1. Introduction
The lysosomal storage diseases (LSDs) are a group of
inherited metabolic disorders caused by mutations in genes encoding
soluble lysosomal hydrolases, integral membrane proteins and
transporters (1). The defective
function of these proteins results in the impaired intracellular
turnover and disposal of a broad range of complex molecules
including sphingolipids, glycosaminoglycans, glycoproteins and
glycogen.
The pathology of LSDs is typically characterized by
intra-lysosomal storage of a variety of substrates in multiple
tissues and organs. Thus, the phenotypes of these disorders are
complex, and characterized by the variable association of visceral,
ocular, hematologic, skeletal and neurological manifestations.
These manifestations are in most instances responsible for physical
and neurological handicaps. In particular, those related to the
involvement of the central nervous system (CNS) may cause
progressive neurodegeneration and severe cognitive impairment.
More than 40 LSDs are currently known and are
traditionally classified according to the chemical properties of
the accumulated substrate. Although each of these disorders is
rare, their overall prevalence is relatively high, compared to
other groups of rare diseases, and is estimated at 1 in 8,000 live
births (2).
For their cumulative prevalence, for the
debilitating effects of their clinical manifestations, and for the
need of a multidisciplinary approach to the care of patients, LSDs
represent a challenge for pediatricians and other physicians, and a
heavy burden in terms of public health and economical costs.
An additional and attractive reason of interest for
these disorders derives from the fact that during the past two
decades the research in the field of LSDs has made significant
progress, particularly with the development of a variety of
innovative therapeutic approaches, that may be potentially extended
to other genetic diseases (3).
The marked advancement in the treatment of LSDs has
been largely stimulated by the improved knowledge on their
molecular bases and pathophysiology. Further impulse to the
development of therapies for LSDs has derived from the availability
of technologies allowing large-scale production, purification and
manipulation of new drugs, recombinant proteins and viral vectors,
and from the orphan drug legislation, that encouraged biotech
companies to invest in the treatments for rare diseases (4).
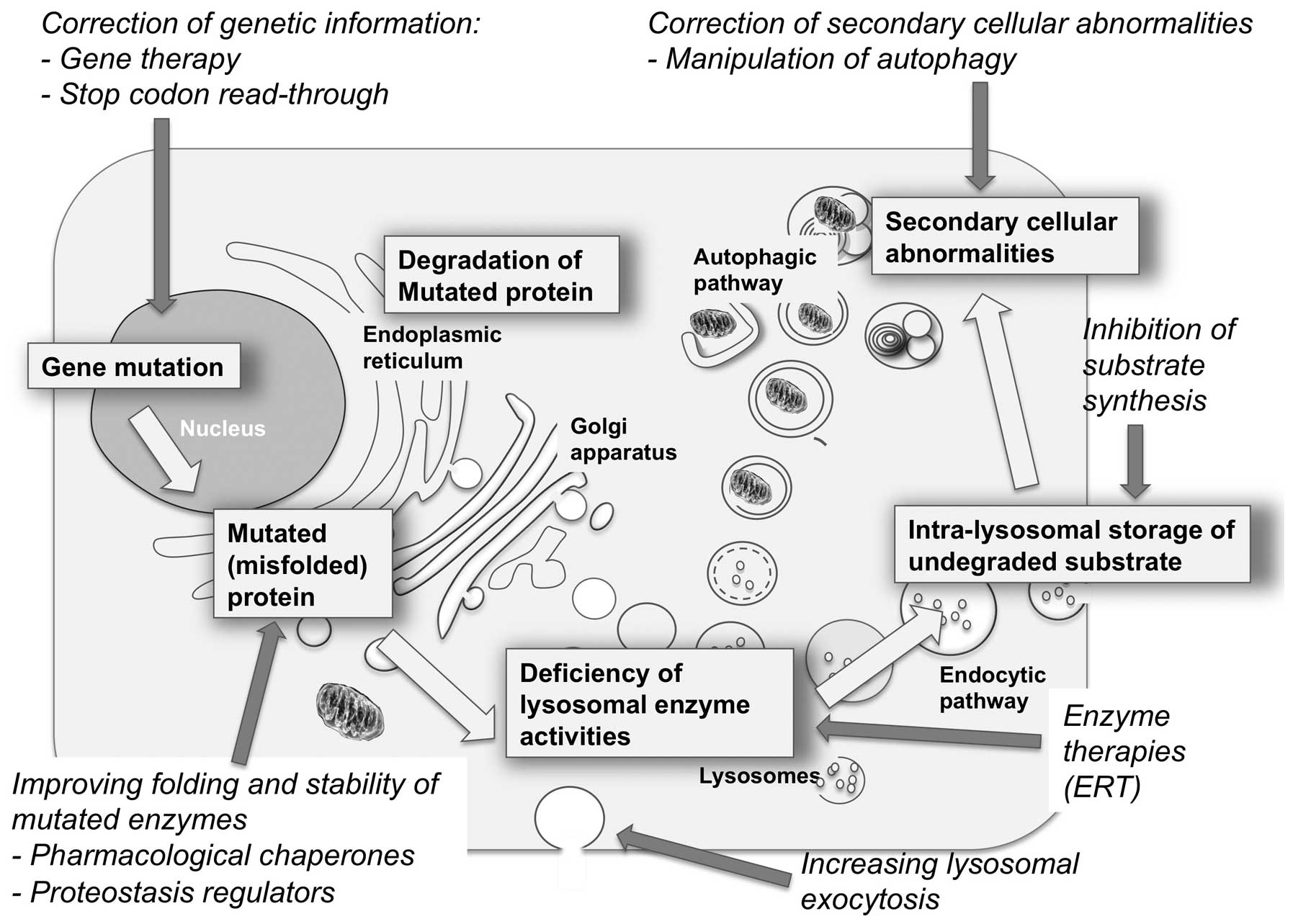
2. Advances in the understanding of LSD
pathophysiology and their therapeutic implications
The understanding of LSD pathophysiology has rapidly
evolved during the past decade and it is now clear that storage of
substrates within the lysosomal compartment triggers a number of
secondary cellular responses that eventually lead to cell death and
tissue damage. Substrate storage acts as a primum movens
that leads to the derangement of housekeeping cellular functions
and pathways, including receptor activation by non-physiologic
ligands, modulation of receptor responses and signal transduction
cascades, activation of inflammatory responses, impaired
intracellular trafficking of vesicles, membranes and membrane-bound
proteins, impairment of autophagy, and others (5).
It is also becoming clear that each of the events in
the pathogenetic cascade of LSDs is a potential target of therapy
and that various approaches, based on different strategies and
rationale, may be exploited to treat these disorders.
Until two decades ago, the treatment of patients
affected by LSDs was exclusively based on palliative or supportive
medical therapies. The first therapeutic strategies directed toward
the correction of the basic defect of these disorders were
introduced in the early 1990s, and since then a number of novel
approaches have been proposed or tested in preclinical studies.
The most obvious approach to correct
loss-of-function diseases, such as LSDs, caused in most cases by
the deficiency of a lysosomal hydrolase, is to restore or replace
the defective enzymatic activity. Thus, the majority of the
approaches thus far developed to treat LSDs are aimed at increasing
the cell and tissue levels of the missing enzyme. These include
hematopoietic stem cell transplantation (HSCT), enzyme replacement
therapy (ERT), pharmacological chaperone therapy (PCT) and gene
therapy (GT).
These types of approaches may be particularly
suitable for disorders such as LSDs, as it is assumed that clinical
symptoms generally develop in patients when the level of residual
enzyme falls below a critical threshold. Thus, residual activities
greater than 10% of the normal level may, in principle, be
sufficient to prevent storage, and it is possible to speculate that
even minor increases of enzyme activity may translate into some
clinical benefit for patients.
An alternative approach, substrate reduction therapy
(SRT), is based on reducing the synthesis of the substrates stored
in the lysosomes.
In recent years, with the improved knowledge on the
pathophysiology of LSDs, other innovative therapeutic strategies
have been proposed and are being evaluated in preclinical studies
(Fig. 1).
