Introduction
Regenerating the heart through cell transplantation
is a promising novel approach in the therapy of myocardial infarct
and various investigations have provided evidence that this
approach has indeed the potential to improve the functionality of
the injured heart (1,2). However, a meta-analysis with nearly
a thousand patients concluded that bone marrow derived cell
transplantation resulted in only a modest 3.66% improvement of the
left ventricular ejection fraction in patients with ischemic heart
disease (3–5), which falls well below the
expectations towards this new therapeutic approach. These
disappointing results could be the consequence of the unclear
underlying mechanism of action of the therapeutic cells which may
involve various mechanisms such as paracrine and direct
cell-to-cell effects (6–10). Understanding the mechanisms of
action could lead to better optimalization of the used protocols.
On the other hand, most of the therapeutically added cells die in
the noxious and aggressive environment of ‘postischemic’ myocardium
(11–13). While some experimental evidence
suggest that the effect is mostly due to apoptotic cells which
secrete factors that could protect cells after myocardial infarct
(14), it is reasonable to
hypothesize that if more cells survived after grafting, then their
actions, either paracrine or cell-to-cell, could be more efficient
and, thus, the treatments could be more effective. Evidence
supporting this concept has already been validated using various
pretreatment methods, such as ‘priming’ with growth factors or
modifying with Akt (15–18). A further possibility to enhance
survival could be to prepare cells for the oxidative stress present
in the reperfused cardiac tissue. Oxidative stress induced pathways
play a major role in the development of ‘postischemic’ injuries in
the heart (19–21) and these involve poly(ADP-ribose)
polymerase (PARP) activation (22,23). PARP is an energy-consuming enzyme
that functions primarily as a DNA damage sensor in the nucleus and
catalyzes the cleavage of NAD+ into nicotinamide and
ADP-ribose, then transfers ADP ribose units to nuclear proteins
such as histons and transcription factors. As a result of this
process, the intracellular NAD+ and ATP levels
remarkably decrease, resulting in cell dysfunction and cell death
(24). Recent studies have also
implicated the importance of mitochondrial dysfunction and
mitochondrial cell death factors, including apoptosis-inducing
factors in the process of oxidant-induced cell death and the
potential role of PARP in regulating these factors in various cell
types including myocardial cells (25–28).
Previous studies have demonstrated the direct
protective effect of PARP inhibition of cells or tissues undergoing
ischemia-reperfusion (I-R) injury (23,29–31). Our aim was to assess the potential
of PARP inhibitor pretreatment in a cell-based therapy setting
where the added therapeutic cells received the pretreatment.
Accordingly, we used a reductionist in vitro model of
cell-based therapy in myocardial infarct where the therapeutically
added cells were pretreated with PARP inhibitor and we investigated
if improved survival of the therapeutic cells could enhance the
viability of cells undergoing simulated I-R injury.
Materials and methods
Cell culture
H9c2 rat cardiomyoblasts were purchased from ATCC
(Wesel, Germany). Cells were cultured in high glucose (4.5 g/l)
DMEM containing 10% fetal bovine serum, 4 mM L-glutamine, 100 U/ml
penicillin and 100 μg/ml streptomycin at 37°C in a humidified
atmosphere of 5% CO2. Cell culture media were changed
every 2–3 days and cells were sub-cultured once they reached 70–80%
confluence. Cells between passages 7 and 13 were used in the
experiments.
Simulated ischemia-reperfusion model
Myocardial I-R was simulated in vitro on H9c2
rat cardiomyoblast cell cultures based on the method of Cselenyák
et al(9) with
modifications. Briefly, to mimic the ischemic conditions, cells
were incubated in glucose-free DMEM in an atmosphere of 0.5%
O2 and 99.5% N2 for 160 min on the stage of a
confocal microscope (PeCon Incubation System, Erbach-Bach,
Germany). Glucose was replaced with fresh high glucose DMEM and the
cells were placed in standard cell culture conditions (37°C, 5%
CO2) until further experimental actions.
Malondialdehyde measurement
Malondialdehyde (MDA) formation was used to quantify
the lipid peroxidation in our simulated I-R model and was measured
as thiobarbituric acid reactive material. According to the
detection limit of the assay protocol 1,000,000 cells were used.
Five hours after the start of simulated reperfusion 50 μl of the
cell culture supernatant was added to a reaction mixture consisting
of 50 μl of 8.1% sodium dodecyl sulfate, 375 μl of 20% acetic acid
(pH 3.5), and 150 μl of distilled water. The mixture was completed
with 375 μl of freshly prepared, boiling hot thiobarbituric acid
(0.8%) and incubated at 95°C for 1 h. After cooling to room
temperature, 200 μl supernatant was transferred into 96-well
microplates and the absorbance was measured at 532 nm (32). Malonaldehyde bis(dimethyl acetal),
thiobarbituric acid and sodium dodecyl sulfate were purchased from
Sigma-Aldrich (St. Louis, MO, USA).
