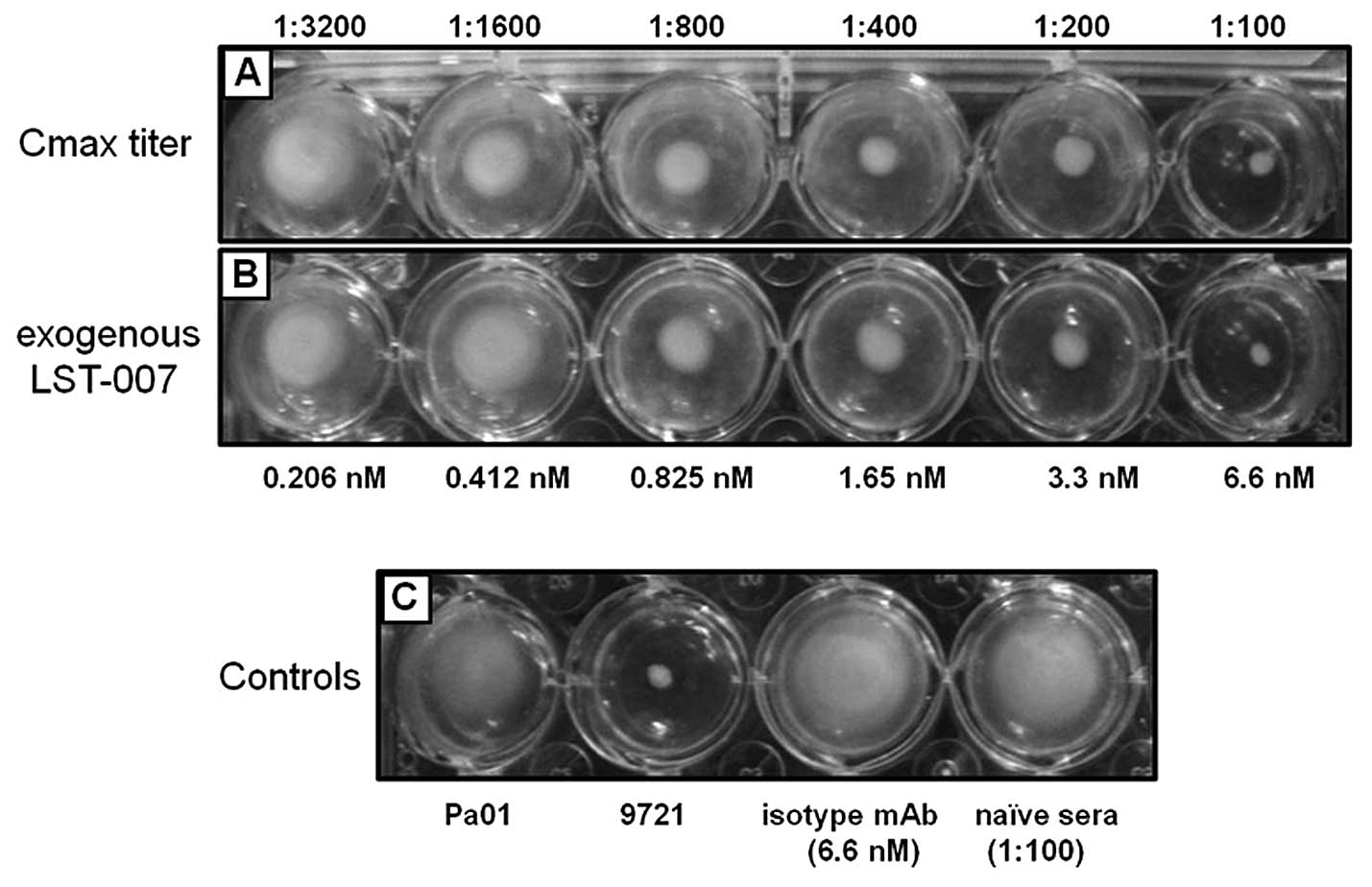
In vitro biological activity of LST-007
on perturbation of PA motility in soft agar assays
Based on the proven, high specific reactivity of
LST-007 towards PA flagellin type b and the quality of the mAb
preparation, bioactivity studies were undertaken by assessing the
capability of LST-007 to perturb PA motility in soft-agar
impregnated with the mAb. In such studies, LST-007 from disparate
sources was employed as follows: i) using Cmax sera from our PK
studies whereby sera were diluted from 1:100-1:3,200, and ii) using
exogenously added LST-007 from our purified mAb preparation at
identical concentrations to the measured serum titer based on the
LST-007 Cmax of 804.86 nM (120.67 μg/ml) (Table I). Concentration-dependent
inhibition profiles on PA motility was observed with either the
ex vivo (Fig. 4A) or
exogenous source (Fig. 4B) of
LST-007 with complete suppression of PA motility observed at 6.6 nM
(∼1 μg/ml) mAb.
 | Table I.Pharmacokinetic analysis of LST-007
in naïve mice blood following the i.v. injection of a single dose
of 5 mg/kg. |
Table I.
