Introduction
Human immunodeficiency virus type 1 (HIV-1) is the
etiologic agent of acquired immune deficiency syndrome. The HIV-I
genome encodes only nine genes, three structural genes and six
accessory genes. Because of its limited genetic capacity, the virus
requires many cellular proteins and cellular pathways to complete
its life cycle. Recent functional genome-scale RNAi screening and
computational analysis have identified more than 300 host
co-factors that are critical for HIV-1 replication in human cells
(1–3). However, cells have evolved
strategies to impede HIV-1 replication, and several innate cellular
restriction factors have been found that may target a number of
steps in the virus’s replication cycle (4,5).
This research suggests that intricate ‘strike-counterstrike’
protein interactions between the virus and the host cell govern the
ultimate outcome of HIV-1 infection.
An essential step in the replication of HIV-1 is the
integration of reverse-transcribed viral cDNA into the chromosome
of the host cell. The key protein responsible for the integration
process is the 32 kDa viral integrase. The catalytic function of
the integrase is essential for the stable maintenance of the viral
genome and the persistence of HIV-1 infection. For these reasons,
the integrase has been the target of intensive pharmacological
research (6). The integration
event is a complex process that is aided by an ever-expanding list
of cellular proteins (7).
Intriguingly, few innate cellular restriction factors that target
the integration process and restrict viral replication have been
reported.
The small ubiquitin-related modifiers are small
polypeptides of approximately 8–11 kDa that were identified as
reversible post-translational protein modifiers. They are
covalently linked as 93–97 amino acid polypeptides to specific
lysine residues of various intracellular proteins (8,9).
The process of SUMOylation is analogous to ubiquitin modification
and occurs in three steps that are catalyzed by enzymatic machinery
including the SUMO-activating enzyme E1, the conjugating E2 enzyme
Ubc9 and various SUMO E3 ligases (10). SUMOylation regulates the function
of the substrates mainly by altering their intracellular
localization or protein-protein interactions or by affecting their
ability to undergo other types of post-translational modifications.
These changes, in turn, affect nuclear trafficking, gene
expression, genomic stability, chromosomal integrity and signal
transduction (11).
In the present study, we report that the
overexpression of SUMO1/SUMO2 and Ubc9 changes the intracellular
localization of HIV-1 integrase (HIV-1 IN). We also identified
SUMO1, SUMO2 and Ubc9 as HIV-1 IN-binding proteins and evaluated
the effects of these proteins on the integration of lentivirus and
HIV-1.
Materials and methods
Cell culture and transfection
293T cells were purchased from American Type Culture
Collection (ATCC) and cultured in Dulbecco’s modified Eagle’s
medium (DMEM) supplemented with 10% fetal calf serum (FBS) at 37°C
in a 5% CO2 humidified atmosphere. Plasmid DNAs or
siRNAs were transfected into 293T cells using Lipofectamine 2000
(Invitrogen) according to the manufacturer’s protocol. H9 cells and
HIV-1/IIIB were obtained from the National Institutes of Health
(NIH) AIDS Research and Reference Reagent Program (USA). H9 cells
were cultured in RPMI-1640 medium with 2 mM L-glutamine (Hyclone),
10% FBS and 100 U/ml of penicillin and streptomycin at 37°C in 5%
CO2 humidified atmosphere. Electrotransfection method
was used for H9 cells.
Plasmid construction
The open reading frames (ORFs) of SUMO1, SUMO2 and
Ubc9 were amplified by PCR from a human fetal brain cDNA library
(Clontech Laboratories). The ORFs were inserted into the plasmid
pCMV-HA (Clontech Laboratories) to generate the three expression
plasmids HA-SUMO1, HA-SUMO2 and HA-Ubc9. Two truncation mutants,
HA-SUMO1ΔC6 and HA-SUMO2ΔC4, were created by PCR-based C-terminal
deletion of 6 amino acids from SUMO1 and 4 amino acids from SUMO2,
respectively. For yeast-mating tests, pB42AD-SUMO1, pB42AD-SUMO2
and pB42AD-Ubc9 were generated by inserting the ORFs of SUMO1,
SUMO2 and Ubc9, respectively, into the pB42AD plasmid (Clontech
Laboratories). The plasmids pB42ADSUMO1ΔC6, pB42AD-SUMO1ΔC4,
pB42AD-SUMO2ΔC4 and pB42AD-SUMO2ΔC2 were constructed by PCR-based
C-terminal deletion as mentioned above. The bait plasmid
pLexA-HIV-1 IN has been described previously (12). To generate pDsRed-SUMO1,
pDsRed-SUMO2 and pDsRed-Ubc9, the ORFs of SUMO1, SUMO2 and Ubc9
were subcloned into pDsRed-C1 (Clontech Laboratories). The
pEGFP-HIV-1 IN has been described previously (13).
Intracellular localization assay
293T cells were plated on coverslips in 6-well
plates and transfected with the indicated plasmids. At 24 h
post-transfection, cells were visualized using an Olympus LX70
microscope. The nuclei were stained with DAPI
(4′,6-diamidino-2-phenylindole).
Yeast two-hybrid assay
The MatchMarker LexA two-hybrid system was purchased
from Clontech Laboratories. Interactions between HIV-1 IN and
SUMO1, SUMO2, Ubc9 and their derivatives were detected using
pB42AD-SUMO1, pB42AD-SUMO1ΔC6, pB42AD-SUMO1ΔC4, pB42ADSUMO2,
pB42AD-SUMO2ΔC4, pB42AD-SUMO2ΔC2 and pB42AD-Ubc9-transferred EGY48
(p8opLacZ) and pLexAHIV-1 IN-transferred YM4271 according to the
standard yeast-mating protocol provided by the manufacturer.
