Introduction
The type I interferon (IFN) receptor consists of 2
subunits, IFN-α receptor 1 (IFNAR1) and IFNAR2, which belong to the
type II cytokine receptor superfamily (1). This heterodimeric complex is able to
interact with IFN-α and IFN-β, resulting in the phosphorylation of
Tyk2 and Jak1 that are bound to IFNAR1 and IFNAR2, respectively
(2). Subsequently, the Stat
proteins, Stat1 and Stat2 are phosphorylated at specific tyrosine
residues, which allows the 2 proteins to form a Stat1/2 heterodimer
based on SH2/phosphotyrosine interactions. The formation of this
heterodimer facilitates its association with IFN regulatory factor
(IRF)9 to form an active heterotrimeric transcription factor called
IFN-stimulated gene factor (ISGF)3. ISGF3 targets specific
sequences such as IFN-stimulated response element and
IFN-γ-activated sequence in the promoters of IFN-stimulated genes,
leading to the establishment of antiviral status (3).
After type I IFN binds to its cognate type I IFN
receptor, the receptor is downregulated by endocytosis mediated by
cargo-specific clathrin machinery, and degraded via the lysosomal
and proteosomal pathways in order to limit the magnitude and
duration of IFN signaling (4,5).
The adaptin protein 2 complex, a component of the cargo-specific
clathrin machinery recognizes the Tyr-based endocytic motif of
IFNAR1, leading to the efficient endocytosis of IFNAR1 (5). The Skip, Cullin, F-box containing
complex β-TrCP E3 ubiquitin ligase mediates the ubiquitination of
IFNAR1 in a phosphorylation-dependent manner, eventually
designating IFNAR1 for lysosomal degradation (5). Catalytic activation of Tyk2 is
required for these events but is not essential for IFNAR1
internalization (6). Conversely,
it has been also reported that Tyk2 is essential for the stable
cell surface expression of IFNAR1 and stabilizes IFNAR1 by its
interaction in the basal condition (in the absence of ligand)
(7). Further studies have
revealed that binding of Tyk2 in the proximity of the Tyr-based
linear motif of IFNAR1 is required to prevent IFNAR1
internalization and to maintain its cell surface expression by
physically shielding the Tyr-based motif from recognition by AP2, a
component of the endocytic cargo machinery (8).
The human hepatitis B virus (HBV) induces acute and
chronic hepatitis and is closely associated with the incidence of
human liver cancer (9). Among the
4 proteins that are derived from the HBV genome, the hepatitis B
virus X (HBX) protein is involved in multiple signaling pathways
associated with cell survival and proliferation. Cell signal
transduction pathways that are activated by HBX include the
Jak1/Stat3, PI-3 kinase pathways (10–13), and the Ras/Raf/MAPK signaling
cascade which leads to NF-κB activation (14,15). HBX expression also increases
reactive oxygen species via calcium signaling and cellular kinases,
resulting in the activation of transcription factors NF-κB and
Stat3 (10). Studies have
revealed that HBV-induced oxidative stress also stimulates the
translocation of Raf-1. Src inhibitors or a dominant negative PAK
mutant abolishes HBX-mediated Raf-1 mitochondrial translocation
(16). Recently, we have shown
that HBX-mediated up-regulation of Foxo4 plays a critical role in
the prevention of oxidative stress-induced apoptosis in a liver
cell line (17).
Based on our observation that HBX induces the
production of type I IFN by the activation of Stat1 (18), we believed it is likely that
secreted type I IFN from HBX-expressing hepatic cells enforces
antiviral signals through its binding to the cognate type I IFN
receptor. We initiated this study to investigate how HBX-expressing
hepatic cells overcome this unfavorable situation. Here, we
reported that HBX expression downregulates type I IFN receptor,
leading to disturbance of extracellular type I IFN signaling.
Materials and methods
Cell cultures, reagent and
antibodies
Chang cells, Chang cells stably expressing vector
(Chang-Vec), Chang cells stably expressing HBX (Chang-HBX), and HEK
293 T cells were cultured in DMEM supplemented with 10% FBS and 1%
penicillin and streptomycin. IFN-α was purchased from R&D
Systems (Minneapolis, MN). Anti-IFNAR1 and anti-IFNγR and rabbit
polyclonal anti-c-Myc antibodies were purchased from Abcam
(Cambridge, MA) and β-tubulin antibodies were obtained from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA). Antibodies against Tyk2
and Jak1 were acquired from Cell Signaling (Danvers, MA) and
anti-Flag antibody was obtained from Sigma-Aldrich (St. Louis,
MO).
siRNA transfection
Cells were trypsinized and incubated overnight to
achieve 60–70% confluence before siRNA transfection. Tyk2 siRNA (60
nM, sense 5′-UCUCACCUCUUCC CAUUCC(dTdT)-3′ and antisense
5′-GGAAUGGGAAGAGGU GAGA(dTdT)-3′) purchased from Bioneer (Daejeon,
Korea) or control siRNA (19)
were mixed with Lipofectamine 2000 (Invitrogen, Carlsbad, CA). The
cells were incubated with the transfection mixture for 6 h and then
rinsed with DMEM containing 10% serum. The cells were incubated for
48 h before harvest.
