Introduction
Hepatocellular carcinoma (HCC) is the fifth-most
common malignant tumor in the world (1). The incidence of HCC has been on the
rise in recent years (1,2), and more than 70% of all newly
diagnosed cases of liver cancer occur in Asia (3). The treatment for HCC usually
involves surgical resection or liver transplantation with curative
options for the patients, when the disease is diagnosed at an early
stage (4). However, only 30% of
the patients are eligible for the curative treatment, and
recurrence is a common concern affecting up to 70% of the patients
after tumor ablation (5). Only a
few non-curative treatment options exist for such patients. Some
effective treatment modalities include: ethanol ablation,
radiofrequency ablation, transarterial chemoembolization, and
selective radiation of lesions (6). Therefore, new therapeutic agents for
the treatment of hepatoma need to be explored.
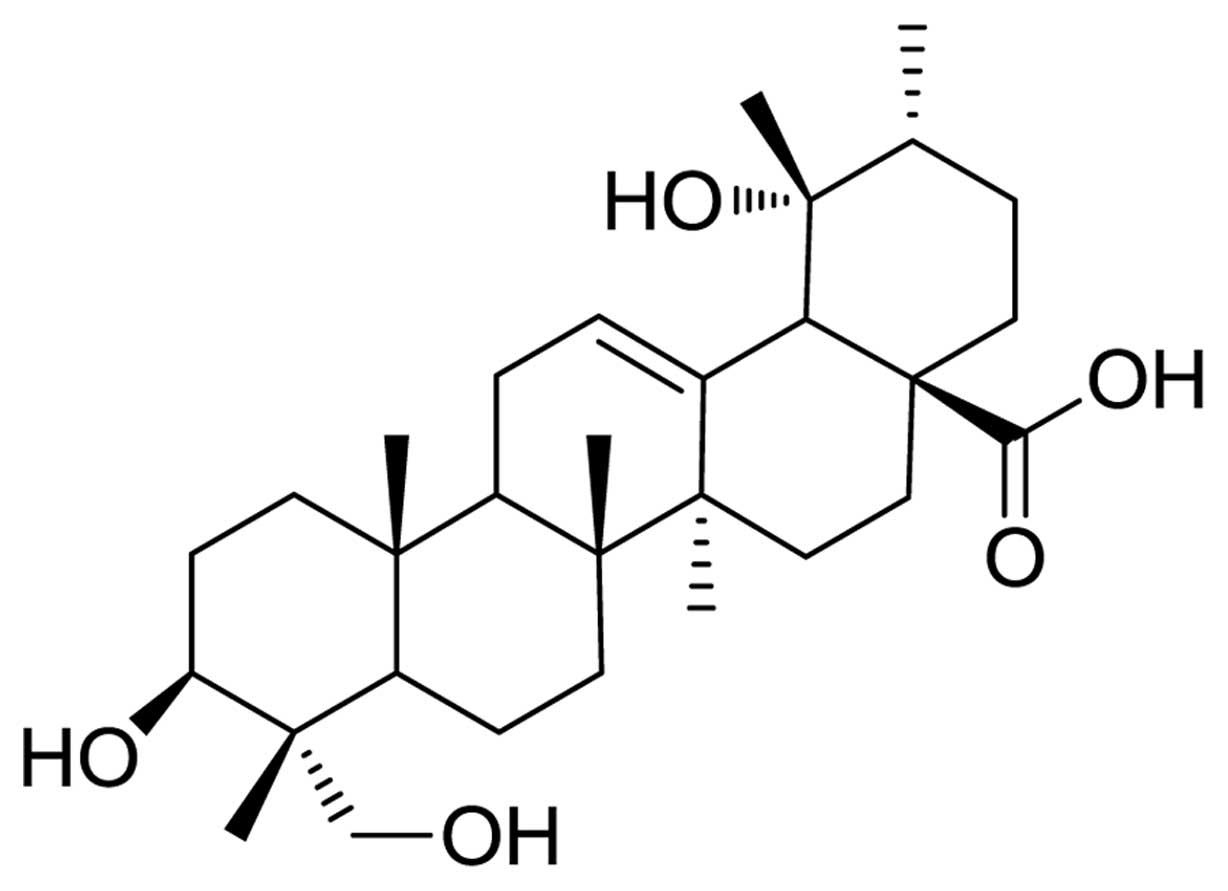
Rotundic acid (RA, 1) (Fig. 1) is one of the pentacyclic
triterpenoids, mainly found in Ilex rotunda, Ilex
purpurea and other Aquifoliaceae plants (7–10).
RA can also be isolated from Mussaenda Pubescens, Olea
europaea and Planchonella duclitan (11–13). RA and its derivatives inhibit cell
growth. Due to its unstable nature during the metabolic processes,
RA induces serious gastrointestinal adverse effects. A considerable
number of patents on RA and its derivatives have been applied by
our research group in the recent years. Our studies explored these
novel compounds extensively and focused on reducing these adverse
effects (14–17). In our previous study, eight amino
acid derivatives of RA at the 28-COOH position were synthesized,
and their in vitro cytotoxic properties were evaluated on
three tumor cell lines, A375 (human malignant melanoma cells),
HepG2 (human hepatoma cells), and NCI-H446 (human small cell lung
cancer cells) (18).
The present study aimed at synthesizing RA
derivatives by making structural modifications at the 28-COOH
position of RA. The synthesized compounds were characterized and
their cytotoxic effects in the three cell lines, HeLa (human
cervical cancer), HepG2 (human hepatoma cell) and SGC-7901 (human
gastric carcinoma), were studied.
Materials and methods
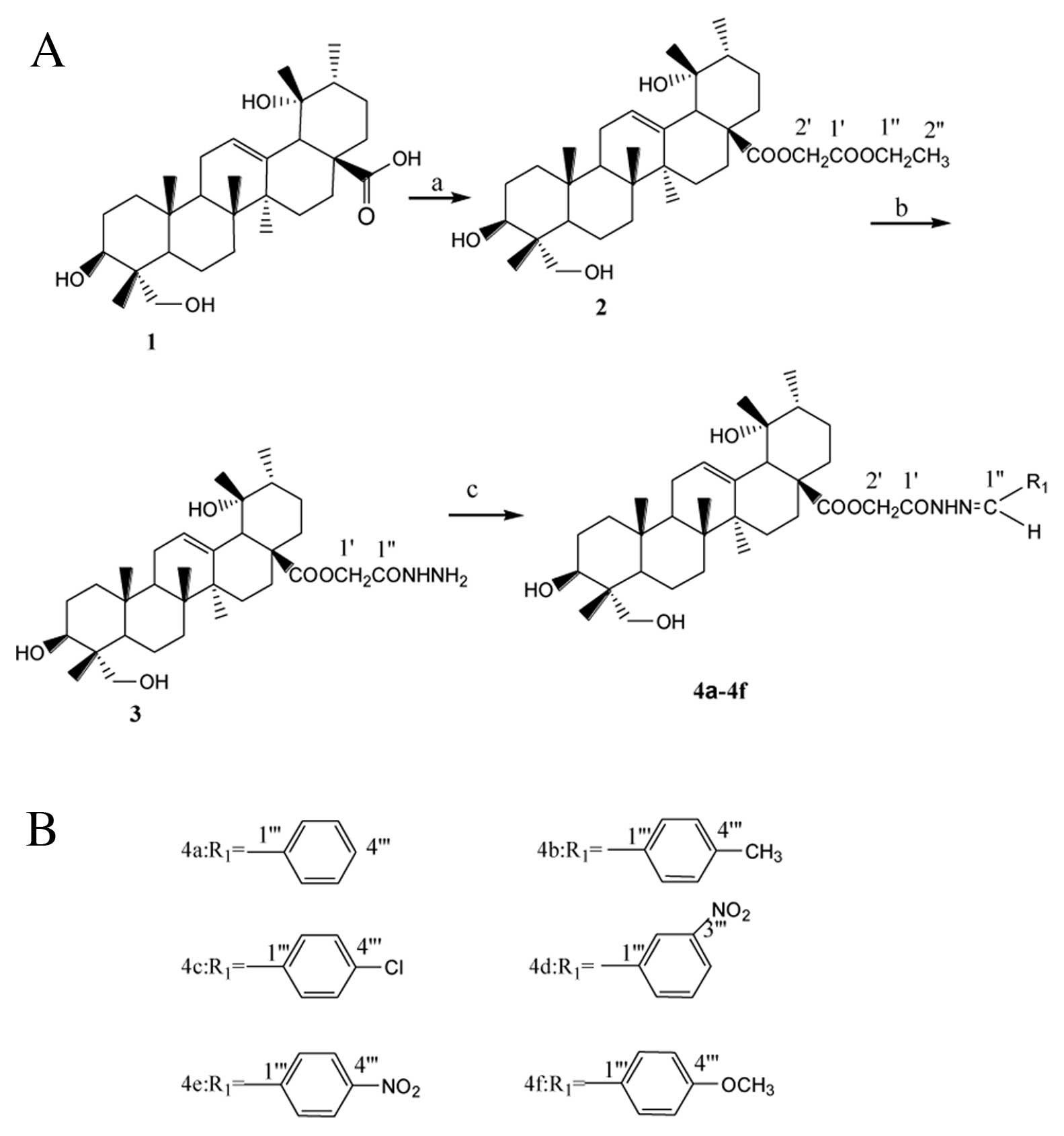
Based on the natural structure of RA, six new
derivatives 4a–4f were designed and synthesized in this study to
improve its bioactivity. Their structures were elucidated on the
basis of spectroscopic assays such as IR, MS, 1H NMR and
13C NMR. The cytotoxicity and antitumor effects of RA
derivatives were assayed by MTT colorimetric assay with the HeLa,
HepG2 and SGC-7901 cell lines. Cell cycle distribution and
measurement of the percentage of apoptotic cells were performed by
flow cytometry, following staining with propidium iodide (PI) and
Annexin V-FITC.