In vitro model of cell based therapy
Based on our previous experiments (33), we used H9c2 cells as therapeutic
cells which were added to the damaged cells 30 min after the start
of simulated reperfusion and set a simulated ischemia so severe
that the untreated therapeutic cells were not able to improve
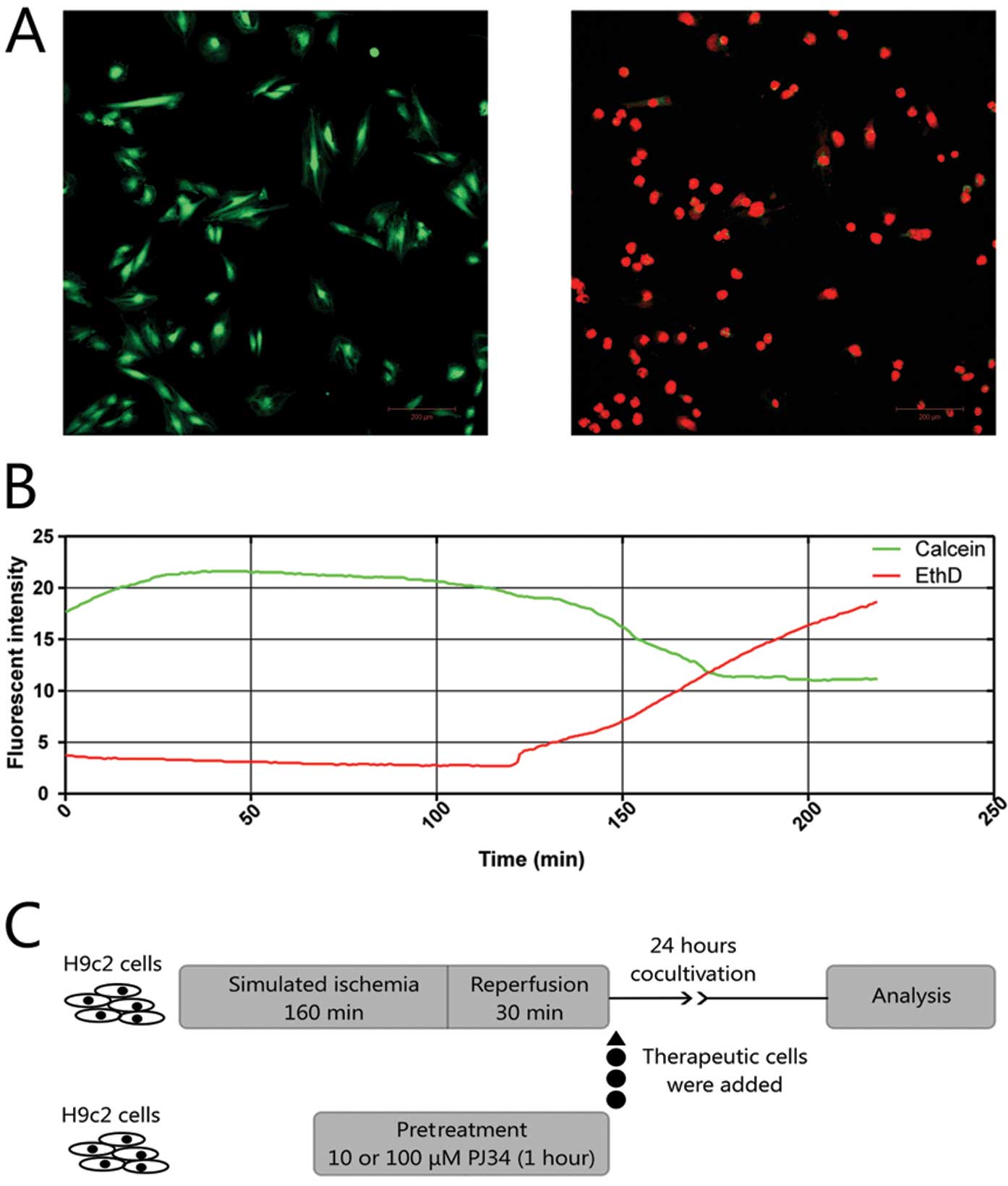
survival. The optimal time of simulated ischemia was set using time
lapse video microscopy to follow the changes in the fluorescent
intensity of calcein-AM/ethidium-homodimer-2 (EthD) viability
staining (Fig. 1A). After ~2 h,
rapid decrease occurred in the calcein-AM intensity and at the same
time the intensity of the dead cell indicator EthD increased
(Fig. 1B). For the experiments we
chose an ischemia length of 160 min. The used therapeutic cells
were divided into 3 experimental groups according to their
pretreatment protocol lasting 1 h: 1) control group treated with
saline vehicle; 2) 10 μM PJ34 (PARP inhibitor; Inotek
Pharmaceuticals Corp., Beverly, MA, USA); 3) 100 μM PJ34. After the
pretreatment, the H9c2 cells were washed twice with PBS to ensure
no PARP inhibitor remained, trypsinized, collected by
centrifugation and 20,000 cells were given the appropriate wells of
the 12-well plate containing 30,000 ‘postischemic’ cells that
underwent simulated I-R. Cells were co-cultivated for 24 h before
further flow cytometric, lactate dehydrogenase (LDH) release and
metabolic activity measurements (Fig.
1C).
Lactate dehydrogenase measurements
We used measurements of LDH release for three
reasons: i) to evaluate the I-R model; ii) to describe the
cytotoxicity and efficacy of PJ34 on our H9c2 cells; iii) to
compare the cumulated necrosis of cells in the experimental groups
of our cell-therapy model. In the evaluation of the model, 100,000
H9c2 cells were used and measured at 24 h after the start of
reperfusion. For the evaluation of the PJ34 PARP inhibitor, we
measured an untreated group and groups pretreated with 10 or 100 μM
PJ34. After incubation with vehicle or PJ34 for 1 h, 10,000 cells
were placed in a well and each group was either treated with
vehicle to evaluate the cytotoxicity of the inhibitor or given 400
μM H2O2 for 2 h to check the inhibitor’s
efficacy. Measurements were standardized using untreated cells and
cells were lysed using 1% Triton X-100 or cell lysis solution to
obtain the background and the total LDH content. In the third case,
the LDH level was estimated in cell culture supernatant 24 h after
reperfusion according to the manufacturer’s instructions (LDH
Cytotoxicity kit I and II; PromoCell, Heidelberg, Germany) and
differences compared to the vehicle treated group (H9c2) were
given. The absorbance was taken at 450 and 650 nm and the
percentage of cytotoxicity was calculated.