Immunoblotting, co-immunoprecipitation
and antibodies
To prepare whole cell extracts, transfected 293T
cells were lysed with ice-cold RIPA buffer (25 mM Tris-HCl pH 7.5,
150 mM NaCl, 1.0% NP-40, 0.5% sodium deoxycholate and 0.1% SDS)
supplemented with PMSF, a protease inhibitor cocktail (Sigma) and
N-ethylmaleimide (Sigma). Lysates were clarified by centrifugation
and stored at −80°C. For co-immunoprecipitation, cell lysates were
prepared in lysis buffer (20 mM Tris-HCl pH 7.5, 100 mM NaCl, 0.5%
NP-40, 0.5 mM EDTA, 0.5 mM PMSF and 0.5% protease inhibitor
cocktail). The lysates were incubated with the indicated antibodies
for 2 h at 4°C. Pre-washed (25 μl) protein A/G Sepharose beads
(Santa Cruz Biotechnology, Inc.) were added to each extract, and
the mixtures were shaken overnight. The beads were washed three
times with lysis buffer and boiled in 2X loading buffer. Protein
samples were then separated on SDS-PAGE and transferred to a
nitrocellulose membrane; the membrane was blocked in 5% skim milk
in TBST and probed with the indicated antibodies. EGFP mouse
monoclonal antibodies and rabbit polyclonal anti-HA antibodies were
purchased from Santa Cruz Biotechnology, Inc.; HRP-conjugated
anti-rabbit IgG and anti-mouse IgG were obtained from Sigma.
Virus stock production and infectivity
assay
Lentiviral particles were produced by
co-transfection of 293T cells using a three-plasmid system as
previously described (12).
Virus-containing cell supernatants were harvested at 48 h
post-transfection, clarified by low-speed centrifugation, filtered
through 0.45 μm pore size filters and treated with DNase I (0.2
U/μl) for 1 h at 37°C. The viral stocks were normalized for
p24CA antigen content by ELISA using the HIV p24
Lentivirus Titer kit (Cell Biolabs, Inc., USA) following the
manufacturer’s instructions. Percentages of EGFP-expressing cells,
which represented the integration rate of the virus, were
determined at 2 days post-infection using a flow cytometer (FASC
Vantage SE).
Replication competent HIV-1/IIIB viruses were
obtained from the NIH AIDS Research and Reference Reagent Program.
For transfection, 107 H9 cells were electroporated with
20 μg plasmid DNA or 2 μM siRNA, using a Bio-Rad gene pulser with a
voltage of 300 V, a capacitance of 250 μF (14,15). After 24 h (for transfection of
plasmid DNA) or 48 h (for transfection of siRNA), cells were washed
with PBS and used for infection with HIV-1/IIIB at moi of 0.01 and
0.1.
Real-time PCR assay
To measure relative levels of lentiviral and
spreading HIV-1 integration, genomic DNA was quantified by Alu-LTR
real-time nested PCR array using a SYBR-Green-based detection kit
(Toyobo code no. QPK-201, 201T) (16). At 48 h post-infection, cellular
genomic DNA was extracted with a genomic DNA purification kit
(Qiagen). The primers used for the first round of PCR were
5′-GGCTAACTAGGGAACCCAC TG-3′, 5′-TCCCAGCTACTGGGGAGGCTGAGG-3′ and
5′-GCCTCCCAAAGTGCTGGGATTACAG-3′. After an initial denaturation at
95°C for 8 min, 12 cycles of denaturing (95°C, 30 sec), annealing
(55°C, 30 sec) and extension (72°C, 170 sec) were carried out. One
aliquot (1/50) of the initial PCR product was subjected to a second
round of PCR amplification. The primers used were
5′-GCTAGAGATTTTCCACACT GACTAA-3′ and 5′-GGCTAACTAGGGAACCCACTG-3′,
and a 100 bp fragment was obtained. A pair of primers for β-actin,
5′-ACGAGGCCCAGAGCAAGAG-3′ and 5′-TCTC CATGTCGTCCCAGTTG-3′, was used
as a control. Ct values were collected, and the relative viral
integration levels of the samples were calculated.
To measure relative levels of total viral cDNA
synthesis and 2-LTR circle formation, total genomic DNA was
extracted using the urea lysis method (17) and quantified by real-time PCR
using previously described primer sets. Primers M667/M661 (18) were used to amplify full-length
reverse transcripts, and primers 9600/515 (19) were used to amplify 2-LTR
circles.
RNA interference
Sense sequences of siRNA duplexes specific for human
SUMO1, SUMO2 and Ubc9 were 5′-CUGGGA AUGGAGGAAGAAG-3′ (20), 5′-GUCAAUGAGGCAGA UCAGA-3′
(21) and
5′-CAAAAAAUCCCGAUGGCAC-3′ (22),
respectively. A nontargeting siRNA was used as a negative control
(NC). The siRNAs were synthesized by RiboBio Co., Ltd. (Guangzhou,
China). The cells were used for viral infectivity assays 48 h after
transfection.
Statistical analysis
Data are described using the mean and standard
deviation of the mean where appropriate. Differences between the
means of experimental groups were analyzed using a two-tailed
Student’s t-test. Differences with a p-value of ≤0.05 were
considered significant.
