Introduction
Several cells that consist of the vasculature
produce reactive oxygen species (ROS), which are a class of
oxygen-derived molecules including hydrogen peroxide
(H2O2), superoxide anion
(O2•−) and hydroxyl radical (•OH).
These elemental molecules have been regarded as deleterious to the
vasculature, leading to pathological processes such as
atherosclerosis, restenosis, hypertension, diabetic vascular
complications and heart failure (1,2).
However, it has become evident that ROS in vascular cells play both
a physiological and pathophysiological role in vascular homeostasis
via the regulation of numerous cellular events including cell
death, differentiation, contraction and cell proliferation
(1,3,4).
They can also act as second messengers, influencing distinct signal
transduction pathways in the cardiovascular and pulmonary systems
(1,5). In particular, vascular endothelial
cells (ECs) are involved in various regulatory responsibilities
such as vascular permeability for gases and metabolites, vascular
smooth muscle tone, blood pressure, blood coagulation, inflammation
and angiogenesis (6). Endothelial
dysfunction has been implicated in the initiation and propagation
of various vascular diseases (7).
Thus, enhanced oxidative stress may contribute to endothelial
dysfunction in vascular diseases via the apoptotic induction of ECs
(1).
ROS are mostly generated as by-products of
mitochondrial respiration or are specifically produced by oxidases
such as nicotine adenine diphosphate (NADPH) oxidase and xanthine
oxidase (8). The major metabolic
pathways embrace superoxide dismutases (SODs), which metabolize
O2•− to H2O2 (9). Further metabolism by catalase or
glutathione (GSH) peroxidase yields O2 and
H2O (10). Among ROS,
H2O2 can diffuse freely through cellular
membranes to a distance of numerous cell diameters before reacting
with specific molecular targets due to its solubility in both lipid
and aqueous environments and its comparatively low reactivity.
Tissue concentrations of H2O2 for the
duration of inflammation are likely to reach near millimolar
levels, whereas minute amounts of H2O2
generated by NADPH oxidase are believed to act only in
microenvironments of the plasma membrane such as lipid rafts
(11,12). Nonetheless, in both cases,
H2O2 may modulate essential cellular
functions of cell growth, proliferation and differentiation or it
can trigger cell death by apoptosis or necrosis.
H2O2 modulates endothelial
cell function via elaborate mechanisms. Ambient production of
O2•− and subsequently
H2O2 at low levels is crucial for endothelial
cell growth and proliferation (2). On the other hand, the mode of action
of H2O2 in provoking endothelial dysfunction
and death has also been extensively investigated. However, the
precise molecular mechanisms underlying these important effects
remain largely unclear. Therefore, it is critical to understand the
different roles ROS play in the physiology and pathophysiology of
ECs. A fuller understanding of how H2O2
affects apoptosis in ECs may aid in the development of novel
strategies to treat or prevent vascular diseases. In the present
study, we evaluated the effects of exogenous
H2O2 on cell growth and death in ECs such as
calf pulmonary artery endothelial cells (CPAECs) and human
umbilical vein endothelial cells (HUVECs) in relation to changes in
intracellular ROS and GSH levels, and investigated its mechanism in
CPAECs.
Materials and methods
Cell culture
CPAECs obtained from the Korean Cell Line Bank
(KCLB, Seoul, Korea) were maintained in a humidified incubator
containing 5% CO2 at 37°C. CPAECs were cultured in
RPMI-1640 supplemented with 10% fetal bovine serum (FBS;
Sigma-Aldrich Chemical Co., St. Louis, MO, USA) and 1%
penicillin-streptomycin (Gibco-BRL, Grand Island, NY, USA). The
primary HUVECs from PromoCell GmbH (Heidelberg, Germany) were
maintained in a humidified incubator containing 5% CO2
at 37°C. HUVECs were cultured in complete endothelial cell growth
medium containing 2% FBS, which was purchased from PromoCell GmbH.
CPAECs and HUVECs were grown in 100-mm plastic tissue culture
dishes (Nunc, Roskilde, Denmark) containing 10 ml media and
harvested with a solution of trypsin-EDTA while in a logarithmic
phase of growth (approximately every 2–3 days). For experiments,
CPAECs were used between passage 40 and 50, and HUVECs were used
between passage four and eight.
Reagents
H2O2 was purchased from
Sigma-Aldrich Chemical Co. The pan-caspase inhibitor
[benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone (Z-VAD-FMK)], the
caspase-3 inhibitor
[benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone
(Z-DEVD-FMK)], the caspase-8 inhibitor
[benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone (Z-IETD-FMK)]
and the caspase-9 inhibitor
[benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethylketone (Z-LEHD-FMK)]
were obtained from R&D Systems, Inc., (Minneapolis, MN, USA)
and were dissolved in dimethyl sulfoxide (DMSO; Sigma-Aldrich
Chemical Co.). N-acetyl cysteine (NAC) was obtained from
Sigma-Aldrich Chemical Co., and was dissolved in the buffer [20 mM
HEPES (pH 7.0)]. Based on previous studies (13,14), cells were pretreated with or
without 15 μM caspase inhibitor or 2 mM NAC for 1 h prior to
H2O2 treatment. DMSO (0.2%) was used as a
control vehicle and it did not appear to affect cell growth or
death.
