Introduction
Studies indicate that angiotensin and angiogenic
factors in the renin-angiotensin system are important early markers
of cardiovascular disease (1–3).
Angiotensin II (Ang II) plays critical roles in the pathogenesis of
pre-eclampsia, acting on endothelial cells, vascular smooth muscle
cells and immune cells to accelerate the development of the
condition at least partly through low grade inflammation. Ang II
stimulates vascular smooth muscle cells to produce monocyte
chemoattractant protein, a major chemokine involved in macrophage
infiltration. Macrophages, which accumulate in cardiac and other
tissues, produce angiotensin-converting enzyme, renin and Ang II
(4–6). Continuous stress induced by Ang II
can lead to damage and apoptosis in vascular endothelial cells,
eventually causing blood vessel damage (1,2).
Thus, preventing continuous Ang II stress is important in
protecting blood vessels.
A number of studies have reported that the
cardiovascular system is a particularly rich source of microRNAs
(miRNAs) with roles in disease, which perhaps reflects the
susceptibility of the heart and blood vessels to injury and the
dependence of mammals on persistent cardiovascular function
(1–3,7).
miRNAs are 20–23-nucleotide (nt) non-coding RNAs that function as
sequence-specific regulators of gene expression through
translational repression and/or transcript cleavage (2,7–11).
They have recently been discovered to play a major role in gene
regulation, including in mammalian cells. Characterization of
miRNAs has gained much attention in biology and medicine, with
implications for the detection and treatment of different
pathologies. miRNAs are abundant in the vascular system, where they
have key roles in development and are likely to be important
mediators of vascular system diseases (2). Thum et al (13) reported that miR-21 is regulated by
mitogen-activated protein kinase (MAPK)/extracellular signal
related kinase (ERK) signaling in response to cardiac stress
(1,12,14). Gao et al (6) suggested that miR-155 overexpression
in vascular cells attenuates Ang II-induced α-smooth muscle actin
expression, and demonstrated that miR-155 regulated adventitial
fibroblast differentiation and contributed to suppression of Ang II
type 1 receptor (AtR1) expression. Liu et al (9) also showed that lack of miR-155
expression in the human umbilical vein endothelial cells (HUVECs)
of puerperant women with pre-eclampsia can lead to high levels of
endogenous AtR1 expression and ERK1/2 activation. In view of this
evidence, we believe that miR-155 and Ang II are involved in
diseases of the blood vessels.
In this study, we isolated and cultured HUVECs from
healthy puerperant women and transfected them with an miR-155
expression vector to determine whether this miRNA can regulate the
cellular response to Ang II stress signaling by targeting AtR1.
Materials and methods
HUVECs
The human umbilical cords of healthy puerperant
women were collected in asepsis after parturition. The HUVECs were
isolated by treatment with 1% Trypsin, as previously described
(7). The HUVECs were grown on 1%
gelatin-coated culture plates in McCoy’s 5A (Sigma-Aldrich, St.
Louis, MO, USA) supplemented with 15% fetal bovine serum (Hyclone),
100 U/ml penicillin and 100 μg/ml streptomycin (Hyclone), at 37°C
in a humidified atmosphere of air containing 5% CO2. The
cells were used to within 2–3 passages and were identified as
endothelial by their cobblestone monolayer appearance in culture
and by the expression of cell-surface antigens CD34 and CD31, as
detected by flow cytometry (FCM) assay.
Vector construction
For expression plasmid pRNAT-CMV32-miR155 (pre-miRNA
of miR-155 expression element), oligonucleotide pairs for pre-miRNA
of miR-155 and linker sequences with BamHI and XhoI
sites were chemically synthesized by GenePharma Co., Ltd.
(Shanghai, China). The sequences of the oligonucleotides were: top
strand, 5′-GT ggatccCTGTTAATGCTAATCGTGATAGGGGTTTTTGC
CTCCAACTGACTCCTACATATTAGCATTAACAGctcgag CC-3′, and bottom
strand, 5′-GGctcgagCTGTTAATGCTAAT
ATGTAGGAGTCAGTTGGAGGCAAAAACCCCTATCAC
GATTAGCATTAACAGggatccAC-3′ (sequences corresponding to
miR-155 seed sequences in capitalized, underlined and bold, and
restriction enzyme sites in lower case and underlined). To built
the expression plasmid the pairs of oligos were annealed and
inserted into the multiple cloning sites between of BamHI
and XhoI sites in the pRNAT-CMV32/Neo vector (GenScript,
Piscataway, NJ, USA). The negative control plasmid
pRNAT-CMV32-miR155-Mut were similarly built, except that 11
nucleotides in sequences corresponding to miR-155 see sequences
were mutated (change TTAATGCTAAT to
TaAtTcCaAtT,
mutations shown in lower-case). Co-transfection of HUVECs from
patients or healthy women were conducted to transfer 0.3 μg miR-155
expressed vector or miR-155 mutant vector, respectively, with
Lipofectamine 2000 reagent according to the manufacturer’s
protocol. The cells were seeded in a 6-well plate in McCoy’s 5A
(Sigma-Aldrich) supplemented with 15% fetal bovine serum, 100 U/ml
penicillin and 100 μg/ml streptomycin (were from Hyclone), at 37°C
in a humidified atmosphere of air containing 5% CO2,
until 80% confluent.
Luciferase report assay
All steps of this assay were performed as previously
described (6,7). NIH-3T3 cells were seeded at
3×104/well in 48-well plates and co-transfected with 400
ng of pRNAT-CMV32-miR-155 or pRNAT-CMV32 or pRNAT-CMV32-miR155-Mut,
20 ng of pGL-ATR1-3UTR-WT or pGL-ATR1-3UTR-Mut, and pRL-TK
(Promega, Madison, WI, USA) using Lipofectamine 2000 reagent
according to the manufacturer’s protocol. After 48 h transfection,
luciferase activity was measured using the dual-luciferase reporter
assay system (Promega).
MTT assay for cell proliferation
Each group of HUVECs was seeded at
2×103/well in 96-well plates and cultured in McCoy’s 5A
supplemented with 15% fetal bovine serum at 37°C with 5%
CO2, until 85% confluent. MTT (Sigma-Aldrich) reagent (5
mg/ml) was added to the maintenance cell medium at different time
points, and incubated at 37°C for an additional 4 h. The reaction
was terminated with 150 μl dimethylsulfoxide (DMSO; Sigma-Aldrich)
per well and the cells were lysed for 15 min; the plates were
gently shaken for 5 min. Absorbance values were determined by using
the enzyme linked immunosorbent assay (ELISA) reader (Model 680;
Bio-Rad) at 490 nm.
RNA extraction and analysis by
quantitative real-time PCR (qRT-PCR)
Total RNA from each cell was isolated using TRIzol
reagent (Invitrogen) according to the manufacturer’s protocol. The
RNA samples were treated with DNase I (Sigma-Aldrich), quantified,
and reverse-transcribed into cDNA using the ReverTra Ace-α First
Strand cDNA Synthesis kit (Toyobo). qRT-PCR was conducted using a
RealPlex4 real-time PCR detection system from Eppendorf Co., Ltd.
