Introduction
Epidemiological studies have demonstrated that
transitional cell carcinoma (TCC) of the bladder is a common
urologic malignancy in men (1–3).
Most of the deaths of patients with muscle invasive bladder cancer
are due to the progression of their disease caused by metastasis
(2,4,5).
Therefore, understanding these metastatic mechanisms is essential
for the design of more effective therapeutic targets. Although many
reports have studied cascading events leading to the metastasis of
muscle invasive bladder tumors (2,6),
the exact molecular mechanisms responsible for bladder cancer
metastasis remain poorly understood.
The metastatic process is associated with the
migration and invasion of tumor cells, which requires the
alteration of the extracellular matrix (ECM) and the basement
membrane by action of proteinases such as the matrix
metalloproteinases (MMPs) (7–9).
MMP-2 (gelatinase A) and MMP-9 (gelatinase B) are type IV
collagenases that degrade the ECM and the basement membrane,
resulting in the migration of tumor cells (7–9).
Previous studies suggest that MMP-9 expression is closely
correlated with the progression of muscle invasive bladder tumors
(10,11). The expression of MMP-9 was found
to be induced by various growth factors, growth factor receptors,
and cytokines via the binding of several transcription factors
including AP-1, Sp-1 and NF-κB (12–15). In addition, intra-cellular
signaling, such as the MAPK and Jak-Stat signaling pathways, is
known to be involved in the regulation of MMP-9 expression
(12,16).
Interleukin-28A (IL-28A) was originally identified
as a human IFN-λ2 protein (17,18), and is a member of the IL-10 gene
family (17,18). Important signaling pathways, such
as MAPK and Jak-Stat, are associated with IL-28A stimulation
(17,19). Moreover, IL-28AR1 is reportedly
expressed in cancers of the bladder, blood, breast, brain, head,
neck and lung (20). IL-28A
interacts with the IL-28AR1 receptor leading to many biological
activities including antiviral activity and inhibition of tumor
growth (17–22). However, the molecular mechanisms
of IL-28A in modulating the migration of human cancer cells remain
elusive.
Thus, we attempted to elucidate the exact roles and
molecular mechanisms of IL-28A in the migration of bladder cancer
cells. In the present study, we demonstrated that IL-28A induces
cell migration through p38 MAPK-dependent MMP-9 expression by
activating transcription factors NF-κB and AP-1 in UMUC-3 bladder
cancer cells.
Materials and methods
Materials
Polyclonal antibodies to ERK, phospho-ERK, p38 MAPK,
phospho-p38 MAPK, JNK, and phospho-JNK were obtained from Cell
Signaling Technology (Danvers, MA). Polyclonal antibodies to Jak1,
Jak2, Jak3, Stat1, Stat2, Stat3, Stat5, phospho-Jak1, phospho-Jak2,
phospho-Jak3, phospho-Stat1, phospho-Stat2, phospho-Stat3, and
phospho-Stat5 were purchased from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA). SB203580, piceatannol, and AG490 were obtained
from Calbiochem (San Diego, CA). The polyclonal MMP-9 antibody was
obtained from Chemicon. Dominant negative p38 MAPK (DN-p38) was
provided by Dr Roger Davis (University of Massachusetts Medical
School, Worcester, MA).
Cell cultures
A human bladder carcinoma cell line (UMUC-3) was
obtained from the American Type Culture Collection. The cells were
maintained in Dulbecco’s modified Eagle’s medium (DMEM) (4.5 g
glucose/l) supplemented with 10% fetal bovine serum (FBS),
L-glutamine, and antibiotics (Biological Industries, Beit Haemek,
Israel) at 37°C in a 5% CO2 humidified incubator.
RNA extraction
Total-RNA was isolated from tissue using TRIzol
reagent (Life Technologies, Grand Island, NY), according to the
manufacturer’s protocol. The quality and integrity of the RNA were
confirmed by agarose gel electrophoresis and ethidium bromide
staining, followed by visual examination under ultraviolet
light.
Real-time polymerase chain reaction
(PCR)
Real-time PCR assays using a Rotor-Gene 3000 PCR
system (Corbett Research, Mortlake, Australia) were performed in
the original and independent cohorts. GAPDH was analyzed in
parallel as an internal control. Real-time PCR reactions containing
primers and SYBR Premix Ex Taq (Takara Bio Inc., Otsu, Japan) were
carried out in micro-reaction tubes (Corbett Research). Spectral
data were captured and analyzed using Rotor-Gene Real-Time Analysis
Software 6.0 Build 14 (Corbett Research). For amplification,
IL-28A, sense, 5′-ACC GCT GAC ACT GAC CCA G-3′ and antisense,
5′-CAG CCA GGG GAC TCC TTT-3′ primers or IL-28AR1, sense, 5′-CAG
AAT GTG ACG CTG CTC TC-3′ and antisense, 5′-ATC CAG GTA TTC GGA CTC
CA-3′ primers were used. GAPDH was analyzed in parallel as an
endogenous RNA reference gene, and data were normalized to the
expression of GAPDH.
Immunoblot analysis
Growth-arrested cells were treated with IL-28A in
the absence of 10% FBS for various durations at 37°C. The cells
were then washed twice with cold phosphate-buffered saline (PBS)
and freeze-thawed in 250 μl lysis buffer (containing, in
mmol/l, HEPES [pH 7.5] 50, NaCl 150, EDTA 1, EGTA 2.5, DTT 1,
β-glycerophosphate 10, NaF 1, Na3VO4 0.1, and
phenylmethylsulfonyl fluoride 0.1 and 10% glycerol, 0.1% Tween-20,
10 g/ml of leupeptin, and 2 μg/ml of aprotinin), and then
scraped into 1.5-ml tubes. The lysates were placed on ice for 15
min and then centrifuged at 12,000 rpm for 20 min at 4°C. The
protein concentration of the supernatant was determined using a
Bradford reagent method (Bio-Rad). Equal amounts of cellular
proteins were resolved by electrophoresis on a 0.1% SDS-10%
polyacrylamide gel (SDS-PAGE) under denaturing conditions. The
proteins were transferred electrophoretically to nitrocellulose
membranes (Hybond; Amersham Corp., Arlington Heights, IL). After
blocking in 10 mmol/l Tris-HCl (pH 8.0), 150 mmol/l NaCl, and 5%
(wt/vol) nonfat dry milk, the membranes were treated with primary
antibodies for 90 min, followed by incubation with
peroxidase-conjugated secondary antibodies for 45 min. The
immunocomplexes were detected using a chemiluminescence reagent kit
(Amersham Corp). For the immunoblotting studies, the experiments
were repeated at least 3 times.
