Introduction
Congenital hypothyroidism (CH), also known as
cretinism, is a disease caused by thyroid hormone deficiency
present at birth (1). CH can lead
to delayed growth and development and mental retardation; 85% of CH
cases are secondary to thyroid developmental abnormalities such as
athyreosis, hypoplastic or ectopic gland, and the remaining 15% of
CH cases are due to genetic defects of thyroid hormone synthesis
within a structurally normal gland (1). Although the molecular mechanisms of
thyroid gland development and thyroid hormone synthesis and
function have been studied extensively, the genetic pathogenesis
remains relatively unclear (2).
A key step in thyroid hormone biosynthesis is iodine
organification, which can only be successfully accomplished in the
presence of H2O2. In thyroid follicular
cells, Duox2 proteins are glycosylated and bound in the follicular
cavity at the apical membrane, catalyzing the formation of
H2O2, which is involved in the key step in
thyroid hormone synthesis - iodine organification (3). Therefore, the normal functioning of
Duox2 is one of the prerequisites for thyroid hormone synthesis.
Several studies have suggested that mutations in the Duox2 gene can
lead to CH (4–9).
DUOXA2, a DUOX maturation factor, plays an important
role in Duox2 maturation and activation, thus determining the
normal functionality of Duox2 (10). Genetic defects in DUOXA2 lead to
defects in DUOX2 protein expression, which in turn lead to CH
(11). Thus, DUOXA2 is an
important entry point for molecular genetic studies of CH. This
study explored the novel mutations of DUOXA2 in CH
patients
Materials and methods
Ethics statement
Approval was obtained from the local Ethics
Committee. All participants provided written informed consent prior
to enrollment in the study.
Patients
All 47 CH cases were first screened at the Fujian
Neonatal Disease Screening Center between 1998 and 2011. The
patients, 27 males and 20 females, were diagnosed and followed up
by the Neonatal Department, Maternal and Child Health Hospital of
Fujian Province. Thyroid agenesis, hypoplasia, or ectopic thyroid
abnormalities were excluded in all objects through ECT or
ultrasound. As controls, 100 unrelated healthy controls were
enrolled in this study in the First Affiliated Hospital of Fujian
Medical University.
Genomic DNA extraction
Venous blood (2 ml) was obtained from the 47 CH
patients, their parents, siblings (if any), and unrelated healthy
controls. Sodium citrate was added as anticoagulant. Genomic DNA
(30–80 ng/μl) was extracted according to the manufacturer’s
instruction (Tiangen Biotech Co.) and stored at −20°C.
Primer design and PCR
All primers were designed to the flanking intron
regions of the exons (1). The
primers were synthesized by Shanghai Sangon Biotechnology Co., Ltd.
The primer sequences and the length of the PCR products are shown
in Table I. The PCR reaction
volume was 50 μl, containing 2 μl DNA template; 2
μl upstream and downstream primers (10
μmol/μl), respectively; 4 μl of dNTP (2.5 mmol
/l); 5 μl buffer solution; 1U Ex Taq DNA polymerase (Takara
Bio Co.). The reaction conditions were: 95°C 5 min, 95°C 30 sec,
55°C 30 sec, 72°C 30 sec, 32 cycles; extension at 72°C for 5 min.
The PCR products were run on 1.5% agarose gel, visualized by
goldview, and analyzed by UV analyzer.
 | Table I.PCR primers and annealing temperatures
for 6 exons of the DUOXA2 gene. |
Table I.
