Introduction
Atherosclerosis occurs in all arteries, inducing
acute arterial occlusion, which may lead to a sudden decreased
blood flow in tissue, resulting in cell necrosis and even death
(1–3). The collateral circulation produced
in the ischemic region may limit infarction size and improve organ
function. However, the slow process of collateral circulation
formation is not sufficient for the needs of ischemic tissue.
Therefore, therapeutic angiogenesis is necessary (4). Toll-like receptor 2 (TLR2), whose
coding gene is located on chromosome 4q32 (5), is an important member of the TLR
family. TLR2, expressed most abundantly in peripheral blood
leukocytes (5), is able to
recognize bacteria and endogenous ligands that activate nuclear
factor-κB (NF-κB) and activator protein-1 (AP-1) (6), playing an important role in
inflammation, immunity response and tumorigenesis (7).
Following ischemic injury, apoptotic cells may
induce a wound healing response, release of inflammatory cytokines
and the recruiting of inflammatory cells, all of which are
important processes in angiogenesis (8). TLR2 deficiency induced renal injury,
in renal ischemia/reperfusion (I/R) (9,10),
and reduced infarction size after I/R (11). Administration of Pam3CSK4 prior to
myocardial I/R reduced infarction size, improved cardiac function,
and decreased leukocyte infiltration to ischemic tissues (12). Treatment of mice with Pam3CSK4
also induced protection against cerebral ischemic injury (13) and attenuated cardiac dysfunction
in septic mice (14).
Helicobacter pylori was reported to activate the
mitogen-activated protein kinase (MAPK) cascade through TLR2,
thereby contributing to cancer cell invasion and angiogenesis
(15). The protective role of
TLR2 against ischemic injury has been clarified, however, the
contribution of TLR2 following the process of recovery from
ischemia was found to be different in various organs (5). Therefore, the manner in which TLR2
impacts on ischemic tissues in vivo remains to be
determined.
This study aimed to clarify the potential role of
TLR2 in angiogenesis following ischemic injury. Human umbilical
vein endothelial cells (HUVECs) were used to assess the role of
TLR2 on cell migration, permeability and lymphocyte invasion in
vitro. TLR2 knockout (TLR2−/−) mice and wild-type
(WT) mice were used to clarify the role of TLR2 in
neovascularization following ischemic injury in a mouse model of
hindlimb ischemia by ligation.
Materials and methods
Cell culture
HUVECs were purchased from the Shanghai Touching
Technology Co., Ltd. (Shanghai, China) and maintained in RPMI-1640
medium (Genom Biopharmaceutical Technology Co., Ltd., Hangzhou,
China) supplemented with 10% fetal bovine serum (FBS) (Sijiqing
Co., Ltd., Hangzhou, China). The cells were grown at 37°C in a
humidified incubator in 5% CO2 and 95% air.
Wound repair assays
HUVECs were seeded into 6-well plates and grown to
confluence. A single scratch wound was made through the middle of
each well with a sterile pipette tip. Cells were cultured with
RPMI-1640 medium without FBS, and stimulated for 30 h with 1 μg/ml
Pam3CSK4 (Invivogen, San Diego, CA, USA) and 2 μg/ml Pam3CSK4,
respectively. Migration of HUVECs across the wound margins from
18–30 h was assessed and photographed by inverted microscopy
(CKX41; Olympus).
Transwell assay on HUVEC permeability
and lymphocyte invasion
The Transwell chamber with 3 μm membranes
(Millipore, USA) was pre-coated with 0.2% gelatin (Sigma, St.