Wound-healing migration assay
Cells were plated on 6-well dishes and grown to 90%
confluence in 2 ml of growth medium. The cells were damaged using a
2-mm-wide tip and were then treated with IL-5. They were allowed to
migrate, and images were captured using an inverted microscope (x40
magnification).
MTT assay
The incorporation of
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich) was used to assess cell proliferation. Briefly,
5,000 cells/well were seeded on a 96-well plate. After serum
starvation for 24 h, the cells were treated with the indicated
concentrations of IL-28A and then further incubated for 4 days. MTT
(5 mg/ml) was added, and the medium was removed 4 h later. The
colored formazan product was resolved in 0.04 N HCl/isopropanol and
quantified by absorbance measurements at 570 nm.
Zymography
Conditioned medium was electrophoresed in a
polyacrylamide gel containing 1 mg/ml gelatin. The gel was then
washed at room temperature for 2 h with 2.5% Triton X-100 and then
soaked overnight at 37°C in a buffer containing 10 mM
CaCl2, 150 mM NaCl and 50 mM Tris-HCl, pH 7.5. The gel
was stained with 0.2% Coomassie blue and photographed on a light
box. Proteolysis was detected as a white zone on a dark blue field
(15).
Transfection
Cells were transfected with DN-p38 using
Lipofectamine 2000 transfection reagent according to the
manufacturer’s protocol (Invitrogen, Carlsbad, CA). After the
indicated incubation with IL-28A, the cells were studied via
immunoblotting, zymography, electrophoretic mobility shift assay
(EMSA), and wound-healing migration assay.
Nuclear extracts and EMSA
Cultured cells were collected by centrifugation,
washed, and suspended in a buffer containing 10 mM
4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES) (pH
7.9), 10 mM KCl, 0.1 mM EDTA, 0.1 mM EGTA, 1 mM DTT and 0.5 mM
PMSF. After 15 min on ice, the cells were vortexed in the presence
of 0.5% Nonidet NP-40. The nuclear pellet was then collected by
centrifugation and extracted in a buffer containing 20 mM HEPES (pH
7.9), 0.4 M NaCl, 1 mM EDTA, 1 mM EGTA, 1 mM DTT and 1 mM PMSF for
15 min at 4°C.
The nuclear extract (10–20 μg) was
preincubated at 4°C for 30 min with a 100-fold excess of an
unlabeled oligonucleotide spanning the −79 MMP-9 cis element
of interest. The sequences were as follows: AP-1,
CTGACCCCTGAGTCAGCACTT; NF-κB, CAGTGGAATTCCCCAGCC; Sp-1, GCCCATTC
CTTCCGCCCCCAGATGAAGCAG. The reaction mixture was then incubated at
4°C for 20 min in a buffer (25 mM HEPES buffer pH 7.9, 0.5 mM EDTA,
0.5 mM DTT, 0.05 M NaCl, and 2.5% glycerol) with 2 μg of
poly dI/dC and 5 fmol (2x104 cpm) of a Klenow
end-labeled (32P-ATP) 30-mer oligonucleotide, which
spanned the DNA binding site in the MMP-9 promoter. The reaction
mixture was electrophoresed at 4°C in a 6% polyacrylamide gel using
a TBE (89 mM Tris, 89 mM boric acid and 1 mM EDTA) running buffer.
The gel was rinsed with water, dried, and exposed to X-ray film
overnight (15).
Statistical analysis
Where it was appropriate, data were expressed as the
mean ± SE. Data were analyzed by factorial ANOVA and Fisher’s least
significant difference test where appropriate. Statistical
significance was set at P<0.05.