Pharmacokinetic analysis of LST-007
in naïve mice blood following the i.v. injection of a single dose
of 5 mg/kg.
| LST-007 PK
analysis |
|---|
| AUC | (0 to 240) | mvc/ml * h | 2463.34 |
| (0 to
infinity) | mcg/ml * h | 2772.88 |
| Tmax | (0 to 240) | h | 0.083 |
| Cmax | (0 to 240) | mcg/ml | 120.67 |
| Cmin | (0 to 240) | mcg/ml | 2 |
| Cavg | (0 to 240) | mcg/ml | 10.26 |
| Ke | 1/h | 0.009565 |
| T1/2 | h | 83.58 |
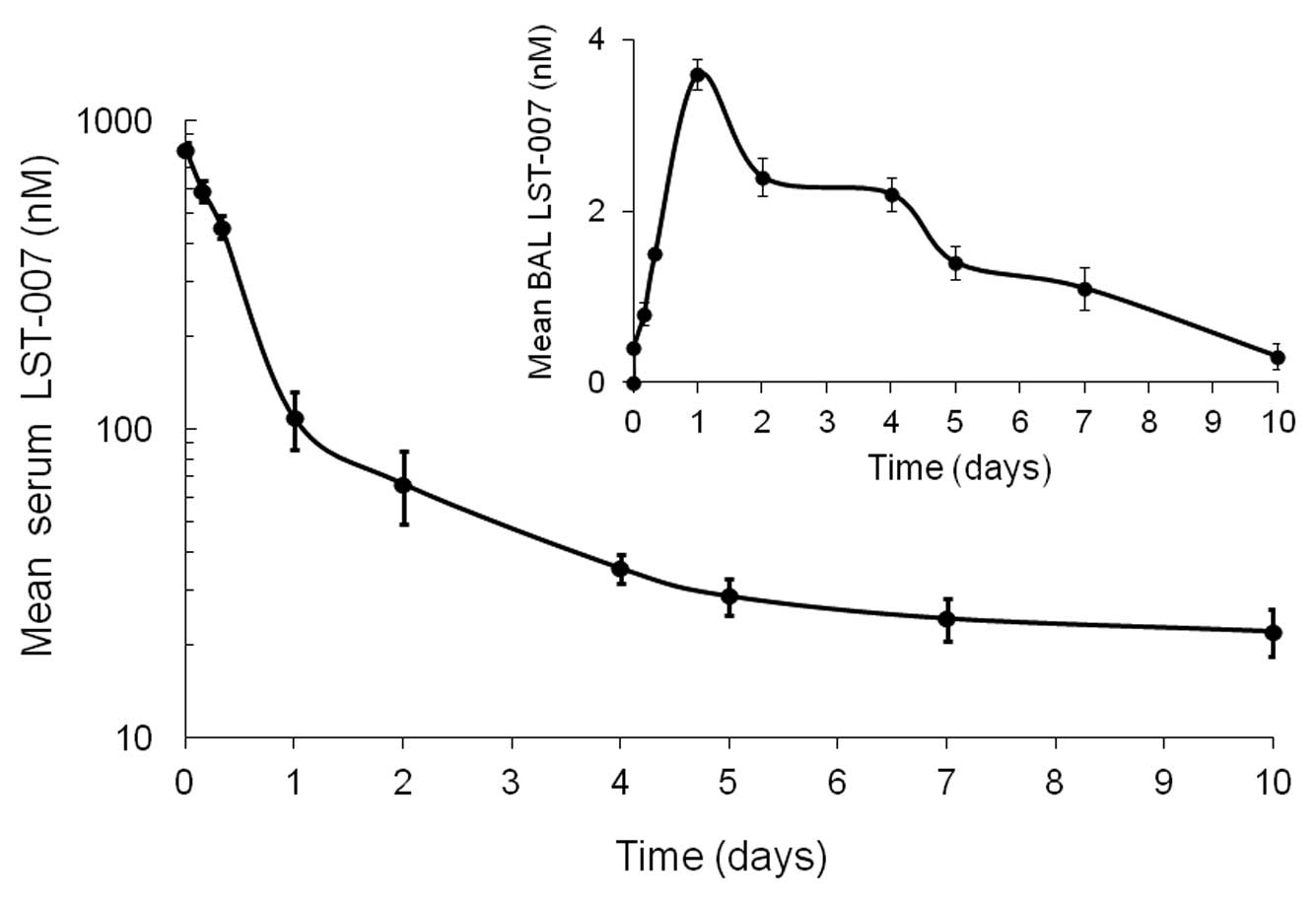
PK profile of LST-007 in blood and
broncholveolar lavage (BAL) fluid from naïve mice
The PK profile of LST-007 in blood and BAL fluid was
investigated following a single i.v. injection of the mAb at a dose
of 5 mg/kg in naïve mice. This dose was chosen based on a PK study
performed with an anti-RAGE mAb (36) and deemed appropriate by us in
terms of our ELISA detection capabilities based on the theoretical
concentrations of LST-007 expected in different compartments. In
sera, LST-007’s Cmax (at 5 min) was 804.86 nM (120.67 μg/ml)
followed by a rapid phase of elimination to a concentration of
111.18 nM (16.67 μg/ml) at 24 h (Fig.