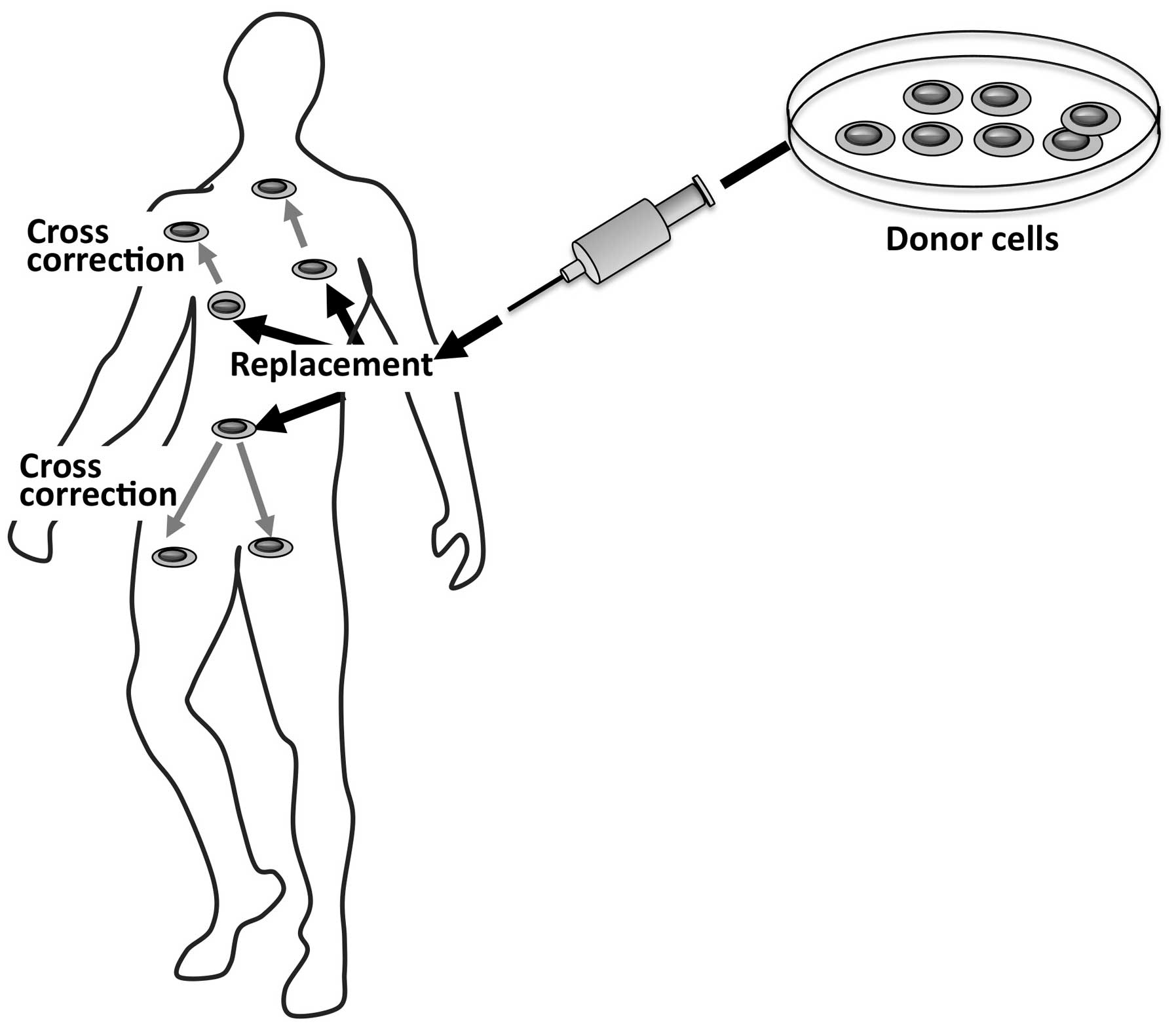
3. Hematopoietic stem cell
transplantation
HSCT was the first therapeutic approach introduced
in the treatment of LSDs. Bone marrow has been traditionally the
graft source for this procedure. However, in recent years, a number
of patients have been treated with unrelated donor umbilical cord
blood transplant, allowing rapid and increased access to
transplantation (6).
HSCT relies on the use of hematopoietic stem cells
derived from a healthy donor as a therapeutic agent. By this
approach, a dual effect may be achieved (Fig. 2); the first is to repopulate
specific tissues by the donor’s healthy cells, and the second and
key effect is related to the secretion of functional lysosomal
hydrolases by the donor’s cells in the extracellular space and into
the blood circulation. The secreted normal enzyme may be taken up
by the recipient cells and may cross-correct the enzyme defect in
these cells.
The clinical experience obtained so far with HSCT,
however, suggests that this procedure is limited to few lysosomal
disorders. HSCT is indicated for the treatment of
mucopolysaccharidosis (MPS) I (7)
and has beneficial effects on the visceral manifestations of MPS
VI, in pre-symptomatic or late-onset Krabbe disease, and in the
attenuated forms of metachromatic leukodystrophy (8). For MPS I indications, optimal
timing, safety and efficacy of HSCT have recently been reviewed by
a European panel of specialists that participated in a modified
Delphi process to develop consensus-based statements on this
approach (9). HSCT is considered
the preferred treatment for patients with severe MPS I diagnosed
before the age of 2.5 years, and may be considered in individual
patients with intermediate phenotypes if there is a suitable
donor.
For Krabbe disease and metachromatic leukodystrophy,
disease phenotype and the extent of disease at the time of
transplantation are of fundamental importance in determining
outcomes (10).
A significant advantage of HSCT is that, as
donor-derived, enzyme-producing cells are able to migrate to the
brain. This procedure has the potential to improve neurocognitive
function and quality of life, particularly when performed early in
the course of the disease. On the other hand, HSCT is burdened by a
significant mortality related to the procedure, by the limited
availability of suitable donors, by the limited number of disorders
that can be treated by this approach and by insufficient
engraftment and correction of pathology in some tissues, such as
bone or heart.
4. Enzyme replacement therapy
The most important advancement and a major
breakthrough in the treatment of LSDs is ERT. This approach is
based on periodic intravenous infusions of human recombinant
lysosomal enzymes produced and purified on large scale from
different sources with recombinant DNA techniques. Once injected,
the recombinant wild-type enzymes are distributed to tissues,
internalized by cells and targeted to the lysosomal compartment,
where they replace the defective enzyme.
