Introduction
γ-catenin (plakoglobin), a member of the Armadillo
proteins (1), plays important
roles in cell adhesion with α-catenin and β-catenin (2), is involved in Wnt signaling
(3), and is translocated into the
nucleus and binds LEF1, but is inefficient in forming a complex
with DNA (4), and even inhibits
the transcriptional activity of the TCF4 complex (5). Despite the homology and the similar
structure of γ-catenin and β-catenin, the differences between them
are rather distinct (6);
β-catenin-null mice and γ-catenin-null mice have different
phenotypes (7). β-catenin is
regarded as an oncogenic protein, while there are different
proposed roles for γ-catenin for different types of tumors. In
renal carcinoma cell KTCTL 60 cells (8), squamous carcinoma cell SCC9 cells
(9), lung cancer (10), mammary carcinoma MCF-7 cells
(11), and bladder carcinomas
(12), γ-catenin acts as a tumor
suppressor. By contrast, γ-catenin acts as an oncogenic factor in
human colonic carcinoma HTC116 cells (13), and γ-catenin can promote the
transformation of RK3E cells (14).
In chronic myeloid leukemia (CML), the reciprocal
translocation of chromosomes 9 and 22 forms the Philadelphia
chromosome, which results in the expression of a fused BCR-ABL
protein. The BCR-ABL protein activates a series of proteins to
promote cell survival and cell growth (15,16). Previous studies have shown that
γ-catenin can be regulated by the acute myeloid leukemia (AML)
fused protein AML1-ETO, PML-retinoic acid receptor-α (RARα) and
PLZF-RARα (17), but the
influence of BCR-ABL on the expression of γ-catenin remains
unclear. It has been shown that β-catenin is crucial for CML cells
(18), and β-catenin siRNA
sensitizes CML cells to imatinib (19). Although γ-catenin and β-catenin
belong to the same family, the role of γ-catenin in CML is elusive.
In a study by Kim et al (20), CML patients in the accelerated
phase and blast crisis (AP/BC) stages had significantly increased
levels of γ-catenin, suggesting that γ-catenin may also play a
crucial role in CML cells.
In this study, we showed that two BCR-ABL positive
CML cell types (K562 cells and KU812 cells) had higher expression
levels of γ-catenin compared to five BCR-ABL-negative leukemia
cells. Suppressing the expression of BCR-ABL with siRNA resulted in
decreased γ-catenin expression. Furthermore, suppression of
γ-catenin by siRNA inhibited proliferation, colony formation and
the β-catenin target genes c-Myc and cyclin D1. Suppression of
γ-catenin also potentiated the effects of imatinib on CML cells and
suppressed the expression of Bcl-xL and survivin (a β-catenin
target gene). Furthermore, suppression of γ-catenin suppressed the
activation of STAT5 and inhibited β-catenin by promoting its
phosphorylation and inhibiting its translocation into the nucleus.
Finally, downregulation of γ-catenin resulted in the increased
total glycogen synthase kinase-3β (GSK3β) and suppression of
phospho-GSK3β. This study helps elucidate the role of γ-catenin in
CML cells and provides a marker or strategy for CML treatment.
Materials and methods
Cells and reagents
The cells were purchased from the Shanghai Cell Bank
(Shanghai, China) and were maintained in a 37°C/5% CO2
atmosphere. IMDM and DMEM medium were purchased from Hyclone, and
fetal bovine serum (FBS) was purchased from Gibco. The human CML
cell lines, K562 and KU812, were maintained in DMEM or IMDM medium
supplemented with 10 or 20% FBS, respectively. Human leukemia
cells, Jurkat cells, U937 cells, CEM cells and Kasumi cells were
maintained in RPMI-1640 medium supplemented with 10% FBS, and HL-60
cells were maintained in IMDM medium supplemented with 20% FBS.
Human embryonic kidney cells HEK293 cells were maintained in DMEM
medium supplemented with 10% FBS. Imatinib, a BCR-ABL inhibitor,
purchased from Selleck Chemicals, was dissolved in DMSO
(Sigma-Aldrich).
Stable cell line construction
The pSES-hus plasmid was used to construct the siRNA
retrovirus. The pSES-hus plasmid was digested with the SfiI
restriction endonuclease and ligated with annealed DNA chains for
siRNA. The BCR-ABL (target sequence, 5′-GCAGAGTTCAAAAGCCCTT-3′) and
γ-catenin (target sequence, 5′-GCTGATCATCCTGGCCAA-3′) genes were
the targets for the siRNA. A randomized sequence that did not
target any gene was used as an siRNA control.
Subsequently, pSES-hus-siBCR-ABL and
pSES-hus-γ-catenin were used to package the retrovirus with
pCL-ampho with co-transfection into HEK293 cells. K562 and KU812
cells were infected with the retrovirus. Stable cell lines were
obtained through Blasticidin (Invitrogen) selection. The cells
infected with the BCR-ABL siRNA retrovirus were labeled
K562-siBCR-ABL and KU812-siBCR-ABL, and the cells infected with the
γ-catenin siRNA retrovirus were labeled K562-siγ-catenin and
KU812-siγ-catenin. The siRNA control cells were labeled
K562-siControl and KU812-siControl cells.