(Germany), with SYBR-Green Realtime PCR Master MIX (Toyobo) used as
the detection dye. qRT-PCR amplification was performed over 40
cycles with denaturation at 95°C for 15 sec and annealing at 58°C
for 45 sec. Target cDNA was quantified using the relative
quantification method. A comparative threshold cycle (Ct) was used
to determine gene expression relative to a control (calibrator) and
steady-state mRNA levels are reported as an n-fold difference
relative to the calibrator. For each sample, the marker gene Ct
values were normalized using the formula ΔCt = Ct_genes-Ct_18sRNA.
To determine relative expression levels, the following formula was
used ΔΔCt = ΔCt_sample_group-ΔCt_control_group. The values used to
plot relative expressions of markers were calculated using the
expression 2−ΔΔCt. The mRNA levels were calibrated based
on levels of 18 sec rRNA. The cDNA of each gene was amplified using
primers as previously described (7,15).
Western blot analysis
Total proteins extracts of each group cells were
resolved by 12% SDS-PAGE and transferred on PVDF (Millipore)
membranes. After blocking, the PVDF membranes were washed four
times for 15 min with TBST at room temperature and incubated with
primary antibody. Following extensive washing, membranes were
incubated with secondary peroxidase-linked goat anti-rabbit IgG
(1:1,000; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) for
1 h. After washing four times for 15 min with TBST at room
temperature once more, the immunoreactivity was visualized by
enhanced chemiluminescence (ECL kit; Pierce Biotechnology), and
membranes were exposed to Kodak XAR-5 film (Sigma-Aldrich).
Northern blot analysis
All steps of northern blotting were carried out as
previously described (7–11). For all groups, 20 μg of good
quality total RNA was analyzed on a 7.5 M ureum 12% PAA denaturing
gel and transferred to a Hybond N+ nylon membrane
(Amersham, Freiburg, Germany). Membranes were crosslinked using UV
light for 30 sec at 1,200 mjoule/cm2. Hybridization was
performed with the miR-155 antisense starfire probe,
5′-CCCCTATCACGATTAGCATTAA-3′ (Integrated DNA Technologies, Inc.,
Coralville, IA, USA), to detect the 22-nt miR-155 fragments
according to the manufacturer’s instructions. After washing,
membranes were exposed for 20–40 h to Kodak XAR-5 film
(Sigma-Aldrich). As a positive control, all membranes were
hybridized with a human U6 snRNA probe,
5′-GCAGGGGCCATGCTAATCTTCTCTG TATCG-3′. Exposure times for the U6
control probe varied between 15 and 30 min.
Flow cytometric (FCM) analysis of cell
cycle by PI staining
Each group cells was seeded at 3×105/well
in 6-well plates and cultured until 85% confluent. Each group cells
was washed by PBS three times and were then collected by
centrifugation (Allegra X-22R; Beckman Coulter) at 1,000 × g for 5
min. The cell pellets were the resuspended in 1 ml of PBS, fixed in
70% ice-cold ethanol, and kept in a freezer >48 h. Prior to FCM
analysis, the fixed cells were centrifuged, washed twice with PBS,
and resuspended in PI staining solution (Sigma-Aldrich) containing
50 μl/ml PI and 250 μg/ml RNase A (Sigma-Aldrich). The cell
suspension, which was hidden from light, was incubated for 30 min
at 4°C and analyzed using the FACS (BD FACSAria; BD Biosciences). A
total of 20,000 events were acquired for analysis using CellQuest
software.
Co-immunoprecipitation assay
All group cells were seeded at 3×105/well
in 6-well plates and cultured until 85% confluent; they were lysed
(500 μl/plate) in a modified cell lysis buffer for western blotting
and IP (20 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100, 1 mM
EDTA, sodium pyrophosphate, β-glycerophosphate,
Na3VO4 and leupeptin) (Beyotime Institute of
Biotechnology). Following lysis, each sample was centrifuged to
clear the lysate of the insoluble debris and pre-incubated with 20
μg protein A agarose beads (Beyotime Institute of Biotechnology) by
rocking for 30 min at 4°C, followed by centrifugation and transfer
to a fresh 1.5 ml tube. The rabbit anti-human ERK1/2 polyclonal
antibody (Cell Signaling Technology, USA) was incubated for 90 min
before re-addition of 20 μg protein A agarose beads to capture the
immune complexes. The pelleted beads were then washed three times
with 500 μl cell lysis buffer, dissolved in 4× SDS-PAGE sample
loading buffer, and heated for 10 min at 95°C.
Immunofluorescence staining analysis of
relative protein expression
The cultured cells were washed three times with PBS
and fixed with 4% paraformaldehyde (Sigma-Aldrich) for 30 min.
After blocking, the cells were incubated first with rabbit
anti-human Angiotensin II polyclonal antibody (1:200;) and rabbit
anti-human ERK1/2 or pERK1/2 polyclonal antibody (1:200; were from
Santa Cruz Biotechnology, Inc.) overnight at 4°C, and then with
FITC- or Cy3-conjugated goat anti-rabbit IgG antibody (1:200;
Abcam, Cambridge, UK) and 5 mg/ml DAPI (Sigma-Aldrich) at room
temperature for 30 min. Then the cells were thoroughly washed with
TBST and viewed through a fluorescence microscope (DMI3000; Leica,
Allendale, NJ, USA).
Mitochondrial membrane potential
(ΔΨm)
JC-1 probe (Beyotime Biotechnology, Shanghai, China)
was employed to measure mitochondrial membrane potential
(ΔΨm) depolarization. All steps of northern blotting were
according to previous reports (15,16). Briefly, cells cultured in 6-well
plates after indicated treatments were incubated with an equal
volume of JC-1 staining solution (5 μg/ml) at 37°C for 20 min and
rinsed twice with PBS. ΔΨm was monitored by determining the
relative amounts of dual emissions from mitochondrial JC-1 monomers
or aggregates using a fluorescence microscope (DMI3000; Leica)
under Argon-ion 488 nm laser excitation. Mitochondrial
depolarization is indicated by an increase in the green/red
fluorescence intensity ratio.