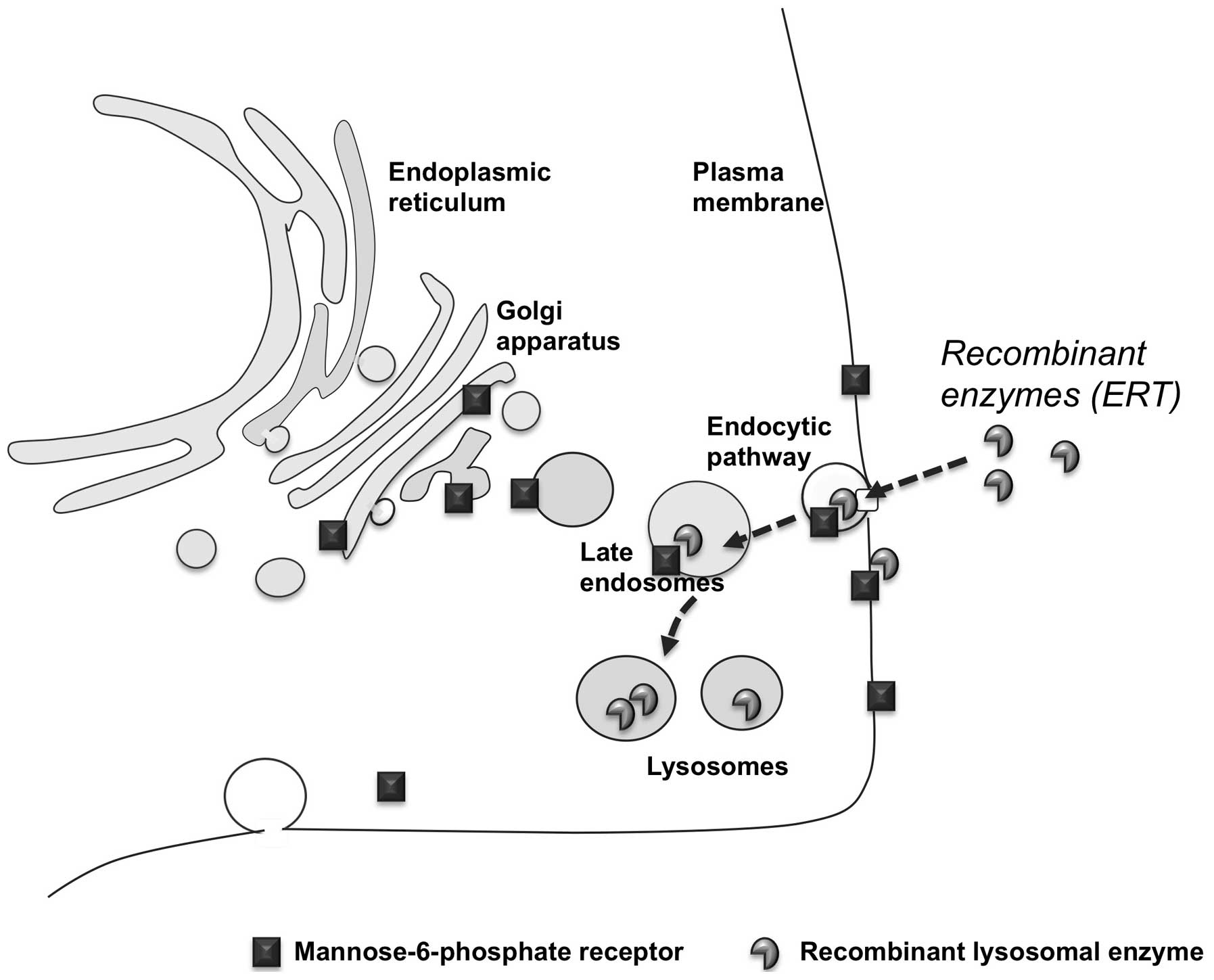
The development of ERT is an excellent example of
how progress in the knowledge of lysosomal biology may lead to the
development of novel therapeutic approaches. The rationale of ERT
evolved from pivotal studies on the molecular and cellular
mechanisms implicated in the sorting of newly synthesized and
secreted lysosomal enzymes. These studies showed that lysosomal
hydrolases are delivered to lysosomes through the mannose or the
mannose-6-phosphate receptor pathways and that these receptors are
also expressed at the plasma membrane of cells (11). Thus, the lysosomal enzymes can be
internalized by cells and follow the endocytic route to be
correctly delivered to lysosomes, by binding to the receptors
exposed on the cellular membrane (Fig. 3). Based on these studies, it has
become evident that the same route could be exploited to deliver
recombinant enzymes to defective cells and tissues and that LSDs
represented the best model of inherited metabolic disorders that
could be treated by providing the missing enzyme from external
sources.
The first attempts to develop an ERT were made in
Gaucher disease. After more than two decades of clinical experience
with thousands of patients treated, the success of ERT has been
clearly documented in this disease. ERT has been firmly proven to
be effective in ameliorating the visceral, hematological and
biochemical manifestations of Gaucher disease and has substantially
improved patient quality of life.
The success of ERT in Gaucher disease (12,13) stimulated the development of this
approach for the treatment of the most prevalent lysosomal
disorders, for which ERT is presently considered the standard of
care.
In Fabry disease, a lysosomal disorder due to the
deficiency of α-galactosidase A and involving vascular endothelia,
heart and the peripheral nervous system, beneficial effects have
been documented on cardiac and renal functions, together with
reduced pain and improved quality of life (14–16). Also, reduction of plasma and
urinary levels of substrate documents ERT efficacy.
In Pompe disease, clinical experience has shown
striking effects on cardiomyopathy, with reduced left ventricular
mass index, prolonged survival in early onset patients (17), improved physical performance,
stabilization or amelioration of skeletal muscle function in late
onset patients (18,19).
In MPS I, II and VI, the general benefits of ERT
include improved walking ability, improved respiration and enhanced
quality of life (7). A
biochemical marker of ERT efficacy is the reduced urinary excretion
of glycosaminoglycans observed in treated patients.
Due to the success of ERT in these disorders,
recombinant enzymes are being tested to treat other LSDs, including
MPS IV (Morquio disease), MPS VII (Sly disease), MPS IIIA
(Sanfilippo disease A), metachromatic leukodystrophy and acid
lipase deficiency.
On the other hand, clinical experience with ERT has
shown that this approach has limitations, mostly related to the
bioavailability of recombinant enzymes and to the high cost of
therapies.
Recombinant enzymes are large molecules that do not
freely diffuse across membranes and that are unable to reach
therapeutic concentrations in some of the target tissues. Thus, a
number of tissues remain refractory to this treatment and some
patients experience limited clinical benefits under ERT treatment
or continue to show signs of disease progression. Examples of the
limitations of ERT are seen in MPS I, II and VI, where correction
of pathology is insufficient in tissues such as bone, cartilage and
heart, that are major sites of pathology. In Pompe disease,
achieving corrective levels of α-glucosidase activity and
correcting pathology in skeletal muscles remains a major challenge
(20).
Of even greater clinical relevance is the inability
of recombinant enzymes to cross the blood-brain barrier (BBB) and
reach the CNS (21,22). As approximately two thirds of LSD
patients have CNS involvement and progressive neurodegeneration,
obtaining corrective enzyme levels in the brain is a major
therapeutic goal.
To circumvent these problems and improve delivery of
the enzymes to target tissues, including CNS, different strategies
are being evaluated. All these strategies are still
experimental.
Attempts to obtain corrective levels of enzyme
activity in brain in neuronopathic LSDs have been made, by
chemically manipulating the recombinant enzymes. β-glucuronidase,
which is deficient in MPS VII, has been modified to increase the
plasma half-life and facilitate its traffic through the BBB
(23). Other approaches that are
currently being explored are based on receptor-mediated penetration
of BBB, such as by coupling recombinant enzymes with transferrin
and using transferrin receptor-mediated endocytosis for transport
across the BBB (24).
In addition to these approaches an invasive
procedure, such as intrathechal ERT administration, has been
evaluated in preclinical studies for several lysosomal disorders
and has been translated into human therapy for MPS I (25).
Attempts have also been made to improve muscle
targeting of ERT in Pompe disease. Pompe disease is a metabolic
myopathy due to the deficiency of α-glucosidase. The recombinant
α-glucosidase (rhGAA) used for the treatment of this disorder has
shown insufficient correction of pathology in skeletal muscles. Zhu
et al (26) developed a
glycoengineered rhGAA (neo-rhGAA) in which additional mannose
6-phosphate moieties were introduced that showed higher affinity
for the mannose-6-phosphate receptor. In a Pompe disease mouse
model, this modified enzyme promoted a greater clearance of
lysosomal glycogen in muscles when compared to the unmodified
enzyme. Other approaches are based on the fusion of a peptide tag
derived from insulin-like growth factor-II (IGF-II), which is also
a ligand for the mannose-6-phosphate receptor, onto rhGAA.
The high cost of ERT is another significant
limitation and a major problem for the treatment of patients with
LSDs. Initial investments in research and costs related to the
production of large amounts of recombinant enzymes according to
good manufacturing practice, contributed to the high cost of ERT.
The treatment of a single LSD patient may require as much as
several hundred thousand euros (or dollars) per year. Such high
costs, that are acceptable in western countries, are barely
affordable in underdeveloped countries, and limit the access of
patients to ERT. The availability of novel molecular technologies
may allow the production of less expensive enzyme preparations. As
an example, recombinant β-glucosidase, the enzyme deficient in
Gaucher disease, has been manufactured in plants or plant-derived
cells (27).
5. Small molecule pharmacological
chaperones
An approach that has recently gained much attention
for the treatment of diseases due to protein misfolding in general
and for LSDs specifically, is enzyme enhancement with small
molecule pharmacological chaperones (28,29). Pharmacological chaperone therapy
is based on the concept that loss-of-function diseases are often
due to missense mutations that cause misfolding (abnormal
conformation) of mutant proteins. The misfolded proteins are
recognized by the quality control systems of the endoplasmic
reticulum and are degraded. Thus, in these so-called ‘misfolding
protein diseases’ the loss of function is not due to the loss of
catalytic activity, but is the result of the degradation of the
aberrant protein. It has been shown that small-molecule ligands
(pharmacological chaperones) can interact with the mutant protein,
favor its correct conformation, and enhance its stability. As a
result, the enzymatic activity of the mutant protein is partially
rescued (Fig. 4). As with ERT and
other approaches directed toward replacing or increasing the
residual activity of the defective enzyme, it is reasonable to
speculate that even minor increases in activity may have a
favorable impact on patient status and rate of disease
progression.