Cell growth assay
Cell growth changes in ECs treated with
H2O2 were indirectly determined by measuring
the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
(MTT; Sigma-Aldrich Chemical Co.,) dye absorbance, as previously
described (15). In brief,
4×104 cells/well were seeded in 96-well plates (Nunc)
for MTT assays. After exposure to the indicated amounts of
H2O2 for 24 h, 20 μl MTT
(Sigma-Aldrich Chemical Co.,) solution (2 mg/ml in PBS) was added
to each well of the 96-well plates. The plates were incubated for
an additional 4 h at 37°C. Media in plates were withdrawn by
pipetting and 200 μl of DMSO was added to each well to
solubilize the formazan crystals. The optical density was measured
at 570 nm using a microplate reader (Synergy™ 2; BioTek Instruments
Inc., Winooski, VT, USA).
Annexin V-FITC staining for cell death
detection
Apoptosis was determined by staining cells with
Annexin V-fluorescein isothiocyanate (FITC; Ex/Em=488/519 nm;
Invitrogen Corporation, Camarillo, CA, USA). In brief,
1×106 cells in 60-mm culture dishes (Nunc) were
incubated with the indicated amounts of H2O2
with or without 15 μM each caspase inhibitor or 2 mM NAC for
24 h. Cells were washed twice with cold PBS and then resuspended in
500 μl of binding buffer (10 mM HEPES/NaOH pH 7.4, 140 mM
NaCl, 2.5 mM CaCl2) at a concentration of
1×106 cells/ml. Five microliters of Annexin V-FITC was
then added to these cells, which were analyzed with a FACStar flow
cytometer (Becton-Dickinson, Franklin Lakes, NJ, USA).
Measurement of mitochondrial membrane
potential (MMP; ΔΨm)
MMP (ΔΨm) levels were measured by a
rhodamine 123 fluorescent dye (Ex/Em=485/535 nm; Sigma-Aldrich
Chemical Co.,) as previously described (16). In brief, 1×106 cells in
60-mm culture dishes (Nunc) were incubated with the indicated
amounts of H2O2 with or without 15 μM
each caspase inhibitor or 2 mM NAC for 24 h. Cells were washed
twice with PBS and incubated with the rhodamine 123 (0.1
μg/ml) at 37°C for 30 min. Rhodamine 123 staining intensity
was determined by a FACStar flow cytometer (Becton-Dickinson).
Rhodamine 123 negative cells indicated the loss of MMP
(ΔΨm) in cells.
Detection of intracellular ROS
levels
Intracellular ROS level including
O2•− was detected by means of an
oxidation-sensitive fluorescent probe dye, dihydroethidium (DHE;
Ex/Em=518/605 nm; Invitrogen/Molecular Probes). In brief,
1×106 cells in 60-mm culture dishes were incubated with
the indicated amounts of H2O2 for 24 h. Cells
were then washed in PBS and incubated with 20 μM DHE at 37°C
for 30 min. DHE fluorescence intensities were detected using a
FACStar flow cytometer (Becton-Dickinson). ROS (DHE) level was
expressed as mean fluorescence intensity (MFI), which was
calculated by CellQuest software (Becton-Dickinson).
Detection of the intracellular GSH
Cellular GSH levels were analyzed using a
5-chloromethylfluorescein diacetate (CMFDA; Ex/Em=522/595 nm;
Invitrogen/Molecular Probes) as previously described (17). In brief, 1×106 cells in
60-mm culture dishes (Nunc) were incubated with the indicated
amounts of H2O2 with or without 15 μM
each caspase inhibitor or 2 mM NAC for 24 h. Cells were then washed
with PBS and incubated with 5 μM CMFDA at 37°C for 30 min.
CMF fluorescence was assessed using a FACStar flow cytometer
(Becton-Dickinson). Negative CMF staining (GSH-depleted) of cells
is expressed as the percentage of (−) CMF cells.
Measurement of cellular SOD and catalase
activities
SOD enzyme activity was measured using the SOD assay
kit-WST (Fluka Chemical Corp., Milwaukee, WI, USA) and catalase
enzyme activity was measured using the catalase assay kit from
Sigma-Aldrich Chemical Co., as previously described (18). In brief, 1×106 cells
were incubated with 30 μM H2O2 for 24
h. The cells were then washed in PBS and suspended in five volumes
of lysis buffer [20 mM HEPES (pH 7.9), 20% Glycerol, 200 mM KCl,
0.5 mM EDTA, 0.5% NP-40, 0.5 mM DTT, 1% protease inhibitor cocktail
(from Sigma-Aldrich Chemical Co.)]. Supernatant protein
concentration was determined by the Bradford method. Supernatant
samples containing 100 μg of total protein were used for
determination of SOD and catalase enzyme activities. These were
added to each well in 96-well microtiter plates (Nunc) with the
appropriate working solutions (according to the manufacturer’s
instructions) at 25°C for 30 min. The color changes were measured
at 450 or 520 nm using a microplate reader (SpectraMax 340;
Molecular Devices Co., Sunnyvale, CA, USA). The value for the
experimental group was converted to the percentage of the control
group.
Statistical analysis
The results represent the means of at least three
independent experiments (means ± SD). The data were analyzed using
Instat software (GraphPad Prism 4; GraphPad Software, San Diego,
CA, USA). The Student’s t-test or one-way analysis of variance
(ANOVA) with post hoc analysis using Tukey’s multiple comparison
test was used for parametric data. P<0.05 was considered to
indicate a statistically significant difference.