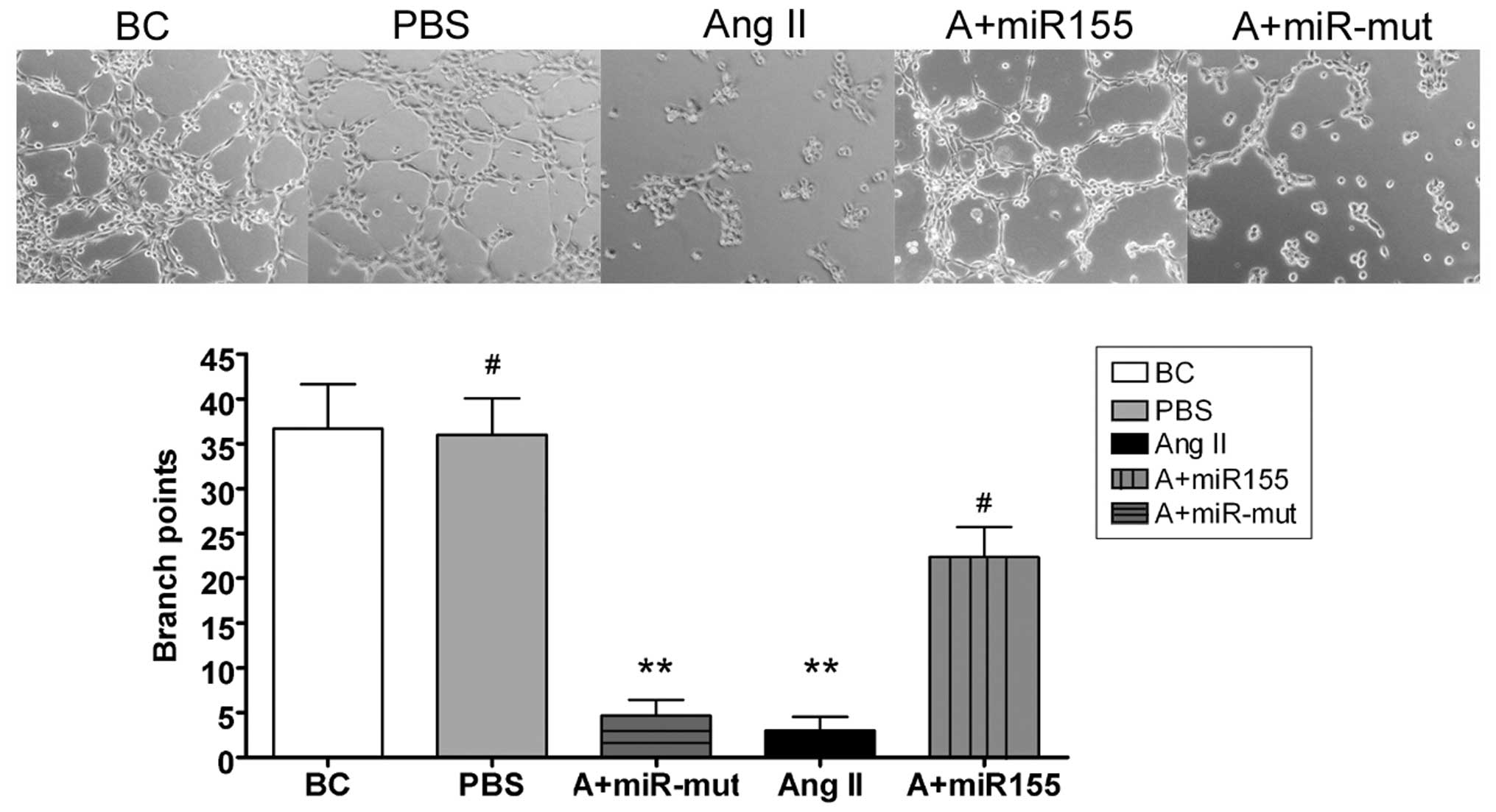
Capillary tubule formation assay
All steps of northern blotting were performed
according to previous reports (16,17). In brief, HUVECs were plated on
matrigel-coated 6 chamber slides (2×105 cells/chamber)
in the presence and absence of various test substances described in
the previous section for the cell migration assay. After 6 h of
incubation in a CO2 incubator, cells were photographed.
To quantitate the data, the number of branch points in four
non-overlapping fields was determined.
Statistical analysis
Each experiment was performed at least three times,
and data are shown as the means ± SE, where applicable, and
differences were evaluated using the Student’s t-test. P<0.05
was considered to indicate statistically significant
differences.
Results
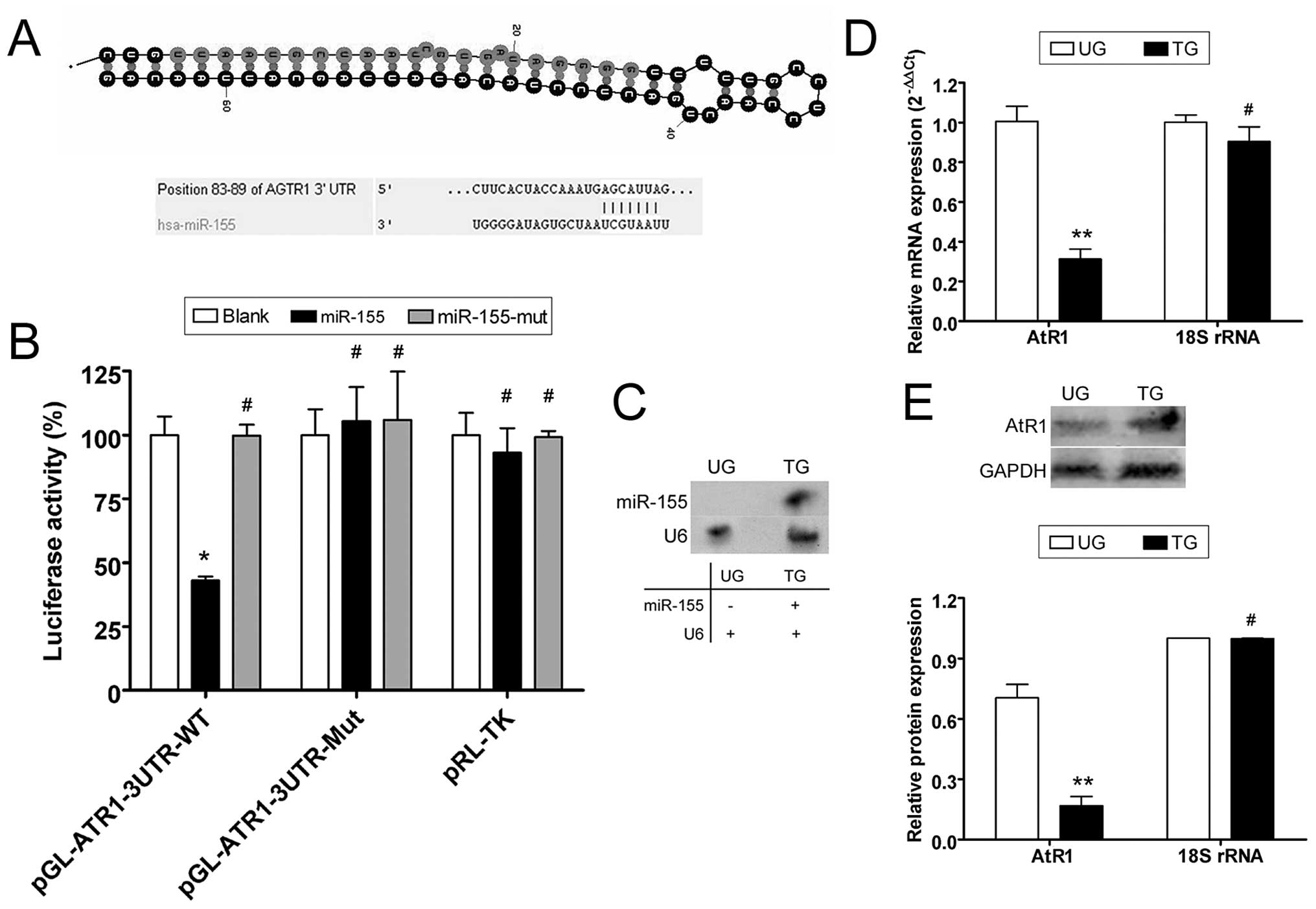
miR-155 interferes with ATR1
expression
According to the miRBase Target database (http://www.mirbase.org), the 3′ untranslated region
(3′-UTR) of AtR1 mRNA harbors 9–16 putative miRNA target sites,
depending on the species analyzed. We focused on human miR-155,
which may target the human AtR1 3′-UTR, since these sites are
conserved to varying degrees across species (Fig. 1). To identify possible miR-155
target genes, the 3′-UTR sites of AtR1 were cloned on a luciferase
activity assay plasmid. Plasmid DNA for wild-type and mutant AtR1
mRNA 3′-UTR and a blank plasmid were co-transfected with a miR-155
expression vector (wild-type, mutant and blank) in NIH-3T3 cells to
determine whether AtR1 gene expression is regulated by mature
miR-155. The luciferase activity of the AtR1 3′-UTR was
significantly inhibited by miR-155 (Fig. 1), whereas that of the mutated AtR1
3′-UTR was not. The data suggest that miR-155 targets AtR1.
