Introduction
Glutamate, an important excitatory neurotransmitter,
plays a key role in learning and memory in the brain, and its
concentration is strictly controlled in the central nervous system
(1). However, sudden and excess
glutamate stimulation induces neuronal damage and even cell death,
known as excitotoxicity (2,3).
This process is caused by massive influx of Ca2+ via
glutamate receptors into cells, leading to initiation of apoptosis
through modulation of several apoptosis-related proteins and
activation of caspase cascade (4,5).
Another insult of excessive calcium influx
attenuates the mitochondrial membrane potential, leading to
enhanced Ca2+ permeability through membrane
depolarization of mitochondria and release of reactive oxygen
species that can confer apoptosis (6). Findings from accumulating studies
have suggested an association of glutamate-induced excitotoxicity
with several neurodegenerative diseases or brain injuries,
including ischemia, Alzheimer’s and Huntington’s disease (7–9).
Therefore, for identification of therapies and prevention of these
diseases, further studies of neuroprotection against
glutamate-induced excitotoxicity is needed.
Uncaria sinensis (Oliv.) Havil, a member of
the Rubiaceae family, is widely used in traditional medicine; in
particular, in East Asian countries. Its hooks with stems are known
to provide relief from various nervous-related symptoms, including
dizziness associated with hypertension, dementia, and epilepsy
(10–12). In recent years, studies reporting
on plants of the Uncaria genus as pharmacological medicine
have been consistent; in particular, several alkaloids and phenolic
compounds of Uncaria species have been reported to have
antihypertensive and neuroprotective activities both in vivo
and in vitro (13,14). The phenolic compound isolated from
U. sinensis has been reported to exert strong protective
effects against glutamate-induced neuronal death (15).
Despite its multiple pharmacological properties,
including neuroprotective activities, its precise molecular
mechanism has not been well studied. We prepared a hexane extract
of U. sinensis (HEUS), which was found to exert protective
effects against cerebral ischemic damage (16). However, these protective effects
of HEUS have not been investigated with regard to the type of
neuronal cell death and its related signaling pathway. Therefore,
we explored the neuroprotective effects of HEUS against
glutamate-induced neurotoxicity and attempted to clarify its
relative mechanism for neuronal death in primary cultured rat
cortical cells.
Materials and methods
Preparation of HEUS
The dried hooks and stems of U. sinensis were
purchased in September 2010 from Hwalim Natural Drugs (Busan,
Korea) and were authenticated by Professor J.W. Hong, Division of
Clinical Medicine I, School of Korean Medicine, Pusan National
University. A voucher specimen (accession number PDRLJG-1) was
deposited at the Plant Drug Research Laboratory of Pusan National
University (Miryang, Korea). The dried hooks and stems of U.
sinensis (1.0 kg) were ground to a fine powder, followed by
successive extraction at room temperature with n-hexane, ethyl
acetate and methanol. Briefly, filtration and evaporation of hexane
extracts of U. sinensis were performed under reduced
pressure at 45°C, followed by lyophilization. A white powder of
HEUS (7.27 g) was obtained. Sequential extraction of the remaining
powder was performed using ethyl acetate and methanol to yield
ethyl acetate extracts of U. sinensis (10.68 g) and methanol
extracts of U. sinensis (31.66 g), respectively. Finally,
the solid form of the extract was dissolved with dimethyl sulfoxide
(DMSO) for use as HEUS in experiments.
Chemicals and antibodies
Cytosine-β-D-arabinose furanoside, Hoechst 33342,
L-glutamate, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium
bromide (MTT), poly-L-lysine, and β-actin antibody were purchased
from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s modified
Eagle’s medium (DMEM), fetal bovine serum (FBS), neurobasal medium,
B27, and other neuronal cell culture reagents were purchased from
Gibco-Invitrogen (Carlsbad, CA, USA). For western blot analysis,
antibodies recognizing DR4, DR5, Bcl-2, Bax, Bid, XIAP, caspases-3,
-8, -9, and poly (ADP-ribose) polymerase (PARP) were supplied by
Santa Cruz Biotechnology (Santa Cruz, CA, USA), and cIAP-1 and
cIAP-2 were supplied by Calbiochem (Cambridge, MA, USA). Secondary
antibodies were supplied by Santa Cruz Biotechnology. Caspase
colorimetric assay kits for caspases-3, 8 and 9 were purchased from
R&D Systems (Minneapolis, MN, USA) and a FITC Annexin V
apoptosis dectection kit was purchased from BD Bioscience (San
Diego, CA, USA). A lactate dehydrogenase (LDH) cytotoxicity assay
kit and terminal deoxynucleotidyl transferase-mediated dUTP nick
end labeling (TUNEL) assay kit were purchased from Promega
(Madison, WI, USA).
Primary cortical cell culture
A previously described protocol with some
modifications was used for preparation of primary cultures of
cortical neurons from fetal rats (17). Briefly, E18-E19 Sprague-Dawley
(SD) rat fetuses (DooYeol Biotech, Seoul, Korea) were sacrificed by
cervical dislocation under anesthesia. All procedures used in these
studies followed the guidelines of protocols approved by the Pusan
National University Animal Care and Use Committee in accordance
with the National Institutes of Health Guidelines. The cerebral
cortex was dissected and chopped into small pieces and digested in
0.25% trypsin for 15 min in Ca2+ and Mg+-free
Hanks’ balanced salt solution (HBSS), followed by mechanical
dissociation using a pasteur pipette. After centrifugation, cells
were re-suspended in DMEM supplemented with 10% FBS, 1 mM pyruvate,
4.2 mM sodium bicarbonate, 20 mM HEPES, 0.3 g/l bovine serum
albumin, and 1% penicillin/streptomycin at a density of
1×106 cells/ml and grown on poly-L-lysine-coated plates
in a 5% CO2 humidified incubator at 37°C. Forty eight
hours after plating, cytosine-β-D-arabinose furanoside was added in
order to inhibit proliferation of non-neuronal cells. After three
days in culture, the medium was replaced with neurobasal medium
containing 2% B27 supplement, 0.5 mM glutamine, and 1%
penicillin/streptomycin. Cortical neurons were maintained for 9–10
days in primary culture for use in the experiments.