Reagents and cell culture
Reagent-grade chemicals and solvents were obtained
from Sigma Chemicals, Munich, Germany. All solvents were freshly
distilled and dried prior to use, according to the standard
procedures. The HeLa, HepG2 and SGC-7901 cell lines were purchased
from the Tumor Center of the Chinese Academy of Medical Sciences,
Beijing, China. Cells were maintained in Dulbecco’s modified
Eagle’s medium (DMEM) containing 10% FBS, penicillin (100 U/ml) and
streptomycin (100 μg/ml) at 37°C in a humidified atmosphere
of 5% CO2.
Preparation of RA
The preparation of RA was reported in our previous
study (18). The purity of RA
used was ≥98% (HPLC assay) and the extraction yield of RA in our
study was up to 100 mg/g, which made it suitable for industry
production. Melting points were determined on a Fisher-Johns
apparatus and were uncorrected. IR spectra were recorded on a
Perkin-Elmer 983G spectrometer. NMR spectra were measured in
C5D5N on a Bruker AM-400 spectrometer, using
tetramethylsilane as an internal standard. Coupling constants
(J values) were given in Hz, and an MS Agilent 1100 Series
LC/MSD ion-trap mass spectrometer was used to record the
HR-ESI-MS.
Structure modification of RA and
synthesis of derivatives
The synthetic routes to the RA derivatives are
outlined in Fig. 2. Firstly, RA
(1) was converted to its 28-ethyl
acetate (2), which was then
treated with hydrazine hydrate to give the 28-acetohydrazide
(3). It was then reacted with the
appropriate aldehyde (benzaldehyde, p-methyl benzaldehyde,
p-chloride benzaldehyde, m-nitryl benzaldehyde, p-nitryl
benzaldehyde, p-methoxy benzaldehyde) in the presence of glacial
acetic acid to give the benzaldehyde
{2-[(3β,19α,23-trihydroxy-urs-12-en-28-oyl)-hydroxy]-acetic}
hydrazone 4a–4f. The structures of these synthesized compounds were
confirmed by IR, MS, 1H NMR and 13C NMR
(25–28). All six compounds obtained were
synthesized in high yields with purities of 98% or better and are
reported for the first time.
Procedure for the preparation of ethyl
2-[(3β,19α,23-trihydroxy-urs-12-en-28-oyl)-hydroxy]-acetate
(2)
RA (10 mmol) was dissolved in acetone (500 ml) and
heated to clarity. Potassium carbonate (100 mmol), potassium iodide
(0.6 mmol), and triethylamine (6 ml) were added to it. Ethyl
chloroacetate (5 ml) was then added drop-wise and refluxed for 24
h. The refluxed content was filtered, and the filtrate was
concentrated to 100 ml. Saturated salt water (100 ml) was added and
was extracted with ethyl acetate five times (30 ml each time). It
was then washed with saturated sodium carbonate and water four
times (25 ml each time). Anhydrous sodium sulfate was added and
filtered. The filtrate was evaporated to dryness. The crude was
purified by column chromatography on a silica gel column with
petroleum ether, ethyl acetate, and formic acid as eluents to give
compound 2 as a white powder. The yield was 83.9%, and its melting
point was between 133 and 135°C. IR(KBr) cm−1: 3545,
3404, 3250, 2929, 2882, 1765, 1715, 1634, 1469, 1455, 1381, 1207,
1148, 1055 and 1005; 1H NMR (400 MHz,
C5D5N) δ: 5.40 (1H, m, H-12), 5.06 (1H, m,
H-3), 4.78 (2H, dd, J=15.6, 27.6 Hz, H-2′), 4.07 (2H, q,
J=7.2Hz, H-1″), 4.06 (1H, d, J=10.4 Hz, H-23a), 3.61
(1H, d, J=10.4 Hz, H-23b), 2.75 (1H, br s, H-18), 1.51 (3H,
s, CH3-25), 1.26 (3H, s, CH3-27), 1.03 (3H,
t, J=7.2 Hz, CH3-2″), 0.97 (3H, d, J=6.64
Hz, CH3-30), 0.96 (3H, s, CH3-29), 0.93 (3H,
s, CH3-26), 0.82 (3H, s, CH3-24);
13C NMR (100 MHz, C5D5N) δ: 177.7
(C-28), 139.4 (C-13), 128.6 (C-12), 73.7 (C-3), 72.7 (C-19), 68.1
(C-23), 54.5 (C-18), 48.8 (C-5), 48.8 (C-9), 47.8 (C-17), 43.0
(C-20), 42.2 (C-14), 42.1 (C-8), 40.5 (C-1), 39.0 (C-4), 38.0
(C-22), 37.3 (C-10), 33.3 (C-7), 29.1 (C-15), 27.8 (C-21), 27.0
(C-29), 26.8 (C-2), 26.3 (C-16), 24.7 (C-27), 24.2 (C-11), 18.8
(C-6), 17.2 (C-25), 16.8 (C-26), 16.2 (C-30), 13.2 (C-24), 61.3
(C-2′), 168.7 (C-1′), 61.0 (C-1″), 14.3 (C-2″). HR-ESI-MS found
575.3953. Calculated 575.3948 for
C34H55O7 [(M+H)+].
Procedure for the preparation of
2-[(3β,19α,23-trihydroxy-urs-12-en-28-oyl)-hydroxy]-acetic
hydrazide (3)
Compound 2 (15 mmol) was dissolved in absolute ethyl
alcohol (EtOH) (90 ml) and 80% hydrazine hydrate (7.5 ml) was
added. The mixture was refluxed for 11 h and the solvent was
removed under reduced pressure using a rotary evaporator to 50 ml.
