Introduction
Degradation and remodeling of extracellular matrix
(ECM) play an important role in both atherosclerosis and aneurysm
disease, two chronic inflammatory diseases (1,2).
Neutrophils and mast cells produce several serine proteases such as
tryptase, chymase, cathepsins and elastase, which are enzymes that
are able to degrade different components of the ECM, a process
involved in both diseases (3–5).
Inhibition of mast cell chymase reduces atherosclerotic plaque
progression and improves plaque stability in Apoe−/−
mice (6) and aneurysmal
dilatation is inhibited in mast cell deficient animals (4,7,8).
Also, neutrophils are major vascular component in atherosclerotic
lesions (9) and an important
source of proteases in the vessel wall of abdominal aortic aneurysm
(AAA) (10). Furthermore,
neutrophil depletion in mice inhibits elastase induced experimental
AAA formation, an effect independent of MMP-2, MMP-8 or MMP-9
(11). The discovery of mediators
of neutrophil recruitment and neutrophil derived factors in
vascular samples, supports the involvement of neutrophils in
vascular inflammatory disease (12).
Serine protease inhibitor A3 (serpinA3) inhibits
several proteases derived from neutrophils and mast cells, such as
human leukocyte elastase, cathepsin G and chymase (13). SerpinA3, also referred to as
α1-antichymotrypsin, was first characterized as an acute phase
plasma protease inhibitor. It is synthesized in a range of tissues
including hepatocytes, bronchial epithelial cells and neuronal
cells (14,15). In mice, the serpinA3 gene has
undergone extensive duplication and diversification resulting in a
family of 13 closely related inhibitors with differing tissue
distribution and protease specificity (16,17). Nevertheless, gene expression and
functional studies suggest that serpinA3n is the closest murine
ortholog of human serpinA3 (13,17).
Because mast cell- and neutrophil-derived proteases
have been implicated in the remodelling of ECM and in the
progression of atherosclerosis and aortic aneurysm formation, we
investigated the putative involvement of serpinA3 in these two
diseases.
Materials and methods
All human and mouse protocols were approved by the
local ethics review board and the study was conducted according to
the Helsinki Declaration.
Human subjects
Patients scheduled for elective surgery for
infrarenal AAA (n=9) at Karolinska University Hospital (Stockholm,
Sweden) where preoperative computer tomography demonstrated an
eccentric intraluminal thrombus were included in the study. The
intima/media and adventitia layers of AAA were separated by
adventicectomy. The Biobank of Karolinska Endarterectomies (BiKE)
study included atherosclerotic tissue collected from asymptomatic
patients and patients with minor stroke or transient ischemic
attack (TIA), undergoing carotid endarterectomy at the Karolinska
University Hospital. Patients were included after informed, written
and signed consent.
Control ascending aorta samples for RNA studies were
obtained from 8 organ donors without clinical or macroscopic signs
of aortic atherosclerosis. Infrarenal control aortic samples for
histology were collected from 14 medicolegal autopsies performed in
the Department of Forensic Medicine, University of Helsinki. The
sections were immediately fixed in 4% formaldehyde for light
microscopy or snap-frozen in liquid nitrogen for RNA isolation. The
use of organ donor and autopsy tissues was approved by The National
Authority for Medicolegal Affairs of Finland.
CaCl2 induced aneurysm model
in mice
CaCl2-induced aneurysm is an inflammatory
driven model and is a frequently used method to experimentally
induce abdominal aortic aneurysm in a controlled fashion (18). Mice were anesthetized with
standard doze of sodium pentobarbital intraperitoneally (6 mg/100
g). The adequacy of anesthesia was monitored by the absence of the
corneal reflex, and adequate levels of anesthesia and analgesia
were ensured with supplemental intraperitoneal injection of
pentobarbital sodium given as required. Body temperature was
maintained using a blanket control unit. A small compress strip
with 0.5 M CaCl2 or NaCl as control was applied to the
isolated abdominal aorta which was covered for 15 min. Then the
compress was removed and the treated area was washed with PBS
twice. Mice were treated with Temgesic (0.1 mg/kg) 30 min before
wakening 2 times/day for 2 days. Operated animals were monitored
daily for any sign of pain or disability. After 2 weeks the mice
were anesthetized with 2% isoflurane over a nose mask, whole blood
was drawn by cardiac puncture and the aorta was removed for further
analysis. Mice were then euthanized with carbon dioxide. The
investigation conforms to the directive 2010/63/EU of the European
Parliament.
Atherosclerotic lesion size
To study the effect of serpinA3n in atherosclerotic
disease development,
Apoe−/− (backcrossed
10 times with C57bl/6) and serpinA3n transgenic mice (on C57bl/6
background) crossed with Apoe−/− were fed a chow
diet and sacrificed at 20 weeks of age. After vascular perfusion
with sterile RNase-free PBS, thoracic aortas and hearts were
dissected and preserved for lesion analysis. Five 10-μm
sections were collected at 100-μm intervals starting at a
100-μm distance from the aortic valves. Formaldehyde-fixed
sections were stained with oil red-O and lesion size was analyzed
using Leica Q500MC image analysis software. For each mouse, a mean
lesion area was calculated from 5 sections, reflecting the
cross-section area covered by atherosclerosis. The descending
thoracic aorta was fixed in 4% formaldehyde, opened longitudinally,
pinned onto black wax plates and stained with Sudan IV (Merck KGaA,
Darmstadt, Germany). The lesion areas and total aortic areas were
calculated using Image J software (NIH, Bethesda, MD, USA).
Cell culture
EA.hy 926 human endothelial cells were maintained in
high-glucose (4.5 g/l) Dulbecco’s modified Eagle’s medium,
supplemented with 10% FCS, sodium bicarbonate solution, 1X HAT
(mixture of sodium hypoxanthine, aminopterin and thymidine)
(Invitrogen Life Technologies), and penicillin-streptomycin. Human
aortic smooth muscle cells (SMCs) were maintained in growth medium
containing supplements (CC-3182, Clonetics, Cambrex, Walkersville,
MD). The cells were used at passages 3–5. All cells were incubated
in 5% CO2 at 37°C and stimulated with lipopolysaccharide
(LPS, 10 μg/ml; Sigma-Aldrich, Stockholm, Sweden), IFN-γ,
IL-1β or TNF-α (20 μg/ml, PeproTech, Inc., London, UK).