Northern blotting detected the hybridization signal of mature
miR-155 in HUVECs transfected with miR-155 but not in untransfected
cells (Fig. 1). Quantitative
real-time PCR (qRT-PCR) and western blotting were used to determine
whether miR-155 interfered with AtR1 mRNA expression. According to
qRT-PCR, AtR1 mRNA expression was inhibited when the miR-155
expression plasmid was transfected into HUVECs (0.313±0.050),
compared with untransfected cells (1.006±0.076). Western blotting
confirmed that AtR1 protein expression was significantly reduced in
miR-155-transfected HUVECs (0.168±0.045) compared with
untransfected cells (0.705±0.066). These data indicate that
exogenous miR-155 downregulated the expression of endogenous AtR1
in HUVECs (Fig. 1).
Ang II influences HUVEC proliferation in
vitro
To determine whether exogenous Ang II can suppress
HUVEC proliferation, inhibition with 0, 5, 10, 20, 40, 80 and 120
ng/μl Ang II was measured by the MTT proliferation assay (Fig. 2). There were no significant
differences between the HUVEC-Ang II group and the HUVEC-PBS group
with Ang II concentrations of 0–20 ng/μl after 24 h. However, with
concentrations of 40–120 ng/μl, the HUVEC-PBS group was
significantly less susceptible to the proliferation inhibitory
effect of Ang II (Table I).
Furthermore, exogenous Ang II suppressed the proliferation of
HUVECs in a concentration-dependent manner, with a half maximal
inhibitory concentration of 68.94 ng/μl according to the MTT cell
proliferation assay.
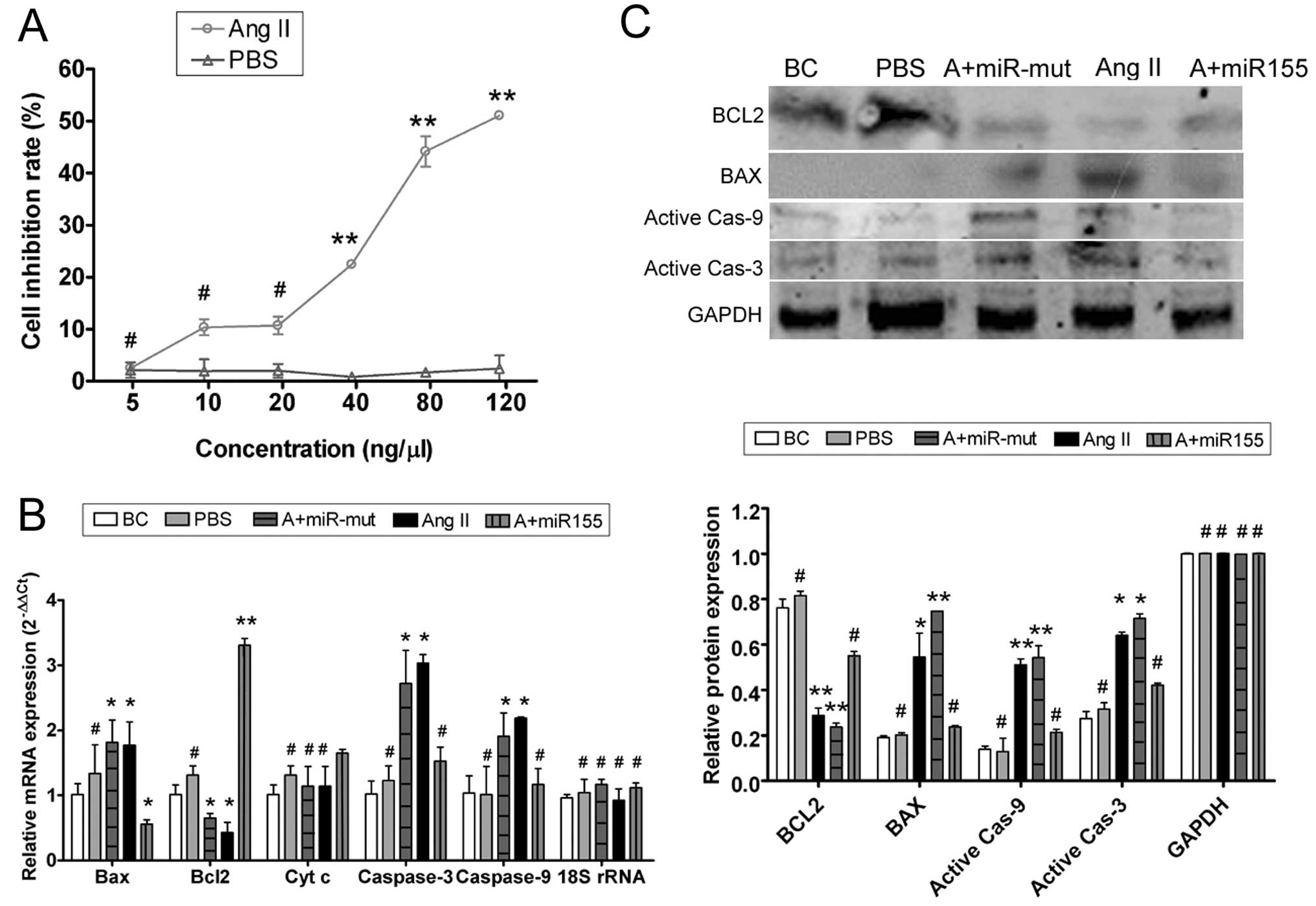 | Figure 2Ang II affects HUVEC proliferation
in vitro and miR-155 attenuates that effect. (A) MTT
proliferation assay was used to show that exogenous Ang II
inhibited HUVEC proliferation in a dose-dependent manner, with a
half maximal inhibitory concentration of 68.94 ng/μl.