PCR primers and annealing temperatures
for 6 exons of the DUOXA2 gene.
| Exon | Primer (5′-3′) | Product length
(bp) | Annealing temp
(°C) |
|---|
| 1 | F:
CAGCCTTGTACGCAAAGAGA
R: CCCCACTCTACCTGCACTA | 289 | 57 |
| 2 | F:
GTCTTGGGGACTCTGGTTT
R: ACCCCAGTTCCCTATTGTCC | 201 | 56 |
| 3 | F:
CAGTGTCCCACCTCCCATA
R: CTCACCTAACCGGGGATCT | 272 | 57 |
| 4 | F:
TTCCTGTCTGAATCCGCTTA
R: CATCCTCCCGCTCATACG | 356 | 58 |
| 5 | F:
GGGGTAGGGATAAAGAAGAGC
R: AATCCTGTCTCCACCCTTAGC | 351 | 56 |
| 6 | F:
GTTTGAGGCCAGAGTTCGAG
R: GGGGAAGGAGTCCAGATTG | 295 | 56 |
PCR amplification
All exons of the DUOXA2 gene were amplified
in patients first. If an exon mutation was identified in a patient,
the target fragment was also amplified in the family members and in
the 100 normal healthy controls.
Sequencing
PCR products were sent to Shanghai Sangon
Biotechnology Co., Ltd., for sequencing using forward and reverse
primers by ABl3700 automatic sequence analyzer. The sequencing