Flow cytometric measurements
Flow cytometric analysis was performed 24 h after
reperfusion (FACSCalibur™; Becton-Dickinson, Franklin Lakes, NJ,
USA). Cells were harvested by trypsinization and resuspended in 500
μl PBS containing 250 nM calcein-AM (ex/em, 494/517 nm; Invitrogen,
Carlsbad, CA, USA) and 350 nM ethidium-homodimer-2 (ex/em, 536/624
nm; Invitrogen). Using flow cytometry we could distinguish the
therapeutically given cells from the ‘postischemic’ cells on the
basis of their Vybrant DiD labeling (ex/em, 633/665 nm; Molecular
Probes, USA) and these cells were gated in or out as appropriate
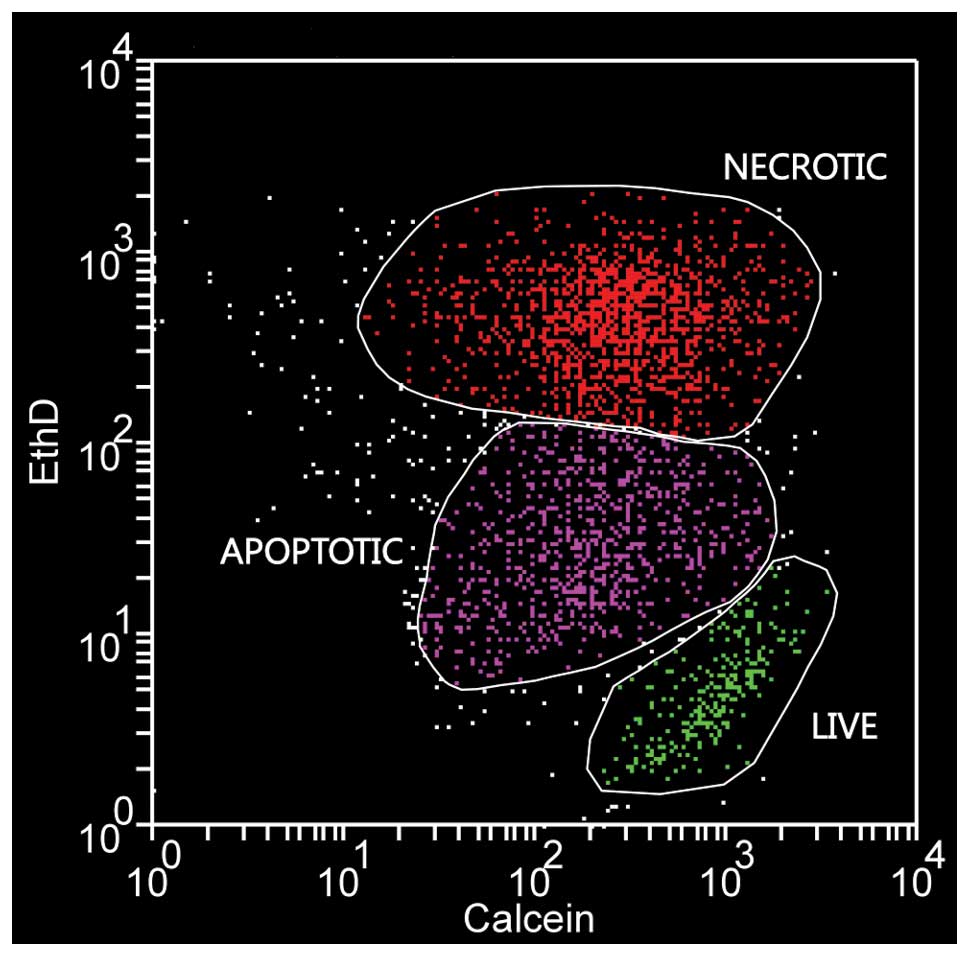
for further analysis. Evaluation of cell death was performed
according to Palma et al(34). Briefly, maximally ethidium
homodimer-2 positive and slightly calcein-AM negative cells were
considered necrotic, ethidium homodimer-2 negative but calcein-AM
positive cells were counted as live and a distinct group of cells
with intermediate staining characteristics was considered as
apoptotic (Fig. 2).
Metabolic activity
For the evaluation of the metabolic activity in the
experimental groups we used PrestoBlue™ cell viability reagent
(Invitrogen). PrestoBlue is a resazurin-based reagent that
functions as a cell viability indicator by using the reducing power
of living cells. Cardiomyoblasts were prepared on a 12-well plate
and therapeutic cells were added after simulated I-R as previously
described. After 24 h of co-cultivation, the culture media was
replaced by phenol red free high glucose DMEM containing 10%
PrestoBlue cell viability reagent. Following a further 24 h of
incubation, 200 μl cell culture supernatant was taken from all
samples and added into the wells of a 96-well plate. The absorbance
was measured at 570 and 600 nm, according to the manufacturer’s
description, and the 570 nm values were normalized to the 600 nm
values.
Statistical analysis
The flow cytometric measurements were evaluated
using Weasel software (WEHI, Australia). Statistical analysis of
data was carried out using one-way analysis of variance with the
Newman-Keuls multiple comparison post hoc test or the Student’s
t-test as appropriate. All data are expressed as the means ± SEM. A
P-value of <0.05 was considered to indicate statistically
significant differences.