Western blotting, co-immunoprecipitation
(Co-IP), and nuclear protein and cytoplasmic protein
extraction
Western blot analysis was carried out as previously
described (21), and the entire
process was performed using the Bio-Rad system. The anti-c-Abl,
anti-c-Myc, anti-cyclin D1, anti-survivin, anti-Bcl-xL,
anti-phospho-STAT5, anti-β-catenin, anti-phospho-β-catenin,
anti-GSK3β, and anti-phospho-GSK3β antibodies were obtained from
Cell Signaling Technology. The anti-β-actin antibody, mouse IgG and
rabbit IgG were obtained from Santa Cruz Biotechnology, Inc., while
the anti-γ-catenin antibody (mouse antibody) was from Abcam.
Co-IP was carried out with a Pierce Co-IP kit. A
Nuclear Protein and Cytoplasmic Protein Extraction kit (Beyotime,
China) was used to extract protein from the cell nucleus or
cytoplasm. An anti-Histone H1 (Santa Cruz Biotechnology, Inc.)
antibody was used as a loading control for the proteins in the
nucleus.
Apoptosis analysis
After washing the cells three times with ice-cold
phosphate-buffered saline (PBS), they were resuspended in binding
buffer, stained with Annexin V-APC (KeyGentec, China) and propidium
iodide (PI; Sigma-Aldrich), and analyzed by flow cytometry (BD
Biosciences).
Cell proliferation analysis and colony
formation assay
Cell proliferation analysis was conducted with a
CCK-8 kit (Dojindo, Japan) according to the manufacturer’s
instructions.
The colony formation assay was performed as follows:
the cells (2×103 cells/well) were plated in
methylcellulose (1% final concentration; Sigma-Aldrich) semi-solid
medium. Seven (K562 cells) or 12 days (KU812 cells) later, the
colonies containing >40 cells each were counted.
Statistical analysis
Statistical tests were performed using the Student’s
t-test with SPSS software. A P-value <0.05 was considered to
indicate statistically significant differences. The tests were
conducted three times, and the results are presented as the means ±
SD.
Results
Downregulation of BCR-ABL suppresses
γ-catenin, but γ-catenin cannot bind BCR-ABL
Kim et al (20) found elevated levels of γ-catenin
in AP/BC stage patients. We evaluated the expression of γ-catenin
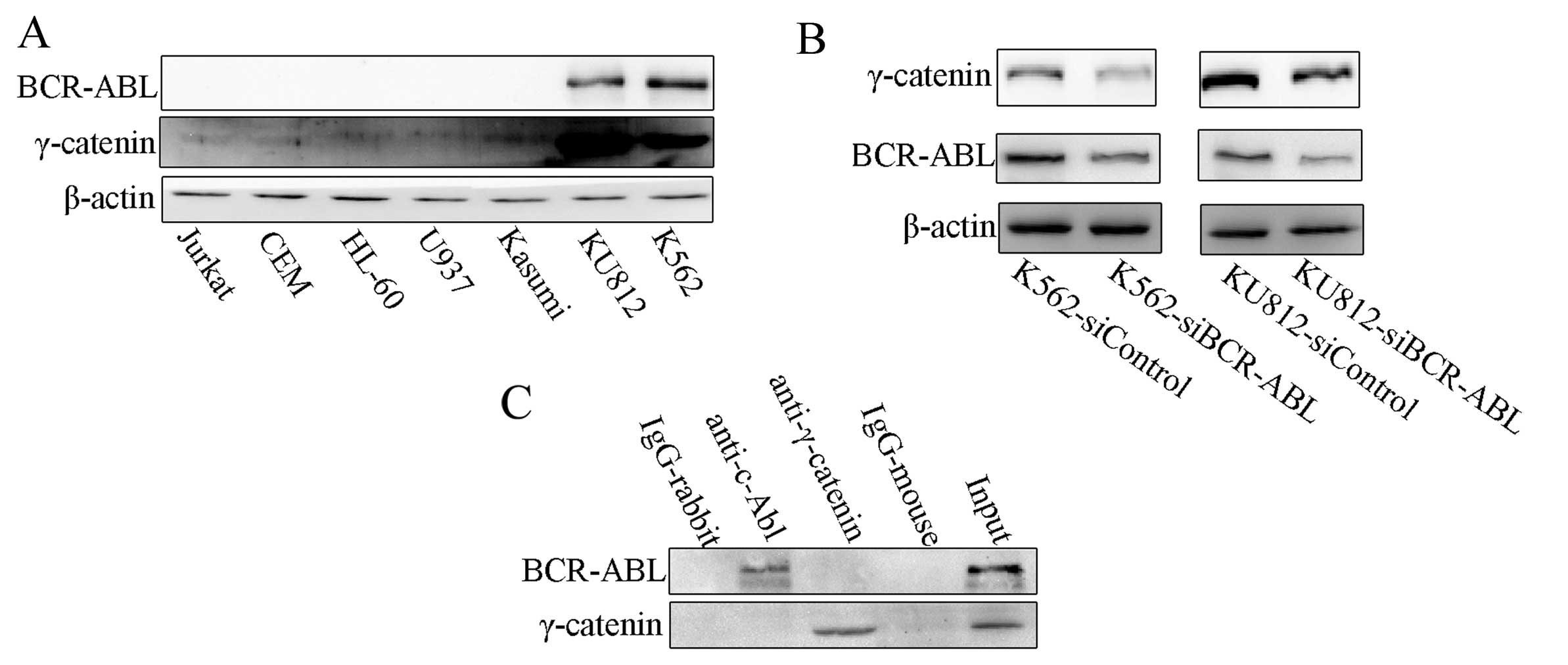
in leukemia cell lines. K562 and KU812 cells had higher levels of
γ-catenin expression than the BCR-ABL-negative cell lines (Fig. 1A). It suggested that BCR-ABL
regulates the expression of γ-catenin. To observe the effects of
BCR-ABL on the expression of γ-catenin, we constructed CML cell
lines with stable BCR-ABL siRNA expression: K562-siBCR-ABL and
KU812-siBCR-ABL cells (Fig. 1A).