Creation of mice transgenic for
serpinA3n
The murine serpinA3n cDNA sequence was
amplified from C57Bl/6 mice and cloned into the chicken β-actin
promoter plasmid pCAGIPuro, in which serpinA3n expression is driven
by a human cytomegalovirus immediate early enhancer (HCMVIEE)
coupled to the chicken β-actin promoter. The sequence of
serpinA3n was confirmed by sequencing. Using DNA
microinjection into C57Bl/6 fertilized oocytes of the β-actin
promoter-HCMVIEE-serpinA3n fragment, we obtained 2
transgene-positive serpinA3n mice. The founder mice were
identified using PCR. Genotyping primers were: F,
5′-GAGGACCTGACCACACCCTA-3′ (exon 3) and R,
5′-TTATCAGGAAAGGCCGATTG-3′ (exon 5). The 2 founder mice were
crossed with C57Bl/6 mice (purchased from Taconic, Denmark).
ELISA of serpinA3
Blood samples were collected in Vacutainer tubes
containing EDTA and then centrifuged to separate plasma, which was
then stored at −70°C until analyzed. EDTA plasma samples were
available from 49 healthy control subjects (69±3.7 years), 40
patients diagnosed with AAA (71±7.4 years) and 28 patients with
myocardial infarction (70±6.2 years). Plasma serpinA3 concentration
was measured using a commercially available ELISA kit (#E-80CYT;
ICL, Inc., Portland, OR, USA). Briefly, EDTA plasma samples were
loaded into the serpinA3 pre-coated ELISA wells and incubated for 1
h. After washing, HRP-conjugated serpinA3 detection antibody was
added, followed by washing and addition of tetramethylbenzidine
substrate. Samples were run in duplicate and the results were
quantified against a standard curve created using the recombinant
standard that was included in the kit.
Protein preparation and western blot
analysis
Aortic tissue from mice was homogenized in ice-cold
lysis buffer containing PBS and proteins in lysates or plasma were
separated under reducing conditions by electrophoresis using 10%
SDS-PAGE (polyacrylamide gel electrophoresis). The blot was
incubated with primary antibodies against murine serpinA3n
(Agrisera, Vännäs, Sweden), overnight and then for 1 h with
horseradish peroxidase-labeled secondary antibody. Signals were
detected with enhanced chemiluminescence western blot detection
reagent (GE Healthcare, Uppsala, Sweden).
Gene expression analysis
Tissue samples were homogenized with FastPrep using
Lysing Matrix D tubes (MP Biomedicals, Illkirch, France). Total-RNA
from tissue or cells in culture was isolated with RNeasy (Qiagen,
Germany) and cDNA (1.0 ng) was amplified by RT-PCR reactions as
described before (19). The
following probe was used for analysis of human serpinA3 expression:
Hs00153674 and murine serpinA3n expression: Mm00776439, and results
were normalized to values of human RPLP0: Hs99999902 or murine β2
microglobulin: Mm00437762. All probes were obtained from Applied
biosystems, Foster City, CA, USA.
The BiKE RNA samples were hybridized and scanned at
the Karolinska Institute Affymetrix core facility. For samples from
the BiKE biobank Affymetrix HG-U133 plus 2.0 arrays and protocols
were used as described before (20).
Immunohistochemistry
Human and mouse abdominal aortas were cryosectioned
and fixed in acetone. Endogenous peroxidase activity was quenched
by treatment with 3% hydrogen peroxide for 5 min followed by
incubation in 5% blocking bovine serum albumin solution. Sections
were then incubated with a polyclonal rabbit anti-human serpinA3
antibody (Abcam) or an isotype control (Abcam) at the same
concentration at 4°C overnight. Consecutive sections were incubated
with rabbit polyclonal anti-human Von Willebrand factor (Dako,
Glostrup, Denmark), mouse monoclonal CD66b (Fitzgerald, North
Acton, MA, USA), mouse monoclonal CD68 (Leica Microsystems,
Newcastle, UK), mouse monoclonal CD163 (Acris, Herford, Germany),
mouse monoclonal CD3 (Santa Cruz Biotechnology, Inc.), mouse
monoclonal mast cell tryptase (Dako) and mouse monoclonal
anti-human α-actin (clone 1A4; Sigma-Aldrich) antibodies at 4°C
overnight followed by secondary biotinylated goat anti-rabbit IgG
or goat anti-mouse IgG (Dako) antibody. Avidin-biotin peroxidase
complexes (Dako) were added, followed by visualization with
3,3′-diaminobenzidine tetrahydrochloride (Dako). All sections were
counterstained with Mayer’s hematoxylin (Histolab Products,
Göteborg, Sweden).
Mast cell chymase activity
For detection of mast cell chymase activity in mouse
serum a chymase assay kit was used according to the manufactures
instruction (Sigma-Aldrich). Briefly, serum was added, followed by
assay buffer and the substrate N-Succinyl-Ala-Ala-Pro-Phe
p-nitroanilide. The absorbance was detected at 405 nm. Serum
without the addition of substrate was used as internal blanking for
each sample.
In situ zymography
In situ zymography was used to analyse
activity of elastase and cathepsin G in mice aortas. Fluorogenic
DQ-substrate for elastin (EnzCheck® Elastase assay kit;
Invitrogen Life Technologies) and cathepsin G
(SensoLyte® 520 cathepsin G assay kit; AnaSpec, Inc.,
Fremont, CA, USA) was mixed with 1% agarose in assay buffer and
DAPI for nuclear localisation and incubated overnight at 37°C.
Fluorescence was detected using a fluorescence microscope.
Statistical analysis
The statistical analysis was performed with the
StatView for Windows software (release 5.0.1; SAS Institute, Inc.,
Cary, NC, USA). P-values <0.05 were considered significant.
Comparisons were done with a Mann-Whitney U test. The Bonferroni
correction was used to adjust for multiple comparisons of
correlations.