Results
Effects of H2O2 on
the growth, death and MMP (ΔΨm) of CPAECs and
HUVECs
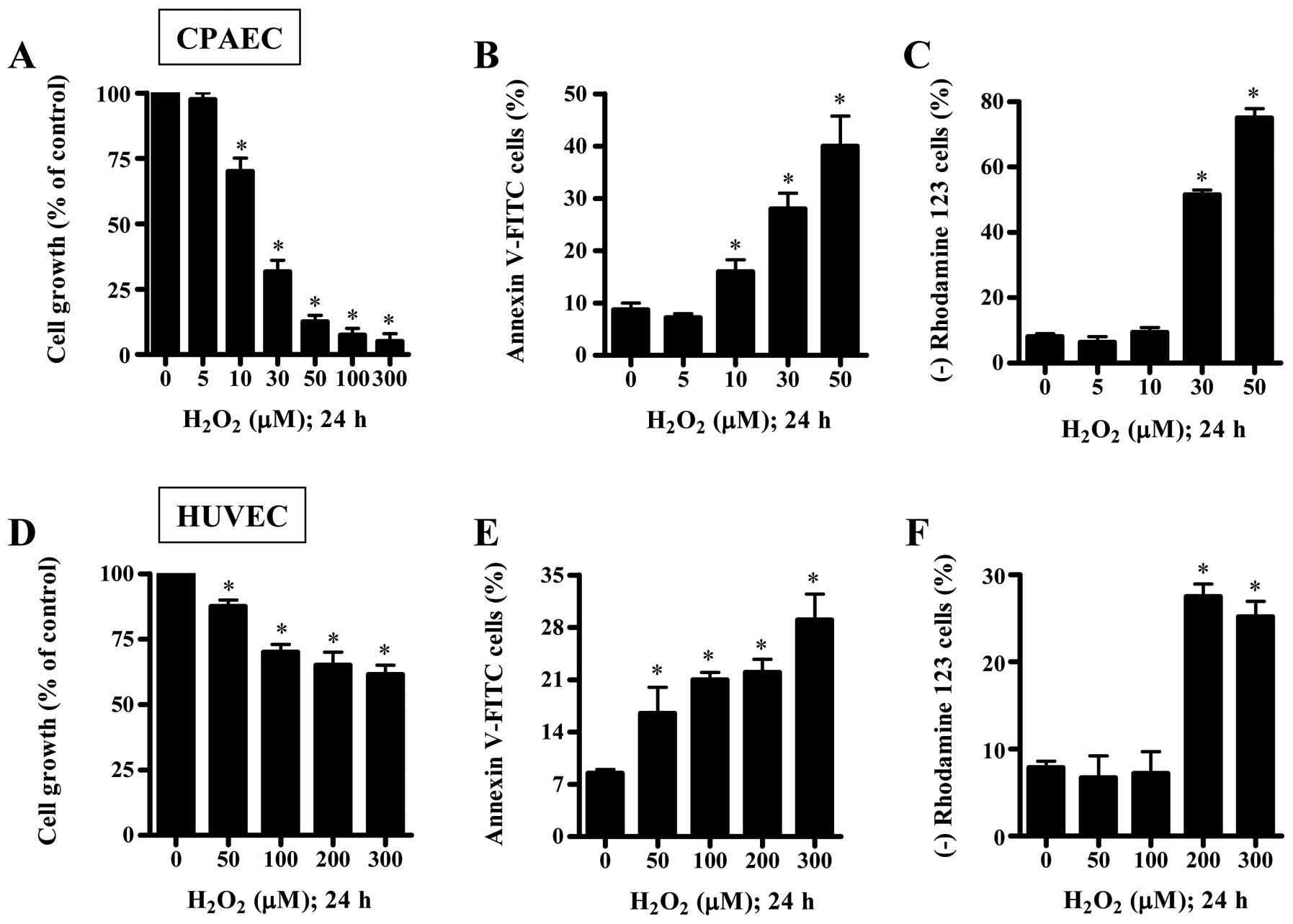
We examined the effect of H2O2
on the growth and death of CPAECs and HUVECs at 24 h. When the
growth of ECs after treatment with H2O2 was
assessed by MTT assays, the reduction of cell growth was observed
in both ECs in a dose-dependent manner, and the IC50
(the half maximal inhibitory concentration) of
H2O2 in CPAECs and HUVECs was ∼20 and 300
μM, respectively (Fig. 1A and
D). When ECs were stained with Annexin V-FITC to evaluate the
induction of apoptosis, the number of Annexin V-staining cells was
increased in H2O2-treated ECs (Fig. 1B and E). At a 50 μM dose of
H2O2, the number of Annexin V-staining cells
in CPAECs increased ∼30% compared with control CPAECs and the
number in HUVECs increased ∼5% compared with control HUVECs
(Fig. 1B and E). Since apoptosis
is closely related to the collapse of MMP (ΔΨm)
(19), we assessed the effect of
H2O2 on MMP (ΔΨm) using rhodamine
123. Although 5 or 10 μM H2O2 did not
induce the loss of MMP (ΔΨm) in CPAECs, 30 or 50
μM H2O2 strongly increased the MMP
(ΔΨm) loss (Fig. 1C).
In HUVECs, 50 or 100 μM H2O2 did not
induce the loss of MMP (ΔΨm), but 200 or 300 μM
H2O2 did (Fig.
1F).