Western blotting
Cells were harvested and treated with lysis buffer
(150 mM NaCl, 1% NP-40, 50 mM Tris-HCl pH 7.5) containing 0.1 mM
Na2VO3, 1 mM NaF and protease inhibitors
(Sigma-Aldrich). For immunoblotting, proteins from whole cell
lysates were resolved by 10 or 12% SDS-PAGE and then transferred to
nitrocellulose membranes. Primary antibodies were used at 1:1,000
or 1:2,000 dilutions, and secondary antibodies conjugated with
horseradish peroxidase were used at 1:2,000 dilutions in 5% nonfat
dry milk. After a final wash, nitrocellulose membranes were exposed
for an enhanced chemiluminescence assay using LAS 3000 (Fuji,
Tokyo, Japan).
Reverse transcription-polymerase chain
reaction (RT-PCR) analysis
Total-RNA was extracted from the cells using the
RNeasy micro kit (Qiagen, Valencia, CA) in accordance with the
manufacturer’s instructions. Three micrograms of total RNA were
converted to cDNA using Superscript II reverse transcriptase
(Invitrogen), and PCR was performed using specific primers
described elsewhere (20). The
cDNAs of each sample were diluted, and PCR was run at the optimized
cycle number. β-actin mRNA was measured as an internal standard.
After amplification, the products were subjected to electrophoresis
on 1.5% agarose and detected by ethidium bromide staining.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde for 15
min, permeabilized with cold acetone for 15 min, blocked with 10%
goat serum for 30 min, and reacted with a 1:100-diluted primary
antibody for 30 min at room temperature. After incubation, the
cells were washed extensively with PBS, incubated with a
1:500-diluted Alexa Fluor 680-conjugated goat anti-rabbit IgG
antibody (Molecular Probes, Eugene, OR), or with a 1:500-diluted
Alexa Fluor 514-conjugated goat anti-mouse IgG antibody (Molecular
Probes) in PBS for 30 min at room temperature, and then washed 3
times with PBS. The stained cells were mounted with PBS containing
10% glycerol and photographed using a LSM510 confocal microscope
(Zeiss, Oberkochen, Germany).
Results
HBX expression induces downregulation of
type I IFN-α receptor 1
We have previously shown that HBX expression mimics
intracellular type I IFN signaling through Stat1 activation in
Chang cells, leading to the production and secretion of type I IFN
into the surrounding microenvironment (18). Although we previously proposed
that HBX may protect HBV-infected hepatic cells from lytic
infection of viruses, we now face a contradiction; the delivery of
an enforced antiviral signal to the cells by the released type I
IFN would exert a disadvantageous effect on HBX-expressing hepatic
cells. How do the HBX-expressing hepatic cells resolve this
detrimental situation? Downregulation of the type I IFN receptor
may well solve this contradiction. To test our hypothesis, we
examined whether the type I IFN receptor consisting of 2 subunits;
IFNAR1 and IFNAR2 is downregulated in the presence of HBX.
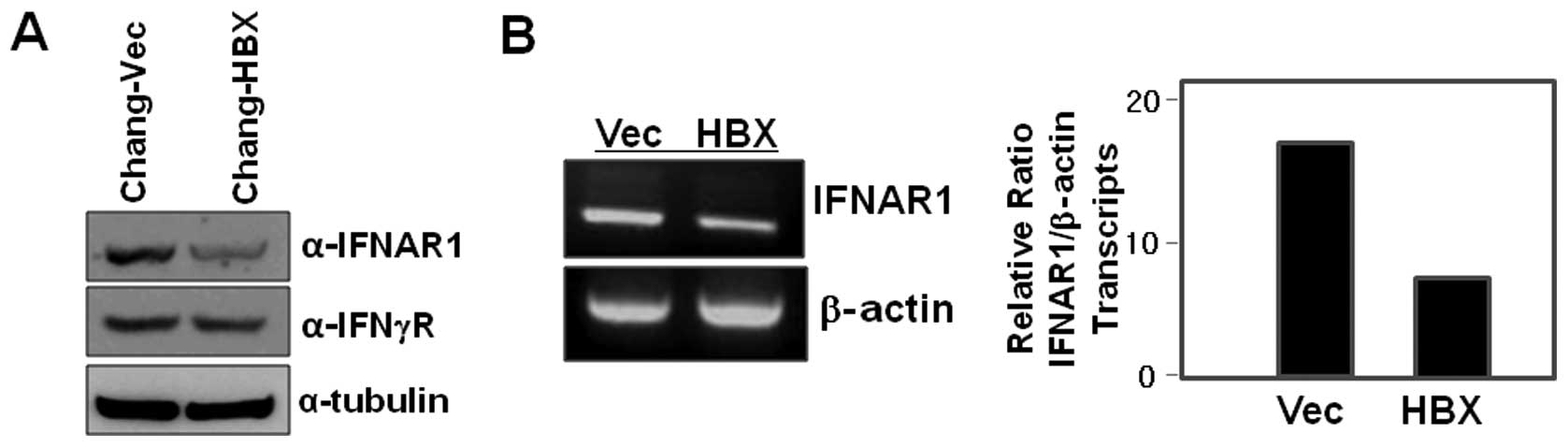
Interestingly, we found that IFNAR1 is less abundant in the cell
lysates of Chang-HBX cells than in those of Chang-Vec cells
(Fig. 1A). We next examined IFN-γ
receptor levels in both Chang-HBX and Chang-Vec cells, and found
that the levels of IFN-γ receptor in Chang-HBX cells are similar to
those in Chang-Vec cells (Fig.