Results
Simulated ischemia-reperfusion
protocol
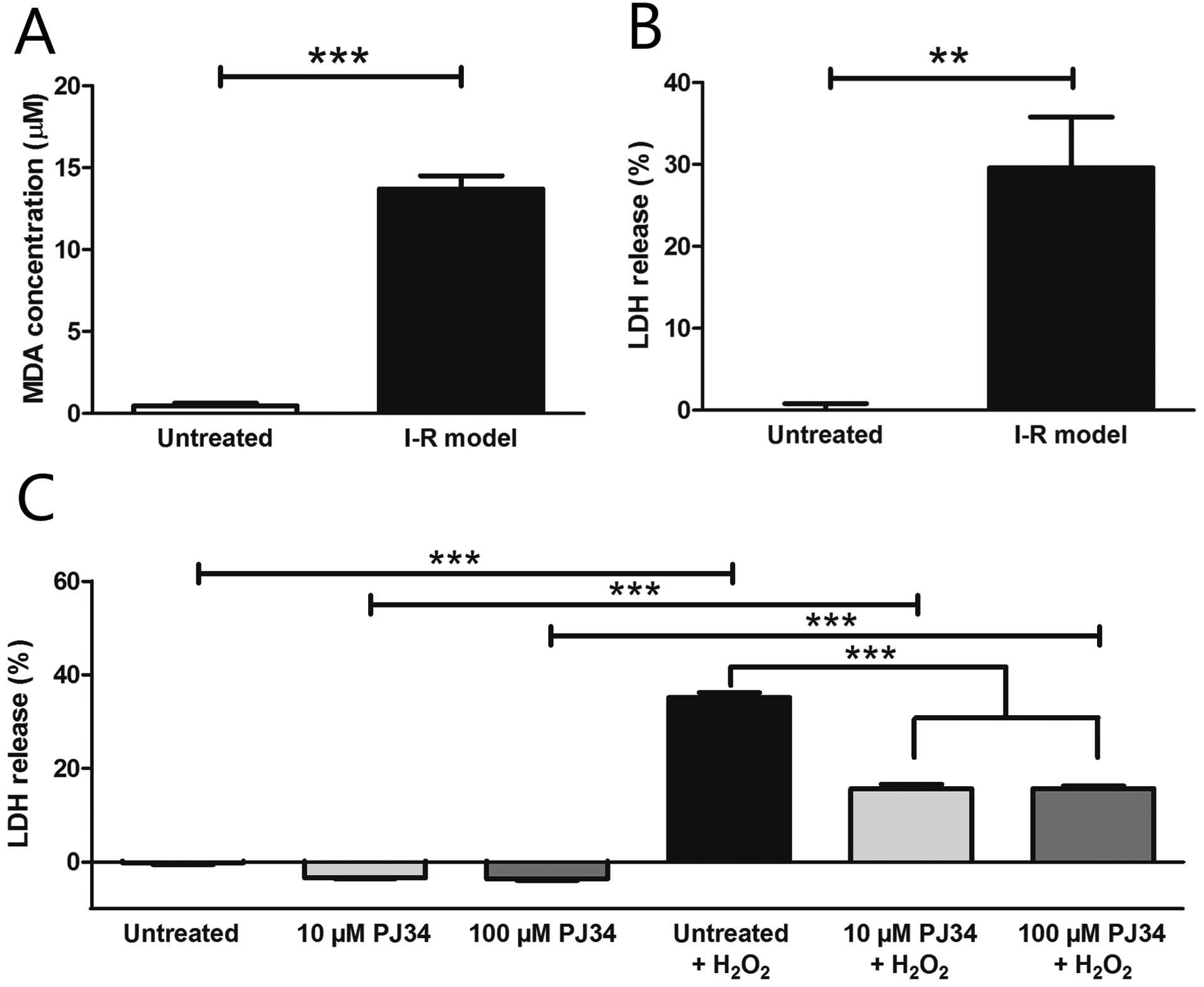
The level of the oxidative stress based on MDA assay
significantly increased in our I-R model (13.70±0.81 μM) compared
to untreated cells (0.47±0.18 μM) (Fig. 3A). Oxygen glucose deprivation led
to a significant increase in the LDH levels in the supernatant
compared to the control conditions (untreated, 0.00±0.81%; I-R
model, 29.58±6.21%) (Fig. 3B).
The PARP inhibitor PJ34 did not exert any cytotoxic effect in the
used concentrations in our H9c2 cells and significantly protected
these cells against H2O2 induced injury in
both concentrations (untreated + H2O2,
35.14±1.01%; 10 μM PJ34 + H2O2, 15.65±0.95%;
100 μM PJ34 + H2O2, 15.69±0.54%) (Fig. 3C).
Flow cytometric measurements
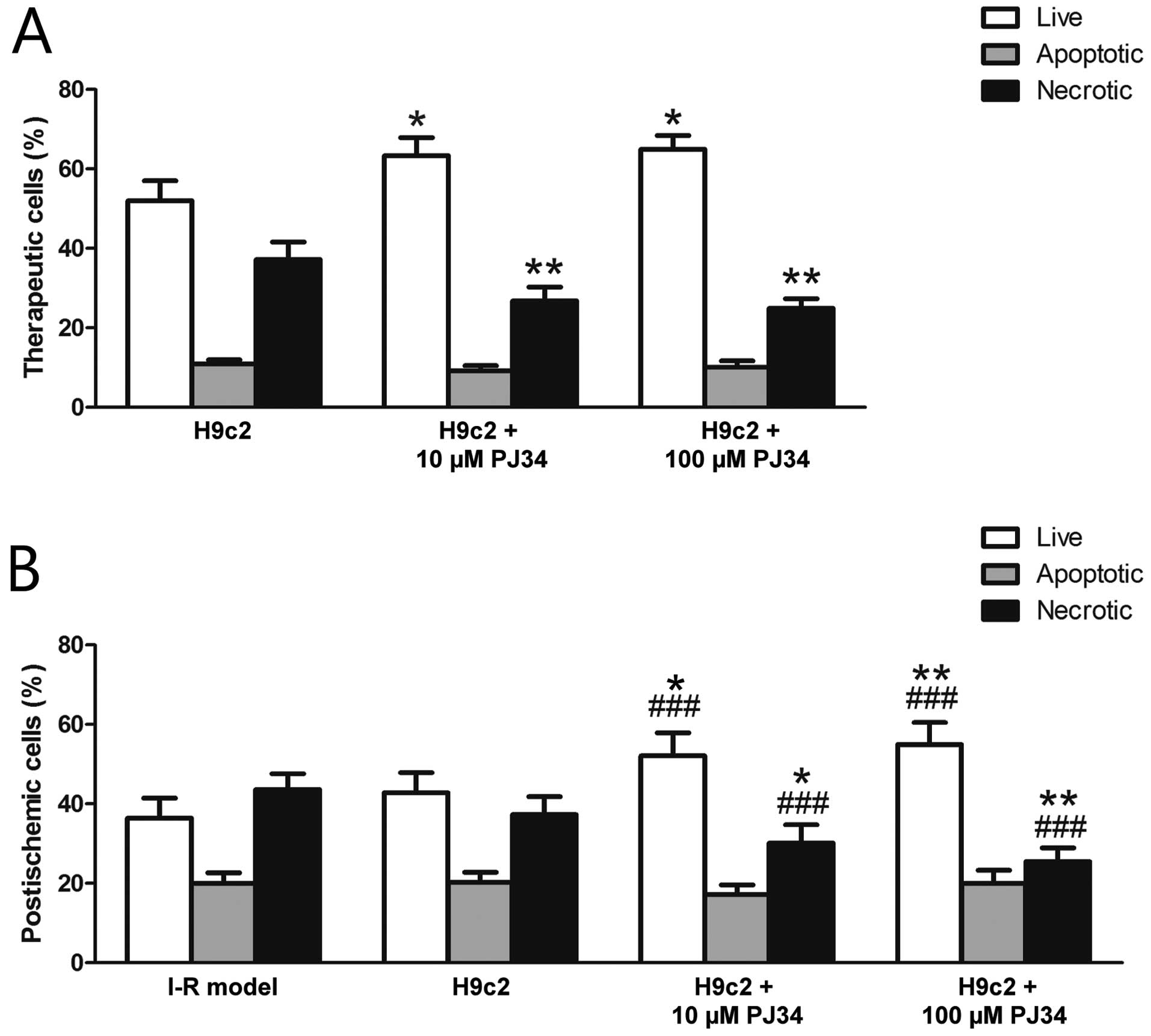
Flow cytometric analysis of live, apoptotic and
necrotic populations of the therapeutically added cells showed
significantly increased survival with PARP inhibition (H9c2,
52.02±5.01%; H9c2 + 10 μM PJ34, 63.38±4.50%; H9c2 + 100 μM PJ34,
64.99±3.47%), while the ratio of necrotic cells decreased when PJ34
pretreatment was used (H9c2, 37.23±4.40%; H9c2 + 10 μM PJ34,
26.83±3.49%; H9c2 + 100 μM PJ34, 24.96±2.43%). No differences were