Results
Expression of IL-28A and IL-28AR1 in
UMUC-3 bladder cancer cells
To determine the expression levels of IL-28A and its
receptor IL-28AR1, real-time PCR assay was performed on bladder
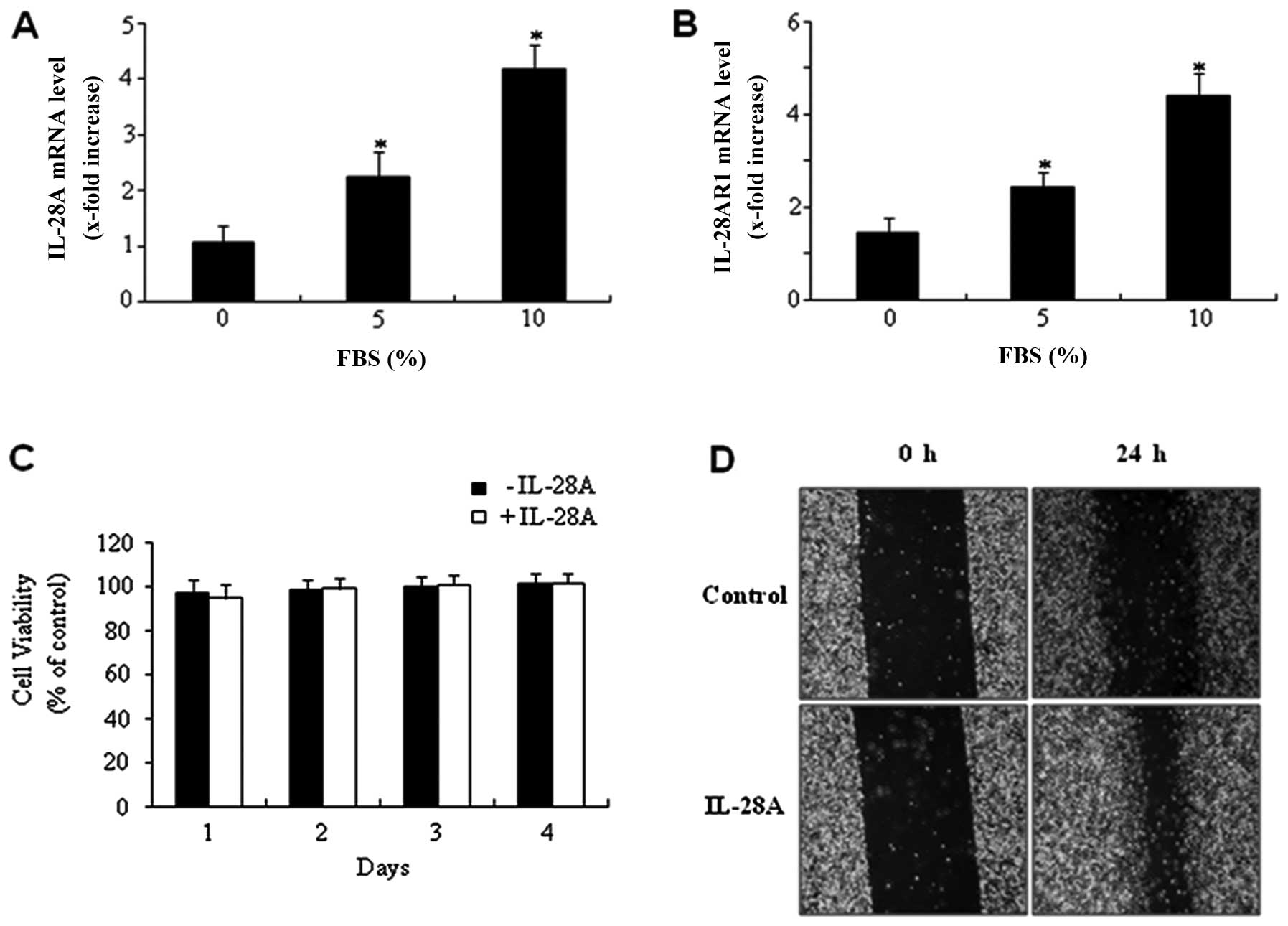
cancer UMUC-3 cells. Expression of IL-28A mRNA was detected in
UMUC-3 cells (Fig. 1A). In
addition, IL-28AR1 mRNA was also found to be expressed in UMUC-3
cells (Fig. 1B). Exposure of
UMUC-3 cells to 10% FBS for 24 h markedly increased the IL-28A mRNA
expression compared with the non-treated cells (Fig. 1A). A similar result was observed
in the expression level of IL-28AR1 mRNA (Fig. 1B). These results indicated that
both IL-28A and IL-28AR1 mRNA were expressed in the UMUC-3 bladder
cancer cells.
IL-28A induces migration of UMUC-3
bladder cancer cells
MTT assay was used to assess the proliferative
ability of IL-28A. IL-28A treatment did not affect the levels of
cell proliferation (Fig. 1C).
Subsequently, a wound-healing migration assay was carried out to
determine whether IL-28A induces cell migration. Monolayer UMUC-3
cells were scratched, and the wounds were allowed to heal for 24 h.
Cell migration ability at the wound front was analyzed and compared
to the initial wound state. UMUC-3 cells treated with IL-28A for 24
h showed a significant increase in cell migration (Fig. 1D). These results suggest that
IL-28A induces migration of UMUC-3 cells in vitro.
IL-28A stimulates MMP-9 expression via
binding activation of NF-κB and AP-1
Since cell migration is associated with matrix
degradation by MMPs (7–9), we next examined the expression of
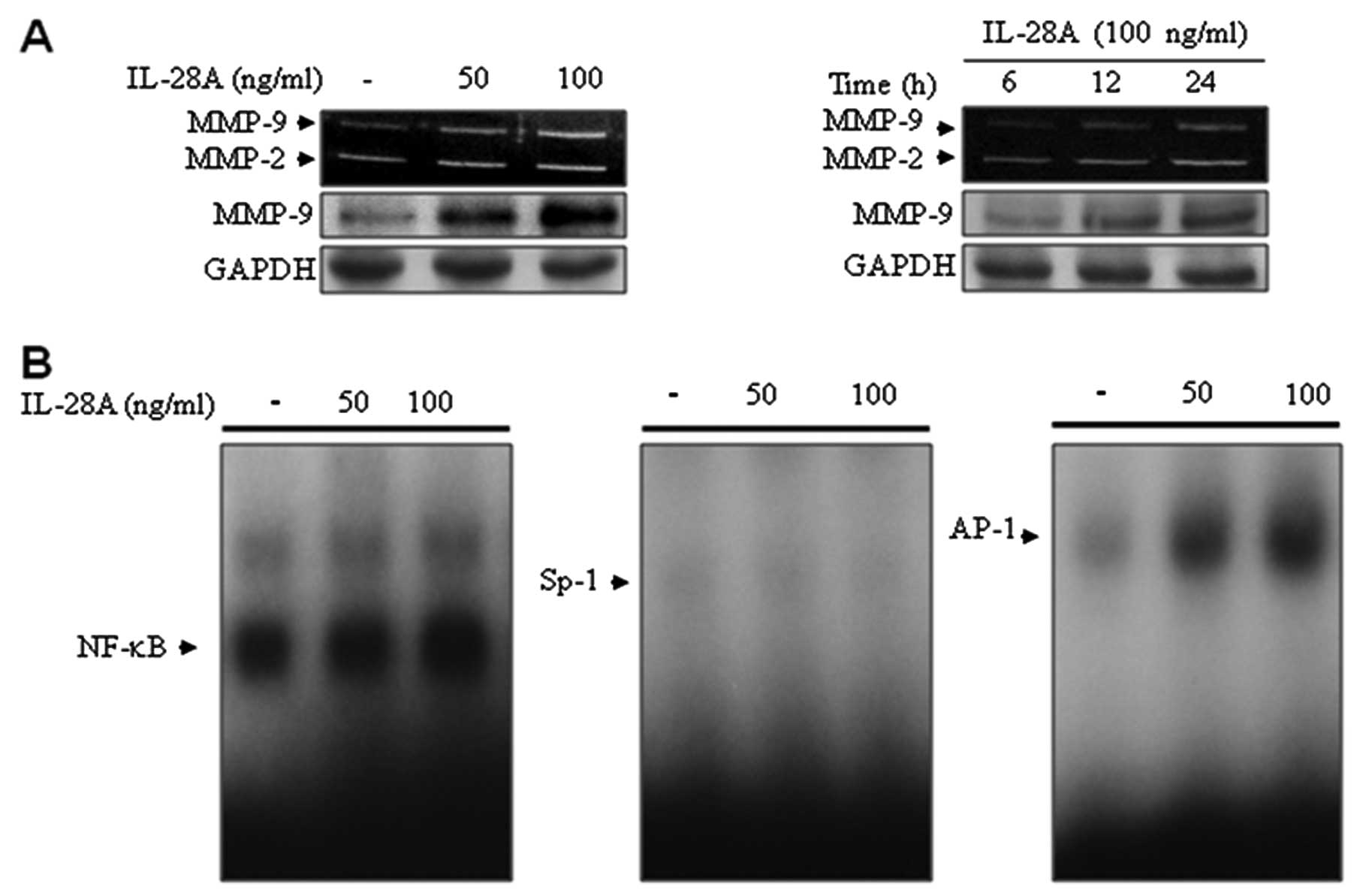
MMPs in the UMUC-3 cell culture supernatants using zymographic
analysis (Fig. 2A). To determine
the effect of IL-28A on the expression of MMPs, cells were treated
with IL-28A at the indicated concentrations for the indicated
times. Treatment with IL-28A induced MMP-9 expression in UMUC-3
cells in a concentration- and a time-dependent manner (Fig. 2A). These results were confirmed by
immunoblot analysis using an antibody that detects a band of MMP-9
(Fig. 2A). In addition, MMP-2
levels were also elevated in UMUC-3 cells following IL-28A
treatment (Fig. 2A). Previous
studies showed that enhanced expression of MMP-9 is directly
correlated with the progression of bladder cancer characterized by
increased tumor grade, invasion and metastasis (10,11). Therefore, we focused on MMP-9
regulation induced by IL-28A in UMUC-3 cells. To do this, we
investigated MMP-9 regulation at the transcriptional level using