5). A much slower rate of elimination occurred over the next 9
days since the LST-007 concentration was measured at 26.6 nM (4
μg/ml) at Day 10 (Fig. 5).
LST-007’s half-life was calculated to be 83.58 h (Table I).
In marked contrast to the blood compartment
kinetics, LST-007 exhibited a different PK profile in BAL fluid
since mAb concentrations increased over the first 24 h, peaking to
3.6 nM (0.54 μg/ml) at the 24 h time point and decreased slowly
over the next 9 days (Fig. 5).
LST-007 was still detectable in BAL fluid at Day 10 at a
concentration of 0.3 nM (0.045 μg/ml).
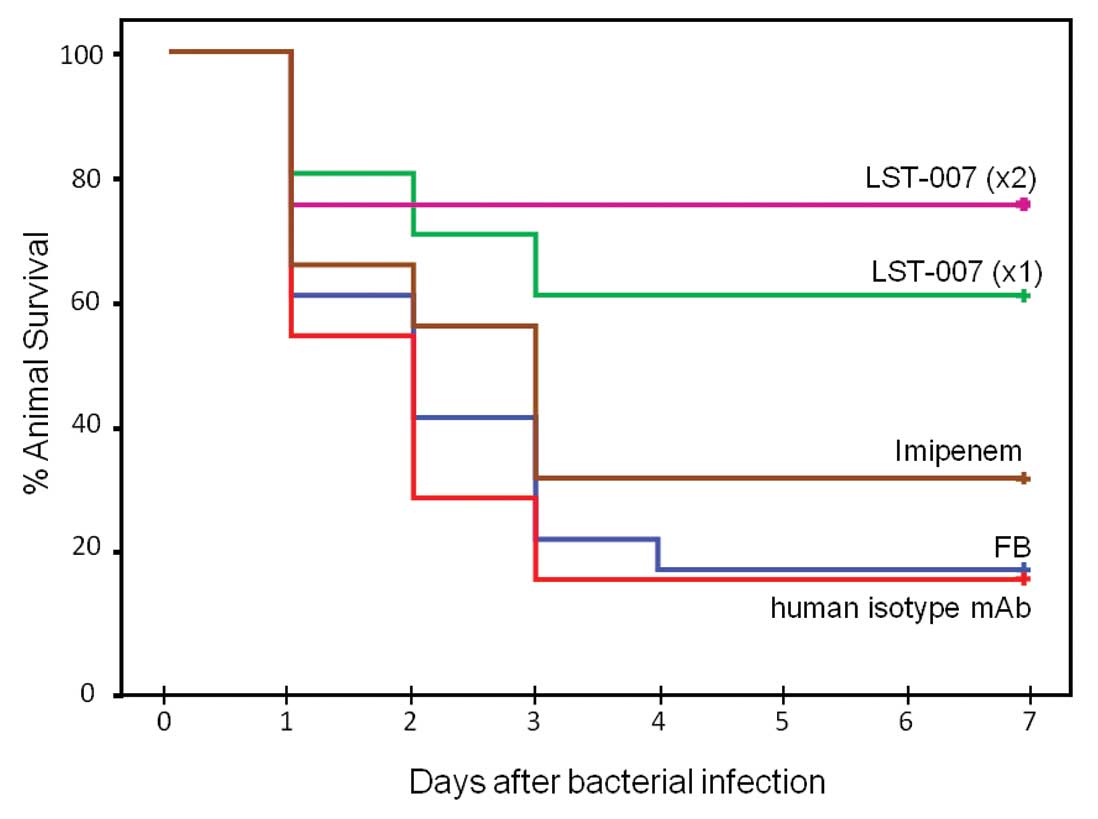
Effect of LST-007 on animal survival in a
lethal MDR PA-induced model of pneumonia
The biological effect of LST-007 on animal survival
was investigated in a lethal mouse model of pneumonia driven by an
MDR PA strain (Ka02). Initial calibration studies demonstrated that
i.t. instillation of 106 cfu Ka02 elicited an LD80 at 3
days (data not shown). This mortality profile was used by us in
subsequent feasibility studies with LST-007. Following lung
infection, mice treated with i.v. FB demonstrated typical lethality
curves with 60, 40 and 20% survival at Days 1, 2 and 3,
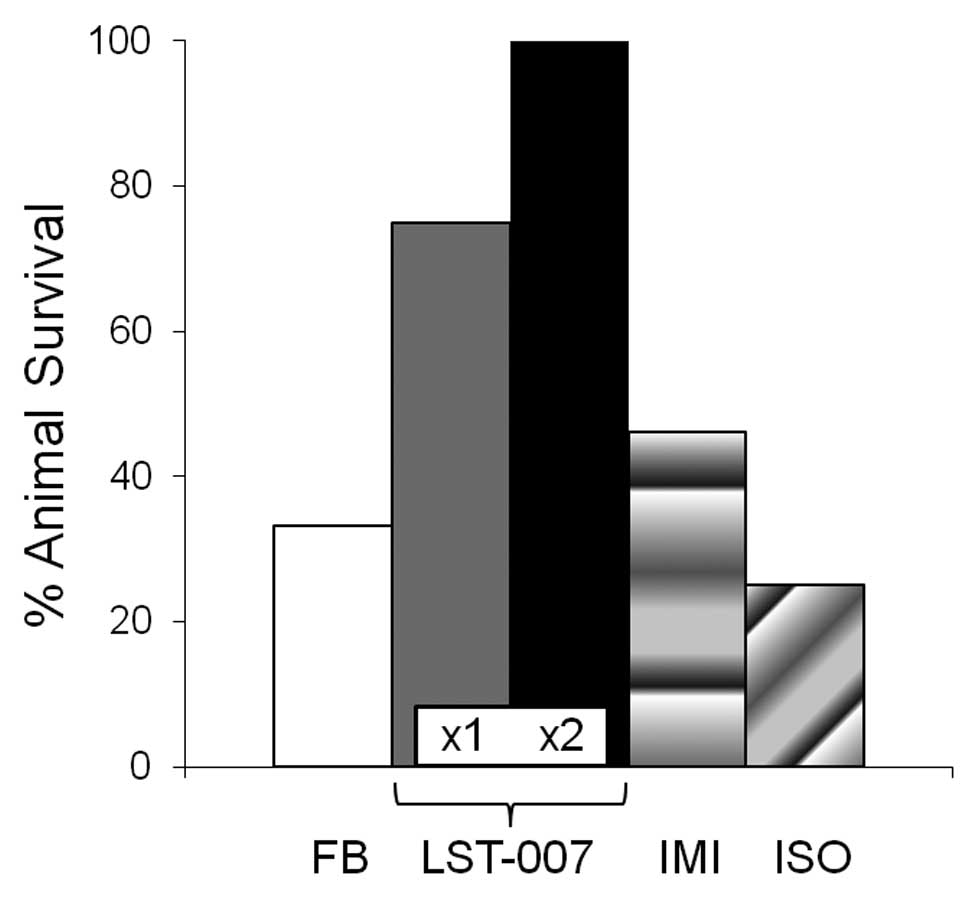
respectively (Fig. 6). In marked
contrast, infected animals treated with LST-007x1 or LST-007x2
demonstrated marked improvements in survival at 1–3 days
post-infection with 60 and 75% survivors at Day 3 respectively
which remained unchanged until Day 7 (Fig. 6). The specificity of this effect
was confirmed since an irrelevant human isotype control mAb
administered at 20 mg/kg twice at +1 and +25 h, failed to curtail
mortality which essentially superimposed on the mortality profile
observed with the FB treatment group (Fig. 6). Importantly, imipenem treatment
at doses and a route of administration known to be protective
against the laboratory strain PAO1 (30,31) was ineffective in preventing
Ka02-induced mortality with survival of only 30% at Day 3 (Fig. 6). The importance and significance
of a second dosing of LST-007 is underscored in Fig. 7 since the mAb completely abrogated