found among the ratios of apoptotic cells (H9c2, 10.87±1.12%; H9c2
+ 10 μM PJ34, 9.22±1.28%; H9c2 + 100 μM PJ34, 10.18±1.55%)
(Fig. 4A).
There was no significant difference between the I-R
model and the H9c2 treated group regarding live (I-R model,
36.44±5.05%; H9c2, 42.81±5.11%) and necrotic (I-R model,
43.64±4.00%; H9c2, 37.29±4.55%) ratios. This was expected as the
experimental damage was set to acquire such data. However, and
importantly, the survival of the ‘postischemic’ cells was higher
when treated with PARP-inhibited therapeutic cells (H9c2 + 10 μM
PJ34, 52.07±5.80%; H9c2 + 100 μM PJ34, 54.95±5.55%) and the ratio
of necrotic cells also decreased (H9c2 + 10 μM PJ34, 30.18±4.60%;
H9c2 + 100 μM PJ34, 25.52±3.47%). The percentages of apoptotic
cells did not show a statistically significant difference among the
groups (I-R model, 19.94±2.75%; H9c2, 20.23±2.62%; H9c2 + 10 μM
PJ34, 17.20±2.42%; H9c2 + 100 μM PJ34, 20.05±3.23%) (Fig. 4B).
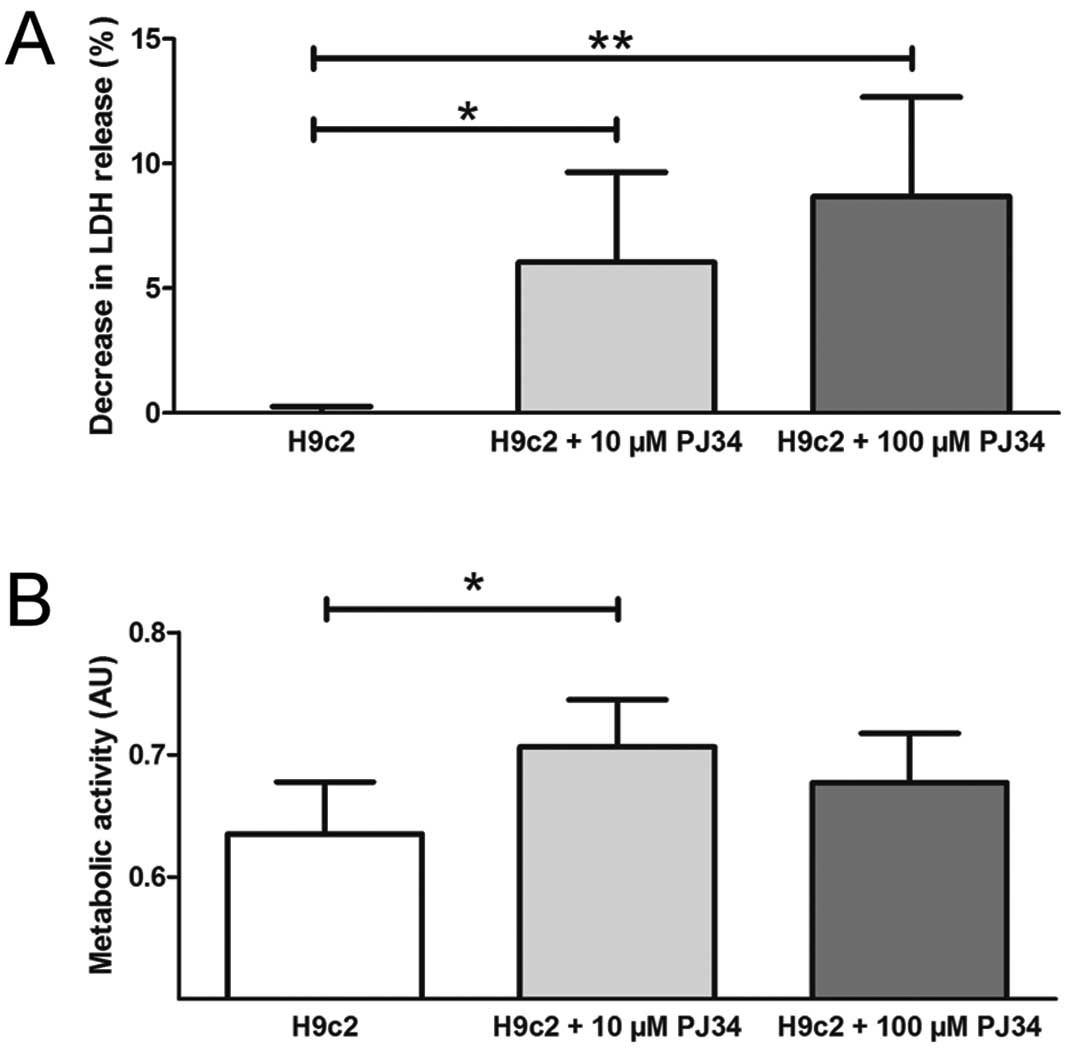
Lactate dehydrogenase cytotoxicity
assay
Cellular necrosis expressed by LDH release decreased
significantly when the therapeutic cells were pretreated with the
PARP inhibitor. Using 10 μM and 100 μM PJ34 the LDH release
decreased with 6.04±3.61% and 8.68±3.98%, respectively (Fig. 5A).