transcription factors NF-κB, AP-1 and Sp-1, all of which were
located in the 5′ flanking region of the MMP-9 gene (12–15). Our results from EMSA showed that
IL-28A treatment enhanced binding activation of NF-κB and AP-1
(Fig. 2B). However, no specific
binding activation of Sp-1 was detected in the IL-28A-induced
UMUC-3 cells (Fig. 2B). These
results demonstrate that IL-28A induced MMP-9 expression through
binding motifs of the transcription factors NF-κB and AP-1.
IL-28A induces the activation of p38 MAPK
and Jak2-Stat2 signaling
To determine the signaling pathway in IL-28A-treated
UMUC-3 cells, we examined the MAPK signaling pathway. Treatment
with IL-28A significantly induced activation of p38 MAPK in UMUC-3
cells (Fig. 3A). Pretreatment
with SB203580 inhibited IL-28A-stimulated activation of p38 MAPK
(Fig. 3D). In contrast,
activation of both ERK1/2 and JNK was not affected in
IL-28A-treated cells (Fig. 3A).
We also observed the effects of IL-28A on the activation of
Jak-Stat signaling. IL-28A treatment increased activation of Jak1,
Jak2, and Stat2. However, activation of Jak3, Stat1, Stat3 and
Stat5 was not affected by IL-28A treatment (Fig. 3B and C). To confirm the activation
of Jak1 and Jak2 signaling, we used Jak1 inhibitor (piceatannol)
and Jak2 inhibitor (AG490). The cells were pretreated with
piceatannol or AG490, respectively, and then further incubated with
IL-28A treatment for 10 min. IL-28A-induced activation of Jak1 was
blocked by piceatannol (Fig. 3D).
AG490 also abolished activation of Jak2 in IL-28A-treated UMUC-3
cells (Fig. 3D). Next, we further
investigated whether Jak1 or Jak2 was involved in IL-28A-induced
Stat2 activation. Inhibition of Jak2 by AG490 suppressed the Stat2
activation that was induced by IL-28A (Fig. 3E). However, piceatannol treatment
had no apparent effect on the IL-28A-stimulated activation of Stat2
(Fig. 3E). These results suggest
that p38 MAPK and Jak2-Stat2 signaling are involved in
IL-28A-treated UMUC-3 cells.
 | Figure 3IL-28A induces activation of p38 MAPK
and Jak2-Stat2 signaling. (A–C) After serum starvation for 24 h,
cells were incubated with IL-28A (100 ng/ml) for the indicated time
intervals, and activation of ERK1/2, p38 MAPK, JNK, Jak1, Jak2,
Jak3, Stat1, Stat2, Stat3 and Stat5 was determined by immunoblot
analysis. (D and E) After serum starvation for 24 h, the cells were
pretreated with p38 MAPK inhibitor SB203580, JAK1 inhibitor
piceatannol, and Jak2 inhibitor AG490 for 40 min prior to treatment
with IL-28A (10 ng/ml), and were then further incubated for 10 min.