MTT assay
The MTT assay was used for assessment of primary
cortical cell viability. Cortical neurons were seeded in 96-well
plates at a density of 5×104/well and incubated for 24 h
prior to experimental treatments. After incubation, cells were
treated with 200 μM of glutamate for 6 h, followed by
treatment with 0.5 mg/ml MTT for 4 h at 37°C. Formazan crystals
formed in cells were solubilized in DMSO and the absorbance was
measured at 570 nm using a SpectraMax 190 spectrophotometer
(Molecular Devices, Sunnyvale, CA, USA). Results were expressed as
a percentage of the control.
LDH assay
Cytotoxicity was measured quantitatively according
to release of LDH in the culture medium of cell samples. After
treatment, cortical neurons were lysed by addition of lysis
solution, followed by incubation at 37°C for 45–60 min. Sample
supernatants were transferred to a 96-well enzymatic assay plate,
followed by addition of substrate to each supernatant sample. The
enzymatic reaction was allowed to proceed for 30 min at room
temperature, protected from light. After the enzymatic reaction was
stopped, the plate was read at 490 nm using a SpectraMax 190
spectrophotometer. Data represent the percentage of LDH released
relative to controls.
Flow cytometric analysis
After treatment, cortical neurons were harvested and
cells were washed twice with cold PBS, followed by re-suspension in
binding buffer at a concentration of 1×106 cells/ml; 100
μl of the solution was transferred to a flow cytometric
tube, followed by double staining with Annexin V-FITC and propidium
iodide (PI) in the dark at room temperature for 15 min.
Subsequently, 400 μl of binding buffer was added and
analysis of the samples was performed using a flow cytometer
(FACSCanto™ II; Becton-Dickinson, San Jose, CA, USA).
Hoechst staining
Cortical neurons were grown on poly-L-lysine-coated
glass coverslips at a density of 5×104 cells/ml. After
treatment, the cells were fixed in 4% paraformalehyde for 20 min at
4°C. Fixed cells were washed with PBS three times, followed by
staining with 10 μg/ml Hoechst 33342 in PBS for 10 min at
37°C. The cells were washed with PBS three times and mounted in the
medium for fluorescence (Vector Laboratories, Inc.), followed by
observation under a laser scanning confocal microscope (LSM 510;
Carl Zeiss, Inc., Oberkochen, Germany). Data are presented as the
ratio of chromosomal condensation and morphological change as a
percentage of total cells.
TUNEL assay
Cortical neurons were grown on Lab-Tek®
chamber slides (Thermo Fisher Scientific, Inc., Rochester, NY, USA)
at a density of 1×105 cells/ml. After treatment, the
cells were fixed in 4% methanol-free formaldehyde solution in PBS
(pH 7.4) for 25 min at 4°C. Fixed cells were then washed with PBS,
followed by incubation with DNA-labeling solution at 37°C for 60
min inside a humidified chamber to allow the tailing reaction to
occur. The DNA-labeling reaction was terminated by 2X SSC, and
followed by washing for removal of unincorporated
fluorescein-12-dUTP. The cells were then counterstained with 1
μg/ml PI solution in PBS for 15 min at room temperature in
the dark. Apoptotic neurons were determined by localization of
green fluorescent cells (fluorescein-12-dUTP) on a red background
using a fluorescence microscope (Carl Zeiss, Inc.). Data are
presented as apoptotic cells as a percentage of total cells.
Western blot analysis
After treatment, cortical neurons were washed in
cold PBS buffer, followed by homogenization in lysis buffer [200 mM
Tris (pH 8.0), 150 mM NaCl, 2 mM EDTA, 1 mM NaF, 1% NP-40, 1 mM
PMSF, 1 mM Na3Vo4, protease inhibitor
cocktail]. Equal amounts of proteins were then separated by 10–12%
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
(SDS-PAGE), after which the resolved proteins were transferred to a
nitro-cellulose membrane (Whatman GmbH, Dassel, Germany). The
membrane was blocked with 5% skim milk, followed by exposure to the
appropriate antibodies. All bands were visualized using horseradish
peroxidase-conjugated secondary antibodies and an enhanced
chemiluminescence system (Pierce Biotechnology, Inc., Rockford, IL,
USA). Results of the western blot assay reported here are
representative of at least three experiments.
Caspase assay
After treatment, quantification and homogenization
of cortical neurons were performed in order to recognize activation
of cellular caspase; 150 μg protein in cortical neurons was
mixed with extraction buffer [40 mM HEPES (pH 7.4), 20% glycerol
(v/v), 1 mM EDTA, 0.2% NP-40, and 10 mM DL-DTT] containing 100
μM fluorogenic peptide substrate, followed by dilution with
extraction buffer, setting a volume of 100 μl/sample in a
microtiter plate. Caspase-3, -8 and -9 substrates were used with
Asp-Glu-Val-Asp (DEVD)-p-nitroaniline (pNA), Ile-Glu-Thr-Asp
(IETD)-pNA and Leu-Glu-His-Asp (LEHD)-pNA, respectively. Caspase
activities were determined by measurement of absorbance (405 nm) of
prepared plates after incubation for a period of 3 h.
Data analysis
All data are expressed as means ± SEM and analyzed
using the SigmaStat statistical program version 11.2 (Systat
Software, Inc., San Jose, CA, USA). The paired Student’s
t-test was used for analysis of data. A P-value <0.05 was
considered to indicate a statistically significant result.