mortality changes noted with LST-007x1 at Day 2 (2/16 deaths) and
Day 3 (2/14 deaths).
Discussion
A call has been heeded by the Infectious Diseases
Society of America (IDSA) and the European Medicines Agency (EMA)
for the urgent development of novel antibacterial strategies
targeting in particular, the ESKAPE group of pathogens (1,37).
In the case of Pseudomonas aeruginosa (PA), a major dearth
exists due to the lack of effective antibacterials targeting MDR PA
strains. Currently, most of the anti-PA drugs in late stage
pre-clinical testing or early clinical evaluation are modifications
of existing drugs, newer combinations of existing drugs or even
newer routes of administration of old drugs (38–40). Clearly, innovative remedies are
urgently required to combat MDR PA which is a major nosocomial
threat, especially in immunocompromised patients.
It is axiomatic that mAbs are proven therapeutics
for cancer and immunologic disorders (41), with increasing recognition that
mAbs may serve as bona fide drugs for a variety of
infectious diseases (42,43) including PA (19,20) by targeting surface-expressed
virulence factors. We developed and expressed in CHO cells, a fully
human mAb (LST-007) against the flagella of PA by targeting
flagellin type b, one of the two major protein forms of this
appendage. LST-007 was purified to homogeneity from CHO cells,
reacted specifically with E. coli-expressed flagellin type b
and exhibited a high affinity of 7.4x10−10 M in SPR
assays. These binding profiles were supported by the capability of
LST-007 to selectively bind its native target in whole PA bacteria.
Since flagellin type b is glycosylated in PA (44) and may have an impact on mAb
binding, integrity of LST-007’s recognition towards PA in the fixed
ELISA format served as an impetus for bioactivity assays.
Previously, anti-PA flagellin mAbs were reported to
inhibit PA motility in vitro which presumably laid the basis
for their beneficial effects in models of PA infection (27–29). Similarly, LST-007 suppressed PA
motility at concentrations close to its KD of the mAbs.