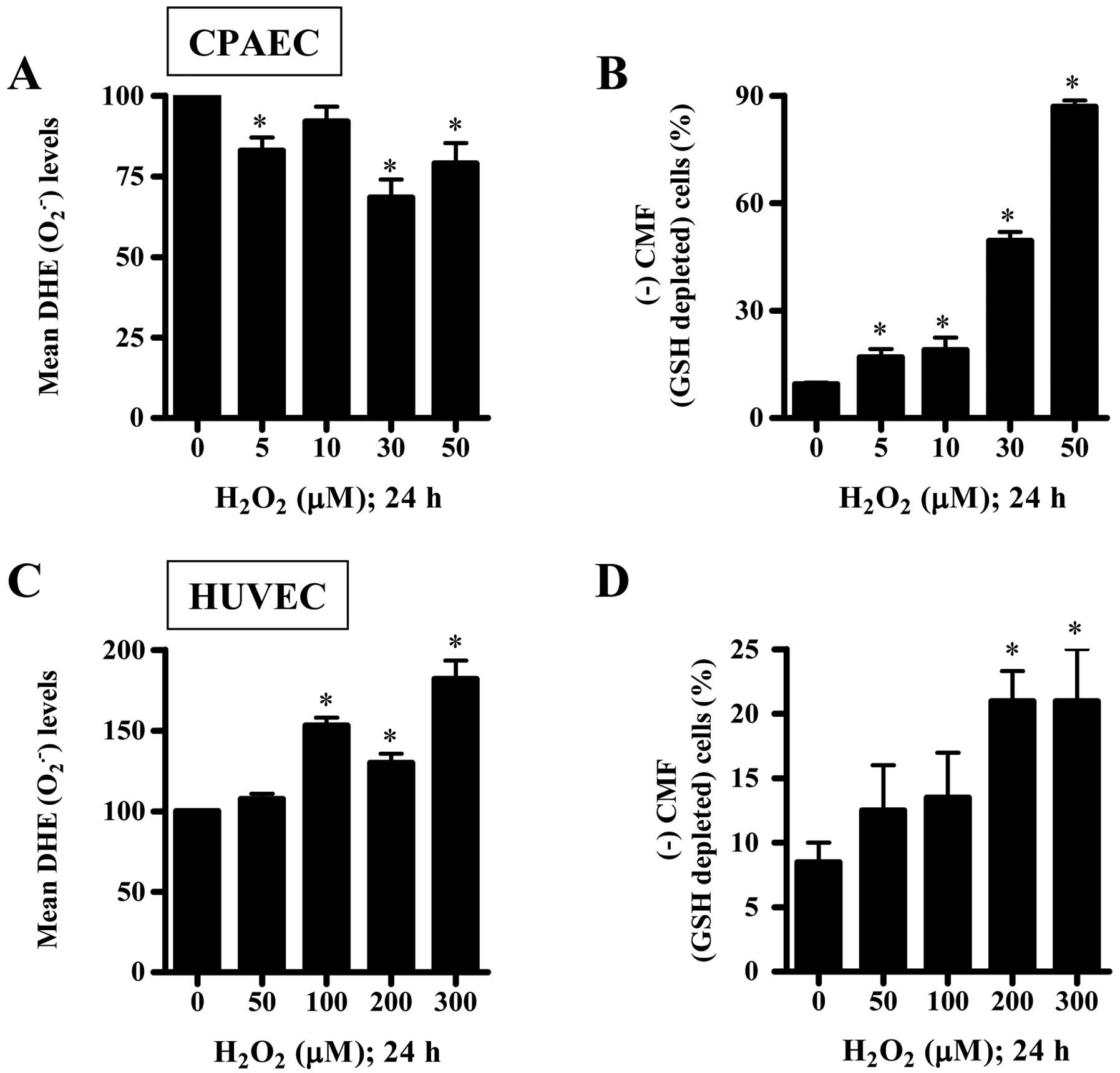
Effects of H2O2 on
intracellular ROS and GSH levels in CPAECs and HUVECs
To assess levels of intracellular ROS including
O2•− in H2O2-treated
ECs at 24 h, we used a DHE fluorescence dye, which specifically
reflects O2•− accumulation in cells. As shown
in Fig. 2A, all the tested doses
of H2O2 decreased DHE
(O2•−) levels in CPAECs. However, 100–300
μM H2O2 significantly increased the
DHE (O2•−) levels in HUVECs (Fig. 2C). Next, we analyzed the changes
of GSH levels in ECs using a CMF fluorescence dye. All the tested
doses of H2O2 significantly increased the
number of GSH-depleted cells in CPAECs (Fig. 2B). The relatively higher doses of
200 or 300 μM H2O2 also increased the
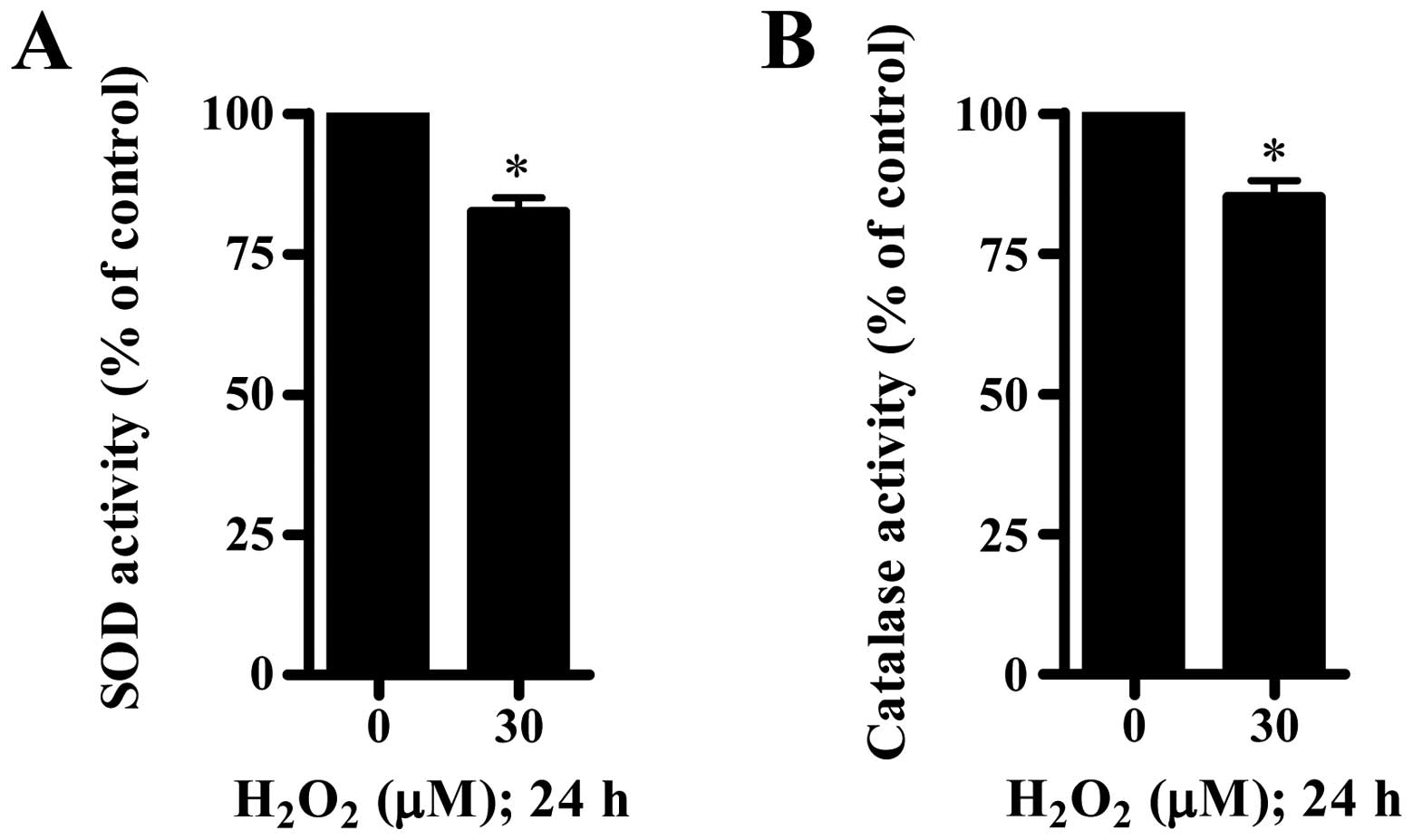