Results
SerpinA3 expression in
atherosclerosis
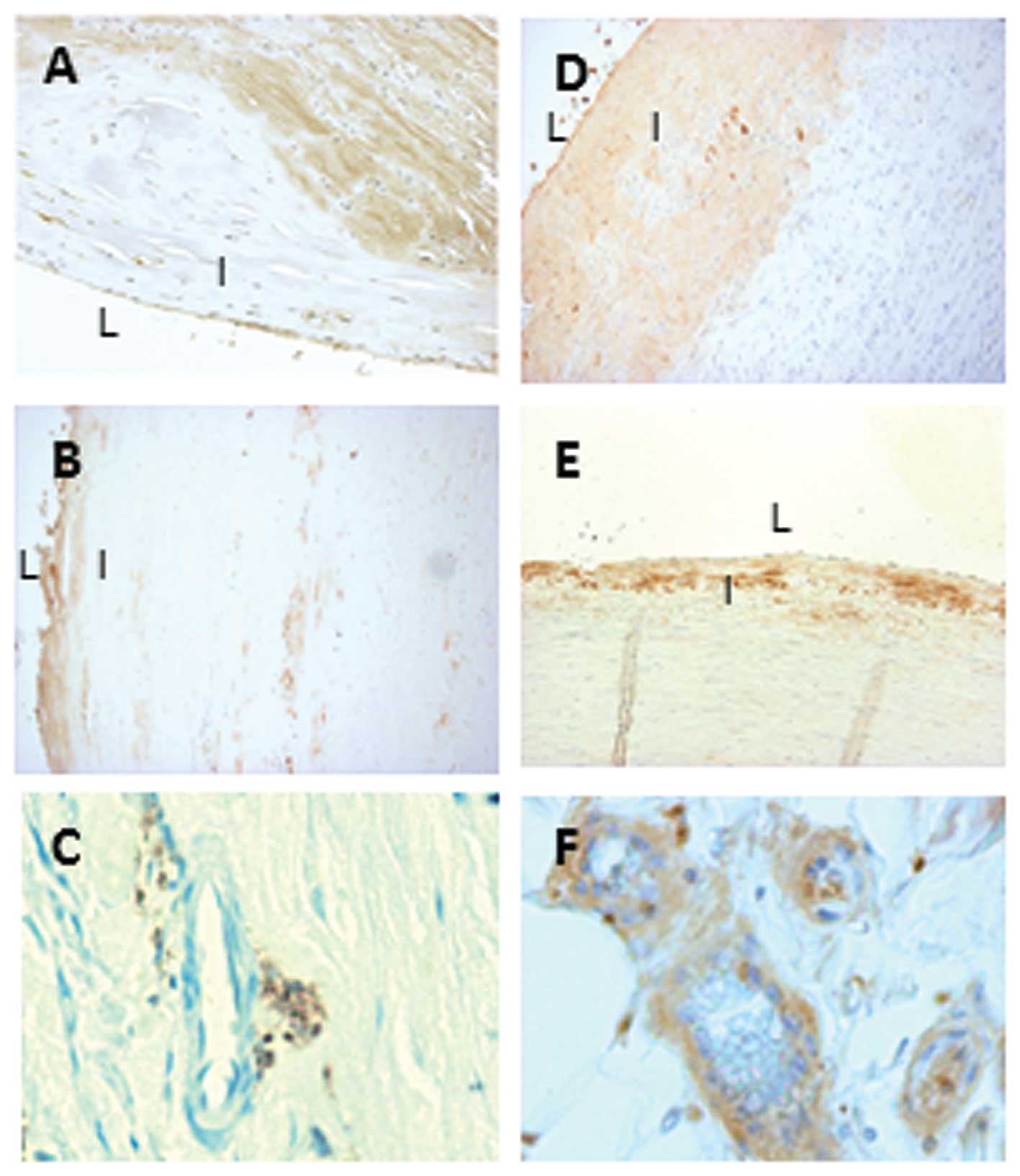
Immunohistochemistry was used to localize serpinA3
expression in human atherosclerotic lesions. As shown in Fig. 1A, serpinA3 was expressed in
endothelial cells and medial smooth muscle cells in the
atherosclerotic lesions. When investigating the mRNA expression of
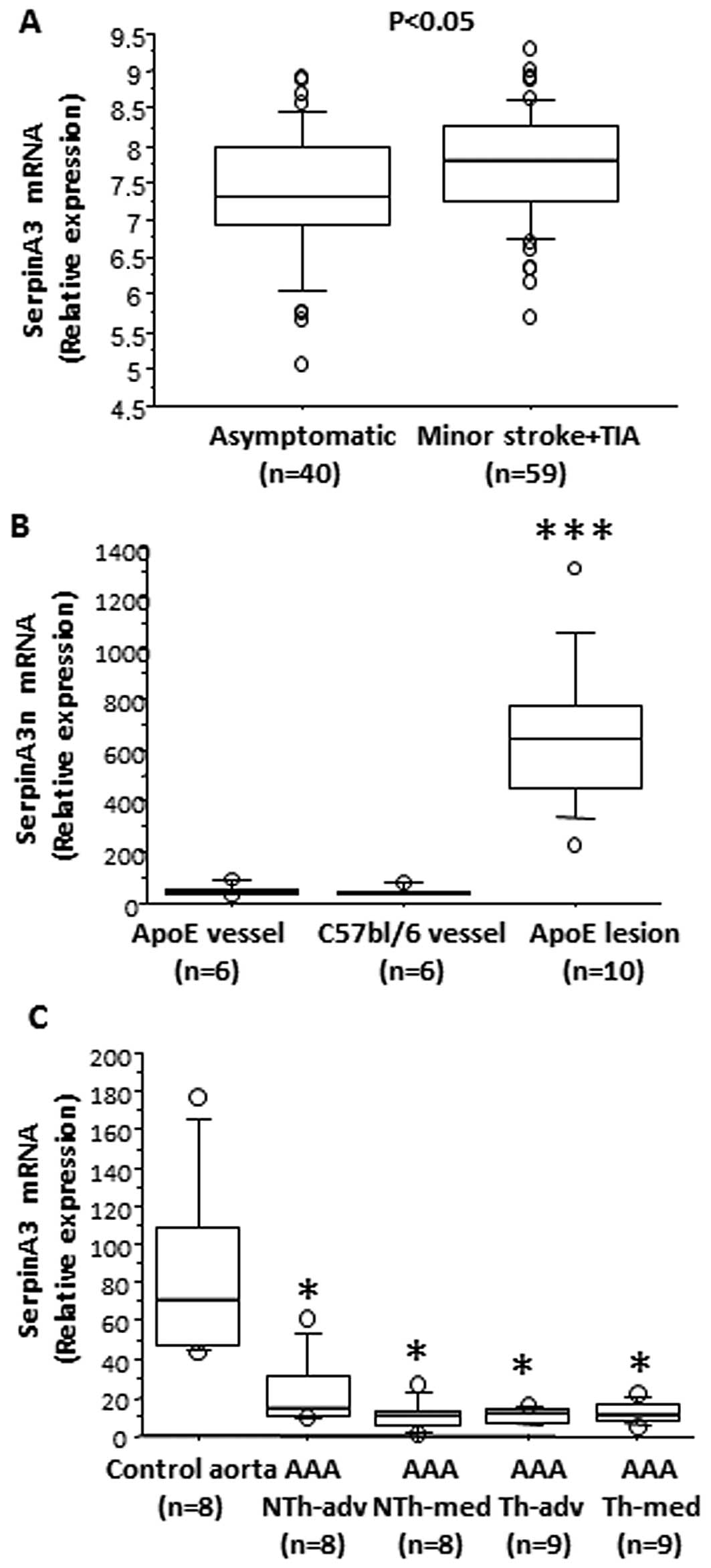
serpinA3 in human carotid lesions from asymptomatic patients and
patients with a minor stroke or TIA we detected a significant
increase in the expression of serpinA3 in the symptomatic patients
(Fig. 2A).