Furthermore, we also demonstrated that hyperimmune sera derived
from PK studies with LST-007 was similarly bioactive to exogenously
added mAb (Fig. 4). This ex
vivo finding was important for 2 reasons: i) it demonstrated
that LST-007 retains bioactivity in vivo indicating its
likely stability within blood, and ii) it provided us with valuable
information regarding LST-007’s target concentration that should
allow effective inhibition of PA motility and positive
proof-of-concept in our pneumonia model. Indeed, a 1:100 dilution
of Cmax sera containing ∼1 μg/ml LST-007 (6.6 nM LST-007) can be
regarded as the minimal inhibitory concentration (MIC) that caused
complete inhibition of PA motility by comparing its phenotype in
soft agar with 9721, the non-motile aflagellated PA strain
(Fig. 4).
The PK profile of LST-007 in the blood from naïve
mice demonstrated a half-life of ∼84 h, reaching a concentration
∼15% of the Cmax at 24 h during a rapid elimination phase. At such
a concentration (16 μg/ml), ∼10X MIC on motility, LST-007 may be
expected to be bioactive in a bloodstream PA infection.
Nevertheless, LST-007 bioactivity in a pneumonia model is governed
by its traversing into the alveolar space from blood. LST-007
concentrations from the same naïve mice were measured in BAL fluid,
peaked at 24 h (Cmax, 0.5 μg/ml) and gradually decreased until Day
10 (Fig. 5). Based on an ∼0.5 MIC
of LST-007 in BAL fluid at 24 h after dosing with 5 mg/kg, we
rationalized that a higher and potential additional dose of LST-007
may be required to allow appropriate pharmacodynamic ‘coverage’ and
a desired biological effect in the pneumonia model. When
administered a single dose of 20 mg/kg, LST-007 improved survival
at each day during the first 3 days during which significant
mortality occurred in both control groups. Importantly, a second
dose of LST-007, 25 h after infection, completely attenuated
mortality changes that occurred at Days 2 and 3. These latter
findings would indicate that LST-007 reached the alveolar space at
concentrations exceeding 1X MIC which are sustained during the 3
days mortality window. Clinical implications from these findings
are 2-fold: i) constant infusion of LST-007 during PA colonization
and infection may be the appropriate mode of administration, and
ii) LST-007 could be given prophylactically or at least during PA
colonization since it traverses into the alveolar space of intact
lung. This latter property is presumably due to LST-007 being an
IgG1 isotype since the accumulation of the IgM mAb panobacumab in
BAL fluid was critically dependent upon the consequences of PA
infection (i.e. lung microvascular permeability defects) (17).
A limitation in the present study is that bacterial
burden in lungs was not measured. However, since the infecting
strain was instilled directly into the lung and there is no
evidence that anti-PA mAbs can exert bactericidal effects,
reduction in lung bacterial burden are unlikely yet still needs to
be verified. Nevertheless, LST-007-mediated suppression of
infection throughout the lung or to other organs may occur as
reported in the suppression of bacterial dissemination to the
spleen in a burn model of PA infection with an anti-type a
flagellin mAb (31). Taken
together, our data indicated that the beneficial effect of LST-007
on survival may be due to factors that not only target PA motility
and accessibility of the bacteria to the TLR5, but also reduced
invasiveness into alveolar epithelial cells as well as potential
toning down of local inflammatory events driven by monomeric
flagellin (23,45). Studies utilizing human alveolar
A549 epithelial cells, recombinant flagellins, appropriate
infecting PA strains as well as the non-motile 9721 strain, could
help to clearly scrutinize the roles of the flagellum in such
processes.
Alanine-scanning mutagenesis has shown that the
structural requirements for flagellar motility are much more rigid
than the permissive nature of TLR5 recognition of flagellin.
(23). Interestingly, mouse mAbs
raised against PA flagella (27)
which impeded motility were shown to bind PA flagellins downstream
from amino acid 161 by SDS-PAGE (31). It may be possible that LST-007
similarly targets PA flagellin type b, especially since mutations
of a number of conserved C’ terminal amino acid residues in
flagellin’s D1 domain resulted in complete abrogation of bacterial
motility (23). Appropriate
epitope mapping using techniques such as a hybrid β-lactamase
display as recently demonstrated for an anti-CD22 immunotoxin
CAT-8015 (46), could be very
useful to localize LST-007’s site of binding within PA flagellin
type b.
From a historical perspective, it is perplexing as
to why therapeutic antibodies targeting PA flagellin failed to come
to fruition yet reached significant stature in the early 1990’s.
Cessation of developmental efforts were probably swayed by the
disappointing clinical studies with anti-sepsis agents including
mAbs (47,48). Additionally, the need to develop
two separate mAbs targeting flagellin type a and b for complete
anti-PA coverage may have been an issue, especially during a period
where many of the methodological aspects concerning the development
of therapeutic mAbs were still in their infancy. Today, such issues
can be overcome, as exemplified by the prior screening of infected
PA patients to ensure compatibility for immunotherapy (19), streamlined mAb manufacturing
approaches (14) or even
potential design of bispecific mAbs (49). Flagellin represents a viable
target for therapeutic intervention which is further fueled by two
additional findings. In translational studies, whereby
polymorphisms in the TLR5 and subsequent flagellin
hyporesponsiveness are associated with improved health indicators
in Crohn’s disease (50), CF
(51) and systemic lupus
erythematosus (52). Secondly,
the highly encouraging clinical data in CF patients using oral
chicken IgY (53) whereby PA
flagellin was proven to be the major immunoreactive antigen bound
by this polyclonal therapeutic from proteomic studies (54).