Water (250 ml) was added and extracted with chloroform five times
(50 ml each time). It was then washed with saturated salt water
four times (50 ml each time). Anhydrous sodium sulfate was added
and filtered. The filtrate was evaporated to dryness to yield
crude, which was then purified by column chromatography on a silica
gel column with petroleum ether and ethyl acetate as eluents to
give compound 3 as colorless needles. The yield was 97.7% and its
melting point was between 225 and 226°C. IR(KBr) cm−1:
3624, 3398, 3335, 2930, 2876, 1731, 1683, 1450, 1387, 1206, 1147,
1046 and 756; 1H NMR (400 MHz,
C5D5N) δ: 9.76 (2H, s, −NH2), 7.45 (1H, s,
−NH), 5.43 (1H, m, H-12), 5.04 (1H, m, H-3), 4.85 (2H, dd,
J=14.2, 33.2 Hz, H-1′), 4.06 (1H, d, J=10.4 Hz,
H-23a), 3.60 (1H, d, J=10.4 Hz, H-23b), 2.71 (1H, br s,
H-18), 1.50 (3H, s, CH3-25), 1.23 (3H, s,
CH3-27), 0.96 (3H, s, CH3-29), 0.94 (3H, d,
J=6.64 Hz, CH3-30), 0.90 (3H, s,
CH3-26), 0.79 (3H, s, CH3-24); 13C
NMR (100 MHz, C5D5N) δ: 177.5 (C-28), 139.7
(C-13), 128.5 (C-12), 73.6 (C-3), 72.7 (C-19), 68.1 (C-23), 54.5
(C-18), 48.8 (C-5), 48.7 (C-9), 47.8 (C-17), 43.0 (C-20), 42.1
(C-14), 42.1 (C-8), 40.4 (C-1), 39.0 (C-4), 38.0 (C-22), 37.3
(C-10), 33.2 (C-7), 29.1 (C-15), 27.8 (C-21), 27.0 (C-29), 26.8
(C-2), 26.2 (C-16), 24.7 (C-27), 24.1 (C-11), 18.8 (C-6), 17.2
(C-25), 16.7 (C-26), 16.1 (C-30), 13.2 (C-24), 62.8 (C-1′), 168.2
(C-1″). HR-ESI-MS found 561.3887. Calculated 561.3898 for
C32H53N2O6
[(M+H)+].
General procedure for the preparation of
benzaldehyde
{2-[(3β,19α,23-trihydroxy-urs-12-en-28-oyl)-hydroxy]-acetic}
hydrazone 4a–4f
Benzaldehyde
{2-[(3β,19α,23-trihydroxy-urs-12-en-28-oyl)-hydroxy]-acetic}
hydrazone (4a,
C39H56N2O6,
R1=C6H5, R2=H)
Compound 3 (2 mmol) was dissolved in absolute EtOH
(30 ml) and benzaldehyde (0.3 ml) was added. Glacial acetic acid
(0.2 ml) was added drop-wise and the mixture was refluxed for 8 h.
Water (100 ml) was then added, stirred and filtered. The crude
obtained was then purified on a silica gel column with petroleum
ether and ethyl acetate as eluents to yield white needles. The
yield was 80.3%, and its melting point was between 169 and 171°C.
IR(KBr) cm−1: 3619, 3425, 2929, 2876, 1693, 1449, 1388,
1204, 1140, 1047, 755 and 695; 1H NMR (400 MHz,
C5D5N) δ: 12.50 (1H, s, −NH), 8.05 (1H, s,
H-1″), 7.64 (2H, d, J=8.0 Hz, H-2‴, 6‴), 7.25 (2H, t,
J=8.0 Hz, H-3‴, 5‴), 7.21 (1H, t, J=8.0 Hz, H-4‴),
5.51 (2H, dd, J=15.9, 26.9 Hz, H-2′), 5.47 (1H, m, H-12),
4.98 (1H, m, H-3), 4.06 (1H, d, J=10.4 Hz, H-23a), 3.61 (1H,
d, J=10.4 Hz, H-23b), 2.90 (1H, br s, H-18), 1.55 (3H, s,
CH3-25), 1.28 (3H, s, CH3-27), 0.97 (3H, s,
CH3-29), 0.96 (3H, d, J=6.64 Hz,
CH3-30), 0.94 (3H, s, CH3-26), 0.90 (3H, s,
CH3-24); 13C NMR (100 MHz,
C5D5N) δ: 177.9 (C-28), 139.6 (C-13), 128.5
(C-12), 73.7 (C-3), 72.8 (C-19), 68.2 (C-23), 54.5 (C-18), 49.0
(C-5), 48.8 (C-9), 47.9 (C-17), 43.0 (C-20), 42.3 (C-14), 42.2
(C-8), 40.5 (C-1), 39.0 (C-4), 38.3 (C-22), 37.3 (C-10), 33.3
(C-7), 30.1 (C-15), 29.3 (C-21), 27.8 (C-29), 27.0 (C-2), 26.4
(C-16), 24.7 (C-27), 24.2 (C-11), 18.8 (C-6), 17.4 (C-25), 16.9
(C-26), 16.2 (C-30), 13.2 (C-24), 61.8 (C-2′), 169.5 (C-1′), 144.0
(C-1″), 135.0 (C-1‴), 129.2 (C-3‴, 5‴), 127.6 (C-2‴, 6‴), 130.3
(C-4‴). HR-ESI-MS found 649.4211. Calculated 649.4212 for
C39H57N2O6
[(M+H)+].
4-methyl-benzaldehyde
{2-[(3β,19α,23-trihydroxy-urs-12-en-28-oyl)-hydroxy]-acetic}
hydrazone (4b,
C40H58N2O6,
R1=CH3C6H4,
R2=H)
Compound 3 was reacted with p-methyl benzaldehyde
using the general procedure to give compound 4b, which was eluted
with petroleum ether/ethyl acetate (V/V)=1:5 to give white powder.
The yield was 87.2% and its melting point was between 210 and
211°C. IR(KBr) cm−1: 3620, 3275, 2928, 2874, 1739, 1688,
1468, 1388, 1229, 1149, 1046, 813 and 514; 1H NMR (400
MHz, C5D5N) δ: 12.43 (1H, s, −NH), 8.04 (1H,
s, H-1″), 7.57 (2H, d, J=8.0 Hz, H-2‴,6‴), 7.08 (2H, d,
J=8.0 Hz, H-3‴, 5‴), 5.54 (2H, dd, J=15.8, 27.0 Hz,
H-2′), 5.46 (1H, m, H-12), 4.97 (1H, m, H-3), 4.07 (1H, d,
J=10.4 Hz, H-23a), 3.61 (1H, d, J=10.4 Hz, H-23b),
2.90 (1H, br s, H-18), 2.10 (3H, s, 4‴-CH3), 1.55 (3H,
s, CH3-25), 1.28 (3H, s, CH3-27), 0.97 (3H,
s, CH3-29), 0.96 (3H, d, J=6.64 Hz,
CH3-30), 0.91 (3H, s, CH3-26), 0.89 (3H, s,
CH3-24); 13C NMR (100 MHz,
C5D5N) δ: 177.9 (C-28), 139.6 (C-13), 128.5
(C-12), 73.7 (C-3), 72.8 (C-19), 68.2 (C-23), 54.5 (C-18), 49.0
(C-5), 48.8 (C-9), 47.9 (C-17), 43.0 (C-20), 42.3 (C-14), 42.2
(C-8), 40.5 (C-1), 39.0 (C-4), 38.3 (C-22), 37.3 (C-10), 33.3
(C-7), 30.1 (C-15), 29.3 (C-21), 27.8 (C-29), 27.1 (C-2), 26.4
(C-16), 24.7 (C-27), 24.2 (C-11), 18.9 (C-6), 17.4 (C-25), 16.8
(C-26), 16.2 (C-30), 13.2 (C-24), 61.8 (C-2′), 169.4 (C-1′), 144.2
(C-1″), 132.3 (C-1‴), 129.9 (C-3‴, 5‴), 127.6 (C-2‴, 6‴), 140.3
(C-4‴), 21.4 (4‴-CH3). HR-ESI-MS found 663.4388.