**P<0.01 vs. HUVEC-PBS group; #P>0.05
vs. HUVEC-PBS group; n=3. (B) qRT-PCR showed that mRNA expression
of the apoptosis inhibitor Bcl-2 was markedly lower in the
HUVEC-Ang II group (Ang II) than in the HUVEC-PBS group (PBS) and
the blank control (BC). mRNA expression of the apoptosis-promoting
factors Bax, cytochrome c, caspases-9 and -3 was markedly
higher in the HUVEC-Ang II group (Ang II) than in the PBS and the
BC groups. However, after transfection with miR-155, mRNA
expression of Bcl-2 in the miR-155-transfected group (A+miR155) was
markedly elevated; Bax, cytochrome c, caspases-9 and -3 were
markedly decreased in this group, but not in the miR-155
mutant-transfected group (A+miR-mut). *P<0.05 vs. BC;
**P<0.01 vs. BC; #P>0.05 vs. BC; n=3.
(C) Western blotting showed that Bcl-2 protein expression was
significantly reduced, but Bax, active caspase-9 and active
caspase-3 were significantly increased in the HUVEC-Ang II group
(Ang II) compared with the PBS and the BC groups. Bcl-2 protein
expression was significantly increased in the miR-155-transfected
group (A+miR155) compared with the HUVEC-Ang II group (Ang II) and
the miR-155 mutant-transfected group (A+miR-mut), and Bax, active
caspase-9 and active caspase-3 in this group were markedly
decreased. *P<0.05 vs. BC; **P<0.01 vs.
BC; #P>0.05 vs. BC; n=3. |
 | Table IDifferent cellular growth inhibition
and exogenous Ang II concentration detected by MTT assay. |
Table I
Different cellular growth inhibition
and exogenous Ang II concentration detected by MTT assay.
| Exogenous Ang II
concentration (ng/μl) | HUVEC-Ang II group
(n=3) | HUVEC-PBS group
(n=3) |
|---|
| 5 | 2.53±1.11 | 2.14±1.47 |
| 10 | 10.36±1.55 | 1.96±2.25 |
| 20 | 10.72±1.72 | 2.03±1.33 |
| 40 | 22.44±0.21 | 0.84±0.76 |
| 80 | 44.17±2.93 | 1.71±0.41 |
| 120 | 51.06±0.70 | 2.47±2.47 |
Exogenous Ang II induces HUVEC damage and
apoptosis
To determine whether exogenous Ang II could induce
apoptosis in HUVECs, qRT-PCR and western blotting were used to
measure the expression levels of apoptosis related factors
(Fig. 2). qRT-PCR showed that
mRNA expression of the apoptosis inhibitor Bcl-2 was markedly lower
in the HUVEC-Ang II group than in the HUVEC-PBS group and the blank
controls (BCs). By contrast, mRNA expression levels of the
apoptosis-promoting factors Bax, cytochrome c, caspases-9
and -3 were markedly higher in the HUVEC-Ang II group than in the
HUVEC-PBS group and the BCs (Table
II). Western blotting confirmed that Bcl-2 protein expression
was significantly reduced in the HUVEC-Ang II group compared with
the HUVEC-PBS group and the BCs. These data indicate that exogenous
Ang II activates the expression of apoptosis related proteins and
thereby induces apoptosis.
| Table IIDetection of mRNA expression of
apoptosis-related genes by qRT-PCR. |
Table II
Detection of mRNA expression of