In summary, LST-007 represents a bona fide
fully human IgG1 mAb targeting PA flagellin type b which was shown
to afford significant improvement in survival against an MDR
PA-induced pneumonia, superceding standard care of the treatment.
In an era of limited therapeutics targeting MDR PA, neutralizing
anti-PA mAbs may not only provide a new monotherapeutic strategy
but could additionally synergize with existing antibiotics. Such a
desired effect could shepherd these precious drugs from resistance
mechanisms. Our current data provide us with significant momentum
in continuing the development of LST-007 with the intent of
evaluating its clinical efficacy. Such patients could include those
within the critical care setting (e.g. ventilator-associated
pneumonia) or CF patients. Alternate indications, especially those
where PA may be the sole infectious culprit (e.g. urinary tract
infections, necrotizing malignant external otitis), could also
represent highly feasible patient populations to demonstrate
clinical proof-of-concept.
Acknowledgements
We are grateful to Dr Aharon Rabinkov
(Department of Biological Services, Weizmann Institute of Science,
Rehovot, Israel) for providing technical support in the Biacore
experiments and Mr. Yariv Shoshany for performing the PK analysis.
Lostam BioPharmaceuticals Ltd. is truly indebted to the Office of
the Chief Scientist (Incubator Program) of the Ministry of
Industry, Trade and Labor, Israel for funding all aspects of this
project with infrastructure company support under the auspices of
the New Generation Technology (NGT) Incubator, Nazareth,
Israel.
References
|
1.
|
HW BoucherGH TalbotJS BradleyBad bugs, no
drugs: no ESKAPE! An update from the Infectious Diseases Society of
AmericaClin Infect Dis481122009
|
|
2.
|
ECDC/EMEA JointTechnical ReportThe
Bacterial Challenge: Time to React. A call to narrow the gap
between multidrug-resistant bacteria in the EU and the development
of new antibacterial agents1422009EMEA doc. ref. EMEA/576176/2009
Stockholm, September 2009.10.2900/2518
|
|
3.
|
JA DriscollSL BrodyMH KollefThe
epidemiology, pathogenesis and treatment of Pseudomonas
aeruginosa
infectionsDrugs67351368200710.2165/00003495-200767030-0000317335295
|
|
4.
|
AS LevinMS OliveiraThe challenge of
multidrug resistance: the treatment of gram-negative rod
infectionsShock303033200810.1097/SHK.0b013e3181819cb818704012
|
|
5.
|
S FujitaniHY SunVL YuJA
WeingartenPneumonia due to Pseudomonas aeruginosa: part I:
epidemiology, clinical diagnosis, and sourceChest1399099192011
|
|
6.
|
A PallettK HandComplicated urinary tract
infections: practical solutions for the treatment of multiresistant
Gram-negative bacteriaJ Antimicrob
Chemother65S25S33201010.1093/jac/dkq29820876625
|
|
7.
|
PA FlumeBP O’SullivanKA RobinsonCystic
fibrosis pulmonary guidelines: chronic medications for maintenance
of lung healthAm J Respir Crit Care
Med176957969200710.1164/rccm.200705-664OC17761616
|
|
8.
|
M Cohen-CymberknohD ShoseyovE
KeremManaging cystic fibrosis: strategies that increase life
expectancy and improve quality of lifeAm J Respir Crit Care
Med18314631471201110.1164/rccm.201009-1478CI21330455
|
|
9.
|
N HoibyRecent advances in the treatment of
Pseudomonas aeruginosa infections in cystic fibrosisBMC
Med9322011
|
|
10.
|
CK StoverXQ PhamAL ErwinComplete genome
sequence of Pseudomonas aeruginosa PAO1, an opportunistic
pathogenNature406959964200010.1038/3502307910984043
|
|
11.
|
CR DeanMA VisalliSJ ProjanPE SumPA
BradfordEfflux-mediated resistance to tigecycline (GAR-936) in
Pseudomonas aeruginosa PAO1Antimicrob Agents
Chemother47972978200310.1128/AAC.47.3.972-978.200312604529
|
|
12.
|
J ChastreJY FagonVentilator-associated
pneumoniaAm J Resp Crit Care
Med165867903200210.1164/ajrccm.165.7.2105078
|
|
13.
|
L CegelskiGR MarshallGR EldridgeSJ
HultgrenThe biology and future prospects of antivirulence
therapiesNature Rev
Microbiol61727200810.1038/nrmicro181818079741
|
|
14.
|
M ChartrainL ChuDevelopment and production
of commercial therapeutic monoclonal antibodies in mammalian cell
expression systems: an overview of the current upstream
methodologiesCurr Pharm
Biotechnol9447467200810.2174/138920108786786367
|
|
15.