Using gene arrays, we identified serpinA3n as one of
the most induced genes in lesions from Apoe−/−
mice as compared to non-atherosclerotic control vessels. When
verifying this result we observed a 14-fold increase in the
serpinA3n mRNA expression in aortic root lesions from 20-week-old
Apoe−/− mice as compared to aortic vessels without
atherosclerosis from 20-week-old Apoe−/− mice and
from 20-week-old C57bl/6 mice (Fig.
2B).
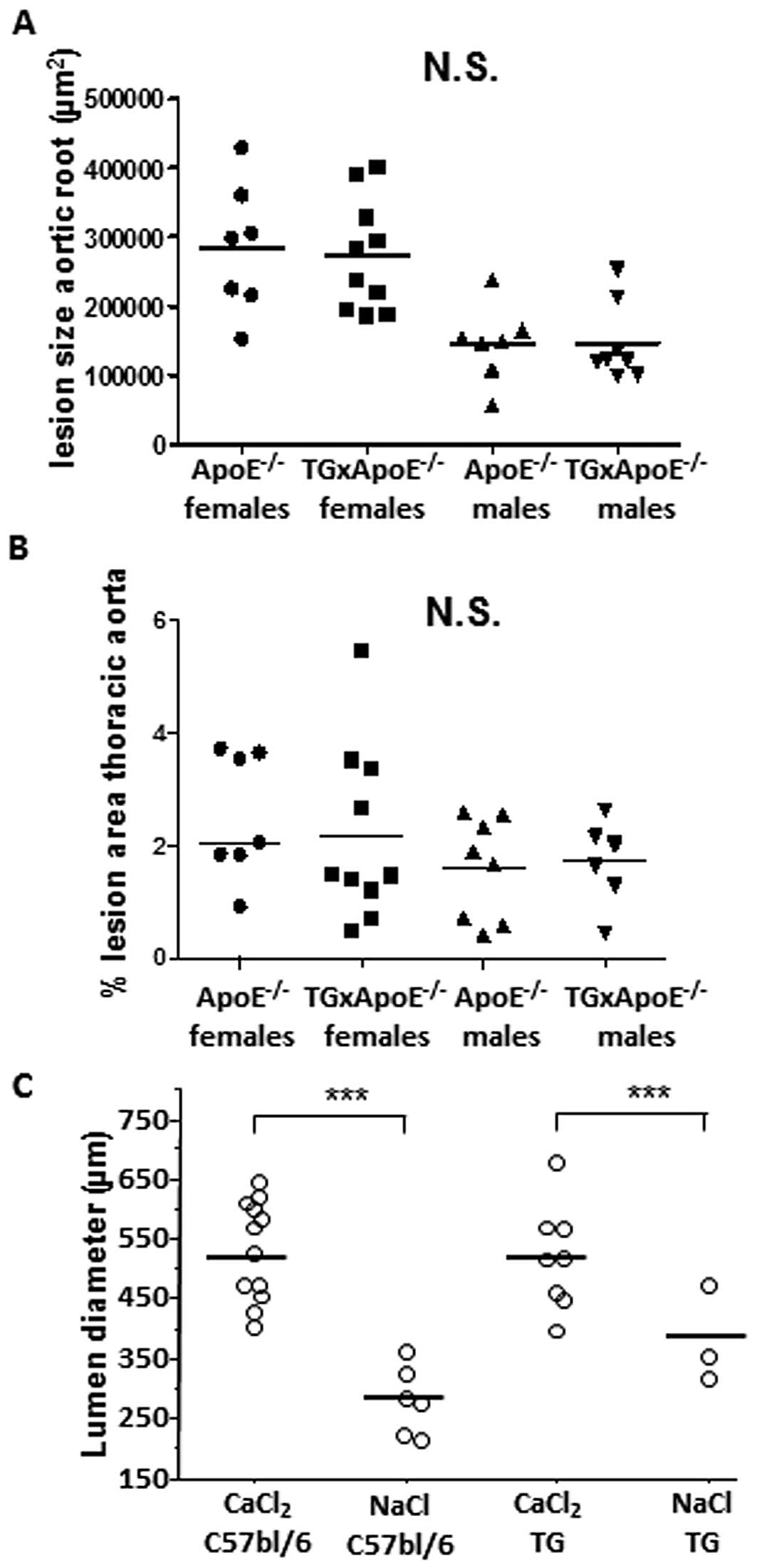
To document the role of serpinA3n in atherosclerosis
we developed transgenic mice overexpressing serpinA3n and crossed
these mice with Apoe−/− mice. In aortas from transgenic
serpinA3n mice, the serpinA3n mRNA level was 10-fold higher
compared to aortas of wild-type C57Bl/6 mice (data not shown).