Calculated 663.4368 for
C40H59N2O6
[(M+H)+].
4-chloride-benzaldehyde
{2-[(3β,19α,23-trihydroxy-urs-12-en-28-oyl)-hydroxy]-acetic}
hydrazone (4c,
C39H55ClN2O6,
R1=p-ClC6H4, R2=H)
Compound 3 was reacted with p-chlorine benzaldehyde
using the general procedure to give compound 4c, which was eluted
with petroleum ether/ethyl acetate (V/V)=1:5 to give white needles.
The yield was 91.0% and its melting point was between 205 and
207°C. IR(KBr) cm−1: 3620, 3440, 2930, 2876, 1699, 1459,
1388, 1259, 1140, 1046, 826 and 515; 1H NMR (400 MHz,
C5D5N) δ: 12.60 (1H, s, −NH), 7.98 (1H, s,
H-1″), 7.57 (2H, d, J=8.8 Hz, H-2‴, 6‴), 7.31 (2H, d,
J=8.8 Hz, H-3‴, 5‴), 5.49 (2H, dd, J=15.8, 28.4 Hz,
H-2′), 5.43 (1H, m, H-12), 5.00 (1H, m, H-3), 4.08 (1H, d,
J=10.4 Hz, H-23a), 3.61 (1H, d, J=10.4 Hz, H-23b),
2.90 (1H, br s, H-18), 1.55 (3H, s, CH3-25), 1.28 (3H,
s, CH3-27), 0.97 (3H, s, CH3-29), 0.96 (3H,
d, J=6.64 Hz, CH3-30), 0.94 (3H, s,
CH3-26), 0.91 (3H, s, CH3-24); 13C
NMR (100 MHz, C5D5N) δ: 177.9 (C-28), 139.6
(C-13), 128.5 (C-12), 73.7 (C-3), 72.8 (C-19), 68.2 (C-23), 54.5
(C-18), 49.0 (C-5), 48.8 (C-9), 47.9 (C-17), 43.0 (C-20), 42.3
(C-14), 42.2 (C-8), 40.5 (C-1), 39.0 (C-4), 38.3 (C-22), 37.3
(C-10), 33.3 (C-7), 30.1 (C-15), 29.3 (C-21), 27.8 (C-29), 27.1
(C-2), 26.4 (C-16), 24.7 (C-27), 24.2 (C-11), 18.9 (C-6), 17.4
(C-25), 16.8 (C-26), 16.2 (C-30), 13.2 (C-24), 61.7 (C-2′), 169.6
(C-1′), 142.6 (C-1″), 129.2 (C-1‴), 129.4 (C-3‴, 5‴), 128.9 (C-2‴,
6‴), 133.7 (C-4‴). HR-ESI-MS found 683.3821. Calculated 683.3821
for C39H56ClN2O6
[(M+H)+].
3-nitryl-benzaldehyde
{2-[(3β,19α,23-trihydroxy-urs-12-en-28-oyl)-hydroxy]-acetic}
hydrazone (4d,
C39H55N3O8,
R1=m-NO2C6H4,
R2=H)
Compound 3 was reacted with m-nitryl benzaldehyde
using the general procedure to give compound 4d, which was eluted
with petroleum ether/ethyl acetate (V/V)=1:5 to give white needles.
The yield was 93.0% and its melting point was between 175 and
177°C. IR(KBr) cm−1: 3423, 2929, 2876, 1702, 1534, 1450,
1388, 1352, 1228, 1140, 1045, 736 and 678; 1H NMR (400
MHz, C5D5N) δ: 12.88 (1H, s, −NH), 8.46 (1H,
s, H-1″), 8.11 (1H, brs, H-2‴), 8.06 (1H, d, J=8.0 Hz,
H-4‴), 8.06 (1H, d, J=8.0 Hz, H-5‴), 7.37 (1H, t,
J=8.0 Hz, H-6‴), 5.52 (2H, dd, J=15.8, 24.3 Hz,
H-2′), 5.46 (1H, m, H-12), 4.97 (1H, m, H-3), 4.08 (1H, d,
J=10.4 Hz, H-23a), 3.61 (1H, d, J=10.4 Hz, H-23b),
2.90 (1H, br s, H-18), 1.55 (3H, s, CH3-25), 1.29 (3H,
s, CH3-27), 0.97 (3H, s, CH3-29), 0.96 (3H,
d, J=6.64 Hz, CH3-30), 0.96 (3H, s,
CH3-26), 0.92 (3H, s, CH3-24); 13C
NMR (100 MHz, C5D5N) δ: 180.0 (C-28), 139.6
(C-13), 128.5 (C-12), 73.7 (C-3), 72.8 (C-19), 68.2 (C-23), 54.6
(C-18), 49.0 (C-5), 48.8 (C-9), 47.9 (C-17), 43.0 (C-20), 42.3
(C-14), 42.2 (C-8), 40.5 (C-1), 39.0 (C-4), 38.3 (C-22), 37.3
(C-10), 33.3 (C-7), 30.1 (C-15), 29.3 (C-21), 27.8 (C-29), 27.1
(C-2), 26.5 (C-16), 24.7 (C-27), 24.2 (C-11), 18.9 (C-6), 17.4
(C-25), 16.8 (C-26), 16.2 (C-30), 13.2 (C-24), 61.7 (C-2′), 169.8
(C-1′), 141.3 (C-1″), 149.1 (C-3‴), 136.9 (C-1‴), 132.6 (C-6‴),
130.2 (C-5‴), 124.5 (C-4‴), 122.3 (C-2‴). HR-ESI-MS found 694.4062.
Calculated 694.4062 for
C39H56N3O8
[(M+H)+].
4-nitryl-benzaldehyde
{2-[(3β,19α,23-trihydroxy-urs-12-en-28-oyl)-hydroxy]-acetic}
hydrazone (4e,
C39H55N3O8,
R1=p-NO2C6H4,
R2=H)
Compound 3 was reacted with p-nitryl benzaldehyde
using the general procedure to give compound 4e, which was eluted
with petroleum ether/ethyl acetate (V/V)=1:5 to give pale yellow
needles. The yield was 85.1% and its melting point was between 230
and 231°C. IR(KBr) cm−1: 3606, 3499, 2925, 2879, 1713,
1699, 1517, 1407, 1341, 1234, 1151, 1043, 835 and 752;
1H NMR (400 MHz, C5D5N) δ: 12.96
(1H, s, −NH), 8.13 (2H, d, J=8.8 Hz, H-2‴,6‴), 8.06 (1H, s,
H-1″), 7.75 (2H, d, J=8.8 Hz, H-3‴, 5‴), 5.52 (2H, dd,
J=15.9, 26.8 Hz, H-2′), 5.47 (1H, m, H-12), 5.10 (1H, m,
H-3), 4.08 (1H, d, J=10.4 Hz, H-23a), 3.61 (1H, d,
J=10.4 Hz, H-23b), 2.90 (1H, br s, H-18), 1.55 (3H, s,
CH3-25), 1.29 (3H, s, CH3-27), 0.97 (3H, s,
CH3-29), 0.96 (3H, d, J= 6.64 Hz,
CH3-30), 0.96 (3H, s, CH3-26), 0.91 (3H, s,
CH3-24); 13C NMR (100 MHz,
C5D5N) δ: 180.0 (C-28), 139.6 (C-13), 128.5
(C-12), 73.7 (C-3), 72.8 (C-19), 68.1 (C-23), 54.5 (C-18), 49.0
(C-5), 48.8 (C-9), 47.9 (C-17), 43.0 (C-20), 42.3 (C-14), 42.2
(C-8), 40.5 (C-1), 39.0 (C-4), 38.3 (C-22), 37.3 (C-10), 33.3
(C-7), 30.1 (C-15), 29.3 (C-21), 27.8 (C-29), 27.1 (C-2), 26.4
(C-16), 24.7 (C-27), 24.2 (C-11), 18.9 (C-6), 17.4 (C-25), 16.8
(C-26), 16.2 (C-30), 13.2 (C-24), 61.7 (C-2′), 169.6 (C-1′), 141.3
(C-1″), 140.9 (C-1″″), 128.0 (C-3‴, 5‴), 124.4 (C-2‴, 6‴), 148.6
(C-4‴). HR-ESI-MS found 694.4061. Calculated 694.4062 for
C39H56N3O8
[(M+H)+].