number of GSH-depleted cells in HUVECs (Fig. 2D). Furthermore, we measured the
activities of SOD and catalase in
H2O2-treated CPAECs. As shown in Fig. 3, 30 μM
H2O2 significantly decreased the activities
of SOD and catalase.
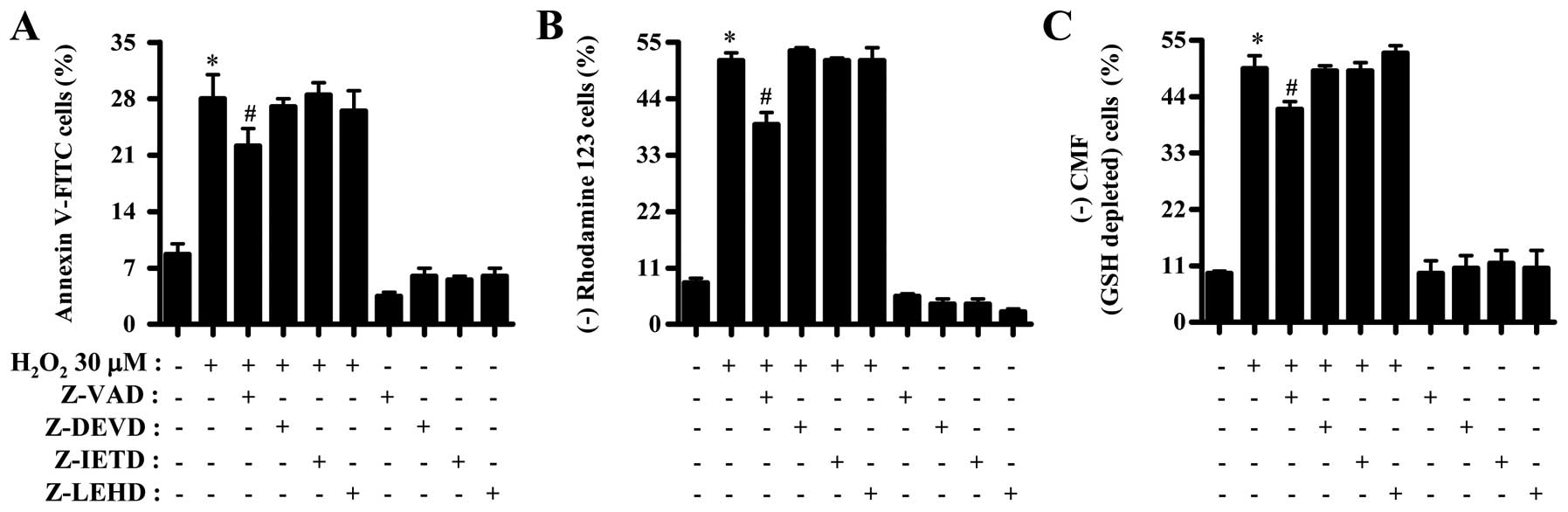
Effects of caspase inhibitors on cell
death, MMP (ΔΨm) and GSH depletion in
H2O2-treated CPAECs
To determine which caspases were involved in
apoptotic cell death in H2O2-treated CPAECs,
cells were pretreated with pan-caspase inhibitor (Z-VAD), caspase-3
inhibitor (Z-DEVD), caspase-8 inhibitor (Z-IETD) or caspase-9
inhibitor (Z-LEHD) prior to treatment with
H2O2. For this experiment, 30 μM
H2O2 was selected as a suitable dose to
differentiate the levels of cell death, MMP (ΔΨm) and
GSH depletion in the presence or absence of each caspase inhibitor.