The use of pharmacological chaperones for the
treatment of lysosomal storage diseases was first proposed for
Fabry disease (30). In cells
from patients with Fabry disease, one of the most potent inhibitors
of α-gal A, 1-deoxygalactonojirimycin (DGJ), was paradoxically
shown to enhance residual α-galactosidase A activity. Oral
administration of DGJ to transgenic mice overexpressing a mutant
α-galactosidase A substantially elevated the enzyme activity in
some organs (31).
The same approach was subsequently investigated for
a restricted number of other disorders of this group (28,29), including Gaucher disease (32) and Pompe disease (33,34).
In principle, chaperones (as with substrate reducing
drugs and small molecule drugs in general) have several advantages,
as compared to ERT, as they can be administered orally, allowing
for a non-invasive treatment, are non-immunogenic and do not need
to be delivered to cells through the mannose-6-phosphate pathway,
which may be secondarily impaired in some of these disorders. In
addition, small molecule chaperones are expected to diffuse freely
across cell membranes and reach therapeutic concentrations in
different tissues and systems, including the CNS. In the animal
model of GM1 gangliosidosis, a severe neurodegenerative
disorder due to the deficiency of β-galactosidase, the use of the
chaperone N-octyl 4-epi-β-valienamine (NOEV) resulted in increased
residual activity in the brain and greater clearance of substrate
(35).
On the other hand, chaperone therapy appears to be
feasible only in patients with responsive mutations mostly located
in specific domains of the enzymatic protein (36).
Concerns have also been raised by the fact that most
chaperones thus far identified for the treatment of LSDs are active
site-directed molecules and, in other words, potential competitive
inhibitors of the target enzymes.
These limitations may be overcome by the discovery
of new molecules interacting with other protein domains. Recently,
allosteric chaperones of α-glucosidase, the enzyme deficient in
Pompe disease, were identified by the combination of biochemical
characterization and a computational analysis of the interactions
of these drugs with the enzyme (37). High throughput screenings of
chemical libraries may also be a time-efficient way to identify
new-generation chaperones (38,39).
After the in vitro studies, suggesting a
potential of pharmacological chaperones for the treatment of LSDs,
research is now moving into clinical translation. Phase I/II
clinical trials are being conducted for Fabry, Gaucher and Pompe
disease. For Fabry disease, a phase II/III clinical trial has been
performed and has shown encouraging results in terms of enhancement
of the residual α-galactosidase A enzyme activity and clearance of
substrate.
6. Combination of chaperones and ERT
Although pharmacological chaperone therapy (PCT) has
been developed as a strategy to rescue mutant enzymes from
degradation, recent studies showed that chaperones are also able to
enhance physical stability and potentiate the therapeutic action of
the enzymes used for ERT (40–43). These studies, performed in cell
systems and in the animal models of Pompe and Fabry disease,
suggested a major change in the use of PCT. In both disorders, when
recombinant enzymes were administered in combination with the
chaperone molecules, the lysosomal trafficking, the maturation and
the intracellular activity of the recombinant enzymes was highly
improved.
Although the mechanism underlying this synergistic
effect of ERT and pharmacological chaperones is not yet fully
understood, this synergy may have important advantages compared to
the ‘traditional’ use of chaperones to rescue mutated enzymes.
First, the therapeutic effect is directed toward the exogenous
wild-type enzyme used for ERT, it is mutation-independent, and may
thus be exploited in any patient on ERT (37,41). Second, while the enhancement of
endogenous defective enzymes by chaperones in most cases results in
minor changes in terms of residual activity (possibly with a modest
impact on patient outcome), the synergy between chaperones and ERT
apparently induces, at least in cellular models, notable increases
in specific activity with complete or near-complete correction of
the enzymatic defect.
Also, these studies suggest that, in principle, the
synergistic effect of chaperone therapy and ERT may be seen in any
LSD for which ERT and a chaperone are available.
Clinical trials based on the combination of
chaperones and ERT are already in progress (see trials NCT00214500
and NCT01196871 at http://clinicaltrials.gov; Telethon foundation trial
GUP09017, http://www.telethon.it/ricerca-progetti/progetti-finanziati).
7. Proteostasis regulators
In addition to chaperone therapy, other approaches
have been proposed that are aimed at rescuing the mutated enzymes
in LSDs. While chaperones are ligands that specifically interact
with mutant proteins (and are thus able to rescue a single
protein), other drugs are able to adjust the cellular mechanisms
controlling the homeostasis of proteins, or proteostasis.
Proteostasis is a complex network that controls protein synthesis,
folding, trafficking, aggregation and degradation. These small
molecule drugs, known as proteostasis regulators, can adjust the
capacity of this network and have the potential to restore the
normal balance between protein folding, trafficking, and
degradation. Two proteostasis regulators have been reported to
restore the function of two mutant lysosomal enzymes in two
distinct LSDs, Gaucher disease and GM2 gangliosidosis
(44). Co-administration of a
pharmacological chaperone and a proteostasis regulator exhibited
synergy and resulted in further enhancement of enzyme activity.
This approach remains largely experimental.
8. Gene therapy
Gene therapy is an attractive strategy that holds
great promise for LSD patients and is commonly viewed as the
approach that has the greatest potential for complete and sustained
correction of the enzymatic defect (45,46). Gene therapy is also an excellent
example of a strategy that is being developed owing to the
combination of improved knowledge of the molecular bases of LSDs
and technical advancements in the optimization and preparation of
therapeutic agents.
Gene therapy, as with the approaches mentioned
above, is aimed at increasing or restoring the activity of the
defective enzyme in the patient’s cells. This is not obtained by
supplying the missing enzymatic protein, but by delivering the
normal copy of the defective gene, that will direct the synthesis
of the normal enzyme by the recipient’s cells.
LSDs appear to be amenable to this therapy for
several reasons. First, these disorders are monogenic and it is
theoretically possible to correct the disease by rectifying the
gene defect; second, if lysosomal enzymes are secreted from a small
number of transduced cells they can be taken up, via the
mannose-6-phosphate receptors, by adjacent affected cells. Such
cross-correction, similar to that documented in HSCT, avoids the
necessity of transferring the gene to all cells, with the advantage
that a small percentage of transduced cells could act
therapeutically with consequent benefits for several organs. Third,
as already mentioned, even minor increases of enzymatic activity
(up to 10%) may be sufficient for clinical benefit and partial
phenotypic correction.