1A).
To explore how IFNAR1 is downregulated in the
presence of HBX (Fig. 1A), we
examined IFNAR1 transcript levels in Chang-Vec and Chang-HBX cells
to assess HBX-mediated transcriptional regulation. The abundance of
IFNAR1 transcripts in Chang-HBX cells was lesser than that in
Chang-Vec cells (Fig. 1B). This
indicates that HBX might play an important role in the
transcriptional regulation of IFNAR1.
HBX induces translocation of IFNAR1 into
the cytoplasm
In addition to the HBX-mediated decrease in IFNAR1
expression, another possible mechanism for the efficient blockage
of type I IFN receptor-mediated antiviral signaling is
translocation of the IFN receptor into the cytoplasm. To test this
theory, we attempted to examine IFNAR1 localization after staining
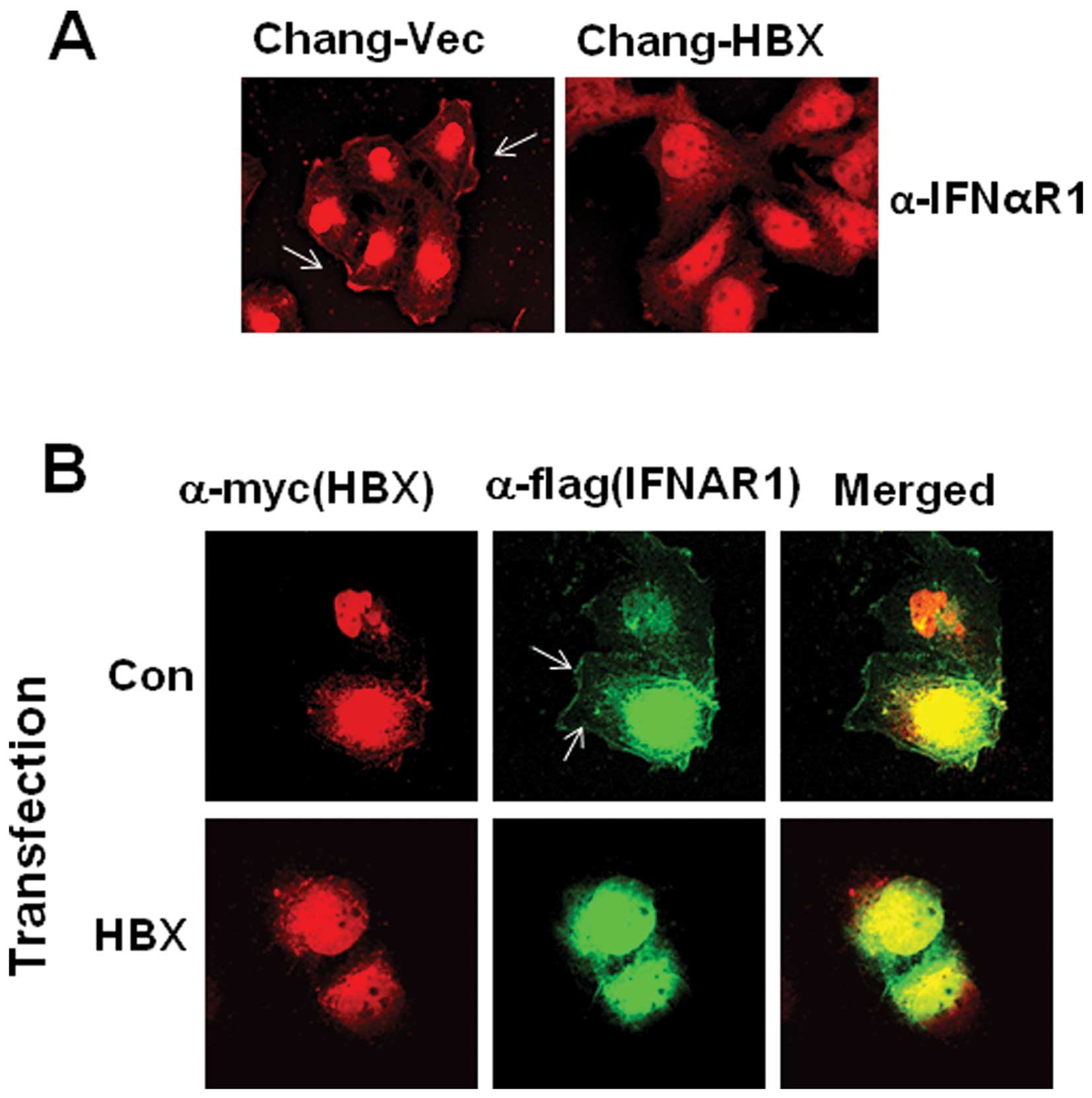
using confocal microscopy. We found that IFNAR1 is localized in the
cytoplasm of Chang-HBX cells, but preferentially localized in the
plasma membrane of Chang-Vec cells (Fig. 2A). To confirm the cytosolic
localization of IFNAR1 in the presence of HBX, exogenous IFNAR1
tagged with a Flag epitope (IFNAR1-Flag) was employed with a
Myc-tagged HBX expression vector. IFNAR1-Flag was detected in the
cytosol rather than in the membrane in the presence of HBX,
similarly to endogenous IFNAR1 in Chang-HBX cells (Fig. 2B). On the other hand, in the