apoptosis-related genes by qRT-PCR.
| Gene name | BC | PBS | A+miR-mut | Ang II | A+miR155 |
|---|
| Bax | 1.01±0.16 | 1.34±0.44 | 1.82±0.34 | 1.77±0.36 | 0.56±0.07 |
| Bcl-2 | 1.01±0.15 | 1.31±0.14 | 0.65±0.07 | 0.43±0.16 | 3.31±0.10 |
| Cyt c | 1.01±0.15 | 1.31±0.14 | 1.14±0.30 | 1.14±0.30 | 1.65±0.05 |
| Caspase-3 | 1.02±0.20 | 1.23±0.22 | 2.72±0.50 | 3.03±0.13 | 1.53±0.22 |
| Caspase-9 | 1.03±0.27 | 1.01±0.43 | 1.91±0.36 | 2.19±0.02 | 1.17±0.25 |
| 18S rRNA | 0.96±0.05 | 1.04±0.20 | 1.17±0.08 | 0.92±0.18 | 1.12±0.07 |
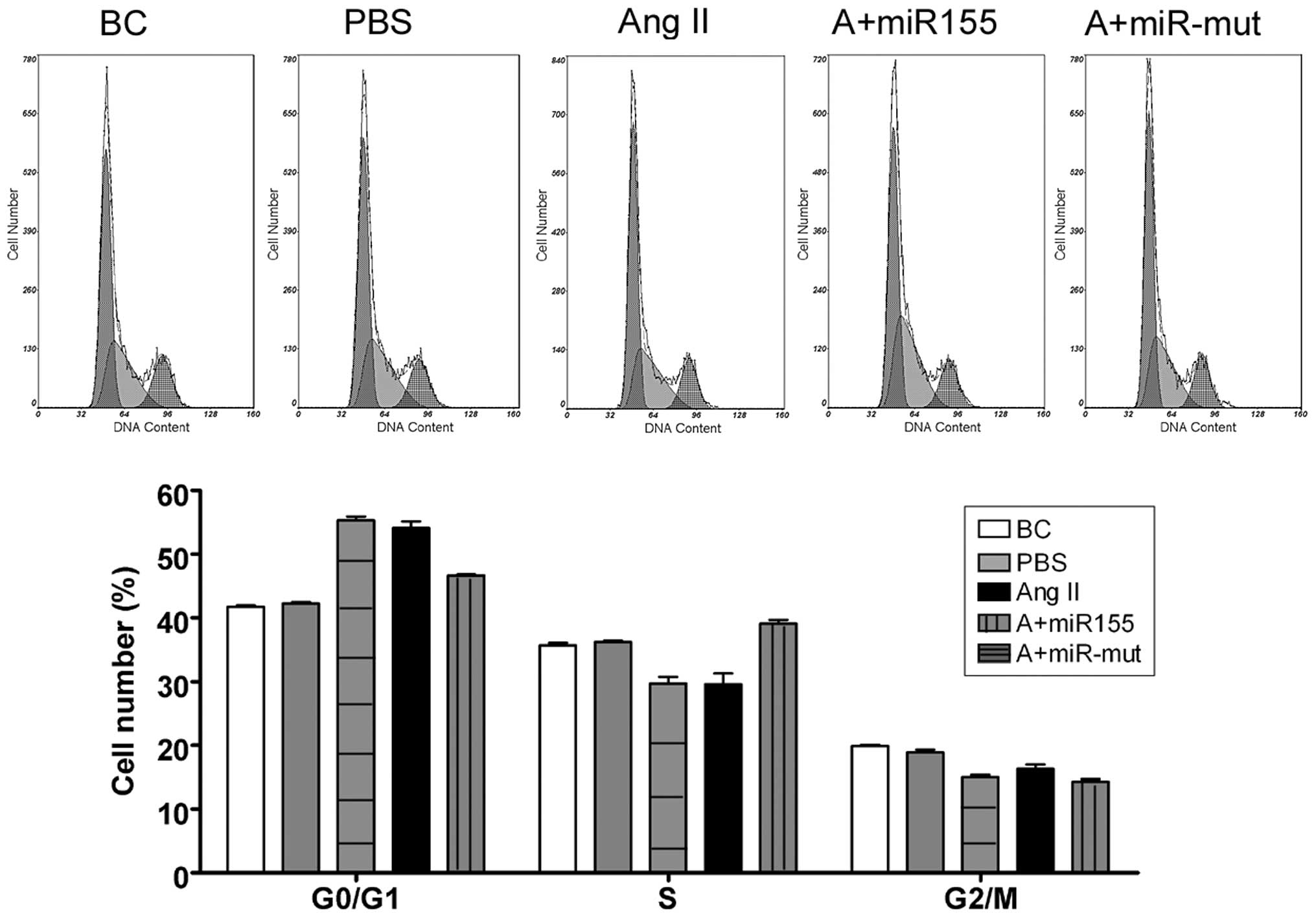
We used FCM to determine whether Ang II influences
the cell cycle in HUVECs. There were no significant changes in the
cell cycle distribution of the BCs and the HUVEC-PBS group
(Fig. 3). By contrast, the
HUVEC-Ang II group was arrested in the G0/G1 phase of the cell
cycle and the percentage of cells in the S phase was significantly
decreased. These results suggest that exogenous Ang II
significantly affects cell cycle regulation in HUVECs in
vitro.
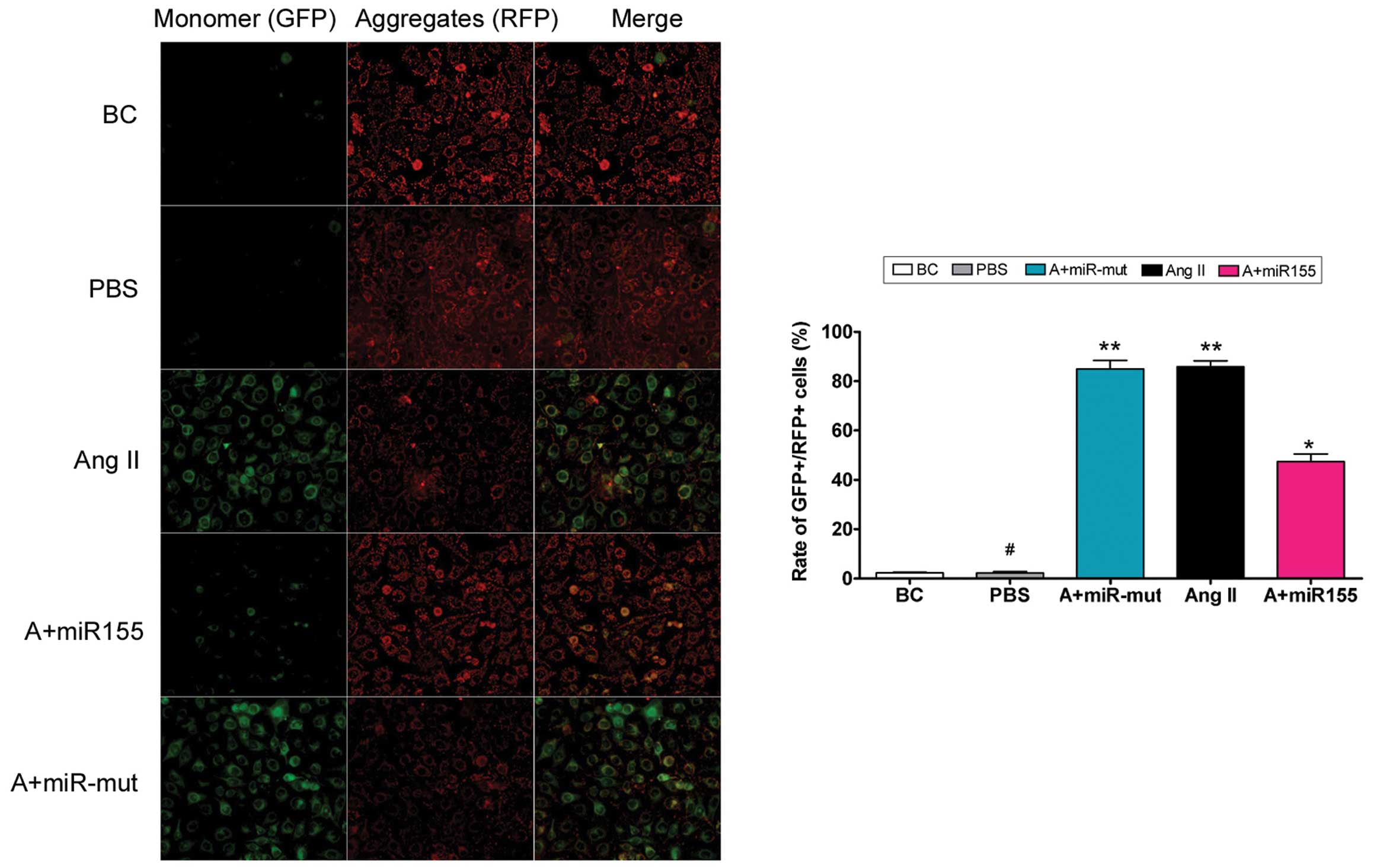
To determine whether exogenous Ang II in the cell
culture medium damages mitochondria, mitochondrial membrane
depolarization was examined. According to previous studies
(15,16), normal HUVECs stained with JC-1
emit mitochondrial orange-red fluorescence with a little green
fluorescence (Fig. 4). However,
compared with the HUVEC-Ang II group, Ang II further decreased the
ΔΨm and produced obvious green fluorescence in the HUVEC-PBS
group. Thus, exogenous Ang II damages mitochondria in HUVECs.
miR-155 attenuates the effect of
exogenous Ang II
To investigate the effect of miR-155 expression,
qRT-PCR and western blotting were used to determine the expression
levels of apoptosis related factors (Fig. 2). qRT-PCR showed that the mRNA
expression of Bcl-2 in miR-155-transfected cells was markedly
elevated, whereas Bax, cytochrome c, caspases-9 and -3 in
this group were markedly decreased (Table II). Western blotting showed that
Bcl-2 protein expression was significantly increased in
miR-155-transfected cells compared with the HUVEC-Ang II group and
the miR-155 mutant-transfected group; Bax, active caspase-9 and
active caspase-3 were markedly decreased. These data indicate that
miR-155 attenuates the expression of exogenous Ang II-induced
apoptosis related proteins. FCM showed no significant differences
in cell cycle distribution between the HUVEC-Ang II group and the
miR-155 mutant-transfected group (Fig. 3). By contrast, the cell cycle in
the miR-155-transfected group was more ameliorated. Compared with
the miR-155-transfected group, Ang II further increased ΔΨm
and produced obvious orange-red fluorescence but not green
fluorescence (Fig. 4). This
difference in ΔΨm between the different groups indicates
that miR-155 attenuates exogenous Ang II-induced mitochondrial
damage in HUVECs.