4-methoxy-benzaldehyde
{2-[(3β,19α,23-trihydroxy-urs-12-en-28-oyl)-hydroxy]-acetic}
hydrazone (4f,
C40H58N2O7,
R1=p-CH3OC6H4,
R2=H)
Compound 3 was reacted with p-methoxy benzaldehyde
using the general procedure to give compound 4f, which was eluted
with petroleum ether/ethyl acetate (V/V)=1:3 to give white needles.
The yield was 89.2% and its melting point was between 173 and
175°C. IR(KBr) cm−1: 3622, 3483, 2932, 2875, 1688, 1607,
1516, 1462, 1377, 1252, 1170, 1034, 831 and 530; 1H NMR
(400 MHz, C5D5N) δ: 12.40 (1H, s, −NH), 8.04
(1H, s, H-1″), 7.63 (2H, d, J=8.8 Hz, H-2‴, 6‴), 6.87 (2H,
d, J=8.8 Hz, H-3‴, 5‴), 5.49 (2H, dd, J=15.9, 28.2
Hz, H-2′), 5.43 (1H, m, H-12), 4.99 (1H, m, H-3), 4.08 (1H, d,
J=10.4 Hz, H-23a), 3.61 (1H, d, J=10.4 Hz, H-23b),
3.56 (3H, s, −OCH3), 2.90 (1H, br s, H-18), 1.55 (3H, s,
CH3-25), 1.28 (3H, s, CH3-27), 0.97 (3H, s,
CH3-29), 0.96 (3H, d, J=6.64 Hz,
CH3-30), 0.94 (3H, s, CH3-26), 0.90 (3H, s,
CH3-24); 13C NMR (100 MHz,
C5D5N) δ: 177.9 (C-28), 139.6 (C-13), 128.5
(C-12), 73.7 (C-3), 72.8 (C-19), 68.2 (C-23), 54.5 (C-18), 49.0
(C-5), 48.8 (C-9), 47.9 (C-17), 43.0 (C-20), 42.3 (C-14), 42.2
(C-8), 40.5 (C-1), 39.0 (C-4), 38.3 (C-22), 37.3 (C-10), 33.3
(C-7), 30.1 (C-15), 29.3 (C-21), 27.8 (C-29), 27.1 (C-2), 26.4
(C-16), 24.7 (C-27), 24.2 (C-11), 18.9 (C-6), 17.4 (C-25), 16.8
(C-26), 16.2 (C-30), 13.2 (C-24), 62.8 (C-2′), 169.3 (C-1′), 143.9
(C-1″), 127.7 (C-1‴), 129.2 (C-3‴, 5‴), 114.8 (C-2‴, 6‴), 161.7
(C-4‴), 55.4 (C-OCH3). HR-ESI-MS found 679.4332.
Calculated 679.4317 for
C40H59N2O7
[(M+H)+].
Cell cytotoxicity assays
The cell survival rate was measured by an MTT assay
(29). Aliquots (200 μl)
of 5×103 cells/ml of HeLa, HepG2 and SGC-7901 cells were
seeded in 96-well plates in DMEM medium containing 10% FBS at 37°C
in a humidified atmosphere of 5% CO2. The test drugs
were dissolved in dimethyl sulfoxide (DMSO). The incubation medium
was replaced with each test medium, making a final concentration of
1.25 to 100 μM of RA and compounds 4a–4f for 48 h and DMSO
at 0.1% in media as a vehicle control. After adding 5 μl of
MTT labeling reagent (MTT Cell Proliferation Assay kit; Trevigen,
USA) to each well, the plates were incubated for 4 h, before each
well was supplemented with 100 μl solubilization solution.
The absorbance at 570 nm was then measured in a microtiter plate
reader (Bio-Tek, Winooski, VT, USA). The drug treatments were
performed separately three times.
Apoptosis determination by DAPI
staining
Apoptotic morphological changes were determined by
DAPI staining. The harvested cells were exposed with compound 4f
for 48 h. The medium was then removed from the culture, and the
cells were washed twice with cold PBS and fixed with 100% EtOH for
20 min at room temperature. It was washed twice again with PBS and
incubated with DAPI solution (0.5 μg/ml) for 30 min. Then,
cell morphology was evaluated by fluorescence microscopy
(IX70-SIF2; Olympus).
Annexin V-FITC and PI double staining
analysis by flow cytometry
The cells were plated at appropriate densities
(∼2.5×104 cells/well) in 3 ml of medium in 6-well
plates. The induction of apoptosis was evaluated in HepG2 cells
plated on 6-well plates overnight with compound 4f dissolved in
DMEM medium. After 48 h in culture, the adherent cells were
harvested, washed twice with cold PBS, and then assayed for
apoptosis by double staining with Annexin V-FITC and PI (Annexin
V-FITC Apoptosis Detection kit; KeyGEN Biotech, Nanking, China).
Briefly, 5×105 cells were re-suspended in 1× binding
buffer and stained with 5 μl of FITC-Annexin V. After 10 min
of incubation, 5 μl of PI was added, and the samples were
incubated again for 15 min. The samples were then immediately
analyzed using a flow cytometer (FACScan; BD Biosciences, Milan,
Italy) with a dedicated software. The cells in the upper right
portion were late-apoptotic cells. The cells in the lower left
portion were viable cells. The cells in the lower right portion
were early apoptotic cells.
Cell cycle analysis
HepG2 cells (2×105/well) were placed in
6-well plates, and then compound 4f was added to the wells and
cultured for 48 h. The vehicle control in the media was DMSO at
0.1%. The cells from each well were harvested individually by
centrifugation. The isolated cells were fixed by 70% EtOH at 4°C
overnight, and then re-suspended in PBS containing 40 μg/ml
PI, 0.1 mg/ml RNase A, and 0.1% Triton X-100 in a dark room for 30
min at 37°C. The DNA content of 20,000 cells for each determination
was measured using EPICS XL-MCL flow cytometer (Beckman Coulter,
Inc., Fullerton, CA, USA), in which an argon laser (488 nm) was
used to excite PI. An emission above 550 nm was collected.
Statistical analysis
The data were analyzed for mean values and standard
deviation for all the experiments. Statistically significant
differences were determined between the control and treated groups
using Student’s t-test. P<0.05 was considered to indicate a
statistically significant difference. Statistical package for the
Social Sciences (SPSS, version 13.0) was used for statistical
analyses.
Results
Preparation of RA
The barks of I. rotunda were shade-dried,
grounded, and extracted with refluxing 80% EtOH. The air-dried and
powdered total saponins from EtOH extract were hydrolyzed by 4%
sodium hydroxide in 30% EtOH and purified by recrystallization to
prepare RA. The purity of RA used was found to be ≥98% in a high
performance liquid chromatography assay.