absence of HBX, exogenous IFNAR1 in Chang cells was found in the
plasma membrane similar to endogenous IFNAR1 in Chang-Vec cells
(Fig. 2B).
Decrease of Tyk2 mediated by HBX
diminishes IFNAR1 levels
Since previous studies have shown that Tyk2 is
essential for the stable cell surface expression of IFNAR1 and
stabilizes IFNAR1 by its interaction in the basal condition (in the
absence of ligand) (7), we
examined the expression levels of Tyk2 in Chang-Vec and Chang-HBX
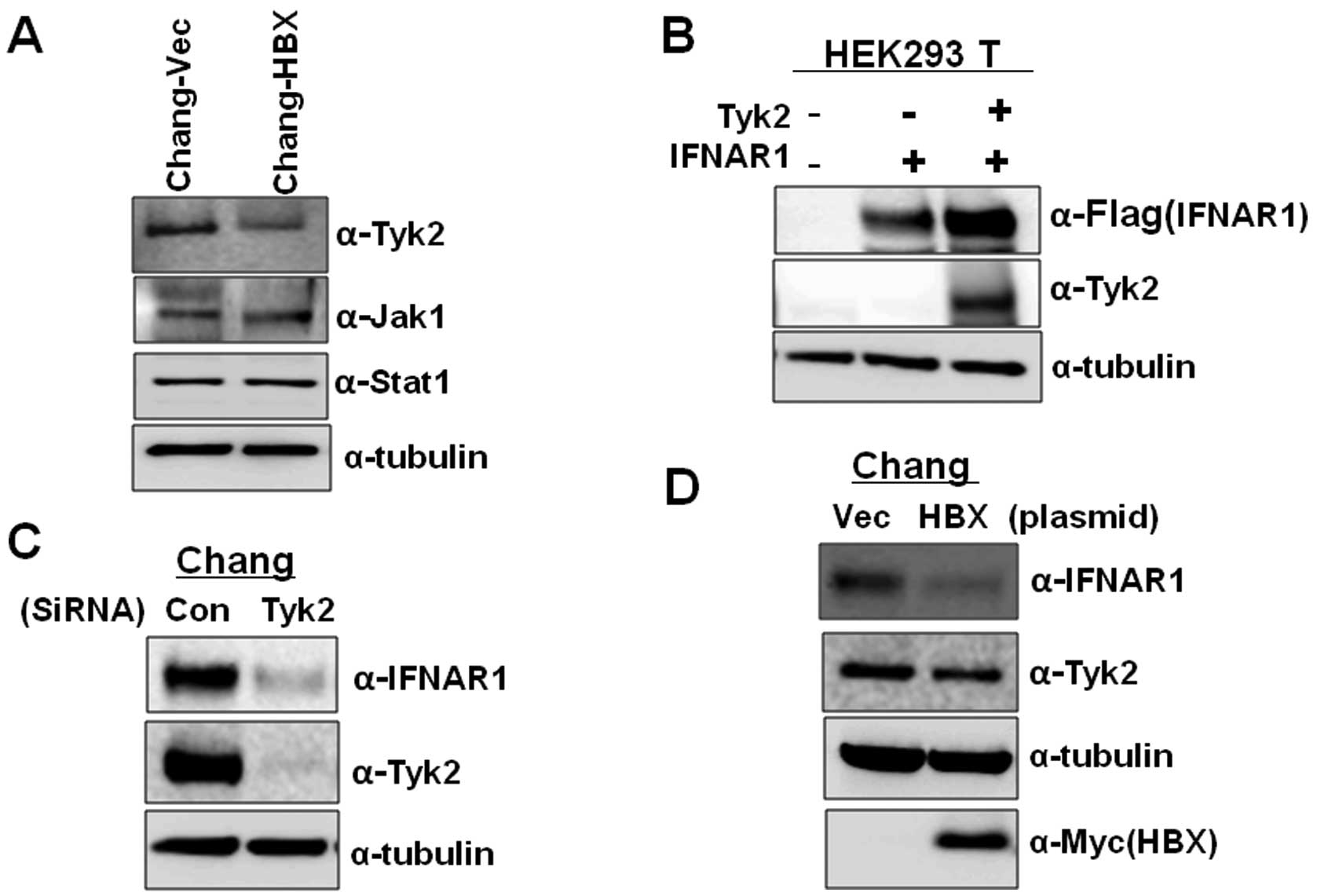
cells. The Chang-HBX cells exhibited a significantly lower
abundance of Tyk2 than Chang-Vec cells (Fig. 3A). However, the levels of Jak1
were similar in both cells. Similar protein quantities of Stat1,
which is associated with the IFN signaling pathway, were also found
in both cells although highly activated Stat1 was found in
Chang-HBX cells (18). To confirm
that the level of Tyk2 determines the IFNAR1 protein level, we
transiently expressed IFNAR1 alone or together with Tyk2 in HEK
293T cells. As seen in Fig. 3B,
compared to the expression of IFNAR1 only, co-expression of IFNAR1
with Tyk2 enhanced the level of IFNAR1 expression. We also examined
the effect of reduced expression of Tyk2 on IFNAR1 protein levels
in Chang cells. When siRNA against Tyk2 was introduced into Chang