primers used were the same as the PCR primers. For samples with
suspected mutations, amplification and sequencing were repeated
three times. If the results were consistent, the mutation was
considered real, otherwise it was considered as experimental
error.
Statistical analysis
The statistical χ2 test was used and
P<0.05 was considered to indicate statistically significant
differences.
Results
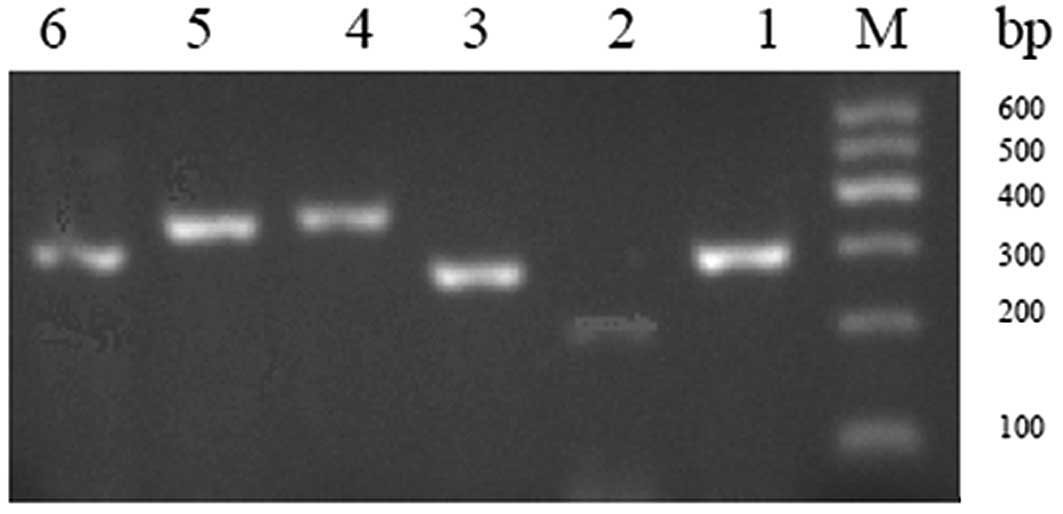
PCR products of 6 exons ran as bright single bands
as visualized by UV analyzers after 1.5% agarose gel
electrophoresis (Fig. 1). These 6
exon sequences were compared with GenBank DUOXA2 standard sequence
(Gene ID, 405 753) using the NCBI BLAST program. The simultaneous
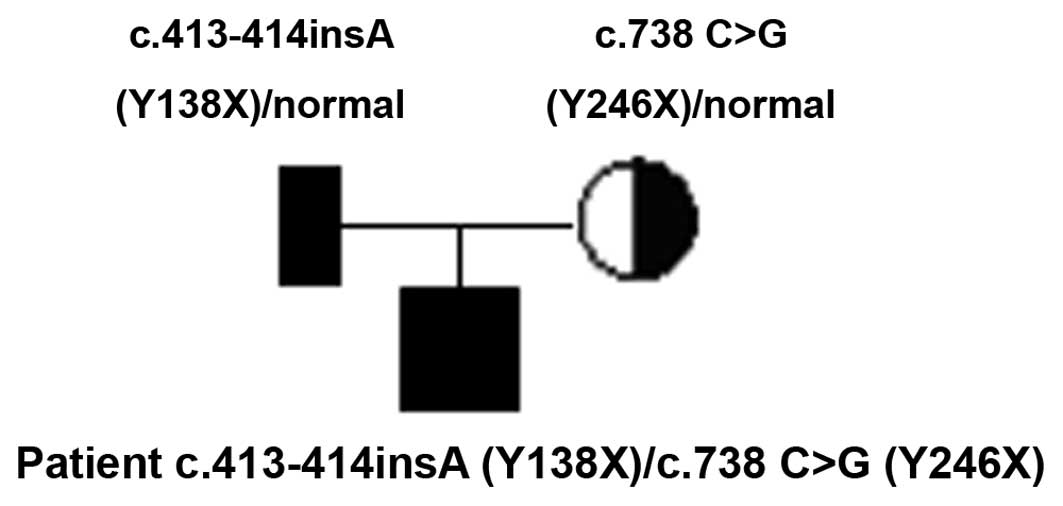
presence of two mutations in the DUOXA2 gene in one CH child was
identified in this study, a compound heterozygous mutation. The
parents of the child were the carriers of the mutated gene
(Fig. 2) and no corresponding