While only Z-VAD significantly prevented apoptotic cell death in
H2O2-treated CPAECs, other caspase inhibitors
did not affect the apoptotic cell death (Fig. 4A). In addition, Z-VAD
significantly attenuated the loss of MMP (ΔΨm) by
H2O2 whereas other caspase inhibitors did not
alter the loss (Fig. 4B). In
relation to GSH depletion, only Z-VAD, no other caspase inhibitor,
significantly decreased GSH depletion in
H2O2-treated CPAECs (Fig. 4C).
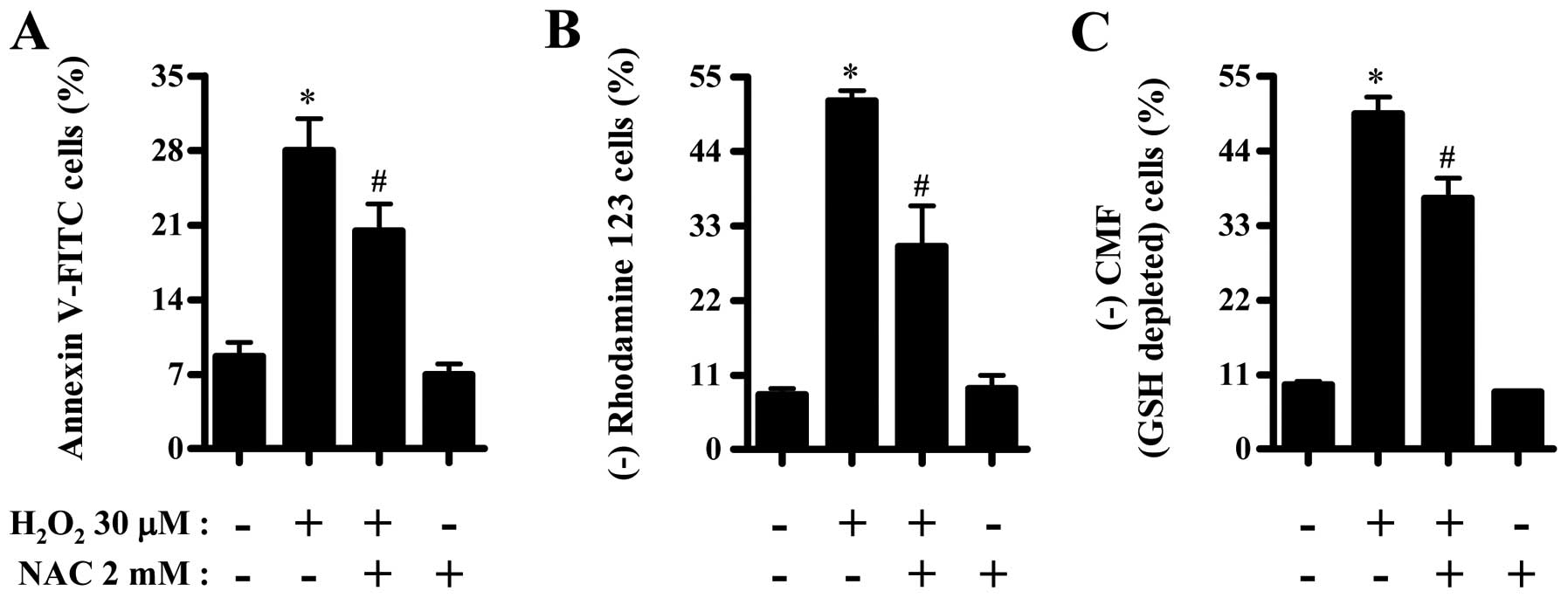
Effects of NAC on cell death, MMP
(ΔΨm) and GSH depletion in
H2O2-treated CPAECs
Next, we investigated the effects of NAC (a
well-known antioxidant or GSH precursor) on cell death, MMP
(ΔΨm) and GSH depletion in
H2O2-treated CPAECs at 24 h. NAC
significantly reduced the number of Annexin V-FITC positive cells
in H2O2-treated CPAECs (Fig. 5A). NAC also significantly
attenuated the loss of MMP (ΔΨm) in these cells
(Fig. 5B). Moreover, NAC
decreased GSH depletion in H2O2-treated
CPAECs (Fig. 5C).
Discussion
ROS are involved in several physiological and
pathophysiological processes in vascular endothelium by influencing
cell proliferation, hypertrophy, migration, inflammation,
contraction and death (1,2,5).
In the present study, we elucidated the cytotoxic effect of
exogenous H2O2 on ECs such as CPAECs and
HUVECs in relation to cell death, ROS and GSH. Other studies have
reported that ROS not only lead to cell death in ECs (20–22) but they are also involved in the
survival of ECs (20). Our
current results demonstrate that H2O2
inhibited the growth of CPAECs and HUVECs with an IC50
of approximately 20 and 300 μM, respectively.