cells expressing endogenous IFNAR1, expression of IFNAR1 was
significantly reduced than that in Chang cells treated with control
siRNA (Fig. 3C). To confirm that
the presence of HBX downregulates the level of Tyk2 protein, which
results in lower IFNAR1 expression, we transiently introduced HBX
into Chang cells. We found that HBX downregulates the expression of
Tyk2, leading to a decrease in IFNAR1 expression while control
vector fails to decrease the Tyk2 level (Fig. 3D). These results indicate that HBX
also modulates IFNAR1 expression via Tyk2.
IFNAR1 does not function normally in
Chang-HBX cells during IFN-α signaling
We have shown that the presence of HBX suppresses
IFNAR1 transcription directly (Fig.
1B), and downregulates IFNAR1 expression via Tyk2 in the
absence of its ligand; IFN-α (Fig.
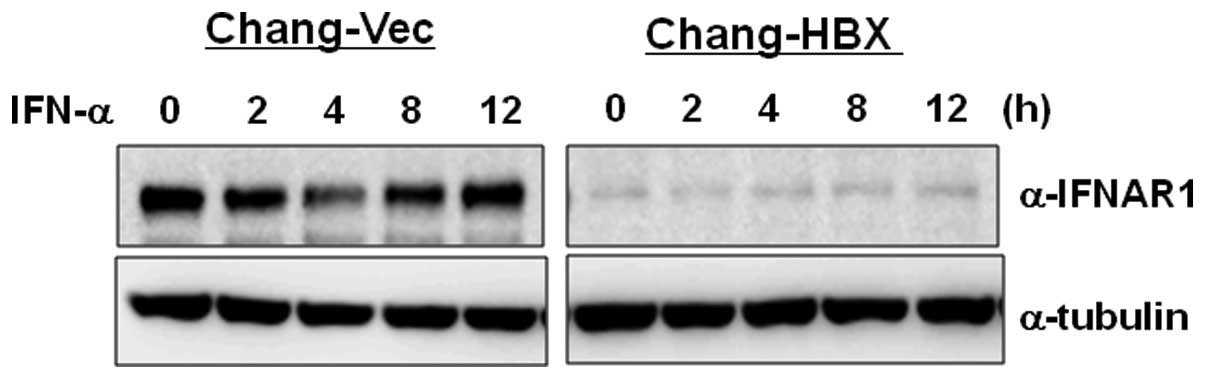
3). To examine how IFNAR1 responds to type I IFN in Chang-Vec
and Chang-HBX cells, both cell lines were treated with IFN-α for 12
h. IFNAR1 levels were reduced at 4 h post-treatment and then
returned to original levels at 8 h post-treatment in Chang-Vec
cells (Fig. 4). However, the
reduced IFNAR1 level was not altered during IFN-α signaling for 12
h in Chang-HBX cells. This result indicates that IFNAR1 may not
function normally in Chang-HBX cells during IFN-α signaling.