Activation of p38 MAPK, Jak1, Jak2 and Stat2 was analyzed by
immunoblotting. The results represent three independent
experiments. |
Involvement of SB203580, a p38 MAPK
inhibitor, in wound-healing migration, MMP-9 expression, and
binding activities of NF-κB and AP-1 in IL-28A-treated UMUC-3
cells
We next investigated which signaling pathway, p38
MAPK and/or Jak2, mediates the enhanced migration of IL-28A-treated
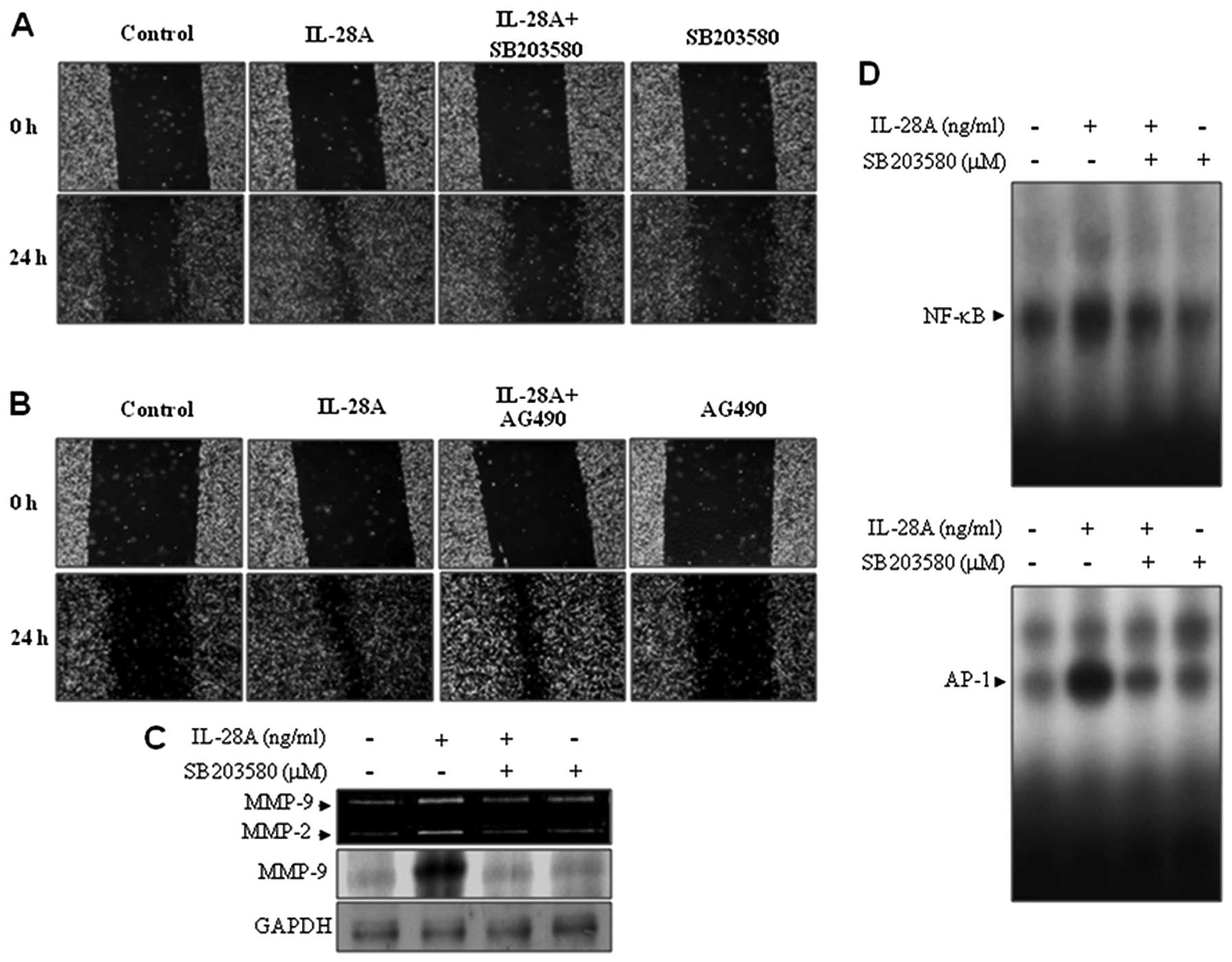
UMUC-3 cells. Pretreatment of cells with SB203580 inhibited the
IL-28A-induced migration of UMUC-3 cells (Fig. 4A). However, the addition of AG490
had no effect on the cell migration induced by IL-28A (Fig. 4B). These results indicate that p38
MAPK signaling may contribute to the induction of UMUC-3 cell
migration. The results obtained above prompted us to examine
whether the p38 MAPK signaling pathway is involved in
IL-28A-mediated MMP-9 regulation. UMUC cells were treated with
SB203580 in the presence or absence of IL-28A. The results from
zymographic and immunoblot analyses showed that pretreatment with
SB203580 significantly attenuated IL-28A-induced MMP-9 expression
(Fig. 4C). To analyze the role of
p38 MAPK in the transcriptional mechanism of MMP-9 in
IL-28A-treated cells, we performed EMSA on the NF-κB and AP-1
binding sites from the MMP-9 promoter using nuclear extracts.
SB203580 treatment inhibited the induction of binding activities of
NF-κB and AP-1 (Fig. 4D). These
results demonstrate that p38 MAPK signaling is involved in cell
migration and MMP-9 expression via the binding activities of NF-κB
and AP-1 in IL-28A-treated UMUC-3 cells.