mutations were found in the 100 normal controls, consistent with
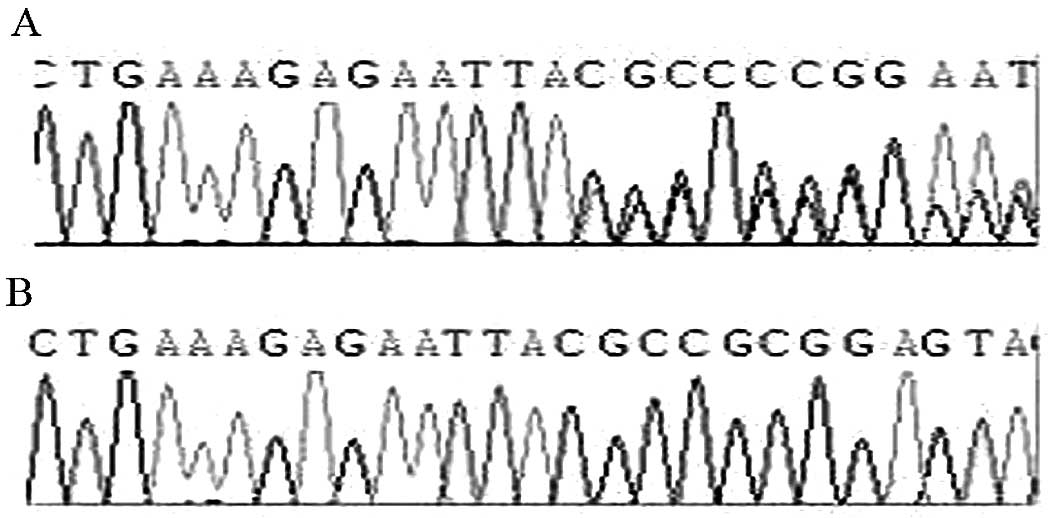
the recessive genetic law. The two mutations are an insertion of A
base between 413 and 414 bp on the 4th exon (Fig. 3), and a C→G substitution on the
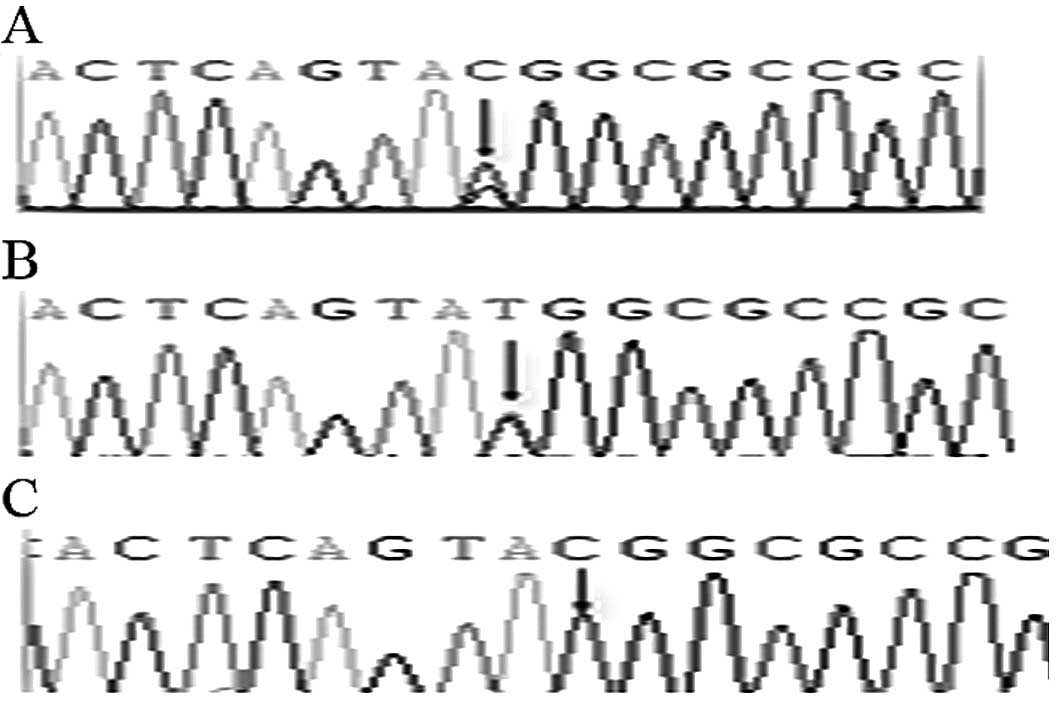
5th exon of 738 bp (p.Y246X). Aside from the C→G substitution on
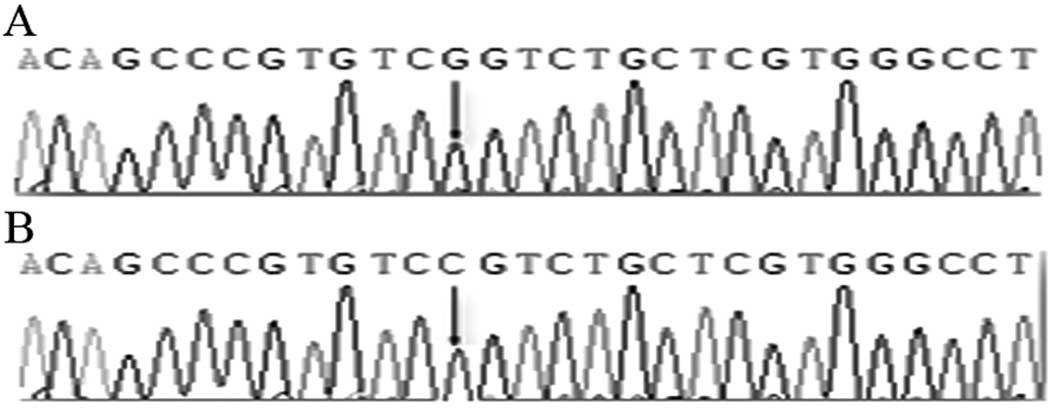
the 5th exon of 738 bp, a C→T substitution was found in 4 patients
and in 11 healthy controls, with a frequency of 4/47 (8.5‰) and
11/100 (11‰), respectively, which was not significantly different
(P>0.05). This substitution did not cause amino acid change
(Fig. 4). In addition, 3 patients
and their family members and 9 normal controls had a C→G
substitution on the 3rd exon of 278 bp (Fig. 5), which is an SNP locus. The
mutation frequency of this locus is 3/47 (6.5‰) in patients and
9/100 (9‰) in healthy controls, without significant difference
between these two groups.