Discussion
HBV infection afflicts more than 400 million people
worldwide and accelerates the development of hepatocellular
carcinoma (21). Regarding the
role of HBX-mediated type I IFN production, we initially proposed
that type I IFN inhibits super-infection by the virus, protecting
the host and eventually maintaining chronic infection of HBV
(18). We additionally propose
that type I IFN mediated by HBX may play a role in inflammation
which is believed to be closely related to liver carcinogenesis on
the basis of mounting evidence from preclinical and clinical
studies that persistent inflammation functions as a driving force
in the development of cancer (22,23). However, the autocrine or paracrine
effects of the released type I IFN from HBV-infected hepatic cells
are yet unknown. Type I IFN enforces antiviral status through its
binding to the cognate receptor; how do the HBV-infected hepatic
cells respond to this unfavorable condition? In this study, we
report the answer to this question; HBX downregulates IFNAR1,
leading to avoidance of extracellular type I IFN-mediated
signaling.
Several viruses have evolved strategies of immune
evasion to impair type I IFN signaling pathways. The E6 protein of
human papillomavirus (HPV) 18 has been shown to selectively
interact with Tyk2 and block its activation (24). Japanese encephalitis virus
infection also selectively impairs Tyk2 phosphorylation and West
Nile virus infection hinders the phosphorylation of both Tyk2 and
Jak1 (25,26); however, the mediators of this
blockade are unknown in both cases. Other viruses affect immune
escape by causing a blockage of IFN signaling at the level of Stat
activation. The V protein of Sendai virus 5 and other
paramyxoviruses target the Stat protein for degradation and the
Sendai virus C protein interferes with Stat phosphorylation
(27,28). In addition, the E7 protein of HPV
impairs the assembly of IRF9 and the E1A protein of adenovirus
impedes the interaction of Stat1 with transcriptional machinery
(29,30). Recently, the hepatitis C virus has
been shown to interfere with Stat1 activation by up-regulation of
protein phosphatase 2A and the RIF protein of Kaposi’s sarcoma
associated herpesvirus forms inhibitory complexes with several
proteins including Tyk2, Jak1, Stat1 and Stat2, leading to the
inhibition of Stat1 and Stat2 (31,32). Reviewing these lines of evidence,
downregulation of Tyk2 mediated by HBX is a unique mechanism
different from dephosphorylation of Tyk2 in Chang cells and,
eventually leads to cytosolic localization of IFNAR1. In this
study, we provide evidence of a novel mechanism for the modulation
of type I IFN signaling by the downregulation of Tyk2. Related to
regulation via Tyk2, another study has reported that expression of
SHP-1 is diminished or abolished in most lymphoma cell lines and in
some colorectal cancer (33–35). Conversely, transient expression of
SHP-1 inhibits tumor cell growth via downregulation of Jak1 and
Tyk2 (33). Our future study will
be directed towards exploring whether protein phosphatases
including SHP-1 are associated with the specific degradation of
Tyk2. Type I IFN signaling pathway involves the binding of type I
IFN to its receptor, and then phosphorylation of Jak1 and Tyk2
takes place followed by the activation of Stat1. We herein suggest
that the phosphorylation of Stat1 is independent of Tyk2 at least
in the Chang-HBX cells.
In addition, we also observed that IFNAR1 is
regulated at the transcriptional level. It is possible that HBX
induces a transcription factor with suppressor activity to bind to
the promoter of IFNAR1 as seen in the case of PTEN promoter, which
is occupied by the p53 tumor suppressor in the presence of HBX
(36). HBX might cause an
instability in IFNAR1 mRNA by inducing the release of a RNA-binding
protein such as HuR from the 3′ untranslated region (37). Moreover, HBX may induce
methylation at a G/C region in the IFNAR1 promoter via the
upregulation of DNA methyltransferase (DNMT) 1 activity; HBX has
similarly been reported to activate DNMT1, resulting in suppression
of p16(INK1a), a cyclin-dependent kinase inhibitor through
hypermethylation of the p16(INK1a) promoter (38). The detailed mechanism of
HBX-mediated suppression of IFNAR1 at the transcriptional level
remains under investigation.
Acknowledgements
This study was supported by the Korea Research
Foundation (KRF-2008-313-E00113) and the World Class University
Program (R31-2008-000-20004-0) through NRF funded by the Korean
government.