Metabolic activity
PARP inhibition significantly enhanced the overall
metabolic activity of the cells when therapeutic cells were treated
with 10 μM PJ34 compared to untreated H9c2 (H9c2, 0.64±0.04%; 10 μM
PJ34, 0.71±0.04%; arbitrary units). There was only a nonsignificant
trend of such increase when a 100 μM concentration PJ34-treated
cardiomyoblasts were used (0.68±0.04%) (Fig. 5B).
Discussion
Herein we report that the pretreatment of
therapeutic cells improves their survival and subsequently
increases the viability of the damaged cardiomyoblasts in an in
vitro reductionist model of cell-based therapy in myocardial
infarct. First, we evaluated our experimental in vitro model
for oxidative stress, necrotic properties and we checked the
cytotoxicity and efficacy of the used PARP inhibitor. We found that
following oxygen and glucose deprivation, the MDA levels are
increased, as it can be observed in in vivo ischemic
conditions. According to our measurements on cell membrane
integrity based on LDH level, the simulated ischemia is followed by
significant membrane damage. Thus the applied model properly
simulates the I-R injury. The earlier data on the PARP inhibitor
were confirmed regarding its cytotoxicity and efficacy in the used
concentrations (29). This
measurement also indicates that our in vitro simulated
ischemia model caused damage similar to a 400 μM
H2O2 treatment for 2 h. The timing of the
cell addition was partly chosen based on the literature that
suggests non-immediate delivery of cells (35) and partly on our own pilot
experiments that also suggested better efficacy if cells were given
30 min after the start of reperfusion (data not shown).
Using flow cytometry we showed that PARP inhibition
of the therapeutic cells could improve the viability of the
‘postischemic’ cells. The mechanism of this beneficial effect seems
to be connected to the increased ratio of surviving therapeutic
cells. It appears, therefore, that the therapeutic cells with PJ34
pretreatment could help damaged cells to survive. Untreated
therapeutic cells had no significant effect on this cell
population. Imaging a real myocardial infarct it may mean that
areas with bigger oxidative damage could also be saved with such
pretreated therapeutic cells. Regarding the exact mechanism of the
therapeutic cells we assume based on our earlier observations
(9,33) and on the results of others
(8,36), that this beneficial effect could
be related partly to cell-to-cell connections and partly to
paracrine factors released from the therapeutic cells.
If we consider the possible mechanisms related to
the improved survival of therapeutic cells we must remember that
reactive oxygen species are believed to play a key role in the
myocardial I-R injury and myocardial cell death in I-R is mediated
mainly by necrosis and the mechanism is based on the oxidative
stress-induced activation of PARP (37). It is important to realize at this
point that the PARP inhibitor treatment occurred before adding the
therapeutic cells to the damaged ones, therefore these latter cells
did not receive any pretreatment. The protective effect of PJ34 is
extended beyond the end of the treatment and pharmacokinetic data
indicate that the prolonged effect of PJ34 is not related to the
continued presence of the inhibitor, but it may be related to the
permanent interruption of positive-feedback cycles of injury.
Earlier studies have demonstrated that PARP inhibitors block
positive-feedback cycles of adhesion-receptor expression and
mononuclear cell infiltration, as well as intracellular oxidant
generation (24). A possible
concern could be the potential genotoxic nature of PARP inhibitors
as these block the DNA-repair mechanisms. Results from earlier
studies disprove this as one study showed that the inhibition of
poly(ADP-ribosyl)ation did not particularly modify genotoxicity
(38) while another, using
bacterial reverse mutation test, in vitro chromosomal
aberration test and bone marrow micronucleus test concluded that
PARP inhibition did not possess genotoxic activity (39).
In our subsequent experiments, the effect of PARP
inhibition pretreatment was examined using LDH and PrestoBlue
measurements. These two methods reflect the necrosis and metabolic
activity of both cell populations after 24 h, respectively. LDH
values significantly decreased with both applied concentrations of
PJ34. Pretreatment with 10 μM PJ34 significantly increased the
metabolic activity in the cell culture compared to untreated H9c2,
but in the case of 100 μM PJ34 we saw only a non-significant trend
towards the increased value. Blocking PARP activation as much as it
is possible should lead, at least in theory, to better protection.
The higher concentration in our experiments, however, could not
improve the beneficial effect further or in the case of PrestoBlue
it was only a non-significant increase. Thus, we suspect that 10 μM
PJ34 fully blocks the PARP enzyme in H9c2 cells after 1 h. The
results that the higher concentration of PJ34 did not significantly
improve the metabolic activity of ‘postischemic’ cells may reflect
that higher concentration of PARP inhibitors could cause metabolic
suppression, but this hypothesis warrants further investigations.
As a limitation to the interpretation of these measurements, it
must be realized that the combined LDH levels and metabolic
activities of the two cell populations were measured. However, as
these factors are frequently used to reflect the viability of cell
groups we believe that the improved combined values provide
valuable data to our study and further strengthen our findings.