The CH patient, of Han ethnicity, with this novel
DUOXA2 gene mutation was born in March 2010 at the Maternal and
Child Health Hospital in Fujian Province. The parents were not
diagnosed with any thyroid-related diseases. Following the initial
screening of newborn diseases after birth, the child was brought to
the hospital regularly for thyroid function tests and was
diagnosed. This patient with the c.413–414insA (Y138X) mutation had
mild CH symptoms.
Discussion
CH is one of the most common endocrine diseases in
pediatrics. CH is caused by abnormal development due to various
reasons such as athyreosis, hypoplastic or ectopic gland, and
defects in thyroid hormone synthesis during embryonic development.
This disease can lead to mental retardation and short stature,
commonly known as cretinism. The average annual incidence rate of
the disease is 1/1750–1/4000, making it the most common congenital
metabolic abnormality (12,13).
In CH cases, approximately 15% are due to genetic
defects of thyroid hormone synthesis, most of which are caused by
iodine organification defects. The successful iodine organification
requires the presence of H2O2, which is
produced by NADPH oxidase. Duox2 is essential for the catalytic
activity of NADPH oxidase (3).
Thus, abnormal Duox2 function leads to the failure of iodine
organification in the thyroid, affecting the synthesis of thyroid
hormones, eventually resulting in reduced thyroid function.
Two genes were cloned and named DUOX maturation
factors (DUOXA1 and DUOXA2) in Chicago in 2006. The DUOXA2 gene is
located on chromosome 15 and consists of 6 exons, encoding a 320
amino acid long transmembrane protein. DUOXA2 is mainly expressed
in the thyroid and is weakly expressed in the epithelium of the
digestive tract. DUOXA2 plays an important role in the process of
Duox2 protein migration from the endoplasmic reticulum to the Golgi
apparatus, in the maturation and transition to the apical membrane.
As the DUOXA2 gene determines the normal functionality of Duox2, it
is used as an important starting point for the molecular genetic
study of CH (10).
In the present study, the 6 exons and their flanking
regions of the DUOXA2 gene were subjected to sequence analysis.
This is the first time that heterozygous mutations were found to
occur simultaneously on the 4th and 5th exons. There was a
heterozygous insertion of an A base between 413–414 bp on the 4th
exon, generating a premature TAA stop codon and resulting in the
early termination of protein translation. The C→G mutation on the
5th exon at 738 bp leads to the generation of a premature stop
codon (TAG). Screening of the 4th and 5th exons in the patient’s
parents and in the 100 healthy controls revealed that the patient’s
father is the carrier of the 413–414 bp insertion and the patient’s
mother is the carrier of the 738 bp C→G mutation; these two
mutations were not found in the normal controls. This pattern is in
line with the recessive inheritance law, thus we consider that this
compound heterozygous mutation is the cause of the disease in this
patient. The insertion mutation on the 4th exon of 413–414 bp was
not found in the NCBI SNP database, therefore this is a novel
finding. Our finding expands the mutation spectrum of the
DUOXA2 gene.
The mutation on the 5th exon of 738 bp was an SNP
locus (rs4774518) found in the NCBI database. It has been indicated
that two mutations, the C→G and the C→T mutation, exist in this
SNP. Among the 47 patients and the 100 normal controls in this
study, we found a C→G heterozygous mutation in one patient only,
and it was not found in the 100 healthy controls. The C→T mutation
was found in patients and healthy controls, including 4 cases of
patients (4/47, 8.5‰) and 11 cases of healthy controls (11/100,
11‰). The difference between the two groups is not statistically
significant (P>0.05), consistent with what was reported by
Zamproni et al (11). In
addition, a C→G homozygous mutation on the 3rd exon of 278 bp in
the DUOXA2 gene was found in 3 patients and in 9 healthy
controls. It was verified as another SNP (rs2576090) in the NCBI
database. Although the mutation led to a CGT→GGT change with a
corresponding amino acid change, the present study suggests that
the mutation frequency between patients and healthy controls was
not statistically different, therefore we do not consider this
mutation the cause of CH.