H2O2 also provoked cell death in both ECs, as
evidenced by Annexin V-staining cells and trypan blue cell counting
(data not shown) and triggered the loss of MMP (ΔΨm). In
addition, H2O2 induced apoptosis in CPAECs in
a caspase-dependent manner. However, the susceptibility of
H2O2 between these ECs was different. HUVECs
were more resistant to H2O2 than CPAECs. The
difference in susceptibility may be due to the dissimilar basal
antioxidant enzymes each cell has. Thus, the cytotoxic effects of
H2O2 may differ depending on various
endothelial cell types, such as artery vs. vein, large vessels vs.
small vessels, human vs. other species, coronary vs. pulmonary. It
is imperative that such effects of ROS, especially
H2O2, be defined and characterized in the
future.
When determining which caspase was involved in
apoptosis in H2O2-treated CPAECs, only
pan-caspase inhibitor Z-VAD significantly prevented apoptotic cell
death in H2O2-treated CPAECs. Other caspase
inhibitors did not affect the apoptotic cell death. In addition,
Z-VAD attenuated the loss of MMP (ΔΨm) in
H2O2-treated CPAECs whereas other caspase
inhibitors did not alter the loss of MMP (ΔΨm). These
results suggest that H2O2-induced CPAEC
apoptosis requires the activation of various caspases containing
both caspase-8, necessary for the death receptor pathway, and
caspase-9, related to the mitochondrial pathway. We observed that
10 μM H2O2 significantly increased the
number of Annexin V-staining cells in CPAECs but this dose did not
induce the MMP (ΔΨm) loss. In addition, 50 and 100
μM H2O2 significantly increased the
number of Annexin V-staining cells in HUVECs but those
concentrations did not induce the MMP (ΔΨm) loss. By
contrast, 30 or 50 μM H2O2 strongly
increased the proportion of MMP (ΔΨm) loss in CPAECs
compared with that of Annexin V-staining cells. Therefore, the
effect of MMP (ΔΨm) loss in
H2O2-induced ECs apoptosis is likely
concentration specific. It appears that relatively higher
concentrations in each EC induce cell death via steadfastly
inducing MMP (ΔΨm) loss.
The main ROS involved in cell signaling pathways
are H2O2 and O2•−. ROS
toxicity is usually mediated by •OH (5). According to our present results,
H2O2 increased DHE
(O2•−) levels in HUVECs.
H2O2 appeared to induce the potential leakage
of electron from mitochondrial respiratory transport chain and/or
activated oxidases such as NADPH oxidase and xanthine oxidase in
HUVECs. By contrast, although H2O2 reduced
the activity of SOD in CPAECs, it did not increase DHE
(O2•−) levels in these cells. Thus,
H2O2 did not affect both mitochondrial
respiratory transport chain and various oxidases to generate
O2•− in CPAECs. Instead,
H2O2 decreased DHE
(O2•−) levels in CPAECs via an unidentified
mechanism. The different effects may be due to different basal
mitochondrial activity and antioxidant enzymes between two ECs. As
H2O2 significantly induced apoptosis and
decreased the activity of catalase in CPAECs, it is possible that
exogenous H2O2 can be efficiently converted
into the toxic ROS of •OH via the Fenton reaction to
kill CPAECs. The intracellular GSH content has a decisive effect on
anticancer drug-induced apoptosis, indicating that apoptotic
effects are inversely proportional to GSH content (23,24). Similarly,
H2O2 increased the number of GSH-depleted
cells in both ECs. At 50 μM
H2O2-treated ECs, the GSH-depleted cell
number in CPAECs was higher than that in HUVECs. These results seem
to be correlated with Annexin V-FITC results from ECs treated with
H2O2. In addition, Z-VAD reduced GSH-depleted
cell numbers in H2O2-treated CPAECs. NAC
showing an anti-apoptotic effect on
H2O2-treated CPAECs significantly decreased
GSH depletion.
In conclusion, H2O2 induced
growth inhibition and death in ECs via GSH depletion. HUVECs were
relatively resistant to H2O2 compared with
CPAECs. H2O2-induced CPAEC death occurs via
apoptosis, which requires the activation of various caspases.
Abbreviations:
|
EC
|
endothelial cell
|
|
CPAEC
|
calf pulmonary arterial endothelial
cell
|
|
HUVEC
|
human umbilical vein endothelial
cell
|
|
ROS
|
reactive oxygen species
|
|
SOD
|
superoxide dismutase
|
|
MMP (ΔΨm)
|
mitochondrial membrane potential;
|
|
Z-VAD-FMK
|
benzyloxycarbonyl-Val-Ala-Asp-fluoromethylketone;MTT,3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium
bromide
|
|
FITC
|
fluorescein isothiocyanate
|
|
Z-DEVD-FMK
|
benzyloxycarbonyl-Asp-Glu-Val-Asp-fluoromethylketone
|
|
GSH
|
glutathione
|
|
DHE
|
dihydroethidium
|
|
Z-IETD-FMK
|
benzyloxycarbonyl-Ile-Glu-Thr-Asp-fluoromethylketone
|
|
CMFDA
|
5-chloromethylfluorescein
diacetate
|
|
FBS
|
fetal bovine serum
|
|
Z-LEHD-FMK
|
benzyloxycarbonyl-Leu-Glu-His-Asp-fluoromethylketone
|
|
NAC
|
N-acetyl cysteine
|
Acknowledgements
This study was supported by a grant
from the Ministry of Science and Technology (MOST)/Korea Science
and Engineering Foundation (KOSEF) through the Diabetes Research
Center at Chonbuk National University (2012-0009323).