Results
Treatment with HEUS reduces
glutamate-induced cytotoxicity
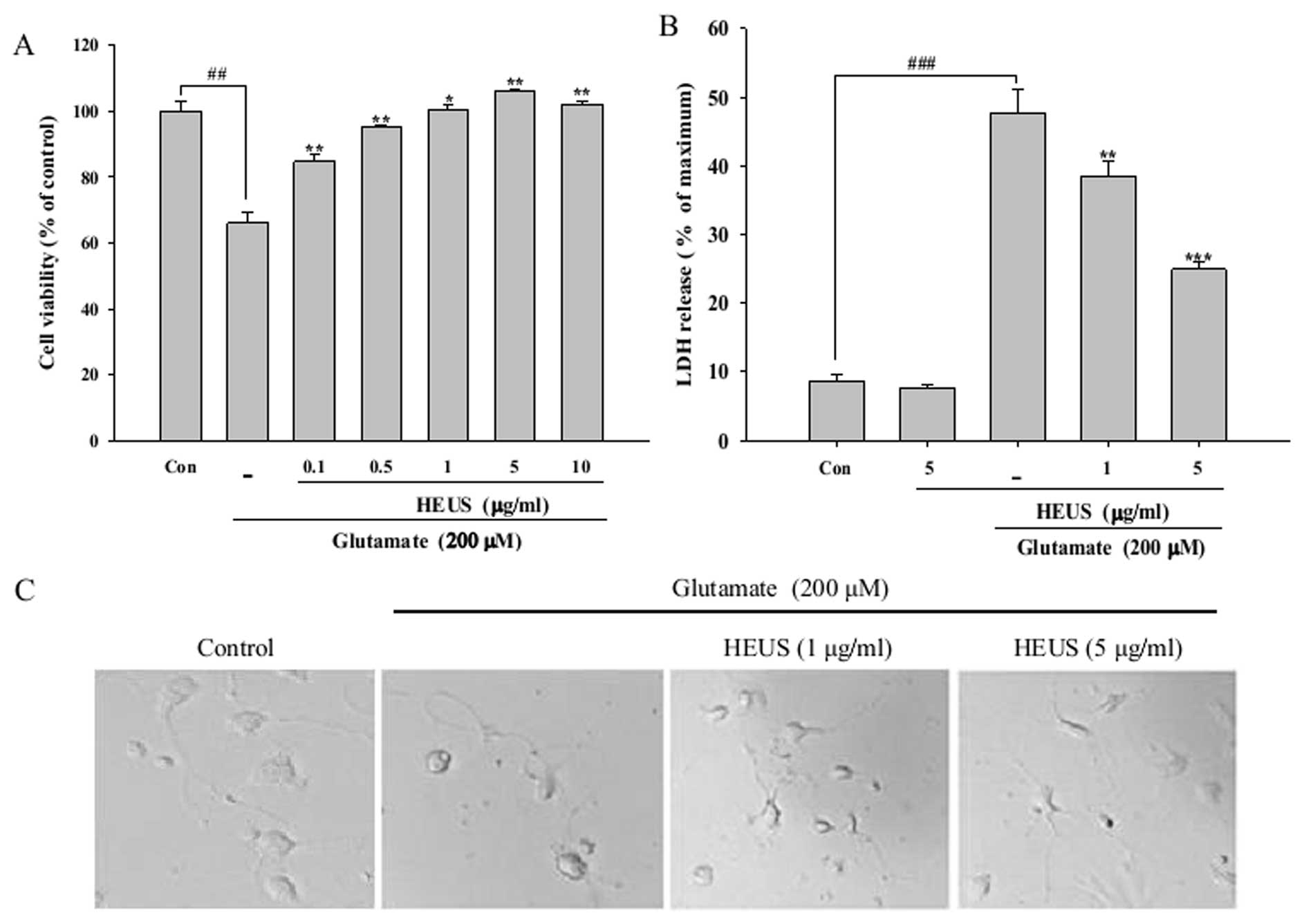
In order to determine whether HEUS is protective
against glutamate-induced toxicity, cell viability was measured by
MTT assay. Cortical neurons were pretreated with various
concentrations of HEUS (0.1–10 μg) for 24 h, followed by
exposure to glutamate at a concentration of 200 μM for a
period of 6 h. Stimulation with glutamate alone resulted in reduced
cell viability, ∼34% of that of the control, however, pretreatment
with HEUS resulted in significantly reduced glutamate-induced
toxicity in a dose-dependent manner (Fig. 1A). Next, the protective effects of
HEUS were also estimated according to changes in the intracellular
release of LDH. The level of LDH release showed a significant
increase, to 47.6%, after exposure to glutamate, while treatment
with HEUS resulted in a marked decrease in the release of LDH, to
38.4 and 24.9%, at a concentration of 1 and 5 μg/ml,
respectively (Fig. 1B). The
protective effects of HEUS were also confirmed by morphological
observation using a phase-contrast microscope. Exposure to
glutamate resulted in significant cell insult to the cortical
neurons, including diminution of cellular processes and decrease in
refraction; these morphological features showed marked recovery
when cells were pretreated with 5 μg/ml of HEUS (Fig. 1C). These results indicated that
pretreatment with HEUS can exert a protective effect against
glutamate-induced toxicity in primary cortical neurons.