Results
Overexpression of SUMOs and Ubc9 changes
the intracellular localization of HIV-1 IN
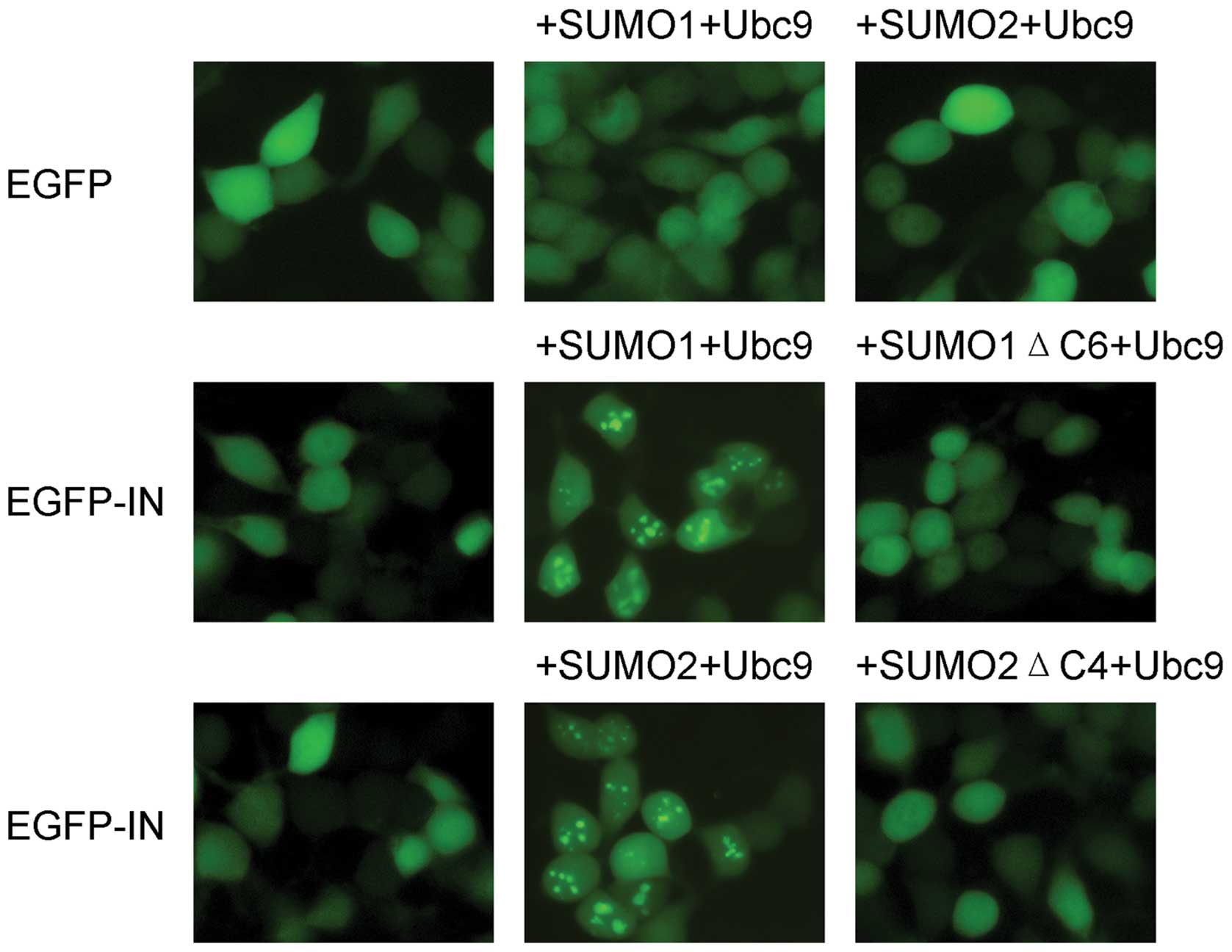
To investigate the subcellular distribution of HIV-1
IN in the presence of SUMO-related proteins, pEGFP-HIV-1 IN,
HA-SUMO1/SUMO2 and HA-Ubc9 were simultaneously introduced into 293T
cells, and the intracellular localization of pEGFP-HIV-1 IN was
observed by fluorescence microscopy. The truncated mutants SUMO1ΔC6
and SUMO2ΔC4, both of which lack SUMO conjugation activity, were
tested in parallel. When expressed alone, the EGFP-HIV-1 IN fusion
protein was diffusely distributed throughout the cell (Fig. 1). However, when EGFP-HIV-1 IN was
co-expressed with SUMO1/SUMO2 and Ubc9, its subcellular
localization changed from diffuse to a distribution that was both
diffuse and distinctly punctate. DAPI staining showed that the
punctate staining associated with EGFP-HIV-1 IN was concentrated in
the nuclei (data not shown). When SUMO1ΔC6 or SUMO2ΔC4 was
co-expressed with EGFP-HIV-1 IN, EGFP-HIV-1 IN remained diffusely
distributed. The EGFP-negative control showed no change in
localization in the presence of SUMOs and Ubc9.
HIV-1 IN interacts with SUMO1, SUMO2 and
Ubc9
The possible interactions between HIV-1 IN and
SUMO-related proteins were investigated. Yeast two-hybrid assays
were employed to identify SUMO-interacting proteins (23). SUMO1ΔC4 and SUMO2ΔC2, both of
which retain the essential double glycine, were tested, and
SUMO1ΔC6 and SUMO2ΔC4, which lack the double glycine essential for
their conjugation activities, were used for comparison. The
interaction between HIV-1 IN and SUMO1/SUMO2 in yeast cells depends
absolutely upon the presence of the C-terminal double-glycine amino
acid residues; when the conjugation-deficiency SUMO1ΔC6 and
SUMO2ΔC4 were used, no interaction was detected (Table I). This result suggests that the
interaction of HIV-1 IN with SUMO1/SUMO2 includes covalent
conjugation of SUMO1/SUMO2 to integrase or, alternatively, that it
requires the intact C-terminus of SUMO1/SUMO2 for protein-protein
binding. Yeast-two-hybrid assays also showed robust binding of
HIV-1 IN to the conjugation enzyme Ubc9.
 | Table I.Yeast two-hybrid screening of HIV-1
IN and full-length or truncated mutants of SUMO1/SUMO2, Ubc9a. |
Table I.
Yeast two-hybrid screening of HIV-1
IN and full-length or truncated mutants of SUMO1/SUMO2, Ubc9a.