We found that K562-siBCR-ABL cells and KU812-siBCR-ABL cells had
decreased levels of γ-catenin compared to control cells (Fig. 1B).
To determine whether BCR-ABL regulates γ-catenin by
directly binding γ-catenin, we performed a Co-IP assay. The results
showed that γ-catenin protein does not bind the BCR-ABL protein
(Fig. 1C).
Downregulation of γ-catenin inhibits the
proliferation and colony formation of CML cells
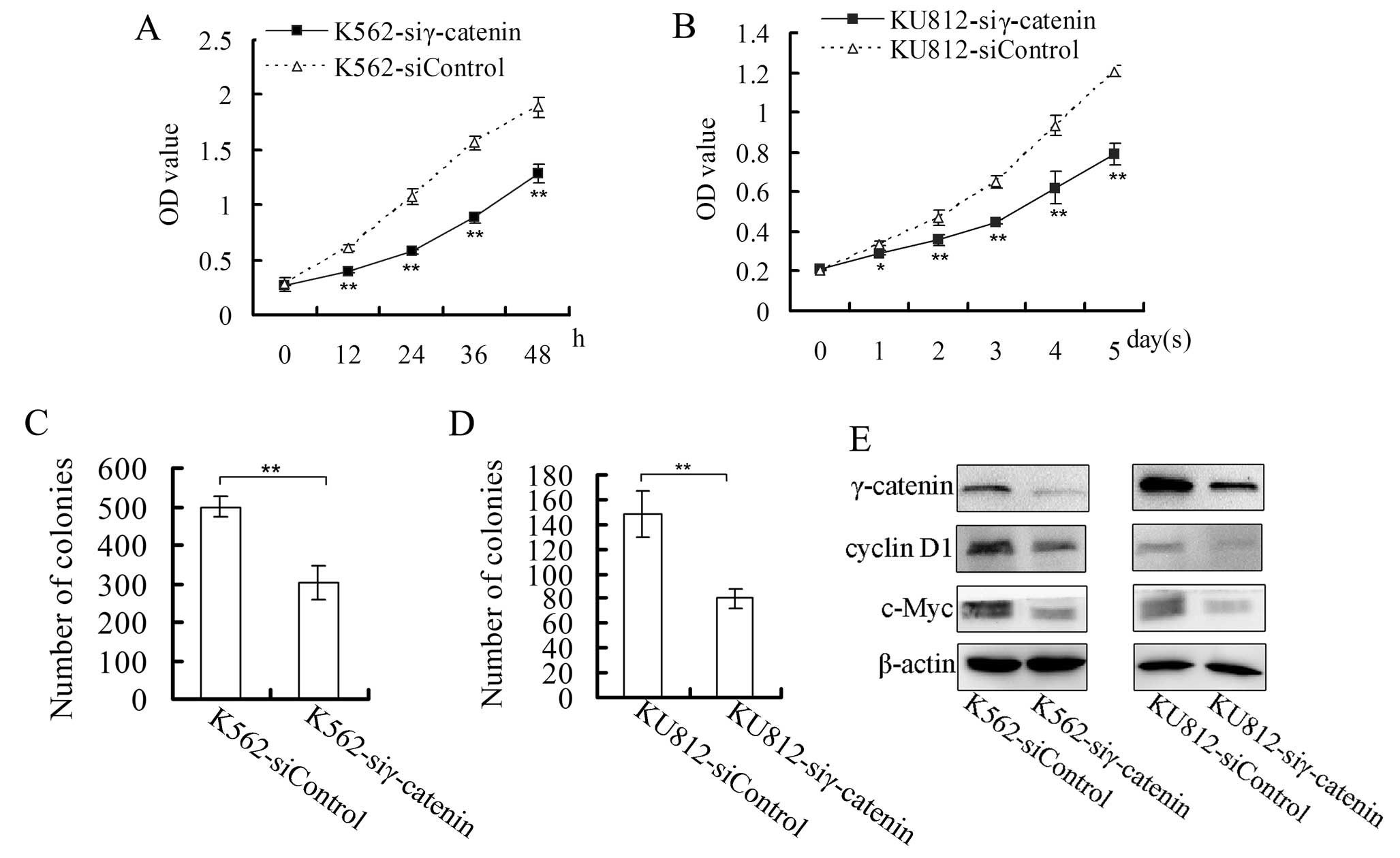
To study the roles of γ-catenin in CML cells, we
constructed the following CML cells with downregulation of
γ-catenin: K562-siγ-catenin cells and KU812-siγ-catenin cells.