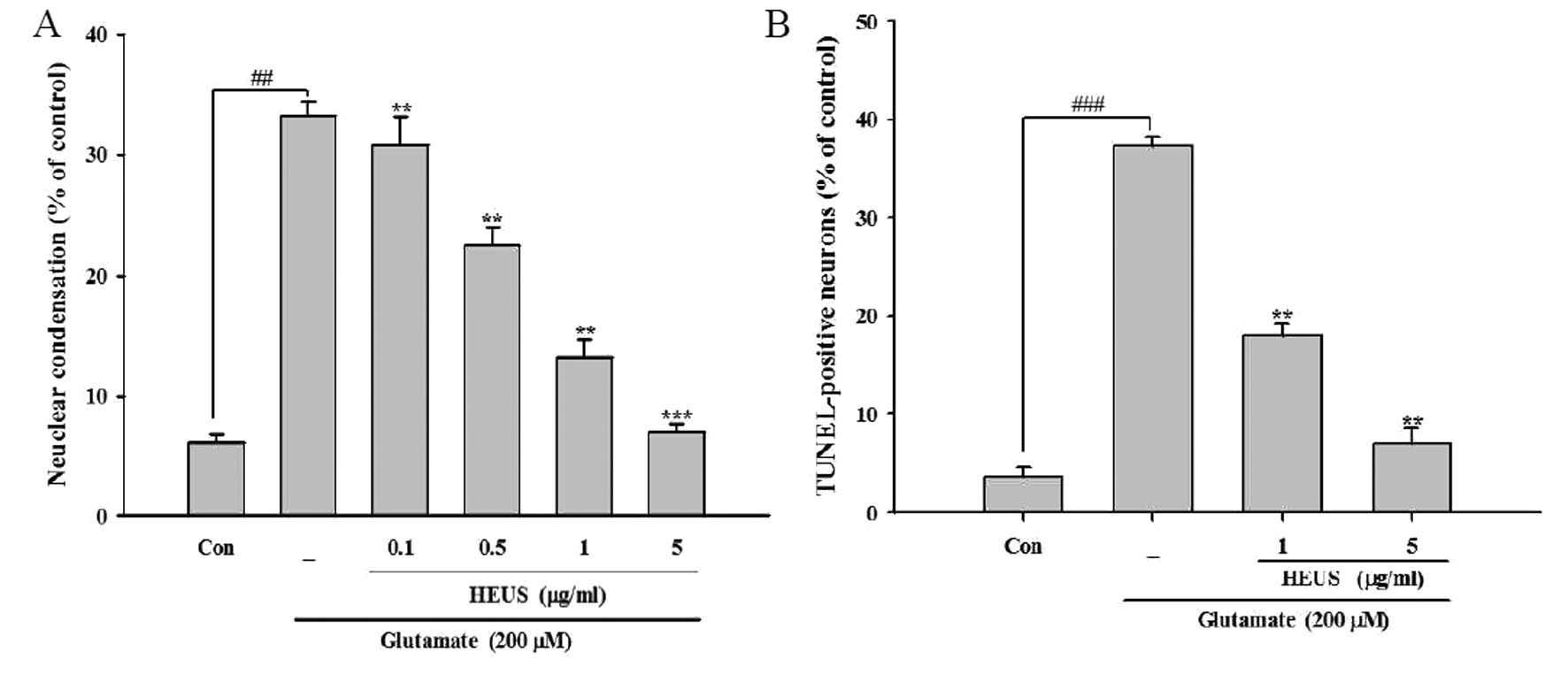
Treatment with HEUS reduces
glutamate-induced apoptotic neuronal death
Findings from recent studies suggest an association
of glutamate-triggered cell death with both apoptotic and necrotic
processes (18). In order to
determine the type of cell death induced by glutamate, we performed
several morphological assessments under our experimental
conditions. Upon nuclear staining with Hoechst 33342, cortical
neurons showed typical morphological signs of apoptosis, including
nuclear and cytoplasmic shrinkage, and chromatin condensation,
after exposure to glutamate, compared with control cells, which had
regular and round-shaped nuclei. However, treatment with HEUS
resulted in a significant reduction in these apoptotic features by
∼30.8, 22.6, 13.2 and 7.0% from 33.2% as observed in the
glutamate-treated cells at a concentration of 0.1, 0.5, 1 and 5
μg/ml, respectively (Fig.
2A). Next, we performed TUNEL staining, which is designed for
detection of DNA strand breaks and quantitation of apoptosis.
Consistent with the results of Hoechst staining, after exposure to
glutamate, cortical neurons showed a ratio of nuclear DNA
fragmentation in total cells to 37.3%. Treatment with HEUS resulted
in a marked reduction in TUNEL-positive cells to 18.0 and 7.0% at a
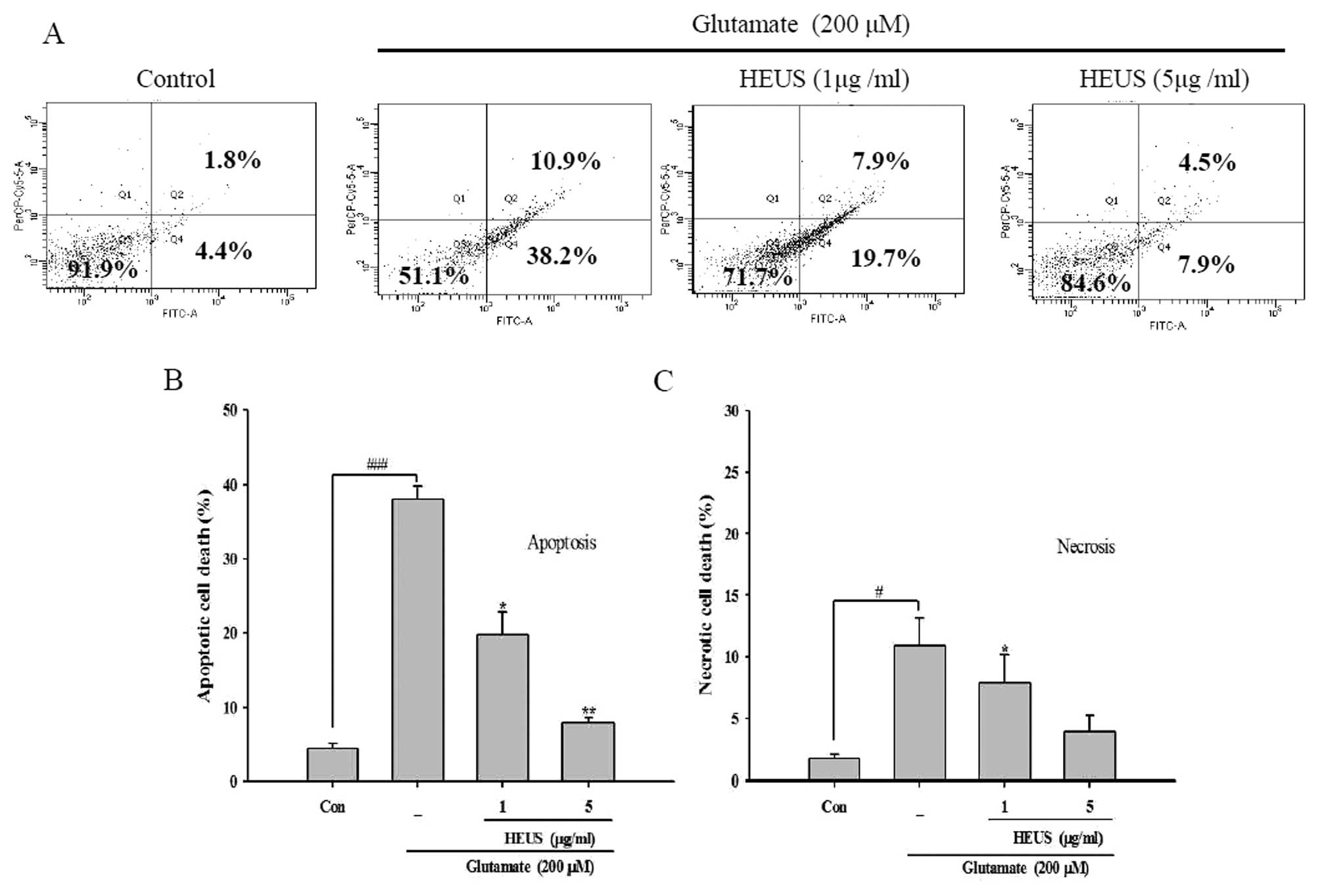
concentration of 1 and 5 μg/ml, respectively (Fig. 2B). We further confirmed our
results by performance of flow cytometry with Annexin V FITC/PI
staining. The percentage of apoptosis in glutamate-treated neurons
was up to 38.2%, while the proportion of necrotic cells showed a
slight increase to 10.9% (Fig.