There are some other limitations to our study as
well. First, the demonstrated protocol is an in vitro
approach to the much more complex issue of stem cell therapy in
myocardial infarct, with all the advantages and disadvantages of
such a model. Obviously, it cannot reflect the complex (e.g.
immunological) events taking place during and after myocardial
infarct but the parameters (time of the ischemia and reperfusion,
the temperature and the O2 concentration) can easily be
controlled and the effects of the added cells on the ‘postischemic’
cells can be quantified. The 24 h co-cultivation time period was
chosen based on preliminary experiments as this time was found to
suffice for the attachment of the newly added cells but was not
enough for proliferation. Second, we used H9c2 cells as a model for
cardiac muscle and although the cell line is frequently used in
similar experiments they resemble skeletal muscle cells just as
much as cardiac muscle (40,41). Thus, conclusions drawn from
results obtained from them should be considered cautiously. Another
concern could be the fact that we used H9c2 cells as therapeutic
cells. Our earlier experimental evidence showed that healthy H9c2
cells could save their oxidatively damaged neighbors from delayed
death (33). Furthermore, since
we obtained similar effectiveness with stem cells and H9c2 cells in
that model, it reflects that multipotency of the grafted cells is
not required for this type of rescue effect, widening the possible
sources for cell therapy.
Our data are in line with other observations on
pretreatments in the literature. Yao et al(15) transplanted mesenchymal stem cells
pretreated with lipopolysaccharide into an in vivo rat
model. They found better vascularisation, lower fibrosis in the
myocardium, and the viability of the therapeutic cells improved.
Hahn et al(16) used a
growth factor combination for pretreatment and checked the survival
of the cells after 0.5% hypoxia for 30 h. They found a 20% increase
in trypan blue exclusion. This effect is quantitatively similar to
the effect of our PJ34 inhibition achieved in the
H2O2 protocol. Mouse BMSCs were pretreated
with H2O2 for 30 min in the Kubo et
al(18) model. The production
of vascular endothelial growth factor was increased and the
differentiation towards endothelial cells was promoted. Mangi et
al(17) pretreated
therapeutic cells with retroviral manipulation for overexpressing
Akt1 which restored the myocardium more effectively than the
control cells. Of note, however, we believe that our study presents
the first quantitative viability data on the effect of therapeutic
cell pretreatment on the ‘postischemic’ cells.
In conclusion, addition of PARP-inhibited cells to
severely injured cardiomyoblasts can improve survival and decrease
cellular necrosis in an in vitro cell therapy model for I-R
injury. This approach, if confirmed in experiments using human
derived cells and in in vivo studies, could lead to a better
efficacy of cell-based therapies with a relatively simple and
economical pretreatment step.
Acknowledgements
The present study was founded by grants from OTKA
83803, TÉT-SIN, TÁMOP 4.2.2-08/1/KMR-2008-0004, TÁMOP-4.2.1/B
09/1/KMR-2010-0001.
References
|
1
|
Dill T, Schachinger V, Rolf A, et al:
Intracoronary administration of bone marrow-derived progenitor
cells improves left ventricular function in patients at risk for
adverse remodeling after acute ST-segment elevation myocardial
infarction: results of the Reinfusion of Enriched Progenitor cells
And Infarct Remodeling in Acute Myocardial Infarction study
(REPAIR-AMI) cardiac magnetic resonance imaging substudy. Am Heart
J. 157:541–547. 2009.
|
|
2
|
Medicetty S, Wiktor D, Lehman N, et al:
Percutaneous adventitial delivery of allogeneic bone marrow derived
stem cells via infarct related artery improves long-term
ventricular function in acute myocardial infarction. Cell
Transplant. Oct 14–2011.(Epub ahead of print).
|
|
3
|
Paul D, Samuel SM and Maulik N:
Mesenchymal stem cell: present challenges and prospective cellular
cardiomyoplasty approaches for myocardial regeneration. Antioxid
Redox Signal. 11:1841–1855. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Penn MS, Dong F, Klein S and Mayorga ME:
Stem cells for myocardial regeneration. Clin Pharmacol Ther.
90:499–501. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Abdel-Latif A, Bolli R, Tleyjeh IM, et al:
Adult bone marrow-derived cells for cardiac repair: a systematic
review and meta-analysis. Arch Intern Med. 167:989–997. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dayan V, Yannarelli G, Billia F, et al:
Mesenchymal stromal cells mediate a switch to alternatively
activated monocytes/macrophages after acute myocardial infarction.
Basic Res Cardiol. 106:1299–1310. 2011. View Article : Google Scholar
|
|
7
|
Ramos GA and Hare JM: Cardiac cell-based
therapy: cell types and mechanisms of actions. Cell Transplant.
16:951–961. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gnecchi M, Zhang Z, Ni A and Dzau VJ:
Paracrine mechanisms in adult stem cell signaling and therapy. Circ
Res. 103:1204–1219. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cselenyák A, Pankotai E, Horváth E, Kiss L
and Lacza Z: Mesenchymal stem cells rescue cardiomyoblasts from
cell death in an in vitro ischemia model via direct cell-to-cell
connections. BMC Cell Biol. 11:292010.PubMed/NCBI
|
|
10
|
Plotnikov EY, Khryapenkova TG, Vasileva
AK, et al: Cell-to-cell cross-talk between mesenchymal stem cells
and cardiomyocytes in co-culture. J Cell Mol Med. 12:1622–1631.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wu KH, Mo XM, Han ZC and Zhou B: Stem cell
engraftment and survival in the ischemic heart. Ann Thorac Surg.
92:1917–1925. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Singla DK, Singla RD, Lamm S and Glass C:
TGF-beta2 treatment enhances cytoprotective factors released from
embryonic stem cells and inhibits apoptosis in infarcted
myocardium. Am J Physiol Heart Circ Physiol. 300:H1442–1450. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Singla DK and McDonald DE: Factors
released from embryonic stem cells inhibit apoptosis of H9c2 cells.
Am J Physiol Heart Circ Physiol. 293:H1590–1595. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lichtenauer M, Mildner M, Hoetzenecker K,
et al: Secretome of apoptotic peripheral blood cells (APOSEC)
confers cytoprotection to cardiomyocytes and inhibits tissue
remodelling after acute myocardial infarction: a preclinical study.