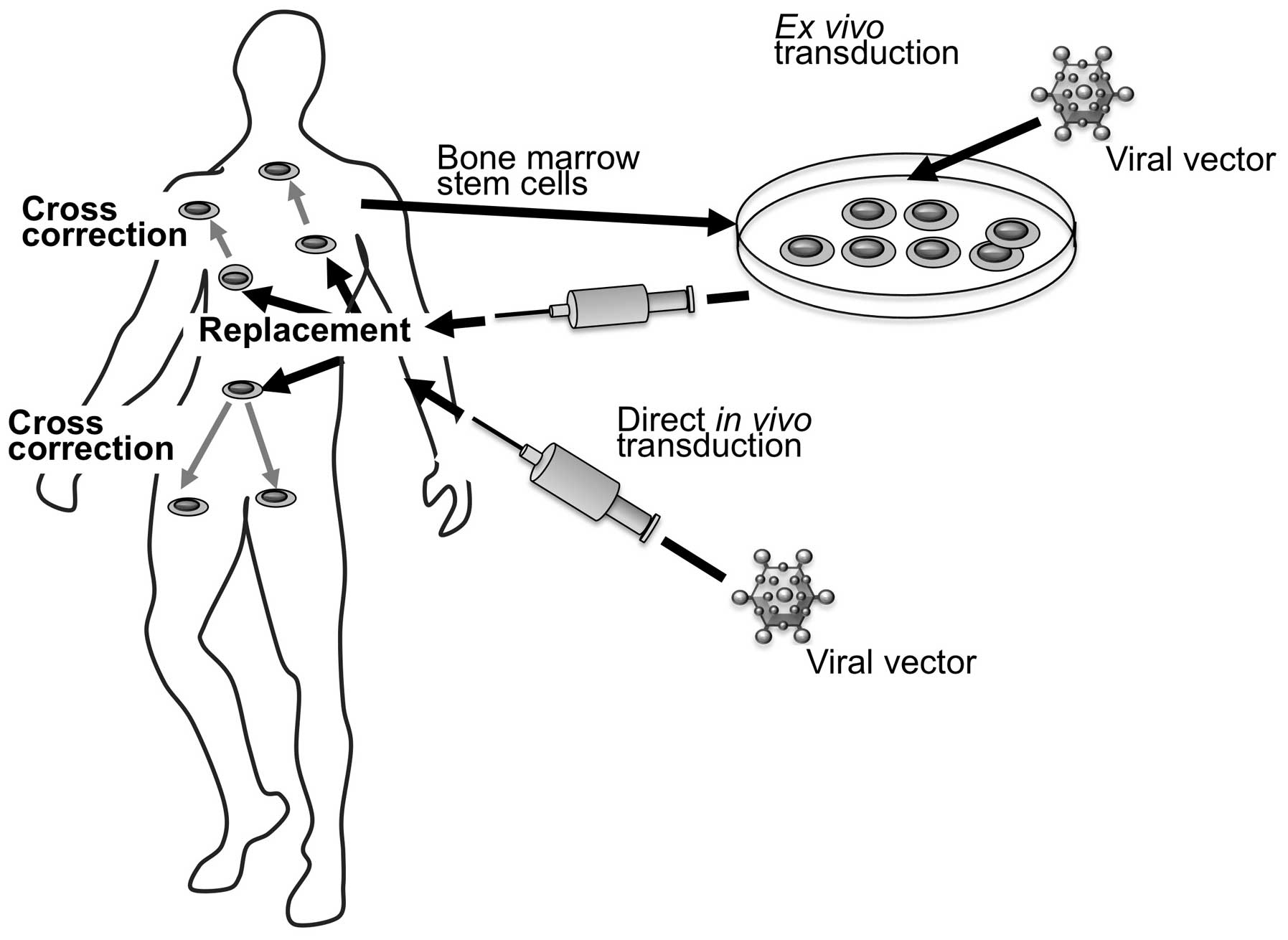
Different viral vectors and different strategies
have been explored to accomplish in vivo gene transfer.
Herpesviruses, lentiviruses, adeno-associated viruses (AAVs),
adenoviruses (Ads) and others have been tested as vectors (45).
Genetic modification may be performed either ex
vivo or in vivo (Fig.
5). The first strategy is based on ex vivo modification
of cells and transplantation of the modified cells into patients.
Cells that are most commonly considered therapeutic targets for
LSDs are hematopoietic progenitor cells.
Another strategy that is being extensively tested is
based on the direct in vivo gene therapy. This approach is
based on the injection of a gene transfer vector directly into a
tissue or into the circulation.
An advantage of a gene therapy approach compared to
ERT is the potential for long-term expression of the therapeutic
protein. Thus, this would be a once-and-for-all procedure, and may
have obvious advantages compared to ERT and small molecule
therapies, that require life-long treatments. Another advantage is
that gene therapy may be available to patients with conditions so
rare that developing enzyme for ERT is not commercially
feasible.
Although gene therapy is a promising treatment for
LSDs, some concerns remain. The major issue is viral vector safety
and the possibility of carcinogenesis following retrovirus- or
adenovirus-mediated gene transfer. Unlike retroviral vectors, AAV
vectors are nonpathogenic and generally do not integrate into the
host genome and are, in this respect, safer. A second consideration
for gene replacement for the LSDs is that the vectors generally
express supra-physiologic levels of enzyme and it is not known
whether this would be safe in humans.
Other issues are related to the doses of vectors to
be used, to the choice of the best vectors to obtain the desired
correction of tissue and organs, and to the possibility of immune
response toward the wild-type enzyme.
Although cross-correction makes it possible to
achieve widespread enzyme distribution while genetically modifying
a minority of cells, effective treatment of CNS disease remains an
unsolved issue. This is due to the presence of the BBB, which
excludes exogenous lysosomal enzymes from the brain. For
neuronopathic LSDs, intracerebral injection of the vector carrying
the wild-type gene has been explored as a possible strategy. In the
animal model of multiple sulfatase deficiency (MSD), a severe
autosomal recessive disease is caused by mutations in the sulfatase
modifying factor 1 gene (Sumf1), the combination of systemic and
intracerebral delivery of the wild-type gene resulted in the global
activation of sulfatases, near-complete clearance of
glycosaminoglycans, and improved behavioral abilities (47).
Gene therapy is under study for a variety of
diseases, mostly in preclinical studies performed in animal models
recapitulating the LSDs, phenotypes. The first clinical trials are
now in progress or are being planned.
9. Substrate reduction therapy
SRT is based on the concept that the inhibition of
specific steps of the biosynthetic pathways of substrates may
reduce their flux to lysosomes and help restore the equilibrium
between their synthesis and catabolism (48,49). This task is generally accomplished
by using small-molecule inhibitors of enzymes involved in the
biosynthesis of substrates.
SRT has already been approved for clinical use for
the treatment of type 1 Gaucher disease, the most prevalent LSD,
due to β-glucocerebrosidase deficiency and characterized by
glycosphingolipid storage, and of Niemann-Pick disease type C
(NPC), a defect of intracellular cholesterol trafficking.
The largest clinical experience on this approach has
been obtained with the use of N-butyldeoxynojirimycin (Miglustat)
in Gaucher disease. In this disorder, Miglustat has been shown to
be effective in improving or stabilizing biochemical, visceral,
hematologic and bone markers of the disease (50), causing limited adverse events
(51).
A novel substrate inhibitor, eliglustat tartrate,
was recently introduced and evaluated in a multisite, open-label,
single-arm phase II clinical trial (52). Treatment with eliglustat tartrate
resulted in improved hemoglobin levels and platelet counts, reduced
spleen and liver volume, and increased lumbar spine bone mineral
density.
In NPC, it has been demonstrated that
glycosphingolipid storage plays a pathogenic role in brain disease
(53). Thus, it was proposed that
Miglustat, the same therapeutic agent used for Gaucher disease,
could also be beneficial in NPC patients. In a controlled trial in
29 adult and juvenile NPC patients, Miglustat stabilized
neurological disease, with improved horizontal saccadic eye
movement, stabilized ambulation, and improved swallowing (54,55). Improved swallowing was also
observed in a study on the efficacy of Miglustat in four pediatric
patients (56).
The approach based on SRT has also been extended to
the treatment of MPS. Preclinical and clinical studies were based
on the use of non-specific inhibitors of glycosaminoglycan
synthesis, the isoflavone genistein and the chemical dye rhodamine
B (57–60). The effects of these drugs,
however, have been quite variable so far. In a two-year follow-up
study of eight MPS III patients (61) treated with genistein, five
patients showed improvement or stabilization of cognitive
functions, whereas three showed further deterioration.
SRT has also been proposed in a few preclinical
studies for the treatment of other LSDs, such as Sandhoff disease
(62), Fabry disease (63), and Pompe disease (64,65).
The use of small-molecule inhibitors of substrate
synthesis has obvious advantages, compared to the use of ERT and
gene therapy, particularly in terms of bioavailability in different
tissues and organs, including the CNS. The efficacy profile in NPC
and other disorders, however, remains to be fully defined.
10. Other experimental approaches
The improvement of the knowledge on the molecular
bases of genetic diseases and on the pathophysiology of LSDs has
recently indicated additional targets of therapy.
As an example, it has been shown that manipulation
of the autophagic pathway may have a beneficial effect in the
animal model of Pompe disease. Genetic suppression of autophagy in
the Pompe mouse resulted in reduced glycogen accumulation in
skeletal muscle by 50–60% compared with mice with genetically
intact autophagy (66). In
addition, in the autophagy-defective Pompe animals, the efficacy of
ERT was greatly improved with a near-complete clearance of glycogen
in the skeletal muscle, a therapeutic goal never attained in PD
mice with genetically intact autophagy.