3). These results indicated that after exposure to glutamate,
cortical neurons underwent extensive apoptotic-like deaths under
our experimental conditions. However, treatment with HEUS resulted
in a significant decrease in the number of glutamate-induced
apoptotic cells, from 19.7 to 7.9%, at a concentration of 1 and 5
μg/ml, respectively (Fig.
3A). These results suggest that pretreatment with HEUS results
mainly in protection of cortical cells against glutamate-induced
neuronal apoptosis.
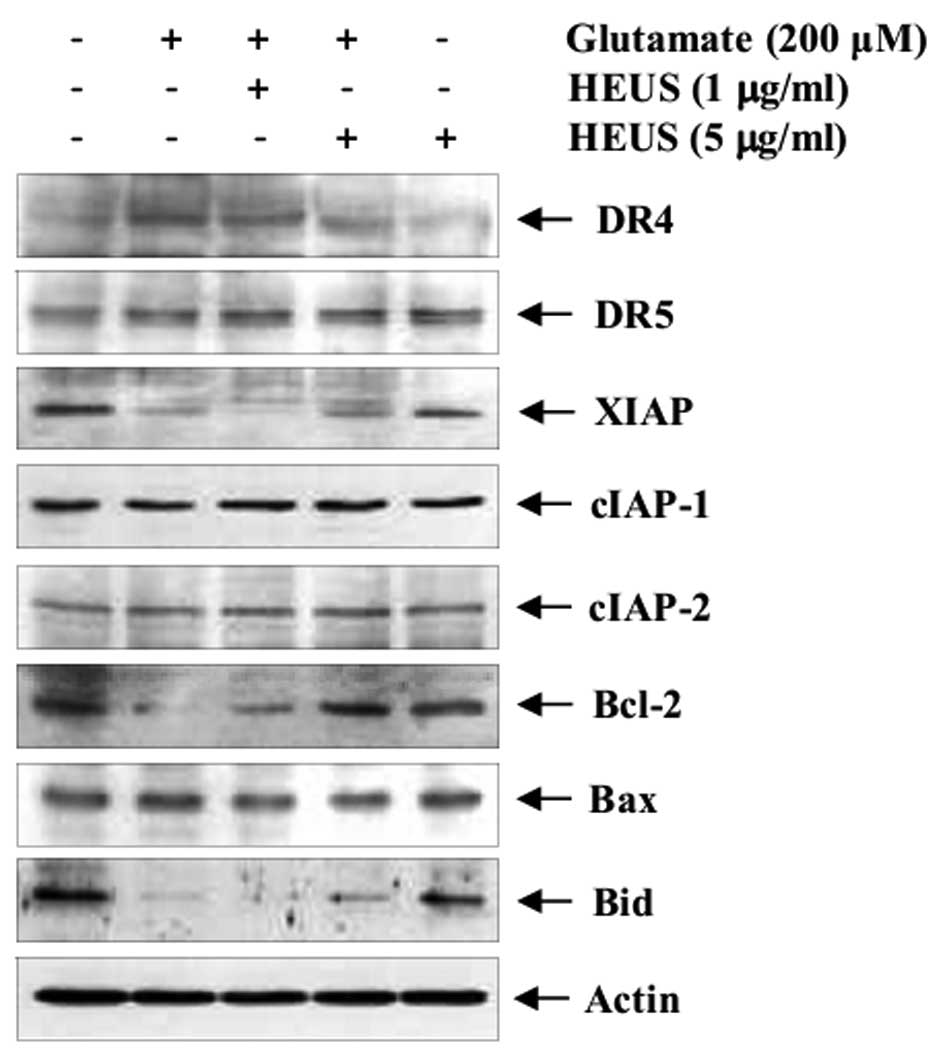
HEUS inhibits neuronal apoptosis via
expression of DR4 and anti-apoptotic proteins
Mediation of apoptosis occurs primarily through
activation of death receptors and/or mitochondrial signaling
(19). Next, using western blot
analysis, we evaluated the levels of DRs and apoptosis-related gene
expression. Exposure to glutamate resulted in markedly enhanced
expression of tumor necrosis factor-related apoptosis-inducing
ligand (TRAIL) receptor DR4, whereas, its expression was reduced by
pretreatment with HEUS (Fig. 4).
Attenuation of TRAIL-induced apoptosis is also known to occur
through upregulation of numerous anti-apoptotic proteins, including
IAPs, which are potent suppressors of apoptosis (19). Thus, we attempted to determine
whether the neuroprotective effect of HEUS was involved in
expression of IAP family members. Pretreatment with HEUS led to
significantly increased expression of glutamate-attenuated XIAP
protein, but not cIAPs. Next, we investigated the effect of HEUS on
members of the pro- and anti-apoptotic Bcl-2 family, a key
regulator of apoptosis. After exposure to glutamate, cortical
neurons showed significantly decreased levels of Bcl-2, compared
with the control. However, treatment with HEUS resulted in markedly
increased glutamate-attenuated anti-apoptotic Bcl-2 expression
without alteration of pro-apoptotic Bax expression. Another
pro-apoptotic member of the Bcl-2 family, Bid, is cleaved to its
truncated active form; tBid induces permeabilization of the outer
mitochondrial membrane, leading to promotion of apoptosis (19). Treatment with HEUS resulted in
slight recovery of glutamate-attenuated whole Bid expression at a
concentration of 5 μg/ml. These results suggest that
pretreatment with HEUS inhibits neuronal apoptosis via both
inhibition of DR4 and activation of anti-apoptotic proteins XIAP
and Bcl-2.