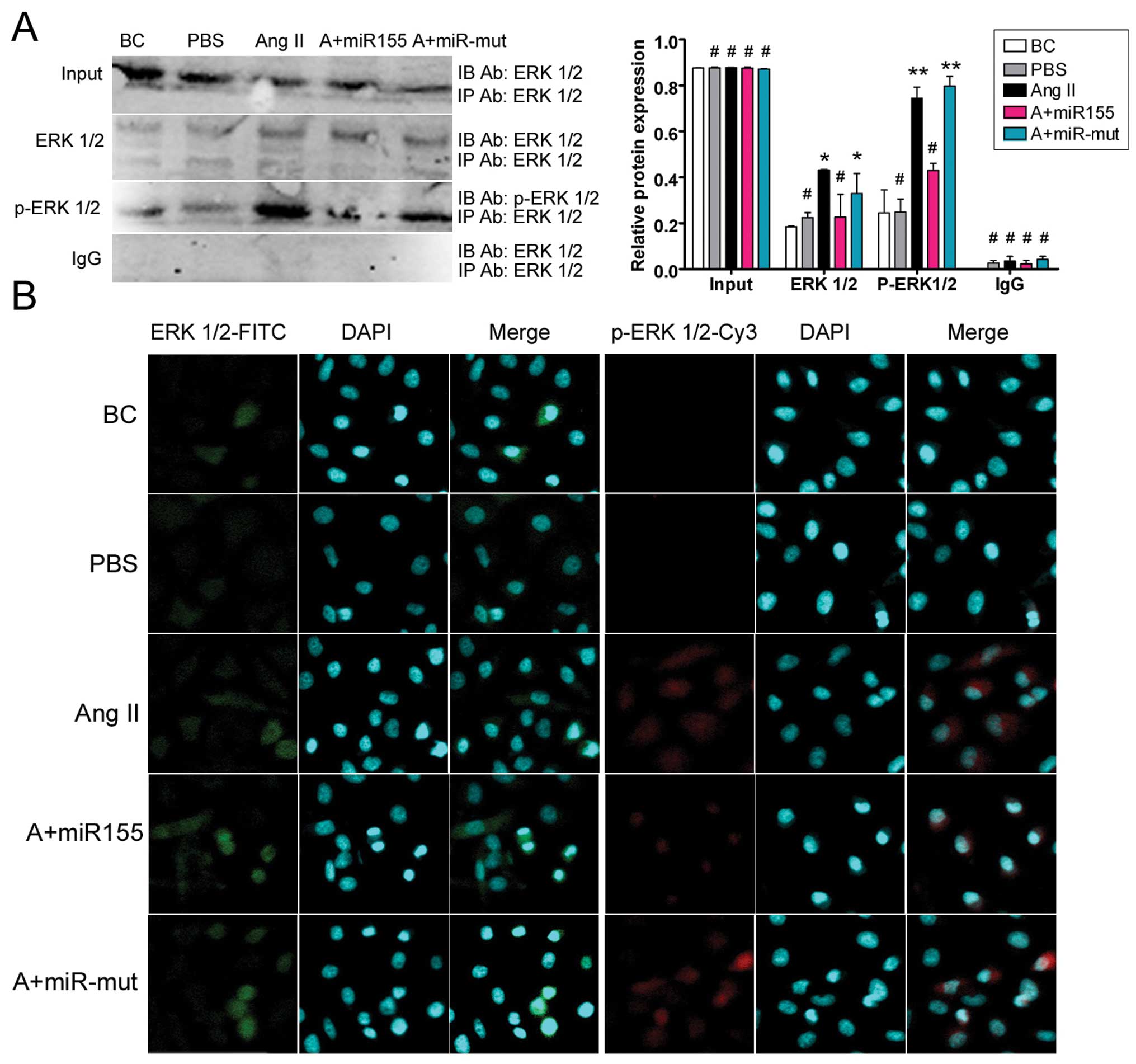
miR-155 reduces Ang II-induced ERK1/2
activation and phosphorylation
In previous studies, Ang II-induced AtR1 activation
was shown to induce ERK1/2 activation and phosphorylation, leading
to cellular hypertrophy and apoptosis (6,7).
To determine whether miR-155 suppression of At1R expression in
HUVECs can reverse the above phenomena, phospho-ERK 1/2 levels were
determined by co-immunoprecipitation (co-IP) western blotting.
There were no obvious differences in the levels of normal ERK1/2
and phospho-ERK1/2 expression between the BCs and the HUVEC-PBS
group (Fig. 5). When exogenous
Ang II was added to the culture medium, the levels of
phospho-ERK1/2 in the HUVEC-Ang II group and the miR-155
mutant-transfected group were 0.745±0.048 and 0.797±0.043 of their
baseline levels, respectively. These values were significantly
higher than those for the miR-155 transfected group, the BCs and
the HUVEC-PBS group (0.430±0.03, 0.244±0.100 and 0.249±0.056 of
baseline, respectively). Immunofluorescence staining showed that
ERK1/2 and phospho-ERK1/2 protein expression were both decreased in
the miR-155-transfected cells compared with the HUVEC-Ang II group
and the miR-155 mutant-transfected cells after stimulation with
exogenous Ang II (Fig. 5). The
results of co-IP western blotting suggest that miR-155
significantly reduces Ang II-induced activation of the MAPK
ERK1/2.
miR-155 maintains HUVEC migration and
capillary tubule formation
We also investigated whether exogenous Ang II can
influence HUVEC migration and capillary tubule formation in
vitro, and whether miR-155 can maintain these abilities. Our
experimental results showed that HUVECs can form capillary tubules
in migration gels under normal conditions (HUVEC-BC, 37±5;
HUVEC-PBS, 36±4). When the cell culture medium contained exogenous
Ang II, HUVEC migration and capillary tubule formation were
markedly reduced (HUVEC-Ang II, 3±2). However, after transfection
with miR-155, the migration and capillary tubule formation
abilities of the HUVEC were maintained in the presence of Ang II
(22±3) (Fig. 6).