References
|
1
|
B Payelle-BrogardS PellegriniBiochemical
monitoring of the early endocytic traffic of the type I interferon
receptorJ Interferon Cytokine
Res308998201010.1089/jir.2009.004420028207
|
|
2
|
TC YehS PellegriniThe Janus kinase family
of protein tyrosine kinases and their role in signalingCell Mol
Life Sci5515231534199910.1007/s00018005039210526570
|
|
3
|
GR StarkIM KerrBR WilliamsRH SilvermanRD
SchreiberHow cells respond to interferonsAnnu Rev
Biochem67227264199810.1146/annurev.biochem.67.1.2279759489
|
|
4
|
JS BonifacinoLM TraubSignals for sorting
of transmembrane proteins to endosomes and lysosomesAnnu Rev
Biochem72395447200310.1146/annurev.biochem.72.121801.16180012651740
|
|
5
|
KG KumarH BarriereCJ CarboneJ LiuG
SwaminathanP XuY LiDP BakerJ PengGL LukacsSY FuchsSite-specific
ubiquitination exposes a linear motif to promote interferon-alpha
receptor endocytosisJ Cell
Biol179935950200710.1083/jcb.20070603418056411
|
|
6
|
J LiuA PlotnikovA BanerjeeKG Suresh KumarJ
RagimbeauZ MarijanovicDP BakerS PellegriniSY
FuchsLigand-independent pathway that controls stability of
interferon alpha receptorBiochem Biophys Res
Commun367388393200810.1016/j.bbrc.2007.12.13718166147
|
|
7
|
J RagimbeauE DondiA AlcoverP EidG UzeS
PellegriniThe tyrosine kinase Tyk2 controls IFNAR1 cell surface
expressionEMBO J22537547200310.1093/emboj/cdg03812554654
|
|
8
|
KG KumarB VargheseA BanerjeeDP BakerSN
ConstantinescuS PellegriniSY FuchsBasal ubiquitin-independent
internalization of interferon alpha receptor is prevented by
Tyk2-mediated masking of a linear endocytic motifJ Biol
Chem2831856618572200810.1074/jbc.M800991200
|
|
9
|
DH NguyenL LudgateJ HuHepatitis B
virus-cell interactions and pathogenesisJ Cell
Physiol216289294200810.1002/jcp.2141618302164
|
|
10
|
G WarisKW HuhA SiddiquiMitochondrially
associated hepatitis B virus X protein constitutively activates
transcription factors STAT-3 and NF-kappa B via oxidative stressMol
Cell Biol2177217730200110.1128/MCB.21.22.7721-7730.2001
|
|
11
|
AS KekuleU LauerL WeissB LuberPH
HofschneiderHepatitis B virus transactivator HBx uses a tumour
promoter signalling
pathwayNature361742745199310.1038/361742a08441471
|
|
12
|
YH LeeY YunHBx protein of hepatitis B
virus activates Jak1-STAT signalingJ Biol
Chem2732551025515199810.1074/jbc.273.39.255109738022
|
|
13
|
YI LeeS Kang-ParkSI DoYI LeeThe hepatitis
B virus-X protein activates a phosphatidylinositol
3-kinase-dependent survival signaling cascadeJ Biol
Chem2761696916977200110.1074/jbc.M01126320011278872
|
|
14
|
P ChirilloM FalcoPL PuriM ArtiniC BalsanoM
LevreroG NatoliHepatitis B virus pX activates NF-kappa B-dependent
transcription through a Raf-independent pathwayJ
Virol7064164619968523586
|
|
15
|
H KimYH LeeJ WonY YunThrough induction of
juxtaposition and tyrosine kinase activity of Jak1, X-gene product
of hepatitis B virus stimulates Ras and the transcriptional
activation through AP-1, NF-kappaB, and SRE enhancersBiochem
Biophys Res Commun286886894200110.1006/bbrc.2001.549611527382
|
|
16
|
J ChenA SiddiquiHepatitis B virus X
protein stimulates the mitochondrial translocation of Raf-1 via
oxidative stressJ
Virol8167576760200710.1128/JVI.00172-0717428866
|
|
17
|
R SrisutteeSS KohEH ParkIR ChoHJ MinBH
JhunDY YuS ParkY Park doMO LeeUpregulation of Foxo4 mediated by
hepatitis B virus X protein confers resistance to oxidative
stress-induced cell deathInt J Mol Med28255260201121567078
|
|
18
|
EH ParkSS KohR SrisutteeIR ChoHJ MinBH
JhunYS LeeKL JangCH KimRN JohnstonYH ChungExpression of HBX, an
oncoprotein of hepatitis B virus, blocks reoviral oncolysis of
hepatocellular carcinoma cellsCancer Gene
Ther16453461200910.1038/cgt.2008.9519096445
|
|
19
|
IR ChoS JeongBH JhunWG AnB LeeYT KwakSH
LeeJU JungYH ChungActivation of non-canonical NF-kappaB pathway
mediated by STP-A11, an oncoprotein of Herpesvirus