Treatment with HEUS reduces activation of
glutamate-induced caspases
Eventually, activation of the caspase cascade was
induced during the process of glutamate-induced neuronal apoptosis
(5,20). Thus we investigated expression and
activities of caspase-8, -9 and -3, which could act either as
initiator caspases or as effector caspases, with involvement in
both death receptor- and mitochondrial-initiated apoptosis. Western
blotting data showed that treatment with HEUS resulted in
significantly decreased expression of the cleaved active form of
caspase-3 (Fig. 5A). Neither of
the active forms of caspase-8 and -9 was detected in our studies,
however, treatment with HEUS induced an increase in their proforms.
To further confirm involvement of caspases, we assessed their
activities in the lysates of the cells. Exposure to glutamate to
cortical neurons resulted in markedly increased activities of
caspase-8, -9 and -3, whereas these activities were significantly
attenuated by treatment with HEUS (Fig. 5B). In addition, pretreatment with
HEUS resulted in significantly decreased glutamate-enhanced
cleavage of PARP, a downstream target of the activated caspase-3
(Fig. 5A). These results suggest
that pretreatment with HEUS results in inhibition of neural
apoptosis via inhibition of caspase-8, -9 and -3 activation.
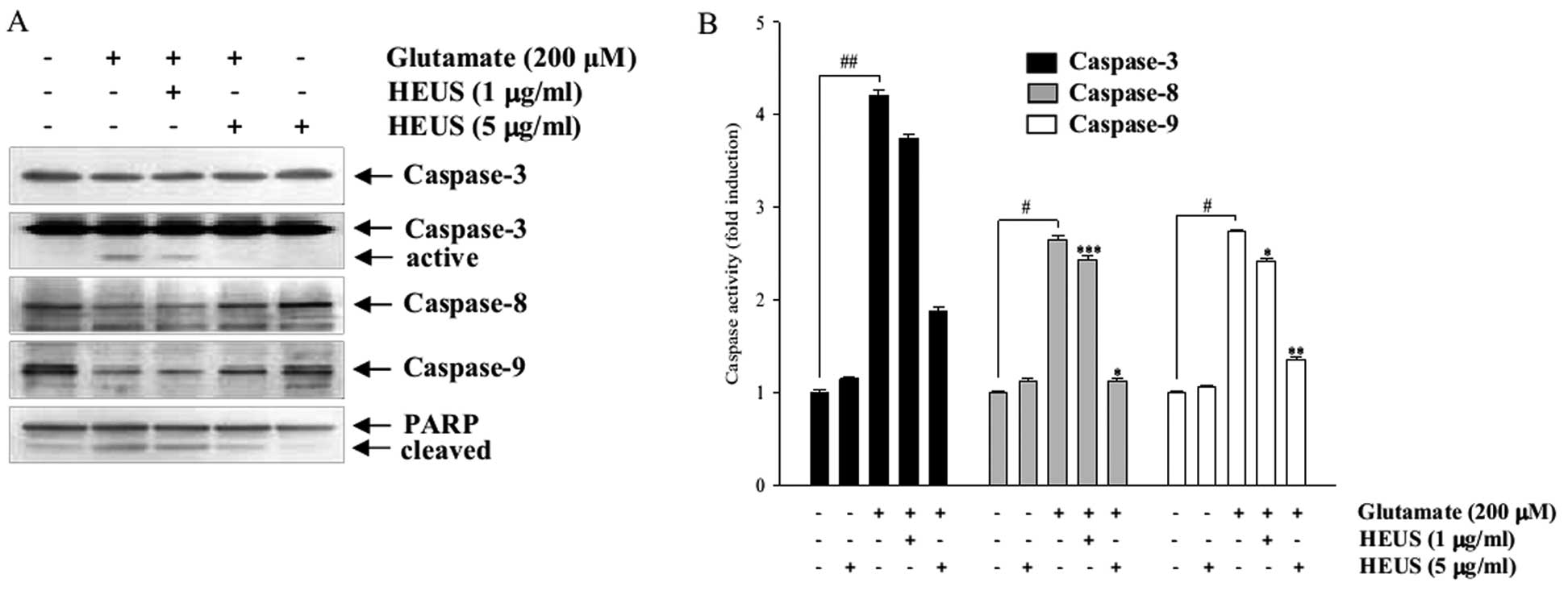 | Figure 5Western blot analysis and caspase
activity assay of caspase-3, -8 and -9 and PARP in
glutamate-treated cortical neurons. Cells were pretreated with HEUS
(1 and 5 μg/ml) for 24 h, followed by exposure to 200
μM glutamate for 6 h. (A) Cells were lysed and equal amounts
of cellular proteins were subjected to western blotting assays
using anti-caspase-3, -8, -9 and PARP antibodies. Equal protein
loading was confirmed by β-actin expression. (B) Quantitative
analysis of caspases-3, -8 and -9 activity in vitro. Cell
lysates were prepared and assayed for in vitro caspase-3, -8
and -9 activity using DEVD-pNA, IETD-pNA and LEHD-pNA,
respectively, as substrates. The concentration of the released
fluorescent products was measured. #P<0.05,
##P<0.01 as compared to the control group;
*P<0.05, **P<0.01 and
***P<0.001 as compared to the glutamate-treated
group. Data are represented as the means ± SEM of three independent
experiments. |
Discussion
The controlling mechanisms of glutamate
excitotoxicity, a pathological process that causes damage leading
to neuronal cell death, have yet to be fully established (2,3,21).
In particular, we observed that Uncaria species have been
intensively studied for use as a pharmacological medicine against
diverse neurological symptoms (13–15). Alkaloids and phenolic compounds
from hooks and stems of U. sinensis have been reported to
exert inhibitory effects against glutamate-induced neuronal death
by inhibition of Ca2+ influx in cultured celebellar
granule cells (22). The main
alkaloid constituent of Uncaria species, rhynchophylline,
has been reported to exert strong antihypertensive and
neuroprotective activities against cerebral ischemia (13).