Discussion
Several studies have shown that angiotensin and
angiogenic factors in the renin-angiotensin system are crucial
markers of cardiovascular function. However, an aberrant or
inadequate response to angiotensin-induced stress can cause
vascular injury and cardiovascular disease (1–7,15–19). Under normal circumstances, the
body regulates excess stress signaling to maintain a dynamic
equilibrium. Previous studies have shown changes in the function of
miRNAs during stress (3). For
example, expression of miR-155 and AtR1 differ between the HUVECs
of severely pre-eclamptic pregnant women and those of normal
pregnant women (7). In view of
this evidence, we investigated whether endogenous miR-155 is
specifically expressed to block AtR1, to avoid cell damage from the
stress caused by overstimulation with exogenous Ang II. We used
HUVECs as a model and simulated them with Ang II. Exogenous Ang II
induced HUVEC proliferation in a concentration-dependent manner.
One group of HUVECs was then transfected with miR-155. We found
that: i) miR-155 maintained cell cycle stability in HUVECs,
ensuring normal division and proliferation via suppression of
endogenous At1R expression; and, ii) with miR-155 overexpression,
HUVEC ΔΨm was almost unchanged following stimulation with
exogenous Ang II. Thus, miR-155 can maintain mitochondrial
stability via suppression of endogenous AtR1 expression to resist
Ang II-induced stress. High concentrations of Ang II hindered the
ability of HUVECs to form capillary tubules in the extracellular
matrix in vitro. However, with overexpression miR-155, HUVEC
migration and capillary tubule formation were not influenced by Ang
II.
Further experiments showed that exogenous miR-155
can reduce Ang II-induced activation and phosphorylation of ERK1/2
in HUVECs to prevent apoptosis. As is well known, ERK1/2 is an
important subfamily of MAPKs that controls a broad range of
cellular activities and physiological processes. ERK1/2 is
activated transiently or persistently by MEK1/2 and by upstream
MAP3Ks in conjunction with scaffolding proteins and phosphatases,
and its activation may have pro-apoptotic functions (1,19).
In this study, we found that miR-155 enhances HUVEC growth by
inhibiting the MAPK pathway through targeting angiogenic signaling,
mainly via AtR1 expression.
In conclusion, HUVECs avoided the damage that may be
caused by continuous Ang II-induced stress by selectively targeting
miR-155 to silence the AtR1 gene and prevent extracellular Ang II
signaling from influencing the intracellular environment. In this
manner, cell response to Ang II-induced stress is blocked via
suppression of AtR1 by miR-155. miR-155 is thus implicated in
pathological angiogenesis in vascular injury and cardiovascular
disease, and represents a potential therapeutic target in these
disorders.
Acknowledgements
This study was supported by a grant from the
Shanghai Municipal Health Bureau Fund (no. 2010225) and for
Traditional Chinese Medicine Scholars (no. 2010L046A) to C.C.
References
|
1
|
Mendell JT and Olson EN: MicroRNAs in
stress signaling and human disease. Cell. 148:1172–1187. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Caporali A and Emanueli C: MicroRNA
regulation in angiogenesis. Vascul Pharmacol. 55:79–86. 2011.
View Article : Google Scholar
|
|
3
|
Leung AK and Sharp PA: MicroRNA functions
in stress responses. Mol Cell. 40:205–215. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Martin MM, Buckenberger JA, Jiang J, et
al: The human angiotensin II type 1 receptor +1166 A/C polymorphism
attenuates microRNA-155 binding. J Biol Chem. 282:24262–24269.
2007.
|
|
5
|
Martin MM, Lee EJ, Buckenberger JA,
Schmittgen TD and Elton TS: MicroRNA-155 regulates human
angiotensin II type 1 receptor expression in fibroblasts. J Biol
Chem. 281:18277–18284. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zheng L, Xu CC, Chen WD, et al:
MicroRNA-155 regulates angiotensin II type 1 receptor expression
and phenotypic differentiation in vascular adventitial fibroblasts.
Biochem Biophys Res Commun. 400:483–488. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cheng W, Liu T, Jiang F, et al:
microRNA-155 regulates angiotensin II type 1 receptor expression in
umbilical vein endothelial cells from severely pre-eclamptic
pregnant women. Int J Mol Med. 27:393–399. 2011.PubMed/NCBI
|
|
8
|
Cheng W, Liu T, Wan X, Gao Y and Wang H:
MicroRNA-199a targets CD44 to suppress the tumorigenicity and
multidrug resistance of ovarian cancer-initiating cells. FEBS J.
279:2047–2059. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Liu T, Cheng W, Gao Y, Wang H and Liu Z:
Microarray analysis of microRNA expression patterns in the semen of
infertile men with semen abnormalities. Mol Med Rep. 6:535–542.
2012.PubMed/NCBI
|
|
10
|
Liu T, Cheng W, Huang Y, Huang Q, Jiang L
and Guo L: Human amniotic epithelial cell feeder layers maintain
human iPS cell pluripotency via inhibited endogenous microRNA-145
and increased Sox2 expression. Exp Cell Res. 318:424–434. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Liu T, Chen Q, Huang Y, Huang Q, Jiang L
and Guo L: Low microRNA-199a expression in human amniotic
epithelial cell feeder layers maintains human-induced pluripotent
stem cell pluripotency via increased leukemia inhibitory factor
expression. Acta Biochim Biophys Sin (Shanghai). 44:197–206. 2012.
View Article : Google Scholar
|
|
12
|
Patrick DM, Montgomery RL, Qi X, et al:
Stress-dependent cardiac remodeling occurs in the absence of
microRNA-21 in mice. J Clin Invest. 120:3912–3916. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Thum T, Gross C, Fiedler J, et al:
MicroRNA-21 contributes to myocardial disease by stimulating MAP
kinase signalling in fibroblasts. Nature. 456:980–984. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
van Rooij E, Quiat D, Johnson BA, et al: A
family of microRNAs encoded by myosin genes governs myosin
expression and muscle performance. Dev Cell. 17:662–673.
2009.PubMed/NCBI
|
|
15
|
Zhang XD, Borrow JM, Zhang XY, Nguyen T
and Hersey P: Activation of ERK1/2 protects melanoma cells from
TRAIL-induced apoptosis by inhibiting Smac/DIABLO release from
mitochondria. Oncogene. 22:2869–2881. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang Z, Tang X, Li Y, et al:
20-Hydroxyeicosatetraenoic acid inhibits the apoptotic responses in
pulmonary artery smooth muscle cells. Eur J Pharmacol. 588:9–17.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Markowska AI, Liu FT and Panjwani N:
Galectin-3 is an important mediator of VEGF- and bFGF-mediated
angiogenic response. J Exp Med. 207:1981–1993. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Henriksen EJ and Prasannarong M: The role
of the renin-angiotensin system in the development of insulin
resistance in skeletal muscle. Mol Cell Endocrinol. May
4–2012.(Epub ahead of print).
|
|
19
|
Lu Z and Xu S: ERK1/2 MAP kinases in cell
survival and apoptosis. IUBMB Life. 58:621–631. 2006. View Article : Google Scholar : PubMed/NCBI
|