|
M De JesusFM WurmManufacturing recombinant
proteins in kg-ton quantities using animal cells in bioreactorsEur
J Pharm Biopharm78184188201121256214
|
|
16.
|
M BaerT SawaP FlynnAn engineered human
antibody fab fragment specific for Pseudomonas aeruginosa
PcrV antigen has potent antibacterial activityInfect
Immun7710831090200910.1128/IAI.00815-0819103766
|
|
17.
|
T SecherL FauconnierA
SzadeAnti-Pseudomonas aeruginosa serotype O11 LPS
immunoglobulin M monoclonal antibody panobacumab (KBPA101) confers
protection in a murine model of acute lung infectionJ Antimicrob
Chemother66110011092011
|
|
18.
|
MP HornAW ZuercherMA ImbodenPreclinical in
vitro and in vivo characterization of the fully human monoclonal
IgM antibody KBPA101 specific for Pseudomonas aeruginosa
serotype IATS-O11Antimicrob Agents
Chemother5423382344201010.1128/AAC.01142-0920308370
|
|
19.
|
Q LuJJ RoubyPF LaterrePharmacokinetics and
safety of panobacumab: specific adjunctive immunotherapy in
critical patients with nosocomial Pseudomonas aeruginosa O11
pneumoniaJ Antimicrob
Chemother6611101116201110.1093/jac/dkr04621398296
|
|
20.
|
CE MillaFJ AccursoJ ChmielModulating
Pseudomonas aeruginosa chronic inflammation with the
anti-PcrV antibody KB001: Results of a pilot clinical and
pharmacodynamic study in subjects with cystic fibrosisAm J Respir
Crit Care Med181A18452010
|
|
21.
|
FA SamateyK ImadaS NagashimaStructure of
the bacterial flagellar protofilament and implications for a switch
for supercoilingNature410331337200110.1038/3506650411268201
|
|
22.
|
K YonekuraS Maki-YonekuraK NambaComplete
atomic model of the bacterial flagellar filament by electron
cryomicroscopyNature424643650200310.1038/nature0183012904785
|
|
23.
|
KD SmithE Andersen-NissenF
HayashiToll-like receptor 5 recognizes a conserved site on
flagellin required for protofilament formation and bacterial
motilityNat Immunol412471253200310.1038/ni101114625549
|
|
24.
|
RA AnsorgME KnocheAF SpiesCJ
KrausDifferentiation of the major flagellar antigens of
Pseudomonas aeruginosa by the slide coagglutination
techniqueJ Clin Microbiol20848819846430957
|
|
25.
|
TJ MontieTR AndersonEnzyme-linked
immunosorbent assay for detection of Pseudomonas aeruginosa
H (flagellar) antigenEur J Clin Microbiol Infect
Dis7256260198810.1007/BF019630972455641
|
|
26.
|
JA MorganNF BellinghamC WinstanleyMA
OusleyCA HartJR SaundersComparison of flagellin genes from clinical
and environmental Pseudomonas aeruginosa isolatesAppl
Environ Microbiol6511751179199910049879
|
|
27.
|
MJ RosokMR StebbinsK ConnellyME LostromAW
SiadakGeneration and characterization of murine anti flagellum
monoclonal antibodies that are protective against lethal challenge
with Pseudomonas aeruginosaInfect Immun58381938281990
|
|
28.
|
H OchiH OhtsukaS YokotaInhibitory activity
on bacterial motility and in vivo protective activity of human
monoclonal antibodies against flagella of Pseudomonas
aeruginosaInfect Immun5955055419911898908
|
|
29.
|
WJ LandspergerKD Kelly-WintenbergTC
MontieInhibition of bacterial motility with human antiflagellar
monoclonal antibodies attenuates Pseudomonas
aeruginosa-induced pneumonia in the immunocompetent ratInfect
Immun624825483019947927761
|
|
30.
|
LF NevilleY BarneaO Hammer-MunzAntibodies
raised against N’-terminal Pseudomonas aeruginosa flagellin
prevent mortality in lethal murine models of infectionInt J Mol
Med161651712005
|
|
31.
|
Y BarneaY CarmeliLF NevilleTherapy with
anti-flagellin A monoclonal antibody limits Pseudomonas
aeruginosa invasiveness in a mouse burn wound sepsis
modelBurns35390396200910.1016/j.burns.2008.08.01418951715
|
|
32.
|
R ErenD LandsteinD TerkieltaubPreclinical
evaluation of two neutralizing human monoclonal antibodies against
hepatitis C virus (HCV): a potential treatment to prevent HCV
reinfection in liver transplant patientsJ
Virol8026542664200610.1128/JVI.80.6.2654-2664.2006
|
|
33.
|
V NeuhoffN AroldD TaubeW EhrhardtImproved
staining of proteins in polyacrylamide gels including isoelectric
focusing gels with clear background at nanogram sensitivity using
Coomassie Brilliant Blue G-250 and
R-250Electrophoresis9255262198810.1002/elps.1150090603
|
|
34.