However, no studies concerning the precise molecular
mechanism of the protective effects of Uncaria species
against glutamate-induced neurotoxicity have been reported. We
prepared a hexane extract of U. sinensis; protection of the
brain from ischemic injury by HEUS has been reported in mice, and
its mechanism may, in part, be attributed to regulation of
Akt/endothelial nitric oxide synthase signaling (16). Findings of our previous studies
suggest that HEUS may have a neuroprotective role; therefore, we
explored the neuroprotective effect of HEUS against
glutamate-induced neurotoxicity in cultured primary cortical cells
and investigated the molecular mechanism underlying these
effects.
First, we demonstrated that glutamate stimulation
resulted in significant induction of toxicity in primary cortical
neurons, while pretreatment with HEUS resulted in significantly
increased cell viability in a dose-dependent manner (Fig. 1A). These results showed a
correlation with those of the LDH release assays (Fig. 1B), and analyses of cell morphology
(Fig. 1C) revealed that
pretreatment with HEUS effectively protects impaired cortical
neurons following glutamate stimulation.
An association of glutamate-induced neuronal death
with necrosis and apoptosis, or both, mixed type, depending on the
timing and severity of stimulation, has been noted (18,23). Thus, we performed morphological
analyses using Hoechst staining, TUNEL assay, and flow cytometric
analysis in order to determine whether pretreatment with HEUS
prevents either glutamate-induced apoptotic or necrotic cell death.
According to our results, glutamate stimulation resulted
predominantly in induction of apoptosis, compared to untreated
control cells, with only slight elevation of necrosis. In addition,
pretreatment with HEUS resulted in effective suppression of
glutamate-induced apoptosis (Figs.
2 and 3). These results serve
to establish that pretreatment with HEUS results in effective
inhibition of glutamate-mediated apoptosis in primary cortical
neurons, thus exerting a neuroprotective effect.
Some reports have demonstrated the involvement of
TRAIL-mediated apoptosis in the pathogenesis of several
neurodegenerative diseases of the central nervous system (24,25). Blockade of TRAIL-induced apoptosis
in the CNS may be a new molecular therapeutic target for treatment
of neurodegenerative diseases. In the present study, we found that
glutamate stimulation resulted in elevated levels of TRAIL-mediated
receptor DR4, while, pretreatment with HEUS resulted in a critical
decrease in its expression (Fig.
4). These results indicated that pretreatment with HEUS
inhibits extrinsic/receptor-mediated apoptosis pathways by
regulating expression of TRAIL/DR4 signaling. Although these
findings do not account for a prominent role in TRAIL-mediated
apoptosis, this could be an indication that the anti-apoptotic
effect of pretreatment with HEUS might be a good model for
investigation of the molecular basis of TRAIL-mediated
apoptosis.
Next, we observed that pretreatment with HEUS
resulted in significantly enhanced levels of expression of
glutamate-attenuated XIAP, Bcl-2, and marked blockade of
glutamate-induced cleavage of Bid (Fig. 4). These results indicated that the
anti-apoptotic effect of pretreatment with HEUS is not a direct
result of altered receptor expression, and that it should induce
potent inhibition of both extrinsic and intrinsic apoptosis
pathways involved in extrinsic/receptor-mediated cascades (i.e., by
downregulation of DR4) and the intrinsic/mitochondrial-mediated
apoptotic pathway (i.e., by upregulation of XIAP, activation of
Bcl-2, and downregulation of Bid cleavage). These findings are
particularly important since Bcl-2 is known to exert protective
action through regulation of mitochondrial membrane potential and
to block release of cytochrome c through caspase cleavage
(26).
Thus, we attempted to determine whether the
anti-apoptotic mechanisms of pretreatment with HEUS were involved
in inhibition of caspase activation. Although the exact mechanism
of glutamate-induced apoptosis has yet to be fully established, an
association of glutamate-induced apoptotic cell death with
upregulation of caspase-3 and its activation has been noted
(20,21). In addition, recent studies have
reported that use of herbal extracts or their bioactive compounds
can result in improvement in the protective activities through
modulation of cell survival genes and signaling or downregulation
of caspase activity, which is one way of providing protection
against neuronal injury (27,28).
Thus, through use of western blot analysis and
caspase activity assay, we extend our observation of downstream
events of cascase cascade activation. Pretreatment with HEUS
resulted in marked attenuation of glutamate-induced caspase-8, -9
and -3 activities, subsequently leading to blocked degradation of
PARP-1, caspase-3 target protein (Fig. 5). These results suggest that
pretreatment with HEUS can result in strong suppression of
glutamate-induced apoptosis through inhibition of a
caspase-dependent pathway, which might contribute to the
neuroprotective effect of pretreatment with HEUS.
Consequently, we demonstrated that pretreatment with
HEUS exerts significant anti-apoptotic effects against
glutamate-induced neurotoxicity in primary cultured cortical
neurons. Futhermore, these effects are mediated by the regulation
of both death receptor- and mitochondrial-mediated apoptotic
pathways, particularly expression of DR4, XIAP and Bcl-2.
Therefore, our findings suggest the potential for HEUS as an
attractive candidate for use as a therapeutic approach to treatment
of various neurodegenerative diseases associated with glutamate
injury.
Acknowledgements
The present study was supported by the
Basic Science Research Program through the National Research
Foundation of Korea (NRF) funded by the Ministry of Education,
Science and Technology (2011-0022151). This study was financially
supported by the 2012 Post-Doctoral Development Program of Pusan
National University.
References
|
1.
|
BS MeldrumGlutamate as a neurotransmitter
in the brain: review of physiology and pathologyJ Nutr130Suppl
41007S1015S200010736372
|
|
2.
|
JT CoyleP PuttfarckenOxidative stress,
glutamate, and neurodegenerative
disordersScience262689695199310.1126/science.79019087901908
|
|
3.
|
Y NishizawaGlutamate release and neuronal
damage in ischemiaLife
Sci69369381200110.1016/S0024-3205(01)01142-011459428
|
|
4.
|
AK StoutHM RaphaelBI KanterewiczE KlannIJ
ReynoldsGlutamate-induced neuron death requires mitochondrial
calcium uptakeNat Neurosci5366373199810196525
|
|
5.
|
Y ZhangBR BhavnaniGlutamate-induced
apoptosis in primary cortical neurons is inhibited by equine
estrogens via down-regulation of caspase-3 and prevention of
mitochondrial cytochrome c releaseBMC Neurosci24613200515730564
|
|
6.