| GAL-BD fusions | GAL-AD fusions | HIV-1 IN
binding |
|---|
| HIV-1 IN | SUMO1 | + |
| SUMO1ΔC6 | − |
| SUMO1ΔC4 | + |
| SUMO2 | + |
| SUMO2ΔC4 | − |
| SUMO2ΔC2 | + |
| Ubc9 | + |
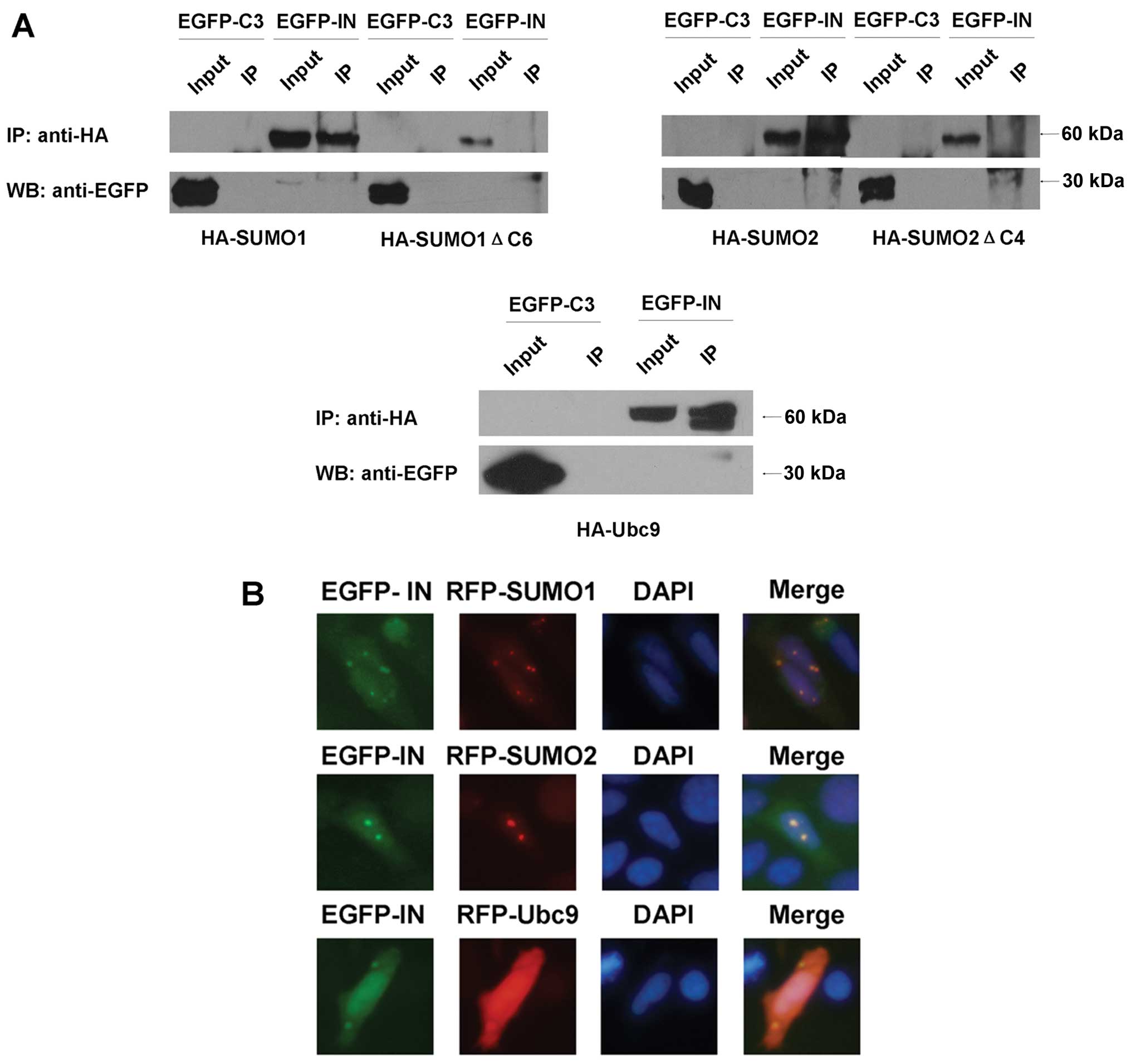
To verify these interactions, co-immunoprecipitation
assays were conducted. Human 293T cells were co-transfected with
pEGFP-HIV-1 IN and HA-SUMO1/HA-SUMO1ΔC6, HA-SUMO2/HA-SUMO2ΔC4 or
HA-Ubc9, respectively. The rabbit anti-HA antibody precipitated
EGFP-HIV-1 IN but not EGFP from extracts of these cells (Fig. 2A). The conjugation-deficiency
forms of the SUMO proteins failed to co-immunoprecipitate with
EGFP-HIV-1 IN (Fig. 2A).
We also examined the subcellular localizations of
HIV-IN and SUMO1/SUMO2, Ubc9. The RFP-SUMO1/SUMO2 and RFP-Ubc9
fusion proteins were mainly found spread throughout the cells and
were comparatively concentrated within the nucleus (Fig. 2B). EGFP-HIV-1 IN fusion protein
presented almost the same distributions as RFP-SUMO1/SUMO2 and
RFP-Ubc9. These results indicated the co-localization of the
corresponding proteins.
Covalent modification of HIV-1 IN by
SUMO1 and SUMO2
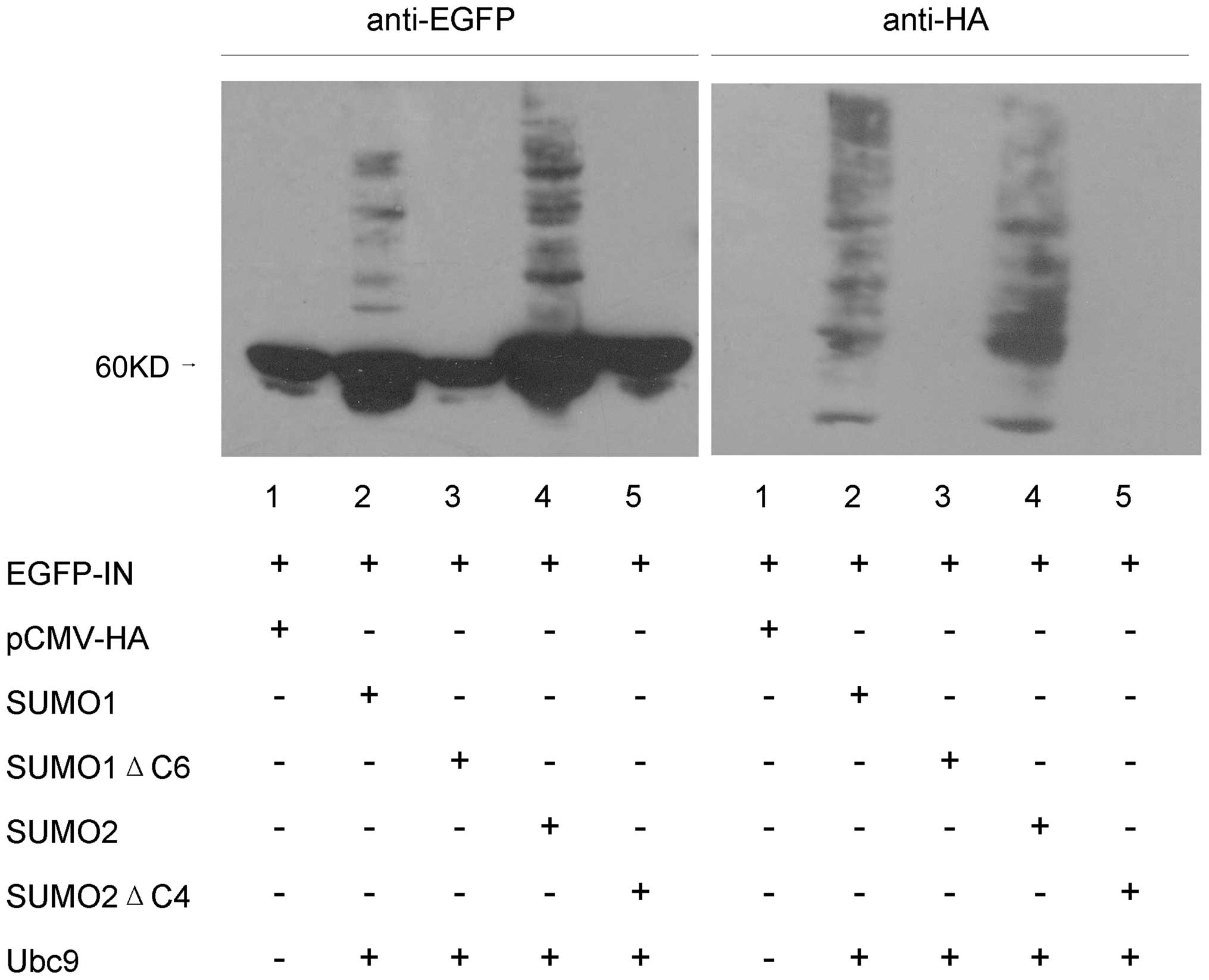
To investigate SUMO conjugation of HIV-1 IN, 293T
cells were co-transfected with pEGFP HIV-1 IN, HA-SUMO1/SUMO2 and
HA-Ubc9. Forty-eight hours after transfection, cells were lysed
with RIPA, proteins were electrophoretically separated on SDS-8%
polyacrylamide gels and anti-EGFP antibody was used for HIV-1 IN
immunoblot analysis. We detected 4 minor immunoreactive bands near
the 60 kDa primary band (Fig. 3,
lanes 2 and 4, left). These minor immunoreactive bands appeared to
represent HIV-1 IN conjugated with various numbers of SUMO
moieties. In control experiments with cells co-transfected with
pEGFP-C3 and HA-SUMO1/SUMO2 or HA-Ubc9, no SUMO conjugation of EGFP
was detected (data not shown). The minor immunoreactive bands were
also not detected in cells that were not subjected to SUMO
transfection (Fig. 3, lane 1,
left) or in SUMO1ΔC6/SUMO2ΔC4 co-transfected cells (Fig. 3, lanes 3 and 5, left). When the
same samples were probed with an anti-HA antibody, the four main
modified forms of HIV-1 IN were readily detected (Fig. 3, lanes 2 and 4, right). The
additional bands detected with the anti-HA antibody presumably
represent cellular proteins that were conjugated with
HA-SUMO1/SUMO2 in the co-transfected cells. Consistent with the
results of anti-EGFP antibody analysis, no bands were detected in
the other three samples (Fig. 3,
lanes 1, 3 and 5, right). These results affirmatively suggest that
a fraction of overexpressed HIV-1 IN protein can be covalently
modified by both SUMO1 and SUMO2.
Upregulations of SUMO1/SUMO2 and Ubc9
inhibit lenti-virus and HIV-1 integration
Because we demonstrated that HIV-1 IN can be