After the cells (0.5×105/ml) were plated
on 96-well plates, their proliferation was monitored at different
time points. As shown in the proliferation curves, the
proliferation rate of K562-siγ-catenin cells was lower than that of
K562-siControl cells, and the proliferation rate of
KU812-siγ-catenin cells was lower than that of KU812-siControl
cells (Fig. 2A and B). The colony
formation assay showed that the γ-catenin siRNA CML cells formed
fewer colonies than the control cells (Fig. 2C and D).
We also examined the expression of c-Myc and cyclin
D1, which are β-catenin target genes (22,23). The expression levels of c-Myc and
cyclin D1 in the γ-catenin siRNA cells were lower than in the
control cells (Fig. 2E).
Downregulation of γ-catenin potentiates
the sensitivity of CML cells to imatinib
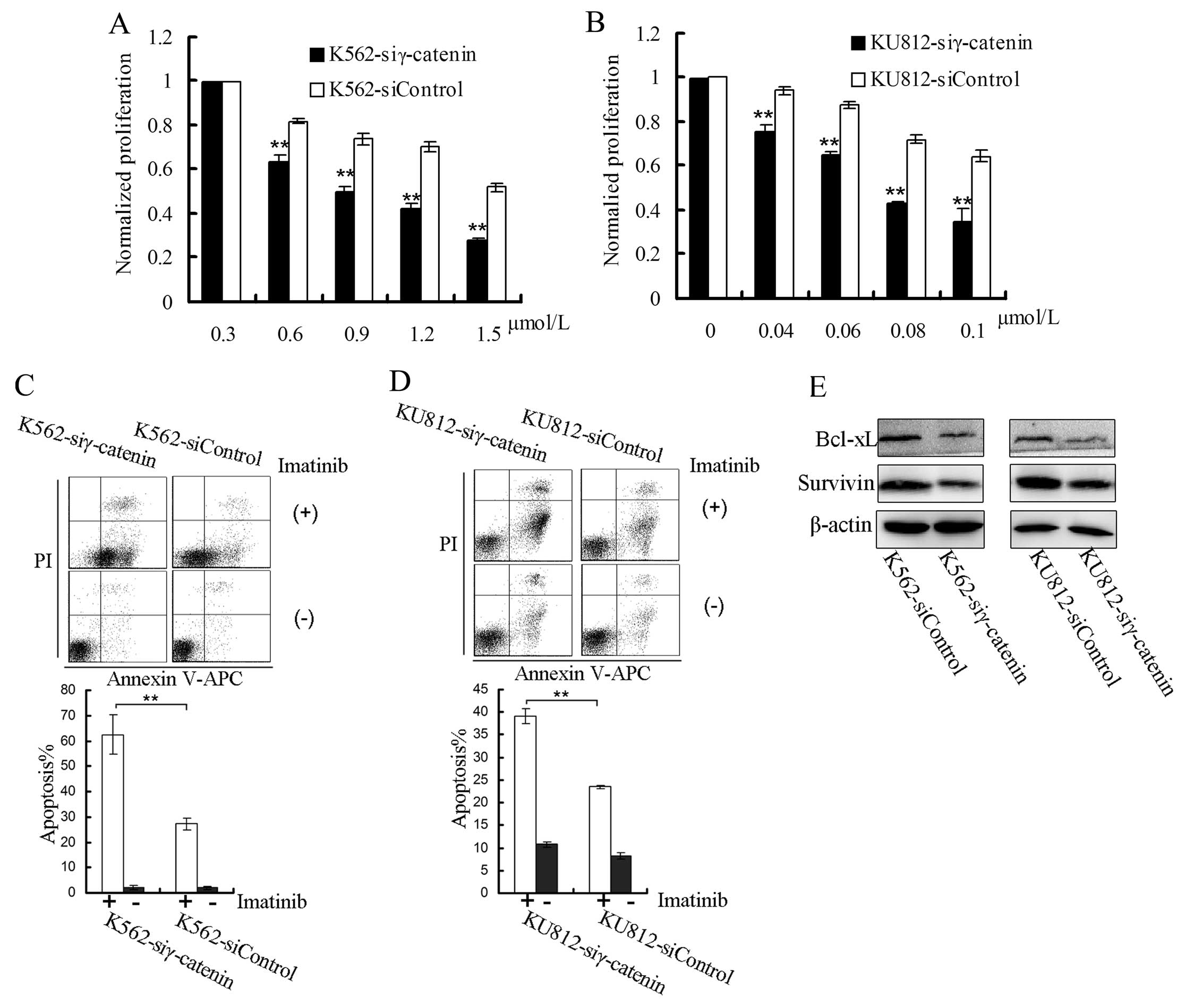
We also examined the influence of γ-catenin on the
sensitivity of CML cells to imatinib. To exclude the influence of
γ-catenin on cell proliferation, the proliferation of the treated
cells was normalized to cells that were not treated with imatinib.