Also, serpinA3n protein was increased in plasma and abdominal
aortas of transgenic mice compared to wild-type mice, which had
barely detectable serpinA3 expression as examined by western blot
analysis (data not shown). However,
Apoe−/− x serpinA3n+/−
mice did not have any impact on the lesion development compared to
Apoe−/− at 20 week of age (Fig. 3) or inflammatory cell content
(data not shown).
SerpinA3 expression in human AAA
SerpinA3 was expressed in the subendothelial layer
of control aortas (Fig. 1D and
E). There was a marked decrease in the staining of serpinA3 in
AAA samples (Fig. 1B) as compared
with abdominal (Fig. 1D) and
thoracic (Fig. 1E) control aorta.
Furthermore, serpinA3 was also expressed in small vessels present
in the adventitia of the abdominal control aorta (Fig. 1F) but less expressed in small
vessels present in the adventitia of AAA (Fig. 1C). Isotypic IgG served as negative
control and showed no staining (data not shown). Serial sections
were stained for T-cells (CD3), macrophages (CD163 and CD68),
neutrophils (CD66b) and mast cells (tryptase) but did not associate
to serpinA3 staining (data not shown). Similar to the
immunohistological analysis, serpinA3 mRNA expression in human AAA
was significantly decreased by 70–80% in the AAA samples compared
to healthy aorta (Fig. 2C).
Since serpinA3 is also produced by the liver as an
acute phase protein and could interfere with the
immunohistochemical analysis we subsequently analyzed human plasma
concentrations of serpinA3 in healthy controls (n=49), AAA patients
(n=40) and patients with myocardial infarction (n=28) by ELISA. We
detected no difference between the 3 groups (data not shown).
Overexpression of serpinA3n inhibits
CaCl2-induced elastinolytic activity
Mice transgenic for serpinA3n (on C57bl/6
background) were also used to study the role of serpinA3 in
aneurysm disease. Since serpinA3 expression was reduced in AAA
samples we speculated whether overexpression of serpinA3n could
inhibit aneurysm development. Abdominal aorta was dilated in
CaCl2-treated littermate wild-type C57bl/6 mice as
compared to NaCl treated controls. However, no inhibition of the
aortic diameter was observed in serpinA3n transgenic animals
(Fig. 3C).
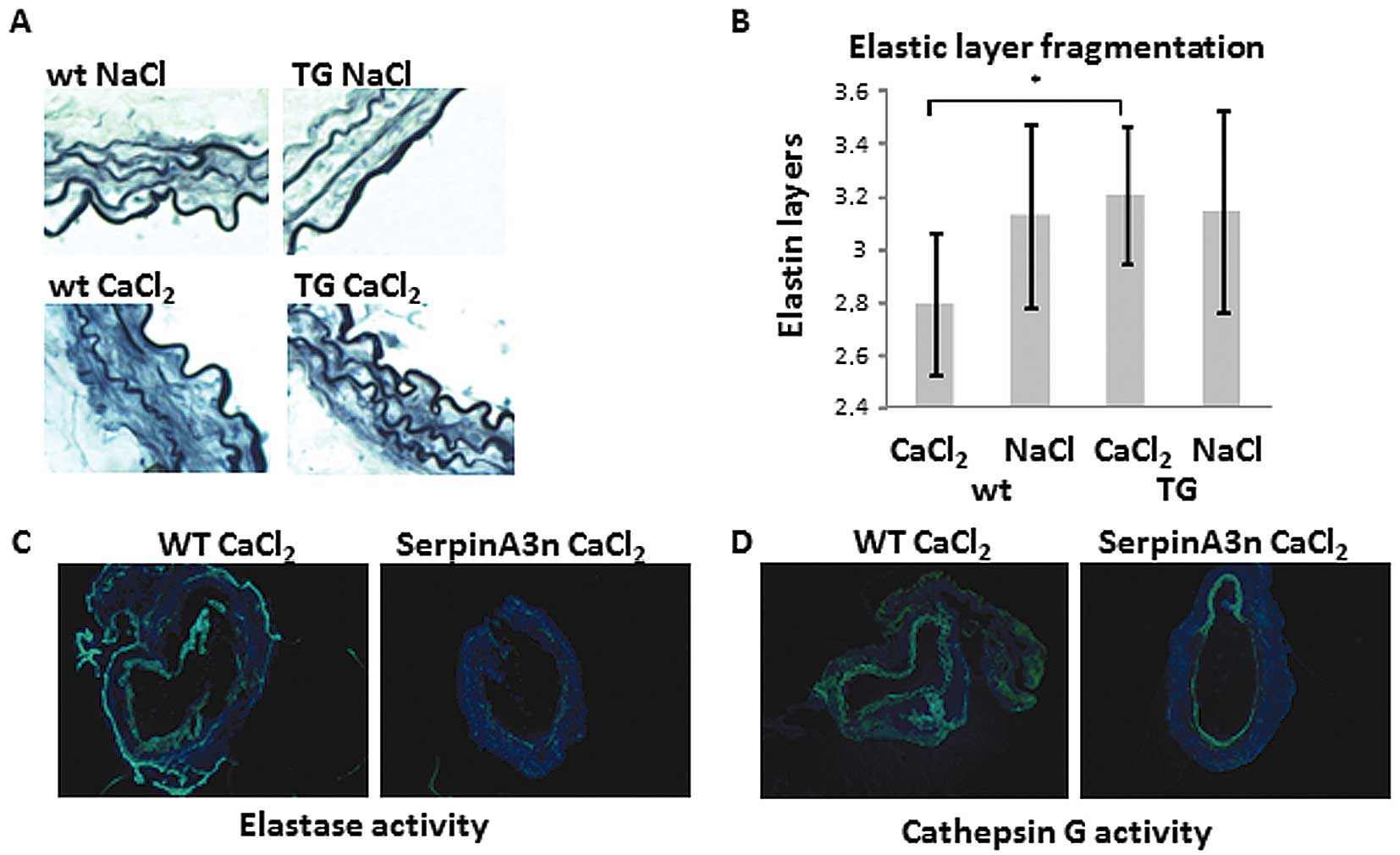
The aortas of wild-type C57bl/6 mice treated with
CaCl2 had lower amounts of elastin compared with the
aortas of NaCl treated controls and serpinA3 transgenic mice
on C57bl/6 background (Fig. 4A and
B). We found a higher mast cell chymase activity (P<0.05) in
the serum from CaCl2-treated wild-type mice (detected in
6/9 animals with a mean of 0.13 mU) as compared to
CaCl2-treated serpinA3n mice (not detected in any of 5
animals). Also, in situ zymography showed a reduced elastase
(Fig. 4C) and cathepsin G
(Fig. 4D) activity in
CaCl2-treated serpinA3n mice as compared to
CaCl2-treated wild-type mice.