saimiriVirology3593745200710.1016/j.virol.2006.09.00117028057
|
|
20
|
H IdeT NakagawaY TeradoY KamiyamaS MutoS
HorieTyk2 expression and its signaling enhances the invasiveness of
prostate cancer cellsBiochem Biophys Res
Commun369292296200810.1016/j.bbrc.2007.08.16017920038
|
|
21
|
PM Mulrooney-CousinsTI MichalakPersistent
occult hepatitis B virus infection: experimental findings and
clinical implicationsWorld J
Gastroenterol1356825686200710.3748/wjg.v13.i43.568217963292
|
|
22
|
SI GrivennikovFR GretenM KarinImmunity,
inflammation, and
cancerCell140883899201010.1016/j.cell.2010.01.025
|
|
23
|
MG BorrelloD Degl’InnocentiMA
PierottiInflammation and cancer: the oncogene-driven
connectionCancer
Lett267262270200810.1016/j.canlet.2008.03.06018502035
|
|
24
|
S LiS LabrecqueMC GauzziAR CuddihyAH WongS
PellegriniGJ MatlashewskiAE KoromilasThe human papilloma virus
(HPV)-18 E6 oncoprotein physically associates with Tyk2 and impairs
Jak-STAT activation by
interferon-alphaOncogene1857275737199910.1038/sj.onc.120296010523853
|
|
25
|
RJ LinCL LiaoE LinYL LinBlocking of the
alpha interferon-induced Jak-Stat signaling pathway by Japanese
encephalitis virus infectionJ
Virol7892859294200410.1128/JVI.78.17.9285-9294.200415308723
|
|
26
|
JT GuoJ HayashiC SeegerWest Nile virus
inhibits the signal transduction pathway of alpha interferonJ
Virol7913431350200510.1128/JVI.79.3.1343-1350.200515650160
|
|
27
|
D GarcinJB MarqL StrahleP le MercierD
KolakofskyAll four Sendai Virus C proteins bind Stat1, but only the
larger forms also induce its mono-ubiquitination and
degradationVirology295256265200210.1006/viro.2001.134212033784
|
|
28
|
B GotohT KomatsuK TakeuchiJ
YokooParamyxovirus strategies for evading the interferon
responseRev Med Virol12337357200210.1002/rmv.35712410527
|
|
29
|
P BarnardNA McMillanThe human
papillomavirus E7 oncoprotein abrogates signaling mediated by
interferon-alphaVirology259305313199910.1006/viro.1999.977110388655
|
|
30
|
DC LookWT RoswitAG FrickY Gris-AlevyDM
DickhausMJ WalterMJ HoltzmanDirect suppression of Stat1 function
during adenoviral
infectionImmunity9871880199810.1016/S1074-7613(00)80652-49881977
|
|
31
|
V ChristenF DuongC BernsmeierD SunM
NassalMH HeimInhibition of alpha interferon signaling by hepatitis
B virusJ Virol81159165200710.1128/JVI.01292-0617065208
|
|
32
|
SA BissonAL PageD GanemA Kaposi’s
sarcoma-associated herpesvirus protein that forms inhibitory
complexes with type I interferon receptor subunits, Jak and STAT
proteins, and blocks interferon-mediated signal transductionJ
Virol83505650662009
|
|
33
|
C WuQ GuanY WangZJ ZhaoGW ZhouSHP-1
suppresses cancer cell growth by promoting degradation of JAK
kinasesJ Cell Biochem9010261037200310.1002/jcb.1072714624462
|
|
34
|
J ChengD ZhangC ZhouWA
MarascoDownregulation of SHP1 and up-regulation of negative
regulators of JAK/STAT signaling in HTLV-1 transformed cell lines
and freshly transformed human peripheral blood CD4+
T-cellsLeuk Res287182200410.1016/S0145-2126(03)00158-914630083
|
|
35
|
C WuM SunL LiuGW ZhouThe function of the
protein tyrosine phosphatase SHP-1 in
cancerGene306112200310.1016/S0378-1119(03)00400-112657462
|
|
36
|
TW ChungYC LeeJH KoCH KimHepatitis B Virus
X protein modulates the expression of PTEN by inhibiting the
function of p53, a transcriptional activator in liver cellsCancer
Res6334533458200312839924
|
|
37
|
K AbdelmohsenR Pullmann JrA LalHH KimS
GalbanX YangJD BlethrowM WalkerJ ShubertDA GillespiePhosphorylation
of HuR by Chk2 regulates SIRT1 expressionMol
Cell25543557200710.1016/j.molcel.2007.01.01117317627
|
|
38
|
JK JungP AroraJS PaganoKL JangExpression
of DNA methyltransferase 1 is activated by hepatitis B virus X
protein via a regulatory circuit involving the p16INK4a-cyclin
D1-CDK 4/6-pRb-E2F1 pathwayCancer
Res6757715778200710.1158/0008-5472.CAN-07-052917575144
|