Structure modification of RA
The 28-COOH position of RA was modified to obtain
six new compounds (Fig. 2). The
cytotoxic activity of RA and its six derivatives were studied.
Cell cytotoxicity assays
The effects of RA and its derivatives 4a–4f on cell
cytotoxicity were measured by MTT assay. The three types of human
cancer cell lines were exposed to 1.25 to 100 μM of RA
derivatives for 48 h. RA showed significant IC50 values
of 18.70, 8.26 and 16.48 μM on the HeLa, HepG2 and SGC-7901
cell lines, respectively (Table
I).
 | Table I.The IC50 values of RA and
its derivatives 4a–4f on human cancer cell lines. |
Table I.
The IC50 values of RA and
its derivatives 4a–4f on human cancer cell lines.
| IC50
(μM)
|
|---|
| Compound | HeLa | HepG2 | SGC-7901 |
|---|
| RA | 18.70±1.61 | 8.26±1.24 | 16.48±2.32 |
| 4a | 86.67±3.86 | >100a | 44.29±4.27 |
| 4b | 20.58±0.79 | 34.60±3.55 | 41.22±2.98 |
| 4c | >100a | >100a | >100a |
| 4d | 9.58±1.14b | 45.36±3.36 | >100a |
| 4e | 18.83±2.26 | 8.74±1.28 | 15.38±1.58 |
| 4f | 8.54±0.97b | 4.16±0.73b | 11.32±1.20b |
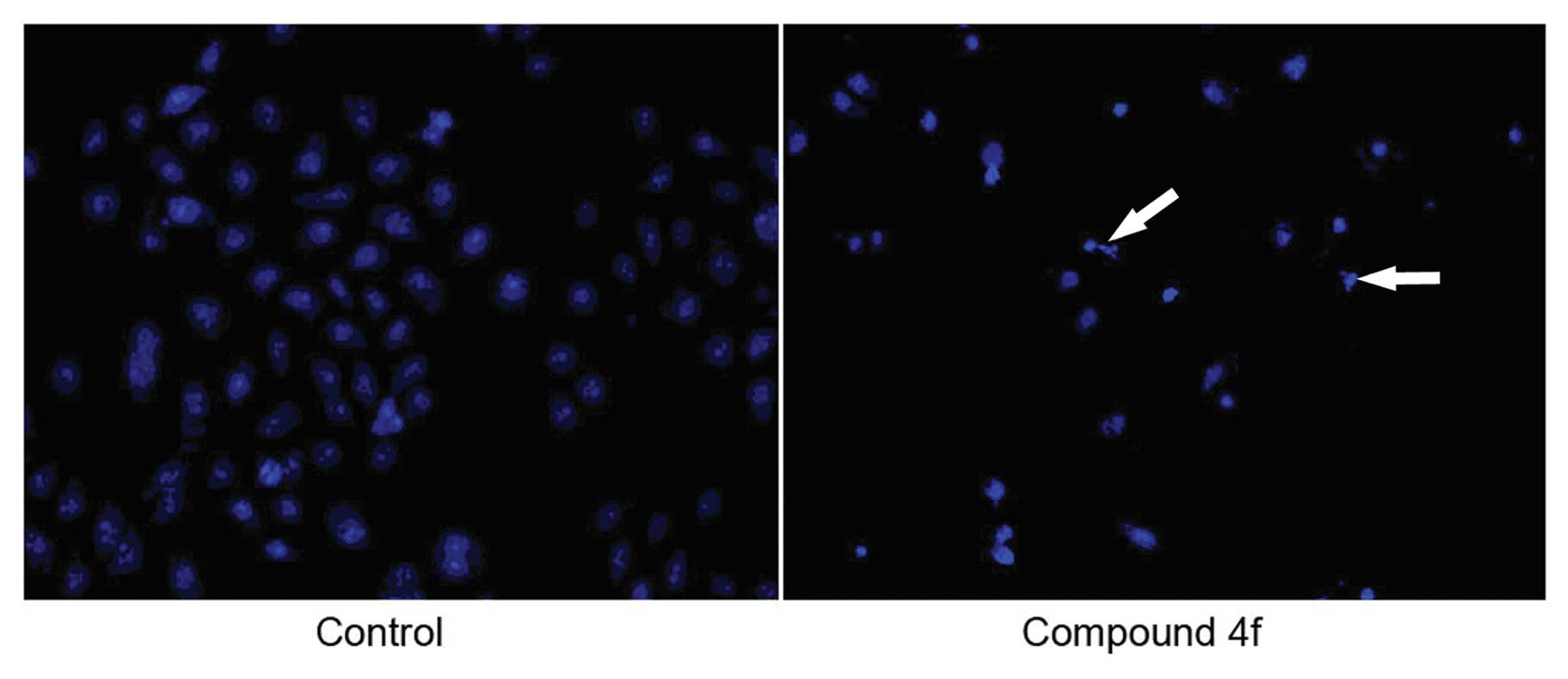
Apoptosis determination by
4′-6-diamidino-2-phenylindole (DAPI) staining
HepG2 cells were exposed to compound 4f in its
IC50 (4.16 μM) for 48 h. The occurrence of
nuclear condensation upon treatment with compound 4f could be
revealed by DAPI staining. Apoptotic bodies, one of the stringent
morphological criteria of apoptosis, were present in the compound
4f-treated HepG2 cells stained with DAPI (Fig. 3).
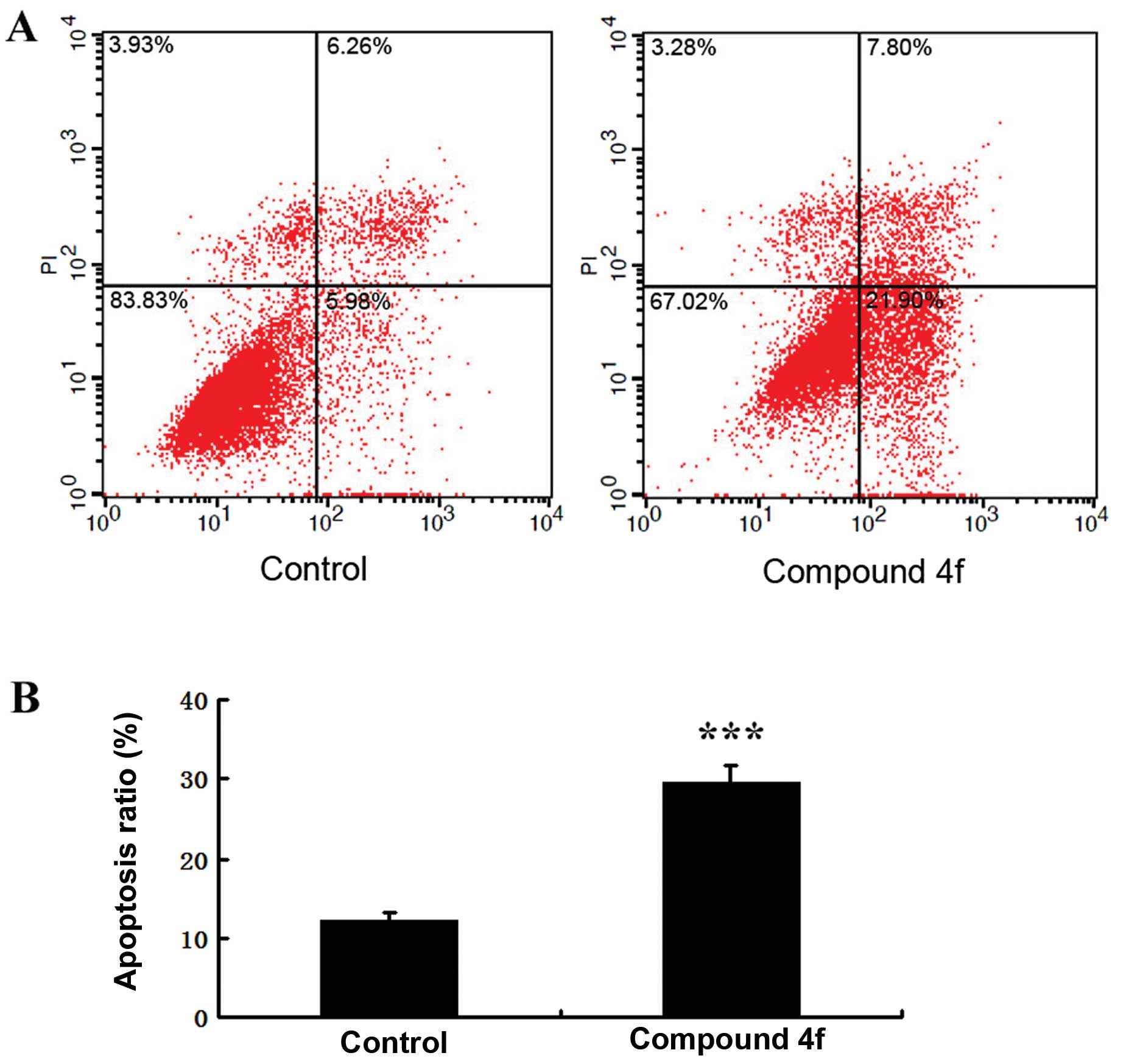
Annexin V-FITC assay
HepG2 cells were treated with compound 4f at a
concentration 4.16 μM for 48 h and were analyzed by flow
cytometry. As shown in Fig. 4A,
the numbers of early and late apoptotic cells were significantly
increased compared to the control group. The proportion of early
and late apoptotic cells in the compound 4f treatment group reached
30.38%, which was higher than the control group (12.5%)
(P<0.001) (Fig. 4B). This
finding suggested that compound 4f suppressed cell proliferation
possibly by inducing apoptosis.