Therapeutic strategies have been developed to induce
ribosomal read-through of nonsense mutations in mRNA and allow
production of a full-length functional protein. Small-molecule
drugs such as aminoglycosides and ataluren (PTC124) have been
developed and are in clinical testing in patients with several
genetic disorders, such as Becker/Duchenne muscular dystrophy,
cystic fibrosis, methylmalonic acidemia and others (67–70). In principle, use of nonsense
mutation suppression may offer the prospect of treating patients
with other genetic diseases due to premature termination of
ribosomal reading, including LSDs. This approach has been explored
in preclinical studies in cells from neuronal lipofuscinoses due to
palmitoyl-thioesterase deficiency (71).
An entirely new and attractive concept of treatment
derived from recent discoveries on the biology of lysosomes. It has
been shown that lysosomal biogenesis and expression is regulated by
the transcription factor EB (TFEB) (72). TFEB also regulates lysosomal
exocytosis (73) by increasing
the pool of lysosomes in the periphery of cells and facilitating
their fusion with the plasma membrane. Induction of lysosomal
exocytosis by TFEB overexpression rescued pathological storage and
restored normal cellular morphology in vitro in cells from
different LSDs (Pompe disease, multiple sulfatase deficiency,
mucopolysaccharidosis type IIIA) and in vivo in the animal
model of multiple sulfatase deficiency. These results indicated an
alternative therapeutic strategy for disorders associated with
intracellular storage, based on stimulation of lysosomal
exocytosis.
11. Conclusion
The past two decades have given rise to significant
progress in the treatment of LSDs. Different therapeutic options
are now available or are being evaluated in preclinical studies.
However, none of these options is applicable to all lysosomal
diseases and most of the approaches have important limitations
related to the bioavailability of therapeutic agents, toxicity,
immune response, and impact on patient quality of life.
Future research is required to address these issues.
Particular attention should be paid to the clear definition of
guidelines for the treatment of patients. In this respect, careful
collection of information in international registries on the
natural history of LSDs and on the effects of therapies will be
beneficial.
The possibility to combine different approaches in
order to obtain the highest therapeutic efficacy and to personalize
treatment protocols for each disorder and for each individual
patient must be explored.
References
|
1
|
Futerman AH and van Meer G: The cell
biology of lysosomal storage disorders. Nat Rev Mol Cell Biol.
5:554–565. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fuller M, Meikle PJ and Hopwood JJ:
Epidemiology of lysosomal storage diseases: an overview. Fabry
Disease: Perspectives from 5 years of FOS. Mehta A, Beck M and
Sunder-Plassmann G: Oxford PharmaGenesis; Oxford: 2006, PubMed/NCBI
|
|
3
|
Beck M: New therapeutic options for
lysosomal storage disorders: enzyme replacement, small molecules
and gene therapy. Hum Genet. 121:1–22. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Beutler E: Lysosomal storage diseases:
natural history and ethical and economic aspects. Mol Genet Metab.
88:208–215. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ballabio A and Gieselmann V: Lysosomal
disorders: from storage to cellular damage. Biochim Biophys Acta.
1793:684–696. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Prasad VK and Kurtzberg J: Transplant
outcomes in mucopolysaccharidoses. Semin Hematol. 47:59–69. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Valayannopoulos V and Wijburg FA: Therapy
for the mucopolysaccharidoses. Rheumatology (Oxford). 50(Suppl 5):
v49–v59. 2011. View Article : Google Scholar
|
|
8
|
Orchard PJ, Blazar BR, Wagner J, Charnas
L, Krivit W and Tolar J: Hematopoietic cell therapy for metabolic
disease. J Pediatr. 151:340–346. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
de Ru MH, Boelens JJ, Das AM, Jones SA,
van der Lee JH, Mahlaoui N, Mengel E, Offringa M, O’Meara A, Parini
R, Rovelli A, Sykora KW, Valayannopoulos V, Vellodi A, Wynn RF and
Wijburg FA: Enzyme replacement therapy and/or hematopoietic stem
cell transplantation at diagnosis in patients with
mucopolysaccharidosis type I: results of a European consensus
procedure. Orphanet J Rare Dis. 6:552011.
|
|
10
|
Orchard PJ and Tolar J: Transplant
outcomes in leukodystrophies. Semin Hematol. 47:70–78. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sly WS: Receptor-mediated transport of
acid hydrolases to lysosomes. Curr Top Cell Regul. 26:27–38. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Barton NW, Brady RO, Dambrosia JM, Di
Bisceglie AM, Doppelt SH, Hill SC, Mankin HJ, Murray GJ, Parker RI,
Argoff CE, et al: Replacement therapy for inherited enzyme
deficiency - macrophage-targeted glucocerebrosidase for Gaucher’s
disease. N Engl J Med. 324:1464–1470. 1991.PubMed/NCBI
|
|
13
|
Barton NW, Furbish FS, Murray GJ, Garfield
M and Brady RO: Therapeutic response to intravenous infusions of
glucocerebrosidase in a patient with Gaucher disease. Proc Natl
Acad Sci USA. 87:1913–1916. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mehta A, Beck M, Elliott P, Giugliani R,
Linhart A, Sunder-Plassmann G, Schiffmann R, Barbey F, Ries M and
Clarke JT; Fabry Outcome Survey investigators. Enzyme replacement
therapy with agalsidase alfa in patients with Fabry’s disease: an
analysis of registry data. Lancet. 374:1986–1996. 2009.
|
|
15
|
Lidove O, West ML, Pintos-Morell G, Reisin
R, Nicholls K, Figuera LE, Parini R, Carvalho LR, Kampmann C,
Pastores GM and Mehta A: Effects of enzyme replacement therapy in
Fabry disease - a comprehensive review of the medical literature.
Genet Med. 12:668–679. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Feriozzi S, Torras J, Cybulla M, Nicholls
K, Sunder-Plassmann G and West M; FOS Investigators. The
effectiveness of long-term agalsidase alfa therapy in the treatment
of Fabry nephropathy. Clin J Am Soc Nephrol. 7:60–69. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van der Ploeg AT and Reuser AJ: Pompe’s
disease. Lancet. 372:1342–1353. 2008.
|
|
18
|
Strothotte S, Strigl-Pill N, Grunert B,
Kornblum C, Eger K, Wessig C, Deschauer M, Breunig F, Glocker FX,
Vielhaber S, Brejova A, Hilz M, Reiners K, Müller-Felber W, Mengel
E, Spranger M and Schoser B: Enzyme replacement therapy with
alglucosidase alfa in 44 patients with late-onset glycogen storage
disease type 2: 12-month results of an observational clinical
trial. J Neurol. 257:91–97. 2010. View Article : Google Scholar
|
|
19
|
van der Ploeg AT, Clemens PR, Corzo D,
Escolar DM, Florence J, Groeneveld GJ, Herson S, Kishnani PS,
Laforet P, Lake SL, Lange DJ, Leshner RT, Mayhew JE, Morgan C,
Nozaki K, Park DJ, Pestronk A, Rosenbloom B, Skrinar A, van Capelle
CI, van der Beek NA, Wasserstein M and Zivkovic SA: A randomized
study of alglucosidase alfa in late-onset Pompe’s disease. N Engl J
Med. 362:1396–1406. 2010.
|
|
20
|
Parenti G and Andria G: Pompe disease:
from new views on pathophysiology to innovative therapeutic
strategies. Curr Pharm Biotechnol. 12:902–915. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Anson DS, McIntyre C and Byers S:
Therapies for neurological disease in the mucopolysaccharidoses.
Curr Gene Ther. 11:132–143. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Begley DJ, Pontikis CC and Scarpa M:
Lysosomal storage diseases and the blood-brain barrier. Curr Pharm
Des. 14:1566–1580. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Grubb JH, Vogler C, Levy B, Galvin N, Tan
Y and Sly WS: Chemically modified beta-glucuronidase crosses
blood-brain barrier and clears neuronal storage in murine
mucopolysaccharidosis VII. Proc Natl Acad Sci USA. 105:2616–2621.