The cells (2×105/ml) were plated in 96-well plates and
treated with different concentrations of imatinib for 24 h.
Imatinib decreased the proliferation of the K562-siγ-catenin and
KU812-siγ-catenin cells more substantially compared to the control
cells (Fig. 3A and B).
Following treatment with imatinib (K562, 6
μmol/l; KU812, 0.3 μmol/l), the percentages of
apoptotic K562-siγ-catenin and KU812-siγ-catenin cells were higher
than for control cells (Fig. 3C and
D).
We then examined the changes in the expression
levels of survivin (a β-catenin target gene) (24) and Bcl-xL and found lower
expression levels of these proteins in K562-siγ-catenin and
KU812-siγ-catenin cells than in the control cells (Fig. 3E).
Downregulation of γ-catenin suppresses
STAT5 phosphorylation, promotes β-catenin phosphorylation and
inhibits β-catenin translocation into the nucleus
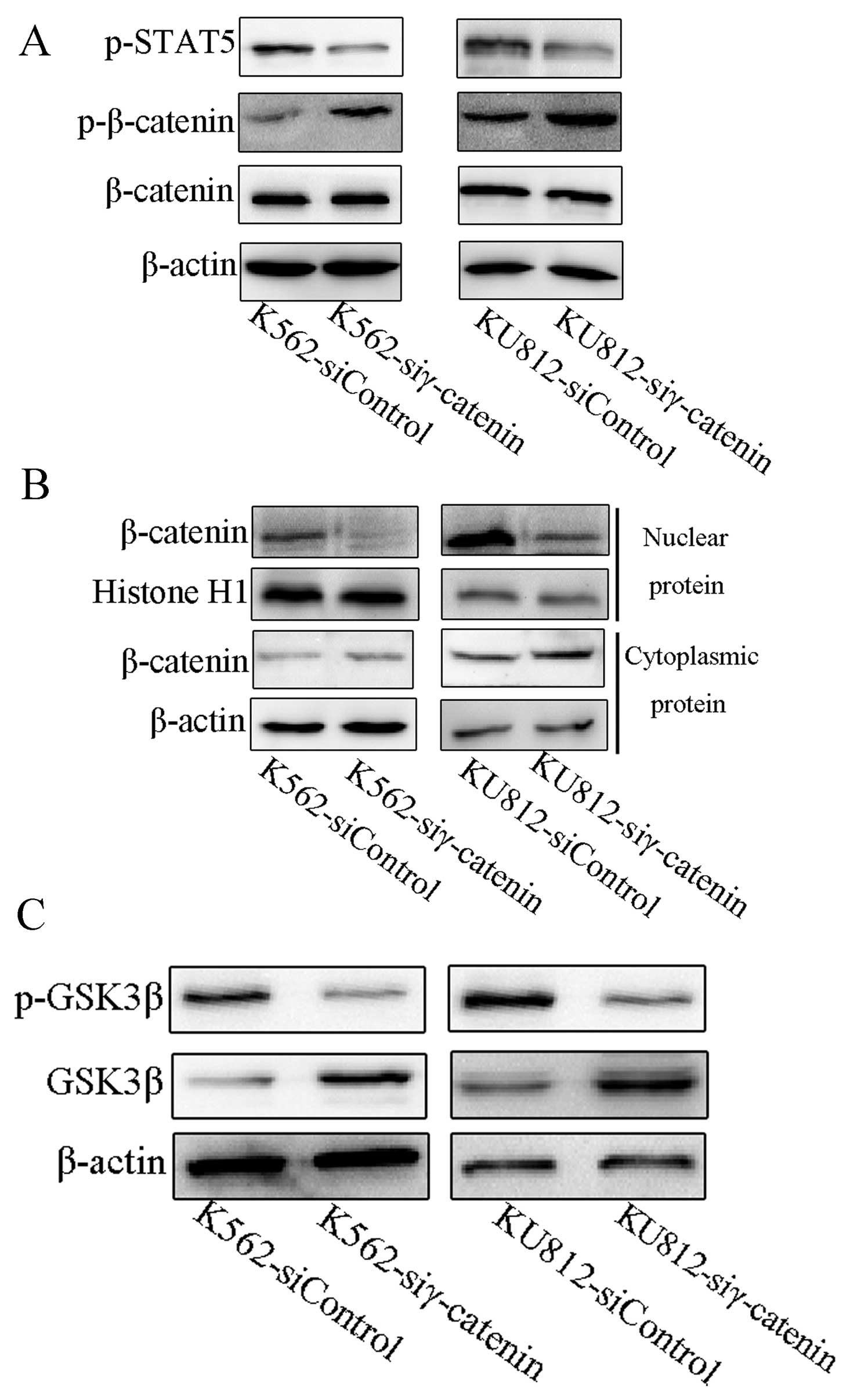
STAT5 has been shown to be activated by BCR-ABL, to
be involved in the signaling of BCR-ABL, and to be important for
growth and apoptosis resistance (15). Bcl-xL and the cyclin D1 have been
shown to be the targets of STAT5 in BCR-ABL-transformed cells
(25). The effects of siRNA
against γ-catenin on the activation of STAT5 were also evaluated.