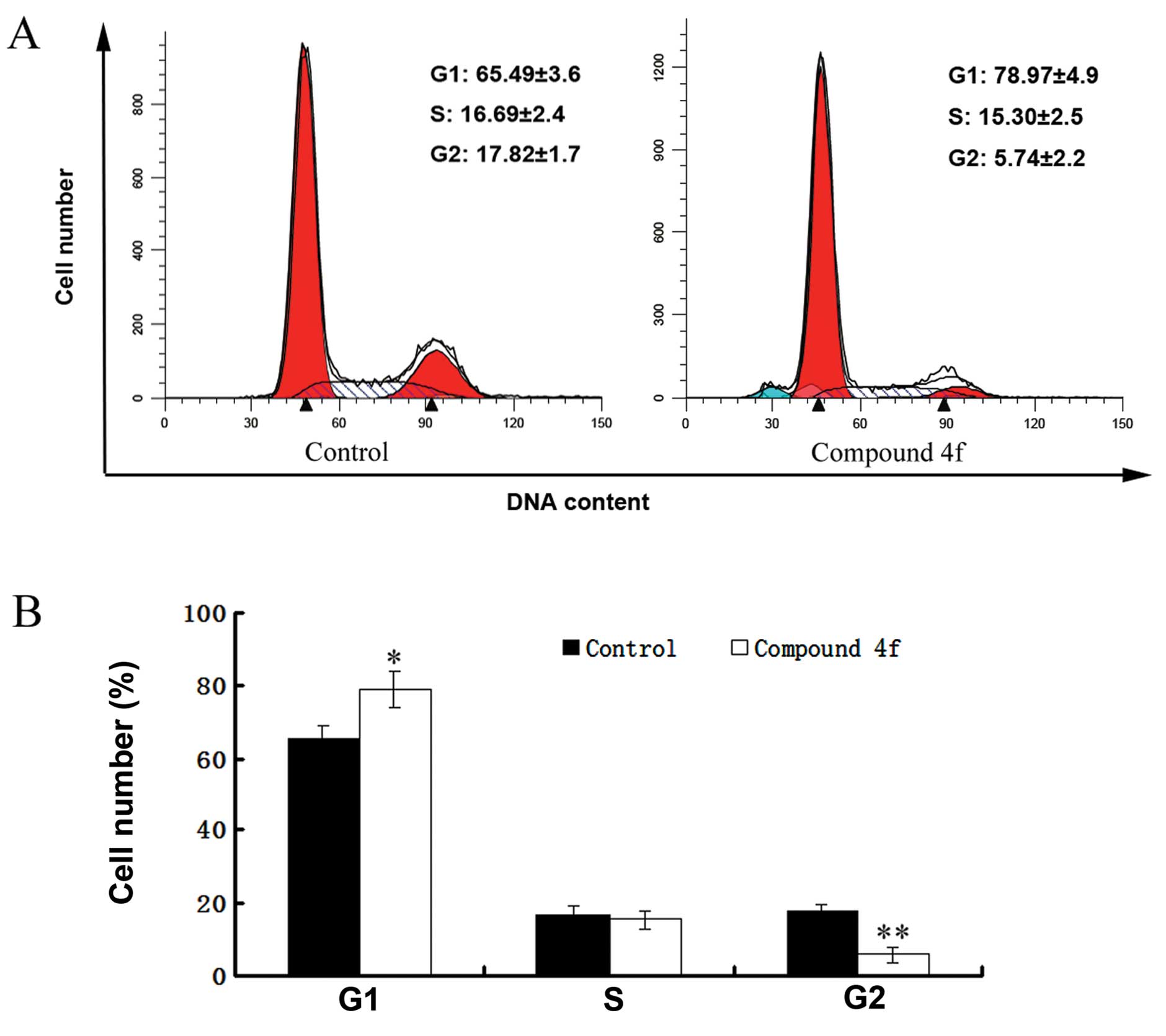
Compound 4f causes G0/G1 cell cycle
arrest in HepG2 cells
As shown in Fig.
5A, compared with the control group, compound 4f resulted in a
significant accumulation of HepG2 cells in the G0/G1 phase and a
decrease in the number of cells in the G2/M phase. Markedly, more
HepG2 cells treated by compound 4f were in the G1 phase compared to
the control (78.97 vs. 65.49%; P<0.05). These results indicated
that compound 4f caused cell cycle arrest in the G0/G1 phase
(Fig. 5B).
Discussion
It has been reported that structural modifications
could enhance the anticancer activities of parent compounds
(19,20). In the present study, the 28-COOH
position of RA was modified, and six new compounds were obtained.
The synthesized derivatives were tested for antitumor activities in
order to test previous evidence that the amino acid modification
may enhance the antitumor activities of the parent (21).
The cytotoxicity results of the RA derivatives,
compounds 4a–4f, demonstrated that the IC50 of compound
4f was significantly less than the groups treated with compounds 4c
and 4e. The results may be explained by the difference of
substituent group in para-position of benzene of R1
group. Antitumor activities of these compounds were enhanced when
they were substituted with electron-donating groups on
para-position of benzene. In addition, the antitumor activity of
compound 4f was found to be better than 4b. Hence, compound 4f was
substituted with a methoxy group (a stronger electron-donation
group) on para-position of benzene of R1 group. On the
other hand, since the compounds 4c, 4d and 4e were substituted with
electron-attracting groups, their antitumor activities were weaker
than RA. Based on the results, the IC50 of compound 4f
was 8.54, 4.16 and 11.32 μM on the HeLa, HepG2 and SGC-7901
cell lines, respectively. This indicated that compound 4f could be
further studied as a novel antitumor agent.