Basic Res Cardiol. 106:1283–1297. 2011. View Article : Google Scholar
|
|
15
|
Yao Y, Zhang F, Wang L, et al:
Lipopolysaccharide preconditioning enhances the efficacy of
mesenchymal stem cells transplantation in a rat model of acute
myocardial infarction. J Biomed Sci. 16:742009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hahn JY, Cho HJ, Kang HJ, et al:
Pre-treatment of mesenchymal stem cells with a combination of
growth factors enhances gap junction formation, cytoprotective
effect on cardiomyocytes, and therapeutic efficacy for myocardial
infarction. J Am Coll Cardiol. 51:933–943. 2008. View Article : Google Scholar
|
|
17
|
Mangi AA, Noiseux N, Kong D, et al:
Mesenchymal stem cells modified with Akt prevent remodeling and
restore performance of infarcted hearts. Nat Med. 9:1195–1201.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kubo M, Li TS, Suzuki R, Ohshima M, Qin SL
and Hamano K: Short-term pretreatment with low-dose hydrogen
peroxide enhances the efficacy of bone marrow cells for therapeutic
angiogenesis. Am J Physiol Heart Circ Physiol. 292:H2582–2588.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hausenloy DJ: Signalling pathways in
ischaemic postconditioning. Thromb Haemost. 101:626–634.
2009.PubMed/NCBI
|
|
20
|
Yellon DM and Hausenloy DJ: Myocardial
reperfusion injury. N Engl J Med. 357:1121–1135. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hori M and Nishida K: Oxidative stress and
left ventricular remodelling after myocardial infarction.
Cardiovasc Res. 81:457–464. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pacher P and Szabo C: Role of
poly(ADP-ribose) polymerase 1 (PARP-1) in cardiovascular diseases:
the therapeutic potential of PARP inhibitors. Cardiovasc Drug Rev.
25:235–260. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sodhi RK, Singh N and Jaggi AS:
Poly(ADP-ribose) polymerase-1 (PARP-1) and its therapeutic
implications. Vascul Pharmacol. 53:77–87. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Virag L and Szabo C: The therapeutic
potential of poly(ADP-ribose) polymerase inhibitors. Pharmacol Rev.
54:375–429. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ullrich O, Diestel A, Eyupoglu IY and
Nitsch R: Regulation of microglial expression of integrins by
poly(ADP-ribose) polymerase-1. Nat Cell Biol. 3:1035–1042. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jagtap P and Szabo C: Poly(ADP-ribose)
polymerase and the therapeutic effects of its inhibitors. Nat Rev
Drug Discov. 4:421–440. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang Y, Kim NS, Haince JF, et al:
Poly(ADP-ribose) (PAR) binding to apoptosis-inducing factor is
critical for PAR polymerase-1-dependent cell death (parthanatos).
Sci Signal. 4:ra202011.PubMed/NCBI
|
|
28
|
Koh DW, Dawson TM and Dawson VL: Mediation
of cell death by poly(ADP-ribose) polymerase-1. Pharmacol Res.
52:5–14. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fiorillo C, Ponziani V, Giannini L, et al:
Protective effects of the PARP-1 inhibitor PJ34 in
hypoxic-reoxygenated cardiomyoblasts. Cell Mol Life Sci.
63:3061–3071. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Oh KS, Lee S, Yi KY, Seo HW, Koo HN and
Lee BH: A novel and orally active poly(ADP-ribose) polymerase
inhibitor, KR-33889 [2-[methoxycarbonyl(4-methoxyphenyl)
methylsulfanyl]-1H-benzimidazole-4-carboxylic acid amide],
attenuates injury in in vitro model of cell death and in vivo model
of cardiac ischemia. J Pharmacol Exp Ther. 328:10–18.
2009.PubMed/NCBI
|
|
31
|
Song ZF, Ji XP, Li XX, Wang SJ, Wang SH
and Zhang Y: Inhibition of the activity of poly(ADP-ribose)
polymerase reduces heart ischaemia/reperfusion injury via
suppressing JNK-mediated AIF translocation. J Cell Mol Med.
12:1220–1228. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Liaudet L, Mabley JG, Soriano FG, et al:
Inosine reduces systemic inflammation and improves survival in
septic shock induced by cecal ligation and puncture. Am J Respir
Crit Care Med. 164:1213–1220. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pankotai E, Cselenyak A, Ratosi O, Lorincz
J, Kiss L and Lacza Z: The role of mitochondria in direct
cell-to-cell connection dependent rescue of postischemic
cardiomyoblasts. Mitochondrion. 12:352–356. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Palma PF, Baggio GL, Spada C, Silva RD,
Ferreira SI and Treitinger A: Evaluation of annexin V and
Calcein-AM as markers of mononuclear cell apoptosis during human
immunodeficiency virus infection. Braz J Infect Dis. 12:108–114.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dimmeler S, Burchfield J and Zeiher AM:
Cell-based therapy of myocardial infarction. Arterioscler Thromb
Vasc Biol. 28:208–216. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Uemura R, Xu M, Ahmad N and Ashraf M: Bone
marrow stem cells prevent left ventricular remodeling of ischemic
heart through paracrine signaling. Circ Res. 98:1414–1421. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Burkle A: Physiology and pathophysiology
of poly(ADP-ribosyl)ation. Bioessays. 23:795–806. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Oliveira NG, Castro M, Rodrigues AS, et
al: Effect of poly(ADP-ribosyl)ation inhibitors on the genotoxic
effects of the boron neutron capture reaction. Mutat Res.
583:36–48. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Vinod KR, Chandra S and Sharma SK:
Evaluation of 5-aminoisoquinoline (5-AIQ), a novel PARP-1 inhibitor
for genotoxicity potential in vitro and in vivo. Toxicol Mech
Methods. 20:90–95. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kimes BW and Brandt BL: Properties of a
clonal muscle cell line from rat heart. Exper Cell Res. 98:367–381.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Sardao VA, Oliveira PJ, Holy J, Oliveira
CR and Wallace KB: Vital imaging of H9c2 myoblasts exposed to
tert-butylhydroperoxide - characterization of morphological
features of cell death. BMC Cell Biol. 8:112007. View Article : Google Scholar : PubMed/NCBI
|