2008. View Article : Google Scholar
|
|
24
|
Osborn MJ, McElmurry RT, Peacock B, Tolar
J and Blazar BR: Targeting of the CNS in MPS-IH using a nonviral
transferrin-alpha-L-iduronidase fusion gene product. Mol Ther.
16:1459–1466. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Munoz-Rojas MV, Vieira T, Costa R,
Fagondes S, John A, Jardim LB, Vedolin LM, Raymundo M, Dickson PI,
Kakkis E and Giugliani R: Intrathecal enzyme replacement therapy in
a patient with mucopolysaccharidosis type I and symptomatic spinal
cord compression. Am J Med Genet A. 146A:2538–2544. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhu Y, Jiang JL, Gumlaw NK, Zhang J,
Bercury SD, Ziegler RJ, Lee K, Kudo M, Canfield WM, Edmunds T,
Jiang C, Mattaliano RJ and Cheng SH: Glycoengineered acid
alpha-glucosidase with improved efficacy at correcting the
metabolic aberrations and motor function deficits in a mouse model
of Pompe disease. Mol Ther. 17:954–963. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zimran A, Brill-Almon E, Chertkoff R,
Petakov M, Blanco-Favela F, Muñoz ET, Solorio-Meza SE, Amato D,
Duran G, Giona F, Heitner R, Rosenbaum H, Giraldo P, Mehta A, Park
G, Phillips M, Elstein D, Altarescu G, Szleifer M, Hashmueli S and
Aviezer D: Pivotal trial with plant cell-expressed recombinant
glucocerebrosidase, taliglucerase alfa, a novel enzyme replacement
therapy for Gaucher disease. Blood. 118:5767–5773. 2011. View Article : Google Scholar
|
|
28
|
Parenti G: Treating lysosomal storage
diseases with pharmacological chaperones: from concept to clinics.
EMBO Mol Med. 1:268–279. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Valenzano KJ, Khanna R, Powe AC, Boyd R,
Lee G, Flanagan JJ and Benjamin ER: Identification and
characterization of pharmacological chaperones to correct enzyme
deficiencies in lysosomal storage disorders. Assay Drug Dev
Technol. 9:213–235. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fan JQ, Ishii S, Asano N and Suzuki Y:
Accelerated transport and maturation of lysosomal
alpha-galactosidase A in Fabry lymphoblasts by an enzyme inhibitor.
Nat Med. 5:112–115. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Germain DP and Fan JQ: Pharmacological
chaperone therapy by active-site-specific chaperones in Fabry
disease: in vitro and preclinical studies. Int J Clin Pharmacol
Ther. 47(Suppl 1): S111–S117. 2009.PubMed/NCBI
|
|
32
|
Sawkar AR, Adamski-Werner SL, Cheng WC,
Wong CH, Beutler E, Zimmer KP and Kelly JW: Gaucher
disease-associated glucocerebrosidases show mutation-dependent
chemical chaperoning profiles. Chem Biol. 12:1235–1244. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Parenti G, Zuppaldi A, Gabriela Pittis M,
Rosaria Tuzzi M, Annunziata I, Meroni G, Porto C, Donaudy F, Rossi
B, Rossi M, Filocamo M, Donati A, Bembi B, Ballabio A and Andria G:
Pharmacological enhancement of mutated alpha-glucosidase activity
in fibroblasts from patients with Pompe disease. Mol Ther.
15:508–514. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Okumiya T, Kroos MA, Vliet LV, Takeuchi H,
Van der Ploeg AT and Reuser AJ: Chemical chaperones improve
transport and enhance stability of mutant alpha-glucosidases in
glycogen storage disease type II. Mol Genet Metab. 90:49–57. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Suzuki Y, Ichinomiya S, Kurosawa M,
Matsuda J, Ogawa S, Iida M, Kubo T, Tabe M, Itoh M, Higaki K, Nanba
E and Ohno K: Therapeutic chaperone effect of N-octyl
4-epi-β-valienamine on murine G(M1)-gangliosidosis. Mol Genet
Metab. 106:92–98. 2012.
|
|
36
|
Flanagan JJ, Rossi B, Tang K, Wu X,
Mascioli K, Donaudy F, Tuzzi MR, Fontana F, Cubellis MV, Porto C,
Benjamin E, Lockhart DJ, Valenzano KJ, Andria G, Parenti G and Do
HV: The pharmacological chaperone 1-deoxynojirimycin increases the
activity and lysosomal trafficking of multiple mutant forms of acid
alpha-glucosidase. Hum Mutat. 30:1683–1692. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Porto C, Ferrara MC, Meli M, Acampora E,
Avolio V, Rosa M, Cobucci-Ponzano B, Colombo G, Moracci M, Andria G
and Parenti G: Pharmacological enhancement of alpha-glucosidase by
the allosteric chaperone N-acetylcysteine. Mol Ther. Sep
18–2012.(Epub ahead of print). View Article : Google Scholar
|
|
38
|
Urban DJ, Zheng W, Goker-Alpan O, Jadhav
A, Lamarca ME, Inglese J, Sidransky E and Austin CP: Optimization
and validation of two miniaturized glucocerebrosidase enzyme assays
for high throughput screening. Comb Chem High Throughput Screen.
11:817–824. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Marugan JJ, Zheng W, Motabar O, Southall
N, Goldin E, Sidransky E, Aungst RA, Liu K, Sadhukhan SK and Austin
CP: Evaluation of
2-thioxo-2,3,5,6,7,8-hexahydropyrimido(4,5-d)pyrimidin-4(1H)-one
analogues as GAA activators. Eur J Med Chem. 45:1880–1897. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Porto C, Cardone M, Fontana F, Rossi B,
Tuzzi MR, Tarallo A, Barone MV, Andria G and Parenti G: The
pharmacological chaperone N-butyldeoxynojirimycin enhances enzyme
replacement therapy in Pompe disease fibroblasts. Mol Ther.
17:964–971. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Porto C, Pisani A, Rosa M, Acampora E,
Avolio V, Tuzzi MR, Visciano B, Gagliardo C, Materazzi S, la Marca
G, Andria G and Parenti G: Synergy between the pharmacological
chaperone 1-deoxygalactonojirimycin and the human recombinant
alpha-galactosidase A in cultured fibroblasts from patients with
Fabry disease. J Inherit Metab Dis. 35:513–520. 2012. View Article : Google Scholar
|
|
42
|
Benjamin ER, Khanna R, Schilling A,
Flanagan JJ, Pellegrino LJ, Brignol N, Lun Y, Guillen D, Ranes BE,
Frascella M, Soska R, Feng J, Dungan L, Young B, Lockhart DJ and
Valenzano KJ: Co-administration with the pharmacological chaperone
AT1001 increases recombinant human α-galactosidase A tissue uptake
and improves substrate reduction in Fabry mice. Mol Ther.
20:717–726. 2012.PubMed/NCBI
|
|
43
|
Khanna R, Flanagan JJ, Feng J, Soska R,
Frascella M, Pellegrino LJ, Lun Y, Guillen D, Lockhart DJ and
Valenzano KJ: The pharmacological chaperone AT2220 increases
recombinant human acid α-glucosidase uptake and glycogen reduction
in a mouse model of pompe disease. PLoS One.
7:e407762012.PubMed/NCBI
|
|
44
|
Mu TW, Ong DS, Wang YJ, Balch WE, Yates JR
III, Segatori L and Kelly JW: Chemical and biological approaches
synergize to ameliorate protein-folding diseases. Cell.
134:769–781. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Sands MS and Davidson BL: Gene therapy for
lysosomal storage diseases. Mol Ther. 13:839–849. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Cardone M: Prospects for gene therapy in
inherited neurodegenerative diseases. Curr Opin Neurol. 20:15–18.
2007. View Article : Google Scholar
|
|
47
|
Spampanato C, De Leonibus E, Dama P,
Gargiulo A, Fraldi A, Sorrentino NC, Russo F, Nusco E, Auricchio A,
Surace EM and Ballabio A: Efficacy of a combined intracerebral and
systemic gene delivery approach for the treatment of a severe
lysosomal storage disorder. Mol Ther. 19:860–869. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Platt FM and Jeyakumar M: Substrate
reduction therapy. Acta Paediatr (Suppl). 97:88–93. 2008.