covalently modified by SUMO1 and SUMO2, it was of interest to
determine whether these modifications affect the fundamental
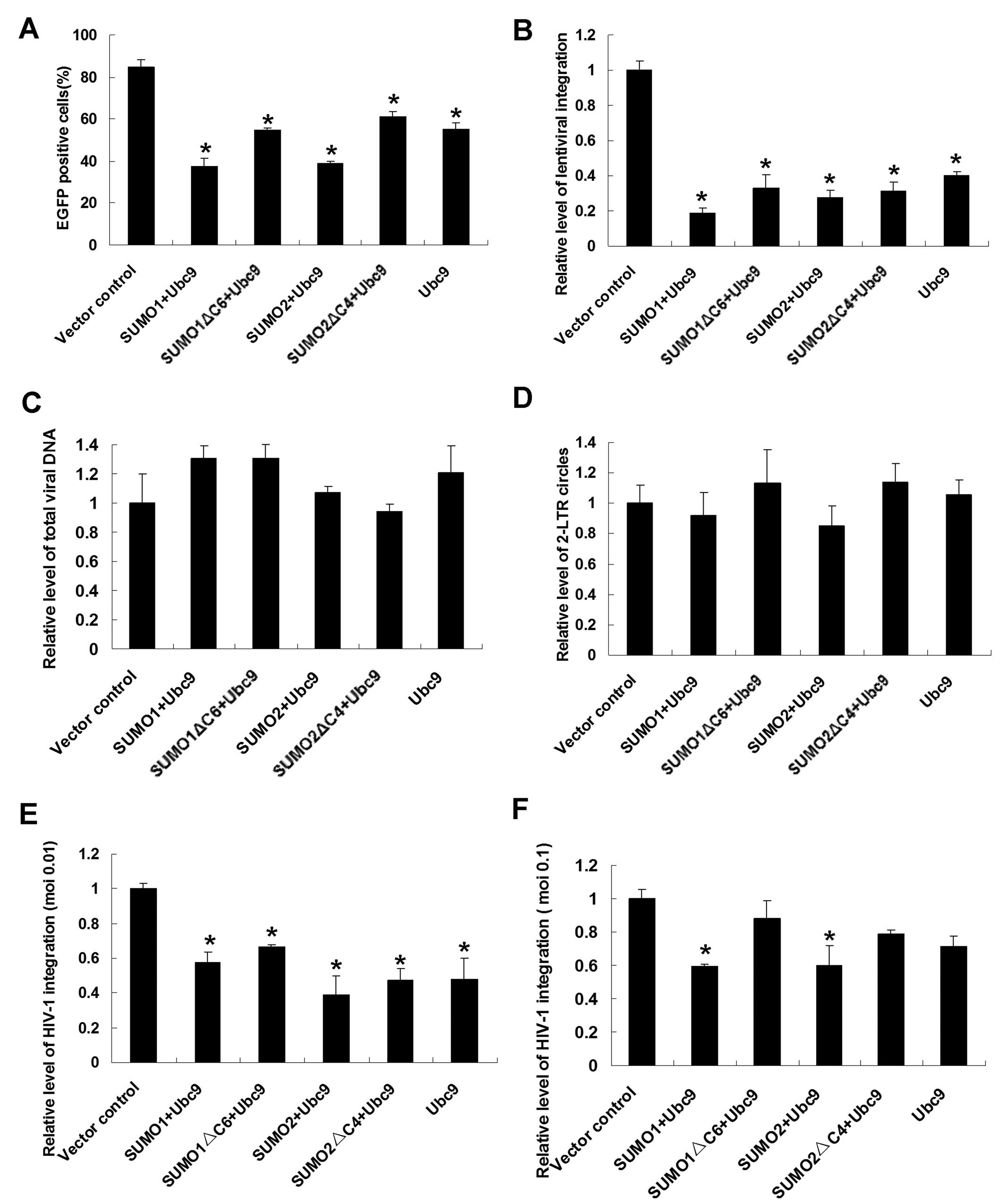
function of the integrase. Firstly, an HIV-1-derived lentiviral
vector system was used, and changes in the integration rate of the
reporter gene (EGFP) were detected by flow cytometry. Transient
transfection of prepared 293T cells with HA-SUMO1/SUMO2 and HA-Ubc9
resulted in upregulation of the cellular expression of SUMO1/SUMO2
and Ubc9. In Ubc9-overexpressing cells, there was a substantial
decline in the percentage of EGFP-positive cells (55.24±3.13%)
compared with the vector control cell group (84.76±3.51%) (Fig. 4A). Interestingly, in populations
co-transfected with HA-SUMO1 and HA-Ubc9, the percentage of
EGFP-positive cells was much lower, 37.42±3.89% (p<0.01). This
result indicates that the presence of SUMO1 and Ubc9 can inhibit
the integration of lentivirus and that these two proteins exert an
additive effect on lentiviral integration. When SUMO2 was
co-expressed with Ubc9, the percentage of EGFP-positive cells
declined to 39.02%±0.88% (p<0.05). However, when mutated SUMO1
or SUMO2 was co-expressed with Ubc9, the percentage of
EGFP-positive cells was almost the same as in populations of
Ubc9-overexpressing cells.
To confirm these results, a more precise
quantitative method, Alu-LTR real-time nested PCR, was employed to
verify changes in integration efficiency. The results were
consistent with the results of flow cytometry analysis (Fig. 4B).
To explore the possible effects of SUMO conjugation
pathway proteins on other stages of HIV-1 early events, including
reverse transcription and preintegration complex (PIC) nuclear
import, real-time PCR was performed to determine the relative level
of total viral DNA synthesis and the formation of 2-LTR circles,
which are used as a marker of PIC nuclear import (24). Similar amounts of late reverse
transcripts and 2-LTR circles were detected in SUMO-overexpressing
cells (Fig. 4C and D).
Having established that SUMO1/SUMO2 and Ubc9 are
presented as cellular restriction factors of the lentivirus, we
next addressed the effects of these SUMO pathway proteins on the
infectivity of spreading HIV-1 viruses at moi of 0.01 and 0.1 and
expanded our conclusion to authentic HIV-1 viruses. For this
purpose, SUMO pathway proteins were overproduced in human
T-lymphoid H9 cells and at 24 h post-transfection, treated cells
were infected with equal amounts of replication competent
HIV-1/IIIB viruses and the relative levels of virus integration
were monitored by Alu-LTR real-time nested PCR. At moi of 0.01,
SUMO pathway proteins overexpressed cells displayed moderate
reductions in the HIV-1 integration rate (between 39 and 66% of
control levels, p<0.05) (Fig.