Regulation of serpinA3 by proinflammatory
cytokines
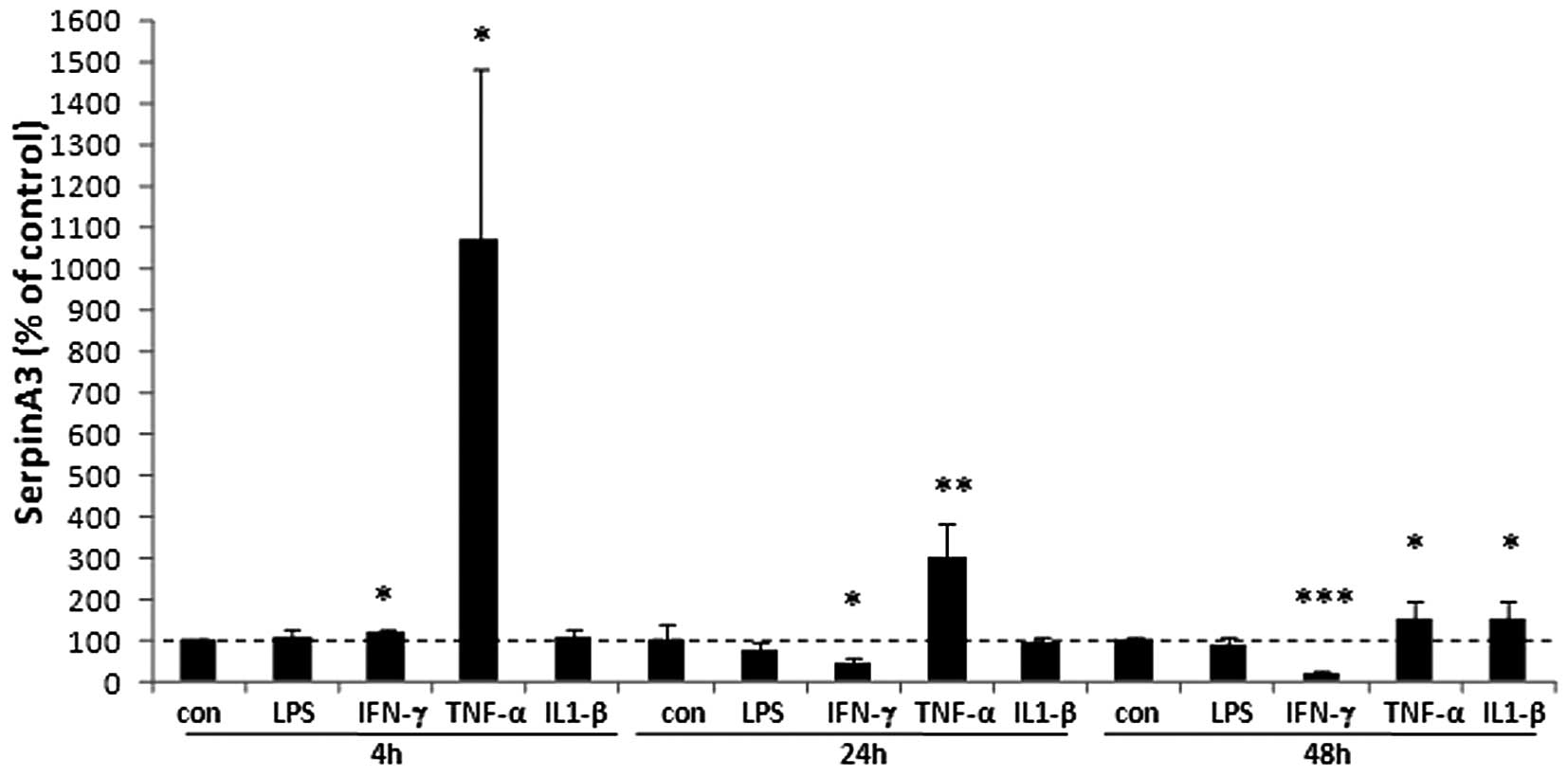
To verify whether endothelial cells can produce
serpinA3 we analyzed the mRNA expression of serpinA3 under
different stimuli. The result showed that the proinflammatory
cytokine TNF-α induced serpinA3 expression more than 10-fold at 4
h, 3-fold at 24 h and 50% at 48 h as compared to non-stimulated
cells (Fig. 5). IFN-γ slightly
induced the expression of serpinA3 at 4 h. At later time points,
IFN-γ reduced the expression of serpinA3 by 55% after 24 h and by
78% at 48 h compared to non-stimulated cells. IL-1β did not have
any dramatic effects on the serpinA3 regulation in the endothelial
cells but resulted in a 55% induction at 48 h. LPS did not have any
effects. SMCs from 2 different donors were also used without any
dramatic effects by any of the used substances. TNF-α reduced the
expression of serpinA3 and LPS slightly induced the expression
(data not shown).