Effect of a dominant-negative p38 MAPK
mutant gene on wound-healing migration, MMP-9 expression, and
binding activities of NF-κB and AP-1 in UMUC-3 cells
To confirm the direct roles of p38 MAPK in
IL-28A-mediated cellular responses, DN-p38, a dominant-negative p38
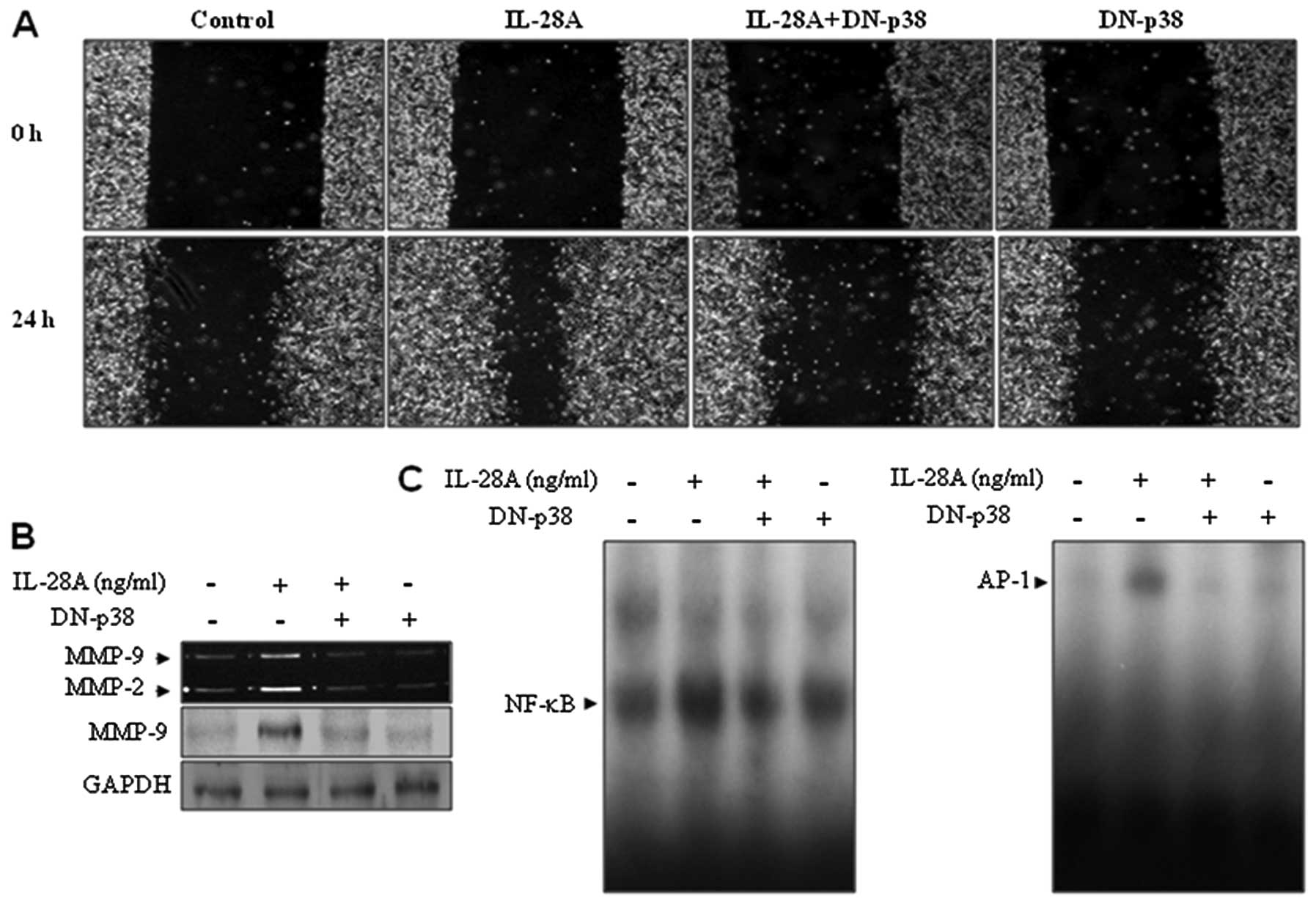
MAPK mutant gene, was tested. To this end, UMUC-3 cells were
transfected with DN-p38 plasmid and then treated with IL-28A.
DN-p38 transfectant inhibited the wound-healing migration of UMUC-3
cells induced by IL-28A (Fig.
5A). In addition, IL-28A-induced MMP-9 expression was
suppressed in the DN-p38-transfected cells (Fig. 5B). Finally, overexpression of
DN-p38 almost abolished increased NF-κB and AP-1 DNA binding
activities in the presence of IL-28A (Fig. 5C). Together, these results
indicate that p38 MAPK signaling is required for cell migration via
MMP-9 expression by inducing activation of NF-κB and AP-1 binding
in IL-28A-treated UMUC-3 cells.
Discussion
There is much evidence linking tumor progression and
development to inflammatory cytokines that are produced by tumor
cells (23,24). The main inflammatory cytokines
include IL-1β, IL-6, IL-23 and TNF-α, which are also associated
with the tumorigenic and metastatic potential of several cancer
cells (23,24). Previous studies have demonstrated
that pro-inflammatory cytokines such as TNF-α, IL-6 and IL-8 are
putative mediators of metastasis in bladder cancer cells (2,25–28). However, there has been no report
of the functional role of IL-28A in tumor migration. In the present
study, we report the role of IL-28A in the migration of UMUC-3
bladder cancer cells.
IL-28A is a human IFN-λ2 protein with
pro-inflammatory and antiviral activity (17,18). Despite all of these functions, the
migratory mechanisms of IL-28A produced by tumor cells are unclear.