4E). Under the 0.1 virus titer condition, the integration
levels of SUMO1/SUMO2 and Ubc9 overproducing cells exhibited
significant reductions (Fig. 4F)
(p<0.05).
Additionally, the relative level of total viral DNA
synthesis and the formation of 2-LTR circles were also monitored
after HIV-1/IIIB viruses’ infection. No significant changes were
detected at moi of both 0.01 and 0.1 (data not shown). Altogether
these findings indicate that SUMO pathway proteins play as innate
cellular restriction factors during HIV-1 replication.
Downregulations of endogenous SUMO1/SUMO2
and Ubc9 increase lentivirus and HIV-1 integration
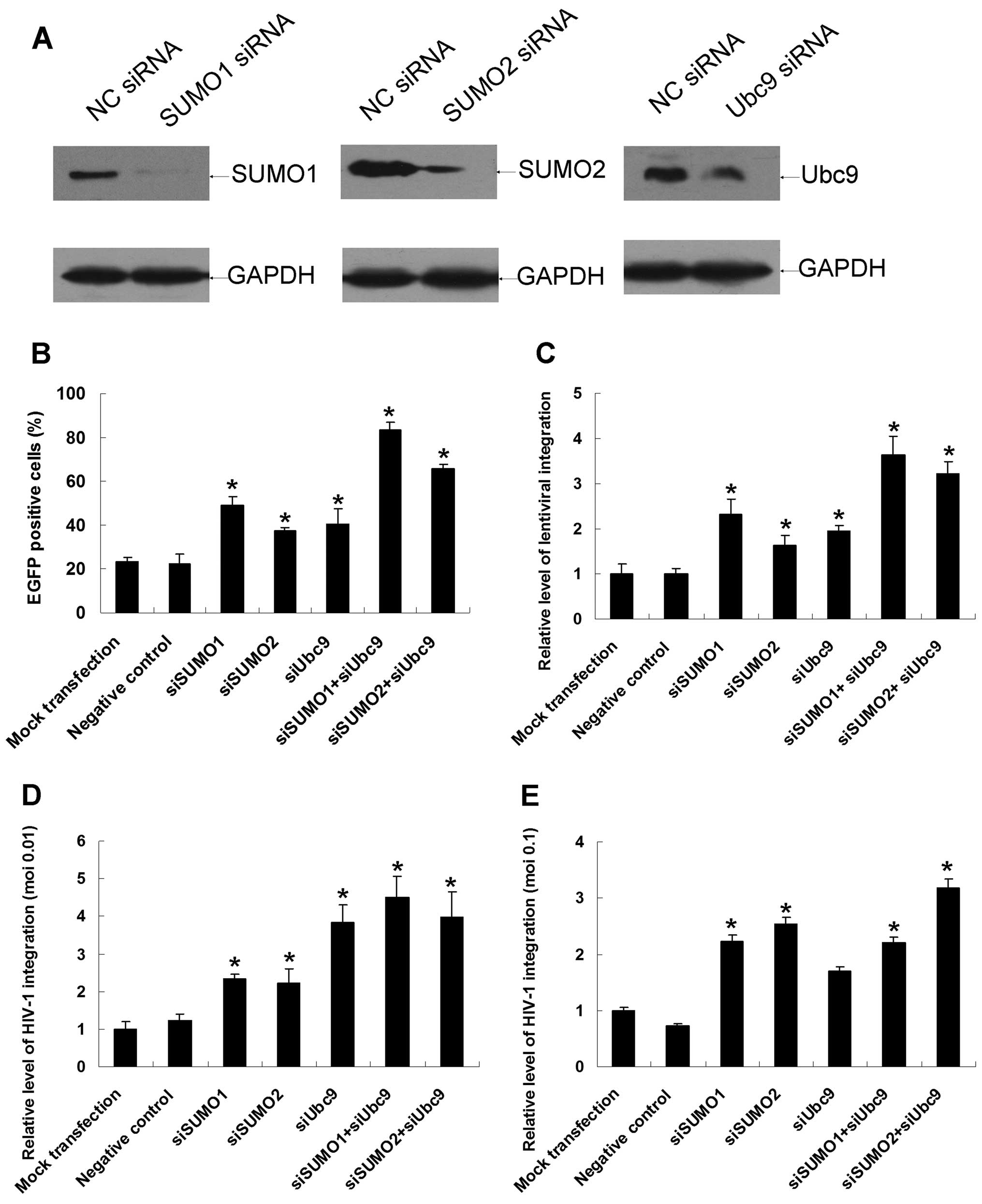
SUMO1-, SUMO2-and Ubc9-specific siRNAs were
introduced into 293T cells to downregulate the corresponding
proteins. Western blotting assays verified that the three siRNAs
dramatically reduced the expression of endogenous SUMO conjugation
pathway proteins in 293T cells 48 h after siRNA transfection
(Fig. 5A). Cells were transfected
with siRNAs for 48 h and then infected with equivalent amounts of
virions. Two days after infection, the percentage of
EGFP-expressing cells was measured by flow cytometry. The
percentages of EGFP-positive cells in the mock transfection and
negative siRNA groups were 23.24±1.95% and 22.40±4.51%,
respectively, whereas in SUMO1/SUMO2 and Ubc9 simultaneous
knockdown cells, the percentage of EGFP-positive cells increased
dramatically to 83.29±3.61% (p<0.01) and 65.57±1.98%
(p<0.01), respectively (Fig.
5B). There was also a noticeable increase in the percentage of
EGFP-positive cells when single siRNAs were introduced into the
cells. As measured by the Alu-LTR real-time nested PCR assay, the
relative levels of lentiviral integration rose by as much as
2.63-fold when SUMO1 and Ubc9 were simultaneously downregulated
(Fig. 5C). The relative level of
total viral DNA synthesis and the formation of 2-LTR circles were
also monitored by real-time PCR under downregulation conditions,
and no significant differences were found (data not shown).
Spreading HIV-1 infection experiments were also
carried out under downregulation conditions, the virus integration
levels increased dramatically to 4.49- and 3.99-fold when SUMO1-
and Ubc9-specific siRNAs or SUMO2- and Ubc9-specific siRNAs were
co-introduced into H9 cells (Fig.
5D). At moi of 0.1, results kept high degree of consistency
with those of lower virus titer except that downregulation of Ubc9
alone did not affect the virus integration (Fig. 5E). Besides, relative level of
total HIV-1 DNA synthesis and the formation of 2-LTR circles were
not affected by these SUMO-related proteins at neither virus titer
(data not shown). In summary, downregulation of endogenous SUMO1,
SUMO2 and Ubc9 is rather advantageous to HIV-1 infection.