We found that the phospho-STAT5 level was lower in the γ-catenin
siRNA CML cells than in the control cells (Fig. 4A).
As shown above, downregulation of γ-catenin reduced
the expression of c-Myc (22),
cyclin D1 (23) and survivin
(24). Since these proteins are
downstream target genes of β-catenin, we posited that γ-catenin
influences β-catenin. There was no change in the expression of
β-catenin in γ-catenin siRNA CML cells (Fig. 4A). However, there was an increased
level of phospho-β-catenin in γ-catenin siRNA CML cells (Fig. 4A), a decreased β-catenin level in
the nucleus, and an increased β-catenin level in the cytoplasm of
the γ-catenin siRNA CML cells (Fig.
4B).
Downregulation of γ-catenin promotes
GSK3β and suppression of phospho-GSK3β
Due to the regulation of β-catenin by GSK3β
(26), we investigated whether
GSK3β was involved in the γ-catenin regulation of β-catenin. We
found that γ-catenin downregulation leads to downregulation of
phospho-GSK3β and upregulation of total GSK3β, compared with the
control cells (Fig. 4C).
Discussion
Our study helps elucidate the roles of γ-catenin in
CML cells. γ-catenin was regulated by the BCR-ABL fused protein,
and downregulation of γ-catenin had an anti-leukemia effect through
the inhibition of β-catenin.
It was previously reported that γ-catenin expression
is amplified in the CML cells of AP/BC stage patients (20) and that BCR-ABL has particularly
increased levels in BC stage patients (27). In this study, two CML cells
expressed higher levels of γ-catenin than BCR-ABL-negative leukemia
cells.
We hypothesized that BCR-ABL can regulate γ-catenin.
To test our hypothesis, we used siRNA to knock down BCR-ABL
expression, and found that γ-catenin is indeed regulated by
BCR-ABL. Additionally, it was reported that BCR-ABL stabilizes
β-catenin through binding β-catenin and its tyrosine
phosphorylating β-catenin (19);
thus, we addressed whether BCR-ABL also binds γ-catenin. However, a
Co-IP assay showed that γ-catenin does not bind BCR-ABL, suggesting
that BCR-ABL regulates γ-catenin in an indirect way, which is
different from the interaction between BCR-ABL and β-catenin.
However, although there are lower levels of γ-catenin in the 5
BCR-ABL-negative leukemia cell lines, high levels of γ-catenin may
still exist in the other types of leukemia. In fact, γ-catenin has
been reported to contribute to the signaling pathway of AML
translocation products (17).
Our study also showed that the suppression of
γ-catenin resulted in the inhibition of CML cell proliferation and
transformation as well as the inhibition of c-Myc and cyclin D1,
the target genes of β-catenin. c-Myc plays important roles in cell
proliferation and in the transformation of BCR-ABL-positive cells
(28), and cyclin D1 also
contributes to BCR-ABL-mediated transformation (25,29). c-Myc has been reported to be
suppressed (30) or enhanced
(13) by γ-catenin. In our study,
downregulation of γ-catenin suppressed the expression of c-Myc.
Some signaling proteins influence the effects of
imatinib on CML cells (31). In
this study, we showed that downregulation of γ-catenin potentiates
the effects of imatinib on CML cells. Based on the report that the
disruption of survivin can sensitize CML cells to imatinib
(32), the report that Bcl-xL is
an apoptosis resistant factor in CML cells (33), and our result that downregulation
of γ-catenin suppressed survivin and Bcl-xL, we speculate that the
survivin and Bcl-xL suppression is the mechanism by which
downregulation of γ-catenin sensitizes CML cells to imatinib.
However, Dusek et al (34)
found that γ-catenin suppresses the expression of Bcl-xL in
keratinocytes, but these differences may be cell-type specific.
It has been reported that Bcl-xL and cyclin D1 are
regulated by STAT5 in BCR-ABL-transformed cells (25). Our study showed that
downregulation of γ-catenin decreased the levels of phospho-STAT5,
which may be a mechanism for the downregulation of
γ-catenin-mediated suppression of Bcl-xL and cyclin D1 in CML
cells. There are differing reports on the influence of γ-catenin on
β-catenin; it has been reported that γ-catenin suppresses β-catenin
(35) and decreases the binding
of β-catenin and TCF4 (5).
However, it has also been reported that γ-catenin can promote
β-catenin translocation into nucleus and increase the levels of
β-catenin and the β-catenin-LEF-DNA complex (4,36).