|
A AtlanteP CalissanoA BobbaA AzzaritiE
MarraS PassarellaCytochrome c is released from mitochondria in a
reactive oxygen species (ROS)-dependent fashion and can operate as
a ROS scavenger and as a respiratory substrate in cerebellar
neurons undergoing excitotoxic deathJ Biol
Chem473715937166200010.1074/jbc.M002361200
|
|
7.
|
W PaschenGlutamate excitotoxicity in
transient global cerebral ischemiaActa Neurobiol Exp
(Wars)5631332219968787192
|
|
8.
|
MR HyndHL ScottPR DoddGlutamate-mediated
excitotoxicity and neurodegeneration in Alzheimer’s
diseaseNeurochem Int455835952004
|
|
9.
|
PC MayPN GrayThe mechanism of
glutamate-induced degeneration of cultured Huntington’s disease and
control fibroblastsJ Neurol Sci701011121985
|
|
10.
|
YW LinCL HsiehOral Uncaria
rhynchophylla (UR) reduces kainic acid-induced epileptic
seizures and neuronal death accompanied by attenuating glial cell
proliferation and S100B proteins in ratsJ
Ethnopharmacol1353133202011
|
|
11.
|
T KuramochiJ ChuT SugaGou-teng (from
Uncaria rhynchophylla Miquel)-induced endothelium-dependent
and -independent relaxations in the isolated rat aortaLife
Sci262061206919948208063
|
|
12.
|
T ItohY ShimadaK TerasawaEfficacy of
Choto-san on vascular dementia and the protective effect of the
hooks and stems of Uncaria sinensis on glutamate-induced
neuronal deathMech Ageing
Dev111155173199910.1016/S0047-6374(99)00062-710656534
|
|
13.
|
J ZhouS ZhouAntihypertensive and
neuroprotective activities of rhynchophylline: the role of
rhynchophylline in neurotransmission and ion channel activityJ
Ethnopharmacol1321527201010.1016/j.jep.2010.08.04120736055
|
|
14.
|
K SukSY KimK LeemYO KimSY ParkJ HurJ
BaekKJ LeeHZ ZhengH KimNeuroprotection by methanol extract of
Uncaria rhynchophylla against global cerebral ischemia in
ratsLife Sci7024672480200212173411
|
|
15.
|
Y ShimadaH GotoT KogureN ShibaharaI
SakakibaraH SasakiK TerasawaProtective effect of phenolic compounds
isolated from the hooks and stems of Uncaria sinensis on
glutamate-induced neuronal deathAm J Chin
Med29173180200110.1142/S0192415X0100019811321476
|
|
16.
|
SH ParkJH KimSJ ParkSS BaeYW ChoiJW HongBT
ChoiHK ShinProtective effect of hexane extracts of Uncaria
sinensis against photothrombotic ischemic injury in miceJ
Ethnopharmacol138774779201122051882
|
|
17.
|
GJ BrewerJR TorricelliEK EvegePJ
PriceOptimized survival of hippocampal neurons in B27-supplemented
Neurobasal, a new serum-free medium combinationJ Neurosci
Res35567576199310.1002/jnr.4903505138377226
|
|
18.
|
P NicoteraM AnkarcronaE BonfocoS
OrreniusSA LiptonNeuronal necrosis and apoptosis: two distinct
events induced by exposure to glutamate or oxidative stressAdv
Neurol729510119978993688
|
|
19.
|
S FuldaKM DebatinExtrinsic versus
intrinsic apoptosis pathways in anticancer
chemotherapyOncogene2547984811200610.1038/sj.onc.120960816892092
|
|
20.
|
Y HirashimaM KurimotoK NogamiS EndoM
SaitohO OhtaniT NagataA MuraguchiA TakakuCorrelation of
glutamate-induced apoptosis with caspase activities in cultured rat
cerebral cortical neuronsBrain
Res849109118199910.1016/S0006-8993(99)02009-010592292
|
|
21.
|
A LauM TymianskiGlutamate receptors,
neurotoxicity and neurodegenerationPflugers
Arch460525542201010.1007/s00424-010-0809-120229265
|
|
22.
|
Y ShimadaH GotoT ItohI SakakibaraM KuboH
SasakiK TerasawaEvaluation of the protective effects of alkaloids
isolated from the hooks and stems of Uncaria sinensis on
glutamate-induced neuronal death in cultured cerebellar granule
cells from ratsJ Pharm
Pharmacol51715722199910.1211/002235799177285310454049
|
|
23.
|
M AnkarcronaJM DypbuktE BonfocoB
ZhivotovskyS OrreniusSA LiptonP NicoteraGlutamate-induced neuronal
death: a succession of necrosis or apoptosis depending on
mitochondrial
functionNeuron15961973199510.1016/0896-6273(95)90186-87576644
|
|
24.
|
Y HuangN ErdmannH PengY ZhaoJ ZhengThe
role of TNF related apoptosis-inducing ligand in neurodegenerative
diseasesCell Mol Immunol2113122200516191417
|
|
25.
|
D UbertiG Ferrari-ToninelliSA BoniniI
SarnicoM BenareseM PizziL BenussiR GhidoniG BinettiP SpanoF
FacchettiM MemoBlockade of the tumor necrosis factor-related
apoptosis inducing ligand death receptor DR5 prevents beta-amyloid
neurotoxicityNeuropsychopharmacology4872880200616936710
|
|
26.
|
J YangX LiuK BhallaCN KimAM IbradoJ CaiTI
PengDP JonesX WangPrevention of apoptosis by Bcl-2: release of
cytochrome c from mitochondria
blockedScience27511291132199710.1126/science.275.5303.11299027314
|
|
27.
|
XY LiJ LiangYB TangJG ZhouYY
GuanGinsenoside Rd prevents glutamate-induced apoptosis in rat
cortical neuronsClin Exp Pharmacol
Physiol37199204201010.1111/j.1440-1681.2009.05286.x19719747
|
|
28.
|
X WangG ZhuS YangX WangH ChengF WangX LiQ
LiPaeonol prevents excitotoxicity in rat pheochromocytoma PC12
cells via downregulation of ERK activation and inhibition of
apoptosisPlanta Med7716951701201110.1055/s-0030-127103321509715
|