Discussion
We investigated the relationship between HIV-1 IN
and three SUMO-conjugation-related proteins and determined how
these associations affect the gene transfer efficiency of
HIV-1-derived lentivirus as well as the infection of authentic
HIV-1 viruses. Firstly, we showed that HIV-1 IN subcellular
localization changes to be punctate in the context of excessive
SUMO1/Ubc9 or SUMO2/Ubc9. The interactions between SUMO1, SUMO2,
Ubc9 and HIV-1 IN were verified by yeast two-hybrid,
co-immunoprecipitation and subcellular localization assays. Further
experimentation revealed that HIV-1 IN could be covalently modified
by SUMO1 and SUMO2. Finally, overexpression of SUMO1/SUMO2 and Ubc9
inhibited viral integration in an additive manner and that
RNAi-mediated downregulation of these proteins promoted viral
integration.
In addition to orchestrating the integration of
viral cDNA into the cellular genome, HIV-1 IN has also been shown
to participate in various steps of the virus life cycle, including
reverse transcription and nuclear localization (25). To rule out possible effects of
SUMO-conjugation-related proteins on processes other than viral
integration, viral cDNA synthesis and 2-LTR circle formation were
monitored. No significant impact on either of these processes was
detected under conditions of upregulation or downregulation of SUMO
pathway proteins.
The SUMO conjugation pathway has been implicated in
a variety of cellular processes and is proving to be as important a
system as ubiquitination. Viruses could manipulate the cellular
SUMOylation system to facilitate viral infection; the system could
also function as an antiviral host response to inhibit viral
functions (26). The precise
biological role of SUMOylation in viral fitness is complicated and
remains to be fully characterized.
A report on a similar topic was published and
somewhat different results were obtained (27). However, we repeated our
experiments in both the lentiviral vector transduction system and
the authentic HIV-1 viruses and verified our data and conclusion.
Further investigations may be needed for more detailed
clarification.
Our study suggests that SUMO-related proteins
function as cellular restriction factors that are detrimental to
HIV-1 infection. These findings merit further investigation for
their potentially significant implications in the cellular
antiviral response to HIV-1 infection.
Acknowledgements
We are very grateful to Dr Carlos Lois
of the California Institute of Technology for providing the
plasmids pFUGW, pCMVΔR8.9 and pVSV-G. This study was supported by
the National Basic Research Program of China, project 973 (grant
no. 2010CB529903), project 863 (grant nos. 2007aa02
1002/2009zx09503-020) and the Nature Science Fund (grant no.
30971617).