The present study showed that downregulation of γ-catenin had no
influence on β-catenin expression, but could inhibit β-catenin
translocation into the nucleus and suppress β-catenin-dependent
genes expression. This observation is a possible mechanism for the
downregulation of γ-catenin to suppress the proliferation and
transformation of CML cells and potentiate the effects of imatinib
on CML cells, due to the reports on the roles of β-catenin
(18,19). Additionally, it has been shown, in
BCR-ABL transformed cells, that the loss of β-catenin can reduce
the levels of phosphorylated STAT5 (18); thus, β-catenin inhibition may be
the mechanism by which γ-catenin downregulation suppressed STAT5
activation.
We also found that the suppression of β-catenin by
down-regulation of γ-catenin occurs through the promotion of GSK3β
activation. This suggests that γ-catenin can stabilize β-catenin by
suppressing GSK3β. It was previously shown that GSK3β regulates
γ-catenin by phosphorylation (37), and, in the present study, we
showed that γ-catenin can regulate GSK3β, indicating that γ-catenin
and GSK3β interact and inhibit each other.
Previously, γ-catenin was considered to play an
important role in cell adhesion and development. It is currently
thought that γ-catenin also plays important roles for tumors. Our
studies suggest that γ-catenin functions as an oncogenic protein in
CML cells and can be regulated by the BCR-ABL fused protein. This
study provides new insight into the BCR-ABL pathways, the roles of
γ-catenin in CML cells, and a potential target for CML
treatment.
Acknowledgements
This study was supported by the
National Natural Science Foundation of China and the Major Program
of Natural Science Foundation of Chongqing, China.
References
|
1.
|
Peifer M, McCrea PD, Green KJ, Wieschaus E
and Gumbiner BM: The vertebrate adhesive junction proteins
beta-catenin and plakoglobin and the Drosophila segment
polarity gene armadillo form a multigene family with similar
properties. J Cell Biol. 118:681–691. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Zhurinsky J, Shtutman M and Ben-Ze’ev A:
Plakoglobin and beta-catenin: protein interactions, regulation and
biological roles. J Cell Sci. 113:3127–3139. 2000.PubMed/NCBI
|
|
3.
|
Barker N and Clevers H: Catenins, Wnt
signaling and cancer. Bioessays. 22:961–965. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Zhurinsky J, Shtutman M and Ben-Ze’ev A:
Differential mechanisms of LEF/TCF family-dependent transcriptional
activation by beta-catenin and plakoglobin. Mol Cell Biol.
20:4238–4252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Miravet S, Piedra J, Miro F, Itarte E,
Garcia de Herreros A and Dunach M: The transcriptional factor Tcf-4
contains different binding sites for beta-catenin and plakoglobin.
J Biol Chem. 277:1884–1891. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Aktary Z and Pasdar M: Plakoglobin: role
in tumorigenesis and metastasis. Int J Cell Biol. 2012:1895212012.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Sadot E, Simcha I, Iwai K, Ciechanover A,
Geiger B and Ben-Ze’ev A: Differential interaction of plakoglobin
and beta-catenin with the ubiquitin-proteasome system. Oncogene.
19:1992–2001. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8.
|
Simcha I, Geiger B, Yehuda-Levenberg S,
Salomon D and Ben-Ze’ev A: Suppression of tumorigenicity by
plakoglobin: an augmenting effect of N-cadherin. J Cell Biol.
133:199–209. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Parker HR, Li Z, Sheinin H, Lauzon G and
Pasdar M: Plakoglobin induces desmosome formation and epidermoid
phenotype in N-cadherin-expressing squamous carcinoma cells
deficient in plakoglobin and E-cadherin. Cell Motil Cytoskeleton.
40:87–100. 1998. View Article : Google Scholar
|
|
10.
|
Winn RA, Bremnes RM, Bemis L, et al:
gamma-catenin expression is reduced or absent in a subset of human
lung cancers and re-expression inhibits transformed cell growth.
Oncogene. 21:7497–7506. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Mukhina S, Mertani HC, Guo K, Lee KO,
Gluckman PD and Lobie PE: Phenotypic conversion of human mammary
carcinoma cells by autocrine human growth hormone. Proc Natl Acad
Sci USA. 101:15166–15171. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Rieger-Christ KM, Ng L, Hanley RS, et al:
Restoration of plakoglobin expression in bladder carcinoma cell
lines suppresses cell migration and tumorigenic potential. Br J
Cancer. 92:2153–2159. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Pan H, Gao F, Papageorgis P, Abdolmaleky
HM, Faller DV and Thiagalingam S: Aberrant activation of
gamma-catenin promotes genomic instability and oncogenic effects
during tumor progression. Cancer Biol Ther. 6:1638–1643. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
Kolligs FT, Kolligs B, Hajra KM, et al:
gamma-catenin is regulated by the APC tumor suppressor and its
oncogenic activity is distinct from that of beta-catenin. Genes
Dev. 14:1319–1331. 2000.PubMed/NCBI
|
|
15.
|
Hazlehurst LA, Bewry NN, Nair RR and
Pinilla-Ibarz J: Signaling networks associated with
BCR-ABL-dependent transformation. Cancer Control. 16:100–107.