References
|
1.
|
Irani K: Oxidant signaling in vascular
cell growth, death, and survival: a review of the roles of reactive
oxygen species in smooth muscle and endothelial cell mitogenic and
apoptotic signaling. Circ Res. 87:179–183. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
2.
|
Cai H: Hydrogen peroxide regulation of
endothelial function: origins, mechanisms, and consequences.
Cardiovasc Res. 68:26–36. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Gonzalez C, Sanz-Alfayate G, Agapito MT,
Gomez-Nino A, Rocher A and Obeso A: Significance of ROS in oxygen
sensing in cell systems with sensitivity to physiological hypoxia.
Respir Physiol Neurobiol. 132:17–41. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4.
|
Baran CP, Zeigler MM, Tridandapani S and
Marsh CB: The role of ROS and RNS in regulating life and death of
blood monocytes. Curr Pharm Des. 10:855–866. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
Perez-Vizcaino F, Cogolludo A and Moreno
L: Reactive oxygen species signaling in pulmonary vascular smooth
muscle. Respir Physiol Neurobiol. 174:212–220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6.
|
Bassenge E: Endothelial function in
different organs. Prog Cardiovasc Dis. 39:209–228. 1996. View Article : Google Scholar
|
|
7.
|
Lum H and Roebuck KA: Oxidant stress and
endothelial cell dysfunction. Am J Physiol Cell Physiol.
280:C719–C741. 2001.PubMed/NCBI
|
|
8.
|
Zorov DB, Juhaszova M and Sollott SJ:
Mitochondrial ROS-induced ROS release: an update and review.
Biochim Biophys Acta. 1757:509–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Zelko IN, Mariani TJ and Folz RJ:
Superoxide dismutase multigene family: a comparison of the CuZn-SOD
(SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures,
evolution, and expression. Free Radic Biol Med. 33:337–349. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Wilcox CS: Reactive oxygen species: roles
in blood pressure and kidney function. Curr Hypertens Rep.
4:160–166. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Rhee SG, Kang SW, Jeong W, Chang TS, Yang
KS and Woo HA: Intracellular messenger function of hydrogen
peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol.
17:183–189. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Vilhardt F and van Deurs B: The phagocyte
NADPH oxidase depends on cholesterol-enriched membrane microdomains
for assembly. EMBO J. 23:739–748. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Han YH, Kim SZ, Kim SH and Park WH:
Pyrogallol inhibits the growth of lung cancer Calu-6 cells via
caspase-dependent apoptosis. Chem Biol Interact. 177:107–114. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
14.
|
You BR and Park WH: Gallic acid-induced
lung cancer cell death is related to glutathione depletion as well
as reactive oxygen species increase. Toxicol In Vitro.
24:1356–1362. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15.
|
Park WH, Seol JG, Kim ES, Hyun JM, Jung
CW, Lee CC, Kim BK and Lee YY: Arsenic trioxide-mediated growth
inhibition in MC/CAR myeloma cells via cell cycle arrest in
association with induction of cyclin-dependent kinase inhibitor,
p21, and apoptosis. Cancer Res. 60:3065–3071. 2000.PubMed/NCBI
|
|
16.
|
Han YH, Kim SZ, Kim SH and Park WH:
Arsenic trioxide inhibits growth of As4.1 juxtaglomerular cells via
cell cycle arrest and caspase-independent apoptosis. Am J Physiol
Renal Physiol. 293:F511–F520. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
17.
|
Han YH, Kim SH, Kim SZ and Park WH:
Caspase inhibitor decreases apoptosis in pyrogallol-treated lung
cancer Calu-6 cells via the prevention of GSH depletion. Int J
Oncol. 33:1099–1105. 2008.PubMed/NCBI
|
|
18.
|
Park WH, Han YH, Kim SH and Kim SZ:
Pyrogallol, ROS generator inhibits As4.1 juxtaglomerular cells via
cell cycle arrest of G2 phase and apoptosis. Toxicology.
235:130–139. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
19.
|
Yang J, Liu X, Bhalla K, Kim CN, Ibrado
AM, Cai J, Peng TI, Jones DP and Wang X: Prevention of apoptosis by
Bcl-2: release of cytochrome c from mitochondria blocked. Science.
275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
20.
|
Deshpande SS, Angkeow P, Huang J, Ozaki M
and Irani K: Racl inhibits TNF-alpha-induced endothelial cell
apoptosis: dual regulation by reactive oxygen species. FASEB J.
14:1705–1714. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
21.
|
Fu YC, Yin SC, Chi CS, Hwang B and Hsu SL:
Norepinephrine induces apoptosis in neonatal rat endothelial cells
via a ROS-dependent JNK activation pathway. Apoptosis.
11:2053–2063. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22.
|
Das A, Gopalakrishnan B, Voss OH, Doseff
AI and Villamena FA: Inhibition of ROS-induced apoptosis in
endothelial cells by nitrone spin traps via induction of phase II
enzymes and suppression of mitochondria-dependent pro-apoptotic
signaling. Biochem Pharmacol. 84:486–497. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23.
|
Estrela JM, Ortega A and Obrador E:
Glutathione in cancer biology and therapy. Crit Rev Clin Lab Sci.
43:143–181. 2006. View Article : Google Scholar
|
|
24.
|
Higuchi Y: Glutathione depletion-induced
chromosomal DNA fragmentation associated with apoptosis and
necrosis. J Cell Mol Med. 8:455–464. 2004. View Article : Google Scholar : PubMed/NCBI
|