Evasion of apoptosis is one of the major hallmarks
of cancer and a target for cancer treatment (22). Previous data suggested apoptosis
as an underlying mechanism, by which various anticancer and
chemopreventive agents exert their antitumor effects (23,24). The current study included the
preliminary research on apoptosis and cell cycle. In the early
stages of apoptosis, phosphatidylserine in the cell membrane was
translocated outside. Compound 4f was able to induce apoptosis of
HepG2 cells, and the apoptosis ratio in the early stage was about
three times higher than in the control group.
The results of fluorescence-activated cell sorting
of HepG2 cells treated with compound 4f at its IC50
showed significant cell cycle arrest at the G0/G1 interface,
suggesting this as a mechanism for the antiproliferative effect of
compound 4f. However, further mechanistic investigation of compound
4f remains to be conducted. These findings will provide new insight
into the cancer chemotherapeutic properties of RA and its
derivatives.
In conclusion, six novel RA derivatives were
synthesized and evaluated for their cytotoxic properties on three
tumor cell lines. Compound 4f had a substantially better antitumor
effect than RA in vitro on HepG2 cells. Compound 4f induced
apoptosis in HepG2 cells and G0/G1 cell cycle arrest. These results
provide further insight into compound 4f-induced apoptosis and
deepen our previous knowledge of the cytotoxicity and anticancer
ability of RA derivatives.
Acknowledgements
The authors thank Professor Richard
Nicholson at the University of Newcastle, Australia, for editing
this manuscript and providing valuable suggestions.
References
|
1.
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Estimating the world cancer burden: Globocan 2000. Int J Cancer.
94:153–156. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
El-Serag HB and Mason AC: Rising incidence
of hepatocellular carcinoma in the United States. N Engl J Med.
340:745–750. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Parkin DM, Bray F, Ferlay J and Pisani P:
Global cancer statistics, 2002. CA Cancer J Clin. 55:74–108. 2005.
View Article : Google Scholar
|
|
4.
|
Hwang JP and Hassan MM: Survival and
hepatitis status among Asian Americans with hepatocellular
carcinoma treated without liver transplantation. BMC Cancer.
9:462009. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Charette N, De Saeger C, Lannoy V,
Horsmans Y, Leclercq I and Starkel P: Salirasib inhibits the growth
of hepatocarcinoma cell lines in vitro and tumor growth in vivo
through ras and mTOR inhibition. Mol Cancer. 9:2562010. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Bruix J, Hessheimer AJ, Forner A, Boix L,
Vilana R and Llovet JM: New aspects of diagnosis and therapy of
hepatocellular carcinoma. Oncogene. 25:3848–3856. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Thang TD, Kuo PC, Yu CS, et al: Chemical
constituents of the leaves of Glochidion obliquum and their
bioactivity. Arch Pharm Res. 34:383–389. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Haraguchi H, Kataoka S, Okamoto S, Hanafi
M and Shibata K: Antimicrobial triterpenes from Ilex integra
and the mechanism of antifungal action. Phytother Res. 13:151–156.
1999. View Article : Google Scholar
|
|
9.
|
Zhao HR, Wang MS and Zhou GP: Studies on
constituents of Ilex chinensis Sims. Zhongguo Zhong Yao Za
Zhi. 18:226–228. 2551993.(In Chinese).
|
|
10.
|
Sun H, Zhang XQ, Cai Y, Han WL, Wang Y and
Ye WC: Study on chemical constituents of Ilex rotunda Thunb.
Chem Indus Forest Prod. 295:111–114. 2009.(In Chinese).
|
|
11.
|
Zhao WM, Wolfender JL, Hostettmann K,
Cheng KF, Xu RS and Qin GW: Triterpenes and triterpenoid saponins
from Mussaenda pubescens. Phytochemistry. 45:1073–1078.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Saimaru H, Orihara Y, Tansakul P, Kang YH,
Shibuya M and Ebizuka Y: Production of triterpene acids by cell
suspension cultures of Olea europaea. Chem Pharm Bull
(Tokyo). 55:784–788. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Lee TH, Juang SH, Hsu FL and Wu CY:
Triterpene acids from the leaves of Planchonella duclitan
(Blanco) Bakhuizan. J Chin Chem Sci. 52:1275–1280. 2005.
|
|
14.
|
Zhao QC, Nan ML, He YF and Chen SW:
Application of rotundic acid in the preparation of lipid-lowering
drugs. CHN. 201010204607.3,. 2010.(In Chinese).
|
|
15.
|
He YF, Zhao QC, Nan ML, et al: Application
of rotundic acid and its derivatives in the preparation of
anticancer drugs. CHN. 201010607515.x,. 2010.(In Chinese).
|
|
16.
|
Zhao QC, He YF, Nan ML, Chen SW, Zhao YW
and Wang LP: Synthesis method of rotundic acid derivatives and
their application in the preparation of cardiovascular disease
prevention Drugs. CHN. 20110030007.4,. 2011.(In Chinese).
|
|
17.
|
He YF, Nan ML, Zhao QC, Zhao YW and Yue
FG: Application of amino acid modified rotundic acid derivatives in
the preparation of anticancer drugs. CHN. 201110351365.5,. 2011.(In
Chinese).
|
|
18.
|
He YF, Nan ML, Sun JM, et al: Synthesis,
characterization and cytotoxicity of new rotundic acid derivatives.
Molecules. 17:1278–1291. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Scrimin F, Becagli L and Ambrosini A:
Malignant melanoma of the vulva. Considerations on 4 cases. Minerva
Ginecol. 38:43–48. 1986.(In Italian).
|
|
20.
|
Wang X, Lu N, Yang Q, et al: Studies on
chemical modification and biology of a natural product, gambogic
acid (III): determination of the essential pharmacophore for
biological activity. Eur J Med Chem. 46:1280–1290. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Xu R: Guangzhou University of Chinese
Medicine. 2009.
|
|
22.
|
Abbott RG, Forrest S and Pienta KJ:
Simulating the hallmarks of cancer. Artif Life. 12:617–634. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Sandur SK, Ichikawa H, Sethi G, Ahn KS and
Aggarwal BB: Plumbagin (5-hydroxy-2-methyl-1,4-naphthoquinone)
suppresses NF-kappaB activation and NF-kappaB-regulated gene
products through modulation of p65 and IkappaBalpha kinase
activation, leading to potentiation of apoptosis induced by
cytokine and chemotherapeutic agents. J Biol Chem. 281:17023–17033.
2006.
|
|
24.
|
Park C, Jin CY, Kwon HJ, et al: Induction
of apoptosis by esculetin in human leukemia U937 cells: roles of
Bcl-2 and extracellular-regulated kinase signaling. Toxicol In
Vitro. 24:486–494. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25.
|
Kim NC, Desjardins AE, Wu CD and Kinghorn
AD: Activity of triterpenoid glycosides from the root bark of
Mussaenda macrophylla against two oral pathogens. J Nat
Prod. 62:1379–1384. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26.
|
Zhang AL, Ye Q, Li BG, Qi HY and Zhang GL:
Phenolic and triterpene glycosides from the stems of Ilex
litseaefolia. J Nat Prod. 68:1531–1535. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27.
|
Xie GB, Zhou SX, Lu YN, Lei LD and Tu PF:
Triterpenoid glycosides from the leaves of Ilex pernyi. Chem
Pharm Bull (Tokyo). 57:520–524. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28.
|
Nakatani M, Hatanaka S, Komura H, Kubota T
and Hase T: The structure of rotungenoside, a new bitter triterpene
glucoside from Ilex rotunda. Bull Chem Soc Jpn. 62:469–473.
1989. View Article : Google Scholar
|
|
29.
|
Cui CZ, Wen XS, Cui M, Gao J, Sun B and
Lou HX: Synthesis of solasodine glycoside derivatives and
evaluation of their cytotoxic effects on human cancer cells. Drug
Discov Ther. 6:9–17. 2012.PubMed/NCBI
|