2009.PubMed/NCBI
|
|
16.
|
Quintas-Cardama A and Cortes J: Molecular
biology of bcr-abl1-positive chronic myeloid leukemia. Blood.
113:1619–1630. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Muller-Tidow C, Steffen B, Cauvet T, et
al: Translocation products in acute myeloid leukemia activate the
Wnt signaling pathway in hematopoietic cells. Mol Cell Biol.
24:2890–2904. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18.
|
Zhao C, Blum J, Chen A, et al: Loss of
beta-catenin impairs the renewal of normal and CML stem cells in
vivo. Cancer Cell. 12:528–541. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Coluccia AM, Vacca A, Dunach M, et al:
Bcr-Abl stabilizes beta-catenin in chronic myeloid leukemia through
its tyrosine phosphorylation. EMBO J. 26:1456–1466. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Kim YM, Ma H, Oehler VG, et al: The gamma
catenin/CBP complex maintains survivin transcription in
beta-catenin deficient/depleted cancer cells. Curr Cancer Drug
Targets. 11:213–225. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Yuan Y, Niu CC, Deng G, et al: The
Wnt5a/Ror2 noncanonical signaling pathway inhibits canonical Wnt
signaling in K562 cells. Int J Mol Med. 27:63–69. 2011.PubMed/NCBI
|
|
22.
|
He TC, Sparks AB, Rago C, et al:
Identification of c-MYC as a target of the APC pathway. Science.
281:1509–1512. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Shtutman M, Zhurinsky J, Simcha I, et al:
The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway.
Proc Natl Acad Sci USA. 96:5522–5527. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24.
|
Zhang T, Otevrel T, Gao Z, Ehrlich SM,
Fields JZ and Boman BM: Evidence that APC regulates survivin
expression: a possible mechanism contributing to the stem cell
origin of colon cancer. Cancer Res. 61:8664–8667. 2001.
|
|
25.
|
de Groot RP, Raaijmakers JA, Lammers JW
and Koenderman L: STAT5-dependent cyclinD1 and Bcl-xL expression in
Bcr-Abl-transformed cells. Mol Cell Biol Res Commun. 3:299–305.
2000.PubMed/NCBI
|
|
26.
|
Polakis P: Wnt signaling and cancer. Genes
Dev. 14:1837–1851. 2000.
|
|
27.
|
Gaiger A, Henn T, Horth E, et al: Increase
of bcr-abl chimeric mRNA expression in tumor cells of patients with
chronic myeloid leukemia precedes disease progression. Blood.
86:2371–2378. 1995.PubMed/NCBI
|
|
28.
|
Sawyers CL, Callahan W and Witte ON:
Dominant negative MYC blocks transformation by ABL oncogenes. Cell.
70:901–910. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
29.
|
Afar DE, McLaughlin J, Sherr CJ, Witte ON
and Roussel MF: Signaling by ABL oncogenes through cyclin D1. Proc
Natl Acad Sci USA. 92:9540–9544. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30.
|
Williamson L, Raess NA, Caldelari R, et
al: Pemphigus vulgaris identifies plakoglobin as key suppressor of
c-Myc in the skin. EMBO J. 25:3298–3309. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31.
|
Diamond JM and Melo JV: Mechanisms of
resistance to BCR-ABL kinase inhibitors. Leuk Lymphoma. 52(Suppl
1): S12–S22. 2011. View Article : Google Scholar
|
|
32.
|
Wang Z, Sampath J, Fukuda S and Pelus LM:
Disruption of the inhibitor of apoptosis protein survivin
sensitizes Bcr-abl-positive cells to STI571-induced apoptosis.
Cancer Res. 65:8224–8232. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33.
|
Amarante-Mendes GP, McGahon AJ, Nishioka
WK, Afar DE, Witte ON and Green DR: Bcl-2-independent
Bcr-Abl-mediated resistance to apoptosis: protection is correlated
with up regulation of Bcl-xL. Oncogene. 16:1383–1390. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
34.
|
Dusek RL, Godsel LM, Chen F, et al:
Plakoglobin deficiency protects keratinocytes from apoptosis. J
Invest Dermatol. 127:792–801. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35.
|
Salomon D, Sacco PA, Roy SG, et al:
Regulation of beta-catenin levels and localization by
overexpression of plakoglobin and inhibition of the
ubiquitin-proteasome system. J Cell Biol. 139:1325–1335. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
36.
|
Li L, Chapman K, Hu X, Wong A and Pasdar
M: Modulation of the oncogenic potential of beta-catenin by the
subcellular distribution of plakoglobin. Mol Carcinog. 46:824–838.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
37.
|
Kodama S, Ikeda S, Asahara T, Kishida M
and Kikuchi A: Axin directly interacts with plakoglobin and
regulates its stability. J Biol Chem. 274:27682–27688. 1999.
View Article : Google Scholar : PubMed/NCBI
|