|
MH RashidA KornbergInorganic polyphosphate
is needed for swimming, swarming, and twitching motilities of
Pseudomonas aeruginosaProc Natl Acad Sci
USA9748854890200010.1073/pnas.06003009710758151
|
|
35.
|
CA BoswellDB TesarK MukhyalaFP TheilPJ
FielderLA KhawliEffects of charge on antibody tissue distribution
and pharmacokineticsBioconjug
Chem2121532163201010.1021/bc100261d21053952
|
|
36.
|
Y VugmeysterD DeFrancoDD PittmanX
XuPharmacokinetics and lung distribution of a humanized anti-RAGE
antibody in wild-type and RAGE−/ −
miceMAbs2571575201010.4161/mabs.2.5.1308920978371
|
|
37.
|
L Freire-MoranB AronssonC ManzCritical
shortage of new antibiotics in development against
multidrug-resistant bacteria - time to react is nowDrug Resist
Updat14118124201110.1016/j.drup.2011.02.00321435939
|
|
38.
|
MG PageJ HeimProspects for the next
anti-Pseudomonas drugCurr Opin
Pharmacol9558565200910.1016/j.coph.2009.08.00619748829
|
|
39.
|
GH TalbotWhat is in the pipeline for
Gram-negative pathogens?Expert Rev Anti Infect
Ther63949200810.1586/14787210.6.1.3918251663
|
|
40.
|
JW MoutonPG AmbroseR CantonConserving
antibiotics for the future: New ways to use old and new drugs from
a pharmacokinetic and pharmacodynamic perspectiveDrug Resist
Updat14107117201110.1016/j.drup.2011.02.00521440486
|
|
41.
|
JM ReichertMonoclonal antibodies as
innovative therapeuticsCurr Pharm
Biotechnol9423430200810.2174/13892010878678635819075682
|
|
42.
|
I LowyDC MolrineBA LeavTreatment with
monoclonal antibodies against Clostridium difficile toxinsN
Engl J Med362197205201010.1056/NEJMoa090763520089970
|
|
43.
|
EL LopezMM ContriniE GlatsteinSafety and
pharmacokinetics of urtoxazumab, a humanized monoclonal antibody,
against Shiga-like toxin 2 in healthy adults and in pediatric
patients infected with Shiga-like toxin-producing Escherichia
coliAntimicrob Agents
Chemother54239243201010.1128/AAC.00343-09
|
|
44.
|
A VermaM SchirmSK AroraP ThibaultSM LoganR
RamphalGlycosylation of b-type flagellin of Pseudomonas
aeruginosa: structural and genetic basisJ
Bacteriol18843954403200610.1128/JB.01642-0516740946
|
|
45.
|
KK ShanksW GuangKC KimEP
LillehojInterleukin-8 production by human airway epithelial cells
in response to Pseudomonas aeruginosa clinical isolates
expressing type a or type b flagellinsClin Vaccine
Immunol1711961202201010.1128/CVI.00167-1020592113
|
|
46.
|
D BannisterB PopovicS SridharanEpitope
mapping and key amino acid identification of anti-CD22 immunotoxin
CAT-8015 using hybrid β-lactamase displayProtein Eng Des
Sel24351360201121159620
|
|
47.
|
RC BoneMonoclonal antibodies to endotoxin.
New allies against
sepsis?JAMA26611251126199110.1001/jama.1991.034700800950381865548
|
|
48.
|
RC BoneWhy sepsis trials
failJAMA276565566199610.1001/jama.1996.035400700610328709407
|
|
49.
|
D HolmesBuy buy bispecific antibodiesNat
Rev Drug Discov10798800201110.1038/nrd358122037028
|
|
50.
|
AT GewirtzM Vijay-KumarSR BrantRH DuerrDL
NicolaeJH ChoDominant-negative TLR5 polymorphism reduces adaptive
immune response to flagellin and negatively associates with Crohn’s
diseaseAm J Physiol Gastrointest Liver
Physiol290G1157G1163200616439468
|
|
51.
|
CJ BlohmkeJ ParkAF HirschfeldTLR5 as an
anti-inflammatory target and modifier gene in cystic fibrosisJ
Immunol18577317738201010.4049/jimmunol.100151321068401
|
|
52.
|
TR HawnH WuJM GrossmanBH HahnBP TsaoA
AderemA stop codon polymorphism of Toll-like receptor 5 is
associated with resistance to systemic lupus erythematosusProc Natl
Acad Sci USA1021059310597200510.1073/pnas.050116510216027372
|
|
53.
|
E NilssonA LarssonHV OlesenPE WejakerH
KollbergGood effect of IgY against Pseudomonas aeruginosa
infections in cystic fibrosis patientsPediatr
Pulmonol438928992008
|
|
54.
|
E NilssonA AminiB WretlindA
LarssonPseudomonas aeruginosa infections are prevented in
cystic fibrosis patients by avian antibodies binding Pseudomonas
aeruginosa flagellinJ Chromatogr B Analyt Technol Biomed Life
Sci8567580200710.1016/j.jchromb.2007.05.029
|