Of note, no mutations were found at the 1st and 2nd
exons of the DUOXA2 gene. Only a point mutation was found in
the flanking intron regions in 2 patients, indicating that segment
of the sequence is relatively stable. Heterozygous or homozygous
single nucleotide substitution polymorphisms were found at the 4th
and 5th intron regions in 11 patients, at different sites. Whether
the polymorphisms affect the RNA transcription process and lead to
genetic instability of the DUOXA2 gene requires further
study.
In conclusion, the present study reports a novel
c.413–414insA (Y138X) mutation for CH, which expands the mutational
spectrum of the DUOXA2 gene. The potential function of this novel
mutation requires further investigation.
References
|
1.
|
Park SM, Clifton-Bligh RJ, Betts P and
Chatterjee VK: Congenital hypothyroidism and apparent athyreosis
with compound heterozygosity or compensated hypothyroidism with
probable hemizygosity for inactivating mutations of the TSH
receptor. Clin Endocrinol (Oxf). 60:220–227. 2004. View Article : Google Scholar
|
|
2.
|
Targovnik HM, Esperante SA and Rivolta CM:
Genetics and phenomics of hypothyroidism and goiter due to
thyroglobulin mutations. Mol Cell Endocrinol. 322:44–55. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
3.
|
Vaisman M, Rosenthal D and Carvalho D:
Enzymes involved in thyroid iodide organification. Arq Bras
Endocrinol Metabol. 48:9–15. 2004.(In Portuguese).
|
|
4.
|
Vigone MC, Fugazzola L, Zamproni I, et al:
Persistent mild hypothyroidism associated with novel sequence
variants of the DUOX2 gene in two siblings. Hum Mutat. 26:3952005.
View Article : Google Scholar : PubMed/NCBI
|
|
5.
|
De Deken X, Wang D, Dumont J, et al:
Characterization of ThOX proteins as components of the thyroid
H(2)O(2)-generating system. Exp Cell Res. 273:187–196.
2002.PubMed/NCBI
|
|
6.
|
Moreno JC, Bikker H, Kempers MJ, et al:
Inactivating mutations in the gene for thyroid oxidase 2 (THOX2)
and congenital hypothyroidism. N Engl J Med. 347:95–102. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
7.
|
Varela V, Rivolta CM, Esperante SA, et al:
Three mutations (p.Q36H, pG418fsX482, and gIVS19-2A>C) in the
dual oxidase 2 gene responsible for congenital goiter and iodide
organification defect. Clin Chem. 52:182–191. 2006.PubMed/NCBI
|
|
8.
|
Hoste C, Rigutto S, Van Vliet G, et al:
Compound heterozygosity for a novel hemizygous missense mutation
and a partial deletion affecting the catalytic core of the
H2O2-generating enzyme DUOX2 associated with
transient congenital hypothyroidism. Hum Mutat. 31:E1304–E1319.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9.
|
Pfarr N, Korsch E, Kaspers S, et al:
Congenital hypothyroidism caused by new mutations in the thyroid
oxidase 2 (THOX2) gene. Clin Endocrinol (Oxf). 65:810–815. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10.
|
Grasberger H and Refetoff S:
Identification of the maturation factor for dual oxidase. Evolution
of an eukaryotic operon equivalent. J Biol Chem. 281:18269–18272.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
11.
|
Zamproni I, Grasberger H, Cortinovis F, et
al: Biallelic inactivation of the dual oxidase maturation factor 2
(DUOXA2) gene as a novel cause of congenital hypothyroidism. J Clin
Endocrinol Metab. 93:605–610. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12.
|
Knobel M and Medeiros-Neto G: An outline
of inherited disorders of the thyroid hormone generating system.
Thyroid. 13:771–802. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13.
|
Zhu W, Wang Z, Chen H, et al: Analysis of
four-year congenital hypothyroidism neonatal screening in Jiudi
City, Fujian Province. Zhong Guo You Sheng You Yu Za Zhi. 12:97–98.
2005.
|