View Article : Google Scholar
|
|
49
|
Schiffmann R: Therapeutic approaches for
neuronopathic lysosomal storage disorders. J Inherit Metab Dis.
33:373–379. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Giraldo P, Alfonso P, Atutxa K,
Fernández-Galán MA, Barez A, Franco R, Alonso D, Martin A, Latre P
and Pocovi M: Real-world clinical experience with long-term
miglustat maintenance therapy in type 1 Gaucher disease: the ZAGAL
project. Haematologica. 94:1771–1775. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hollak CE, Hughes D, van Schaik IN,
Schwierin B and Bembi B: Miglustat (Zavesca) in type 1 Gaucher
disease: 5-year results of a post-authorisation safety surveillance
programme. Pharmacoepidemiol Drug Saf. 18:770–777. 2009.PubMed/NCBI
|
|
52
|
Lukina E, Watman N, Arreguin EA, Dragosky
M, Iastrebner M, Rosenbaum H, Phillips M, Pastores GM, Kamath RS,
Rosenthal DI, Kaper M, Singh T, Puga AC and Peterschmitt MJ:
Improvement in hematological, visceral, and skeletal manifestations
of Gaucher disease type 1 with oral eliglustat tartrate
(Genz-112638) treatment: 2-year results of a phase 2 study. Blood.
116:4095–4098. 2010.PubMed/NCBI
|
|
53
|
Lachmann RH, te Vruchte D, Lloyd-Evans E,
Reinkensmeier G, Sillence DJ, Fernandez-Guillen L, Dwek RA, Butters
TD, Cox TM and Platt FM: Treatment with miglustat reverses the
lipid-trafficking defect in Niemann-Pick disease type C. Neurobiol
Dis. 16:654–658. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Patterson MC, Vecchio D, Prady H, Abel L
and Wraith JE: Miglustat for treatment of Niemann-Pick C disease: a
randomised controlled study. Lancet Neurol. 6:765–772. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Wraith JE, Vecchio D, Jacklin E, Abel L,
Chadha-Boreham H, Luzy C, Giorgino R and Patterson MC: Miglustat in
adult and juvenile patients with Niemann-Pick disease type C:
long-term data from a clinical trial. Mol Genet Metab. 99:351–357.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Fecarotta S, Amitrano M, Romano A, Della
Casa R, Bruschini D, Astarita L, Parenti G and Andria G: The
videofluoroscopic swallowing study shows a sustained improvement of
dysphagia in children with Niemann-Pick disease type C after
therapy with miglustat. Am J Med Genet A. 155A:540–547. 2011.
View Article : Google Scholar
|
|
57
|
Piotrowska E, Jakóbkiewicz-Banecka J,
Barańska S, Tylki-Szymańska A, Czartoryska B, Wegrzyn A and Wegrzyn
G: Genistein-mediated inhibition of glycosaminoglycan synthesis as
a basis for gene expression-targeted isoflavone therapy for
mucopolysaccharidoses. Eur J Hum Genet. 14:846–852. 2006.
View Article : Google Scholar
|
|
58
|
Roberts AL, Thomas BJ, Wilkinson AS,
Fletcher JM and Byers S: Inhibition of glycosaminoglycan synthesis
using rhodamine B in a mouse model of mucopolysaccharidosis type
IIIA. Pediatr Res. 60:309–314. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Roberts AL, Fletcher JM, Moore L and Byers
S: Trans-generational exposure to low levels of rhodamine B does
not adversely affect litter size or liver function in murine
mucopolysaccharidosis type IIIA. Mol Genet Metab. 101:208–213.
2010. View Article : Google Scholar
|
|
60
|
Malinowska M, Wilkinson FL, Langford-Smith
KJ, Langford-Smith A, Brown JR, Crawford BE, Vanier MT, Grynkiewicz
G, Wynn RF, Wraith JE, Wegrzyn G and Bigger BW: Genistein improves
neuropathology and corrects behaviour in a mouse model of
neurodegenerative metabolic disease. PLoS One. 5:e141922010.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Piotrowska E, Jakobkiewicz-Banecka J,
Maryniak A, Tylki-Szymanska A, Puk E, Liberek A, Wegrzyn A,
Czartoryska B, Slominska-Wojewodzka M and Wegrzyn G: Two-year
follow-up of Sanfilippo disease patients treated with a
genistein-rich isoflavone extract: assessment of effects on
cognitive functions and general status of patients. Med Sci Monit.
17:196–202. 2011.PubMed/NCBI
|
|
62
|
Ashe KM, Bangari D, Li L, Cabrera-Salazar
MA, Bercury SD, Nietupski JB, Cooper CG, Aerts JM, Lee ER, Copeland
DP, Cheng SH, Scheule RK and Marshall J: Iminosugar-based
inhibitors of glucosylceramide synthase increase brain
glycosphingolipids and survival in a mouse model of Sandhoff
disease. PLoS One. 6:e217582011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Marshall J, Ashe KM, Bangari D, McEachern
K, Chuang WL, Pacheco J, Copeland DP, Desnick RJ, Shayman JA,
Scheule RK and Cheng SH: Substrate reduction augments the efficacy
of enzyme therapy in a mouse model of Fabry disease. PLoS One.
5:e150332010. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Douillard-Guilloux G, Raben N, Takikita S,
Batista L, Caillaud C and Richard E: Modulation of glycogen
synthesis by RNA interference: towards a new therapeutic approach
for glycogenosis type II. Hum Mol Genet. 17:3876–3886. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Douillard-Guilloux G, Raben N, Takikita S,
Ferry A, Vignaud A, Guillet-Deniau I, Favier M, Thurberg BL, Roach
PJ, Caillaud C and Richard E: Restoration of muscle functionality
by genetic suppression of glycogen synthesis in a murine model of
Pompe disease. Hum Mol Genet. 19:684–696. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Raben N, Schreiner C, Baum R, Takikita S,
Xu S, Xie T, Myerowitz R, Komatsu M, Van der Meulen JH, Nagaraju K,
Ralston E and Plotz PH: Suppression of autophagy permits successful
enzyme replacement therapy in a lysosomal storage disorder - murine
Pompe disease. Autophagy. 6:1078–1089. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Jones AM and Helm JM: Emerging treatments
in cystic fibrosis. Drugs. 69:1903–1910. 2009. View Article : Google Scholar
|
|
68
|
Nelson SF, Crosbie RH, Miceli MC and
Spencer MJ: Emerging genetic therapies to treat Duchenne muscular
dystrophy. Curr Opin Neurol. 22:532–538. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Sermet-Gaudelus I, De Boeck K, Casimir GJ,
Vermeulen F, Leal T, Mogenet A, Roussel D, Fritsch J, Hanssens L,
Hirawat S, Miller NL, Constantine S, Reha A, Ajayi T, Elfring GL
and Miller LL: Ataluren (PTC124) induces CFTR protein expression
and activity in children with nonsense mutation cystic fibrosis. Am
J Respir Crit Care Med. 182:1262–1272. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Finkel RS: Read-through strategies for
suppression of nonsense mutations in Duchenne/Becker muscular
dystrophy: aminoglycosides and ataluren (PTC124). J Child Neurol.
25:1158–1164. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Sarkar C, Zhang Z and Mukherjee AB: Stop
codon read-through with PTC124 induces palmitoyl-protein
thioesterase-1 activity, reduces thioester load and suppresses
apoptosis in cultured cells from INCL patients. Mol Genet Metab.
104:338–345. 2011. View Article : Google Scholar
|
|
72
|
Medina DL, Fraldi A, Bouche V, Annunziata
F, Mansueto G, Spampanato C, Puri C, Pignata A, Martina JA,
Sardiello M, Palmieri M, Polishchuk R, Puertollano R and Ballabio
A: Transcriptional activation of lysosomal exocytosis promotes
cellular clearance. Dev Cell. 21:421–430. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Sardiello M, Palmieri M, di Ronza A,
Medina DL, Valenza M, Gennarino VA, Di Malta C, Donaudy F, Embrione
V, Polishchuk RS, Banfi S, Parenti G, Cattaneo E and Ballabio A: A
gene network regulating lysosomal biogenesis and function. Science.
325:473–477. 2009.PubMed/NCBI
|