In this study, we found that UMUC-3 bladder cancer cells produced
IL-28A and its receptor IL-28AR1 as determined by real-time PCR. We
next examined whether IL-28A is involved in the migration of
bladder cancer cells. The exogenous IL-28A enhanced in vitro
migration of UMUC-3 cells without altering cell proliferation,
suggesting that IL-28A induces bladder cancer cell migration. These
results contradict other reports which have suggested that IL-28A
exhibits an antitumor effect against B16 melanoma and murine
fibrosarcoma (21,22). Further study is required to verify
the in vivo efficacy of the IL-28A gene in bladder tumor
migration.
MMPs degrade various components of the extracellular
matrix (ECM) and basement membrane proteins that are considered to
be essential in tumor migration (7–9).
Gelatinases (MMP-2 and -9) have been connected to the metastatic
potential of malignant tumors (7–9).
The expression of MMP-9 is directly correlated with bladder tumor
progression characterized by increased tumor grade, invasion and
metastasis (10,11). Our results showed that exposure to
IL-28A induced MMP-9 expression in UMUC-3 cells. The MMP-9 promoter
contains a proximal AP-1 element, an upstream Sp-1, and a NF-κB
binding motif in tumor cell lines (12–15). Previous reseach has demonstrated
that the transcription factors NF-κB, AP-1 and Sp-1 are associated
with the regulation of TNF-α-stimulated MMP-9 in bladder cancer
5637 and HT1376 cells (27,28). However, the identification of
cis elements in the induction of MMP-9 in response to IL-28A
in bladder cancer is unclear. The results from EMSA indicated that
NF-κB and AP-1 binding sites within the MMP-9 promoter region are
crucial for IL-28A-stimulated MMP-9 expression. These data suggest
that IL-28A may promote cell migration through the induction of
MMP-9 by activating NF-κB and AP-1 binding to the promoter.
Inflammatory cytokine signals of IL-28A have been
shown to induce activation of Stat1, Stat2 and Stat3 in HT-29 cells
expressing IL-28AR1 (17). In
addition, IL-28A treatment was found to decrease cell proliferation
in colon cancer cell lines, which was followed by activated
signaling of ERK1/2, JNK, AKT and Stat-1 (19). We examined the signaling pathway
induced by IL-28A in UMUC-3 bladder cancer cells. The addition of
IL-28A induced activation of p38 MAPK and Jak2-Stat2 signaling in
UMUC-3 cells. Several signaling pathways, ncluding p38 MAPK and
Jak-Stat signaling, are known to be involved in cell migration and
MMP-9 regulation in various cell types (12,16). Our results showed that
pretreatment with the p38 MAPK inhibitor, SB203580, significantly
blocked IL-28A-induced migration and MMP-9 expression. However,
unexpectedly, inhibition of cell migration by Jak2 inhibitor AG490
was not observed in IL-28A-treated UMUC-3 cells. We also found that
the involvement of p38 MAPK in IL-28A-induced migration and MMP-9
expression was revealed by transfection with DN-p38. Finally, we
identified the transcription factors that are involved in the p38
MAPK-mediated control of MMP-9 expression in UMUC-3 cells in the
presence of IL-28A using SB203580 and the DN-p38 plasmid. The
present study revealed that the ability of SB203580 and DN-p38 to
reduce MMP-9 expression in IL-28A-treated UMUC-3 cells was
accomplished by suppressing NF-κB and AP-1 binding. Collectively,
our results demonstrated that p38 MAPK signaling by IL-28A
participated in MMP-9 expression by activating NF-κB and AP-1
binding resulting in the degradation of ECM and leading to the
migration of bladder cancer cells.
Cytokines have the potential to stimulate the
proliferation and migration of malignant cells. However, the
relationship between cytokines and tumor progression has been
controversial (23,24). In recent years, many researchers
have studied molecular mechanisms by which cytokines may act to
promote tumor development. In the present study, we demonstrated
that IL-28A induces the migration of bladder cancer cells via the
degradation of ECM components by proteolytic enzymes such as type
IV collagenase. Previous studies have suggested an opposing role
for IL-28A, i.e., that it exhibits an antitumor effect in tumor
systems in vitro and in vivo (21,22). Future study is required to clarify
the biological action of IL-28A on different tumor cells.
In conclusion, we introduced a novel mechanism by
which p38 MAPK is responsible for IL-28A-induced migration of
UMUC-3 bladder cancer cells, which is associated with MMP-9
expression via the binding activities of NF-κB and AP-1. Therefore,
IL-28A is a potential therapeutic target for suppressing the
metastasis of bladder tumor cells.
Acknowledgements
This research was supported by the
Basic Science Research Program through the National Research
Foundation of Korea (NRF), funded by the Ministry of Education,
Science and Technology (2012-0000482). This research was also
supported by a grant from the Academic Research Program of Korea
National University of Transportation in 2012.
References
|
1.
|
A JemalR SiegelE WardE MurrayT XuMJ
ThunCancer statisticsCA Cancer J Clin5743662007
|
|
2.
|
PC BlackCP DinneyBladder cancer
angiogenesis and metastasis - translation from murine model to
clinical trialCancer Metastasis
Rev26623634200710.1007/s10555-007-9084-917726580
|
|
3.
|
MC MettsJC MettsSJ MilitoCR Thomas
JrBladder cancer: a review of diagnosis and managementJ Natl Med
Assoc92285294200010918764
|
|
4.
|
JM CrawfordThe origins of bladder
cancerLab Invest88686693200810.1038/labinvest.2008.4818475256
|
|
5.
|
NM HeneyK ProppeGR ProutPP GriffinWU
ShipleyInvasive bladder cancer: tumor figuration, lymphatic
invasion and survivalJ Urol13089589719836632095
|
|
6.
|
AP MitraCC BartschRJ CoteStrategies for
molecular expression profiling in bladder cancerCancer Metastasis
Rev28317326200910.1007/s10555-009-9196-519997771
|
|
7.