Discussion
In the present study we investigated the expression
and role of serpinA3 in atherosclerosis and AAA. The results
suggest that AAA in humans is associated with a local decrease in
serpinA3 expression in the aorta, whereas a local increase was
found in atherosclerotic lesions. The presence of serpinA3 in the
subendothelial layer of healthy aortas suggests that serpinA3 may
be derived from endothelial and smooth muscle cells. The
subendothelial protein expression of serpinA3 is not likely to be
reflected by serpinA3 expression in plasma, since there was no
difference in plasma levels between patients and controls. Cell
culture experiments using vascular aortic SMCs and endothelial
cells showed that these cells have the capacity to synthesize
serpinA3.
The role of serpinA3 in atherosclerosis was
investigated using mice transgenic for serpinA3 on an Apoe
deficient background. To our surprise, we did not find any
difference in lesion development in these mice compared to pure
Apoe deficient mice at 20 weeks of age. Speculatively,
serpinA3 could influence the formation of a more stable plaque
phenotype in longer terms due to the protease inhibitory
properties. Prolonged effects were however not investigated in this
study.
In contrast to atherosclerosis, serpinA3 expression
was decreased in AAA. Human AAA samples covered by thrombus are
devoid of endothelial cells, and endothelial cell erosion is also
increased in samples without thrombus (3,21).
SMC apoptosis is one of the hallmarks of AAA and serpinA3
expression was absent in the major part of the media in AAA samples
but increased in the media in carotid lesions. Conversely, it is
unlikely that serpinA3 is expressed by leukocytes because these
cells did not co-localize with areas of serpinA3 staining. No or
very few inflammatory cells could be detected in the healthy
aortas. Also, the increased leukocyte accumulation associated with
aneurysm formation argues against inflammatory cells being
responsible for the decreased serpinA3 expression in AAA. Taken
together, these findings support that loss of endothelial cells and
SMCs may be responsible for the decrease in serpinA3 expression in
the intimal/medial part of aneurysmal tissue.
The role of serpinA3 in AAA was investigated using
serpinA3n mice transgenic on a C57bl/6 background. To our surprise,
we did not find any difference in AAA formation in transgenic mice
compared to wild-type mice. A recent work by Ang et al
(22) showed that administration
of recombinant serpina3n reduced aortic rupture in Angiotensin
II-induced AAA in Apoe−/− mice by the inhibition of
Granzyme B mediated decorin degradation leading to enhanced
collagen remodeling. Our experiments where primarily performed on
serpinA3n transgenic mice on C57bl/6 background but also when
crossing these mice with Apoe−/− mice we obtained the
same results (data not shown). In addition, rupture is very
uncommon in the CaCl2 induced aneurysm model.
There may be several mechanisms whereby low
expression of serpinA3 is associated with aneurysm formation. The
most plausible mechanism may be that proteases inhibited by
serpinA3 are involved in the degradation of elastin fibers, since
serpinA3 is known to inhibit a broad range of mast cell- and
neutrophil-derived proteases, such as chymase, cathepsin G and
elastase. Cathepsin G released from both neutrophils and mast cells
is a potent elastinolytic protease with ∼20–30% activity compared
to elastase (23). We have
previously found an increased expression of mast cell and
neutrophil derived cathepsin G in human AAA as compared to control
aorta (3) but its role in AAA
formation has not yet been investigated. The role of chymase in AAA
has been demonstrated by reduced aneurysm formation in mast cell
deficient mice compared with wild-type mice (7,23).
In the present study, we demonstrate a decreased chymase activity
in serum from serpinA3n transgenic mice as well as a suppressed
activity of cathepsin G and elastase in CaCl2-induced
transgenic mice compared to the CaCl2-treated wild-type
mice. Taken together, this may explain the preserved elastin
content in the CaCl2-treated transgenic animals. Our
results therefore suggest that serpinA3 could be involved in a
phenotypic stabilization of the aorta that might inhibit rupture in
longer perspectives, as seen in the study by Ang et al
(22). A direct effect on
elastinolytic activity could also have an effect on the recruitment
of inflammatory cells. Decreased elastin degradation probably
results in a decreased release of elastin-derived peptides (EDPs),
which may influence the migration of inflammatory cells. Soluble
EDPs released within human AAA tissue were proven to attract
mononuclear phagocytes through ligand-receptor interactions with
the elastin-binding protein, providing a plausible molecular
mechanism explaining the inflammatory response that accompanies
aneurysmal degeneration (24).
In summary, differential vascular expression of
serpinA3 is clearly associated with atherosclerosis and AAA.
SerpinA3 has no major effect on experimentally induced
atherosclerosis or AAA development in mouse. However, serpinA3
might be involved in a phenotypic stabilization of the aorta that
could prevent AAA rupture. These results further support the
influence of neutrophils and mast cells in AAA.
Acknowledgements
We thank Dr Marianne Gervais-Taurel
from Université Paris Est (Créteil Cedex, France) for assistance
with the CaCl2 aneurysmal model. This study was
supported by the Swedish Research Council (4203, 12660, 14231 and
Linnaeus support 8703), the Swedish Heart-Lung Foundation, the
European Commission (FAD, Health-F2-2008-200647 and 201668), the
Stockholm County Council (20080413, 20090077 and 560183), the
Foundation for Strategic Research, Foundation for Medical Research,
the Fredrik and Ingrid Thuring Foundation and the Magnus Bergvall
Foundation.
References
|
1.
|
GK HanssonA HermanssonThe immune system in
atherosclerosisNat Immunol12204212201121321594
|
|
2.
|
E ChokeG CockerillWR WilsonS SayedJ
DawsonI LoftusMM ThompsonA review of biological factors implicated
in abdominal aortic aneurysm ruptureEur J Vasc Endovasc
Surg30227244200515893484
|
|
3.
|
MI MayranpaaJA TrosienV FontaineM
FolkessonM KaziP ErikssonJ SwedenborgU HedinMast cells associate
with neovessels in the media and adventitia of abdominal aortic
aneurysmsJ Vasc
Surg50388396200910.1016/j.jvs.2009.03.05519515525
|
|
4.
|
T TsurudaJ KatoK HatakeyamaK KojimaM YanoY
YanoK NakamuraF Nakamura-UchiyamaY MatsushimaT ImamuraAdventitial
mast cells contribute to pathogenesis in the progression of
abdominal aortic aneurysmCirc Res10213681377200818451339
|
|
5.