References
|
1.
|
AL BrassDM DykxhoornY BenitaN YanA
EngelmanRJ XavierJ LiebermanSJ ElledgeIdentification of host
proteins required for HIV infection through a functional genomic
screenScience319921926200810.1126/science.115272518187620
|
|
2.
|
O TastanY QiJG CarbonellJ
Klein-SeetharamanPrediction of interactions between HIV-1 and human
proteins by information integrationPac Symp
Biocomput14516527200919209727
|
|
3.
|
H ZhouM XuQ HuangAT GatesXD ZhangJC
CastleE StecM FerrerB StruloviciDJ HazudaAS EspesethGenome-scale
RNAi screen for host factors required for HIV replicationCell Host
Microbe4495504200810.1016/j.chom.2008.10.00418976975
|
|
4.
|
K StrebelJ LubanKT JeangHuman cellular
restriction factors that target HIV-1 replicationBMC
Med748200910.1186/1741-7015-7-4819758442
|
|
5.
|
D WolfSP GoffHost restriction factors
blocking retroviral replicationAnnu Rev
Genet42143163200810.1146/annurev.genet.42.110807.09170418624631
|
|
6.
|
V NairHIV integrase as a target for
antiviral chemotherapyRev Med
Virol12179193200210.1002/rmv.35011987143
|
|
7.
|
B Van MaeleK BusschotsL VandekerckhoveF
ChristZ DebyserCellular co-factors of HIV-1 integrationTrends
Biochem Sci31981052006
|
|
8.
|
RT HaySUMO: a history of modificationMol
Cell18112200510.1016/j.molcel.2005.03.01215808504
|
|
9.
|
ES JohnsonProtein modification by SUMOAnnu
Rev Biochem73355382200410.1146/annurev.biochem.73.011303.074118
|
|
10.
|
G GillSUMO and ubiquitin in the nucleus:
different functions, similar mechanisms?Genes
Dev1820462059200410.1101/gad.121460415342487
|
|
11.
|
J ZhaoSumoylation regulates diverse
biological processesCell Mol Life
Sci6430173033200710.1007/s00018-007-7137-417763827
|
|
12.
|
L HuangGL XuJQ ZhangL TianJL XueJZ ChenW
JiaDaxx interacts with HIV-1 integrase and inhibits lentiviral gene
expressionBiochem Biophys Res
Commun373241245200810.1016/j.bbrc.2008.06.01718558084
|
|
13.
|
JQ ZhangJJ WangWJ LiL HuangL TianJL XueJZ
ChenW JiaCellular protein TTRAP interacts with HIV-1 integrase to
facilitate viral integrationBiochem Biophys Res
Commun387256260200910.1016/j.bbrc.2009.06.15319580783
|
|
14.
|
MF FragaM BerdascoE BallestarS RoperoP
Lopez-NievaL Lopez-SerraJI Martin-SuberoMJ CalasanzI Lopez de
SilanesF SetienEpigenetic inactivation of the Groucho homologue
gene TLE1 in hematologic malignanciesCancer
Res6841164122200810.1158/0008-5472.CAN-08-008518519670
|
|
15.
|
CD NovinaMF MurrayDM DykxhoornPJ
BeresfordJ RiessSK LeeRG CollmanJ LiebermanP ShankarPA
SharpsiRNA-directed inhibition of HIV-1 infectionNat
Med8681686200212042777
|
|
16.
|
A BrusselO DelelisP SonigoAlu-LTR
real-time nested PCR assay for quantifying integrated HIV-1
DNAMethods Mol Biol304139154200516061972
|
|
17.
|
T IkedaH NishitsujiX ZhouN NaraT OhashiM
KannagiT MasudaEvaluation of the functional involvement of human
immunodeficiency virus type 1 integrase in nuclear import of viral
cDNA during acute infectionJ
Virol781156311573200410.1128/JVI.78.21.11563-11573.200415479797
|
|
18.
|
JA ZackSJ ArrigoSR WeitsmanAS GoA
HaislipIS ChenHIV-1 entry into quiescent primary lymphocytes:
molecular analysis reveals a labile, latent viral
structureCell61213222199010.1016/0092-8674(90)90802-L2331748
|
|
19.
|
EA LusiP GuarascioC PresuttiR VillaniA
PellicelliF SoccorsiOne-step nested PCR for detection of 2 LTR
circles in PBMCs of HIV-1 infected patients with no detectable
plasma HIV RNAJ Virol
Methods1251113200510.1016/j.jviromet.2004.11.01615737411
|
|
20.
|
V DorvalMJ MazzellaPM MathewsRT HayPE
FraserModulation of Abeta generation by small ubiquitin-like
modifiers does not require conjugation to target proteinsBiochem
J404309316200710.1042/BJ2006145117346237
|
|
21.
|
WZ GuoS SugayaM SatohT TomonagaF NomuraT
HiwasaM TakiguchiK KitaN SuzukiNm23-H1 is responsible for
SUMO-2-involved DNA synthesis induction after X-ray irradiation in
human cellsArch Biochem
Biophys4868187200910.1016/j.abb.2009.03.01119332021
|
|
22.
|
X LinM LiangYY LiangFC BrunicardiXH
FengSUMO-1/Ubc9 promotes nuclear accumulation and metabolic
stability of tumor suppressor Smad4J Biol
Chem2783104331048200310.1074/jbc.C30011220012813045
|
|
23.
|
MB KroetzM HochstrasserIdentification of
SUMO-interacting proteins by yeast two-hybrid analysisMethods Mol
Biol497107120200910.1007/978-1-59745-566-4_719107413
|
|
24.
|
SP GoffRetroviridae: The retroviruses and
their replicationFields VirologyDM KnipePM Howley4th
editionLippincott Williams and WilkinsPhiladelphia,
PA187119392001
|
|
25.
|
A EngelmanG EnglundJM OrensteinMA MartinR
CraigieMultiple effects of mutations in human immunodeficiency
virus type 1 integrase on viral replicationJ
Virol692729273619957535863
|
|
26.
|
R BoggioS ChioccaViruses and sumoylation:
recent highlightsCurr Opin
Microbiol9430436200610.1016/j.mib.2006.06.00816815735
|
|
27.
|
A ZamborliniA CoifficG BeauclairO DelelisJ
ParisY KohF MagneML GironJ Tobaly-TapieroE DeprezImpairment of
human immunodeficiency virus type-1 integrase SUMOylation
correlates with an early replication defectJ Biol
Chem2862101321022201110.1074/jbc.M110.18927421454548
|