|
WG Stetler-StevensonS AznavoorianLA
LiottaTumor cell interactions with the extracellular matrix during
invasion and metastasisAnnu Rev Cell
Biol9541573199310.1146/annurev.cb.09.110193.0025458280471
|
|
8.
|
LM MatrisianMetalloproteinases and their
inhibitors in matrix remodelingTrends
Genet6121125199010.1016/0168-9525(90)90126-Q2132731
|
|
9.
|
J WestermarckVM KahariRegulation of matrix
metalloproteinase expression in tumor invasionFASEB
J13781792199910224222
|
|
10.
|
B DaviesJ WaxmanH WasanP AbelG WilliamsT
KrauszD NealD ThomasA HanbyF BalkwillLevels of matrix
metalloproteases in bladder cancer correlate with tumor grade and
invasionCancer Res535365536919938221672
|
|
11.
|
FJ BiancoDC Gervasi JrR TiguertDJ
GrignonJE PontesJD CrissmanR FridmanDP Wood JrMatrix
metalloproteinase-9 expression in bladder washes from bladder
cancer patients predicts pathological stage and gradeClin Cancer
Res43011301619989865914
|
|
12.
|
OR MookWM FrederiksCJ Van NoordenThe role
of gelatinases in colorectal cancer progression and
metastasisBiochim Biophys Acta17056989200415588763
|
|
13.
|
M BondP RosalindP FabunmiAH BakerAC
NewbySynergistic upregulation of metalloproteinase-9 by growth
factors and inflammatory cytokines: an absolute requirement for
transcription factor NF-kappa BFEBS
Lett4352934199810.1016/S0014-5793(98)01034-59755853
|
|
14.
|
H SatoM SeikiRegulatory mechanism of 92
kDa type IV collagenase gene expression which is associated with
invasiveness of tumor cellsOncogene839540519938426746
|
|
15.
|
SK MoonBY ChaCH KimERK1/2 mediates
TNF-alpha-induced matrix metalloproteinase-9 expression in human
vascular smooth muscle cells via the regulation of NF-kappaB and
AP-1: Involvement of the ras dependent pathwayJ Cell
Physiol198417427200410.1002/jcp.1043514755547
|
|
16.
|
S KimJH ChoiHI LimSK LeeWW KimS ChoJS
KimJH KimJH ChoeSJ NamEGF-induced MMP-9 expression is mediated by
the JAK3/ERK pathway, but not by the JAK3/STAT-3 pathway in a SKBR3
breast cancer cell lineCell
Signal21892898200910.1016/j.cellsig.2009.01.03419385051
|
|
17.
|
SV KotenkoG GallagherVV BaurinA
Lewis-AntesM ShenNK ShahJA LangerF SheikhH DickensheetsRP
DonnellyIFN-λs mediate antiviral protection through a distinct
class II cytokine receptor complexNat Immunol469772003
|
|
18.
|
P SheppardW KindsvogelW XuK HendersonS
SchlutsmeyerTE WhitmoreR KuestnerU GarriguesC BirksJ RorabackIL-28,
IL-29 and their class II cytokine receptor IL-28RNat
Immunol46368200310.1038/ni87312469119
|
|
19.
|
S BrandF BeigelT OlszakK ZitzmannST
EichhorstJM OtteJ DieboldH DiepolderB AdlerCJ AuernhammerIL-28A and
IL-29 mediate antiproliferative and antiviral signals in intestinal
epithelial cells and murine CMV infection increases colonic IL-28A
expressionAm J Physiol Gastrointest Liver
Physiol289G960G968200510.1152/ajpgi.00126.2005
|
|
20.
|
L YangY LuoJ WeiS HeIntegrative genomic
analyses on IL28RA, the common receptor of interferon-λ1, -λ2 and
-λ3Int J Mol Med25807812201020372826
|
|
21.
|
A LasfarA Lewis-AntesSV SmirnovS AnanthaW
AbushahbaB TianK ReuhlH DickensheetsF SheikhRP
DonnellyCharacterization of the mouse IFN-λ ligand-receptor system:
IFN-λs exhibit antitumor activity against B16 melanomaCancer
Res66446844772006
|
|
22.
|
M NumasakiM TagawaF IwataT SuzukiA
NakamuraM OkadaY IwakuraS AibaM YamayaIL-28 elicits antitumor
responses against murine fibrosarcomaJ
Immunol17850865098200710.4049/jimmunol.178.8.508617404291
|
|
23.
|
LM CoussensZ WerbInflammation and
cancerNature420860867200210.1038/nature0132212490959
|
|
24.
|
A MantovaniP AllavenaA SicaF
BalkwillCancer-related
inflammationNature454436444200810.1038/nature07205
|
|
25.
|
M OkamotoK HattoriR OyasuInterleukin-6
functions as an autocrine growth factor in human bladder carcinoma
cell lines in vitroInt J
Cancer72149154199710.1002/(SICI)1097-0215(19970703)72:1%3C149::AID-IJC21%3E3.0.CO;2-D9212236
|
|
26.
|
K InoueJW SlatonSJ KimP PerrotteBY EveM
Bar-EliR RadinskyCP DinneyInterleukin 8 expression regulates
tumorigenicity and metastasis in human bladder cancerCancer
Res6022902299200010786697
|
|
27.
|
SJ LeeSS ParkYH ChoK ParkEJ KimKH JungSK
KimWJ KimSK MoonActivation of matrix metalloproteinase-9 by TNF-α
in human urinary bladder cancer HT1376 cells: The role of MAP
kinase signaling pathwaysOncol Rep19100710132008
|
|
28.
|
SJ LeeSS ParkUS LeeWJ KimSK MoonSignaling
pathway for TNF-alpha-induced MMP-9 expression: mediation through
p38 MAP kinase, and inhibition by anti-cancer molecule magnolol in
human urinary bladder cancer 5637 cellsInt
Immunopharmacol818211826200810.1016/j.intimp.2008.08.01818801463
|