|
DF KetelhuthM BackThe role of matrix
metalloproteinases in atherothrombosisCurr Atheroscler
Rep13162169201110.1007/s11883-010-0159-721271310
|
|
6.
|
I BotM BotSH van HeiningenPJ van
SantbrinkIM LankhuizenP HartmanS GruenerH HilpertTJ van BerkelJ
FingerleEA BiessenMast cell chymase inhibition reduces
atherosclerotic plaque progression and improves plaque stability in
apoe−/− miceCardiovasc
Res89244252201110.1093/cvr/cvq26020693162
|
|
7.
|
J SunJ ZhangJS LindholtGK SukhovaJ LiuA
HeM AbrinkG PejlerRL StevensRW ThompsonCritical role of mast cell
chymase in mouse abdominal aortic aneurysm
formationCirculation120973982200919720934
|
|
8.
|
K TsunemiS TakaiM NishimotoD JinM
SakaguchiM MuramatsuA YudaS SasakiM MiyazakiA specific chymase
inhibitor,
2-(5-formylamino-6-oxo-2-phenyl-1,6-dihydropyrimidine-1-yl)-n-[[3,4-dioxo-
1-phenyl-7-(2-pyridyloxy)]-2-heptyl]acetamide (nk3201), suppresses
development of abdominal aortic aneurysm in hamstersJ Pharmacol Exp
Ther3098798832004
|
|
9.
|
P RotziusS ThamsO SoehnleinE KenneCN
TsengNK BjorkstromKJ MalmbergL LindbomEE ErikssonDistinct
infiltration of neutrophils in lesion shoulders in
apoe−/− miceAm J
Pathol177493500201010.2353/ajpath.2010.09048020472897
|
|
10.
|
JR CohenL KeeganI SarfatiD DannaC IlardiL
WiseNeutrophil chemotaxis and neutrophil elastase in the aortic
wall in patients with abdominal aortic aneurysmsJ Invest
Surg4423430199110.3109/089419391091411721777436
|
|
11.
|
JL EliasonKK HannawaG AilawadiI SinhaJW
FordMP DeograciasKJ RoelofsDT WoodrumTL EnnisPK HenkeNeutrophil
depletion inhibits experimental abdominal aortic aneurysm
formationCirculation112232240200510.1161/CIRCULATIONAHA.104.51739116009808
|
|
12.
|
X HouardZ TouatV OllivierL LouedecM
PhilippeU SebbagO MeilhacP RossignolJB MichelMediators of
neutrophil recruitment in human abdominal aortic
aneurysmsCardiovasc Res82532541200910.1093/cvr/cvp04819201759
|
|
13.
|
AJ HorvathJA IrvingJ RossjohnRH LawSP
BottomleyNS QuinseyRN PikePB CoughlinJC WhisstockThe murine
orthologue of human antichymotrypsin: A structural paradigm for
clade a3 serpinsJ Biol
Chem2804316843178200510.1074/jbc.M50559820016141197
|
|
14.
|
T ChandraR StackhouseVJ KiddKJ RobsonSL
WooSequence homology between human alpha 1-antichymotrypsin, alpha
1-antitrypsin, and antithrombin
iiiBiochemistry2250555061198310.1021/bi00291a0016606438
|
|
15.
|
NM SchechterLM JordanAM JamesBS
CoopermanZM WangH RubinReaction of human chymase with reactive site
variants of alpha 1-antichymotrypsin. Modulation of inhibitor
versus substrate propertiesJ Biol Chem268236262363319938226889
|
|
16.
|
S ForsythA HorvathP CoughlinA review and
comparison of the murine alpha1-antitrypsin and
alpha1-antichymotrypsin multigene clusters with the human clade a
serpinsGenomics81336345200310.1016/S0888-7543(02)00041-112659817
|
|
17.
|
AJ HorvathSL ForsythPB CoughlinExpression
patterns of murine antichymotrypsin-like genes reflect evolutionary
divergence at the serpina3 locusJ Mol
Evol59488497200410.1007/s00239-004-2640-915638460
|
|
18.
|
AC ChiouB ChiuWH PearceMurine aortic
aneurysm produced by periarterial application of calcium chlorideJ
Surg Res99371376200110.1006/jsre.2001.620711469913
|
|
19.
|
D WagsaterC ZhuHM BjorckP ErikssonEffects
of pdgf-c and pdgf-d on monocyte migration and mmp-2 and mmp-9
expressionAtherosclerosis202415423200910.1016/j.atherosclerosis.2008.04.05018573494
|
|
20.
|
L FolkersenF van’t HooftE ChernogubovaHE
AgardhGK HanssonU HedinJ LiskaAC SyvanenG Paulsson-BerneA
Franco-CerecedaAssociation of genetic risk variants with expression
of proximal genes identifies novel susceptibility genes for
cardiovascular diseaseCirc Cardiovasc
Genet3365373201010.1161/CIRCGENETICS.110.94893520562444
|
|
21.
|
M KaziJ ThybergP ReligaJ RoyP ErikssonU
HedinJ SwedenborgInfluence of intraluminal thrombus on structural
and cellular composition of abdominal aortic aneurysm wallJ Vasc
Surg3812831292200310.1016/S0741-5214(03)00791-214681629
|
|
22.
|
LS AngWA BoivinSJ WilliamsH ZhaoT AbrahamK
Carmine-SimmenBM McManusRC BleackleyDJ GranvilleSerpina3n
attenuates granzyme B-mediated decorin cleavage and rupture in a
murine model of aortic aneurysmCell Death
Dis2e209201110.1038/cddis.2011.8821900960
|
|
23.
|
C BoudierG GodeauW HornebeckL RobertJG
BiethThe elastolytic activity of cathepsin G: An ex vivo study with
dermal elastinAm J Respir Cell Mol
Biol4497503199110.1165/ajrcmb/4.6.4971711351
|
|
24.
|
KA HanceM TatariaSJ ZiporinJK LeeRW
ThompsonMonocyte chemotactic activity in human abdominal aortic
aneurysms: Role of elastin degradation peptides and the 67-kd cell
surface elastin receptorJ Vasc
Surg35254261200210.1067/mva.2002.120382
|