Louis, MO, USA). Lymphocytes were separated from peripheral blood
of mice using the density gradient centrifugation method. To assess
the effect of Pam3CSK4 on the permeability of HUVECs to
lymphocytes, HUVECs were seeded in the upper chamber at a density
of 5×104/well, and half of the cells were then treated
with Pam3CSK4 (1 μg/ml) for 24 h at 37°C. Lymphocytes, separated
from WT mice, were placed in the upper chambers at a density of
5×104/well with 100 μl 2% FBS RPMI-1640 medium, while
the lower chamber contained 600 μl 10% FBS RPMI-1640 medium. To
evaluate the effect of Pam3CSK4 on lymphocyte invasion, HUVECs were
seeded at a density of 1×105/well in the upper chamber
for 24 h at 37°C. Lymphocytes, separated from the peripheral blood
of WT and TLR2−/− mice, were placed in different upper
chambers at a density of 5×105/well with 100 μl 2% FBS
RPMI-1640 medium, while the lower chamber contained 600 μl 10% FBS
RPMI-1640 medium and 20 ng/ml vascular endothelial growth factor.
Migration was carried out for 24 h at 37°C and 5% CO2.
The medium was then removed. Transwell membranes of the upper
chambers were fixed in 4% paraformaldehyde and stained with crystal
violet. The HUVECs were removed and migrated cells on the membrane
were quantified using an inverted microscope (CKX41; Olympus).
Animals
WT mice (C57BL/6, 18–25 g, 8–10 weeks old) were
obtained from the Laboratory Animal Center at the Zhejiang Chinese
Medical University and TLR2−/− mice
(B6.129-Tlr2tm1kir/J Strain) were obtained from the
Jackson Laboratory. The two types of mice were maintained under
specific pathogen-free conditions and received a standard diet and
water ad libitum in the Laboratory Animal Center at the
First Affiliated Hospital of the College of Medicine, Zhejiang
University. Experimental studies were carried out in accordance
with the Guide for the Care and Use of Laboratory Animals published
by the National Institutes of Health (NIH publication no.
85-23).
Hindlimb ischemia by ligation
(16) and blood flow
monitoring
Under sterile conditions, proximal and distal
portions of the left femoral artery were exposed and ligated,
followed by the excision of the artery between ligation points. The
superficial branch of the femoral artery was also ligated. A sham
procedure (dissection of vessels without ligation) was performed on
the right leg. Blood flow in the two limbs was measured using a
Laser Doppler blood perfusion monitor (PeriFlux System 5000,
Perimed AB) prior to ligation, and 1, 3, 7 and 14 days after
ligation (TLR2−/− vs. WT mice, respectively).
Measurement was performed on six different locations on the leg,
and the mean value of blood perfusion was used to evaluate the
blood flow of each hindlimb. The gastrocnemius and serum were also
harvested for analysis.
Immunohistochemistry
Ischemic gastrocnemiuses were collected 1, 3, 7 and
14 days after ligation, and fixed in 4% paraformaldehyde. Optimal
cutting temperature (OCT) sections (5 μm) were sliced and placed on
glass slides coated with polylysine. The streptavidin-biotin
complex technique was used for immunohistochemistry assay.
Endogenous peroxidase activity was blocked using 3%
H2O2. The sections were incubated with
primary antibody against the mouse cluster of differentiation 31
(CD31) (Abcam, USA) at room temperature for 1 h. Samples were then
washed with phosphate-buffered saline (PBS) and incubated with
secondary goat anti-rabbit antibody and
streptavidin-biotin-peroxidase complexes. The samples were
visualized using a microscope (DM 2500; Leica, Germany).
Enzyme-linked immunosorbent assay
(ELISA)
Mouse blood was collected 1, 3, 7 and 14 days after
ligation using pyrogen-free tubes (BD Biosciences, San Jose, CA,
USA) and centrifuged at 3,000 rpm for 20 min at 4°C. The resulting
serum was diluted 100-fold with double-distilled water. Mouse tumor
necrosis factor-α (TNF-α) and interleukin-6 (IL-6) were measured by
ELISA according to the manufacturer’s instructions (Boster, China),
respectively. ELISA standards ranged from 15.6 to 1,000 pg/ml. The
absorbance was measured at 450 nm.
Statistical analysis
Data were presented as the mean ± standard
deviation. Comparisons were made by the two-tailed Student’s t-test
for independent samples or one-way analysis of variance (ANOVA) and
post hoc Scheffe’s test as appropriate. P<0.05 was considered
statistically significant.
Results
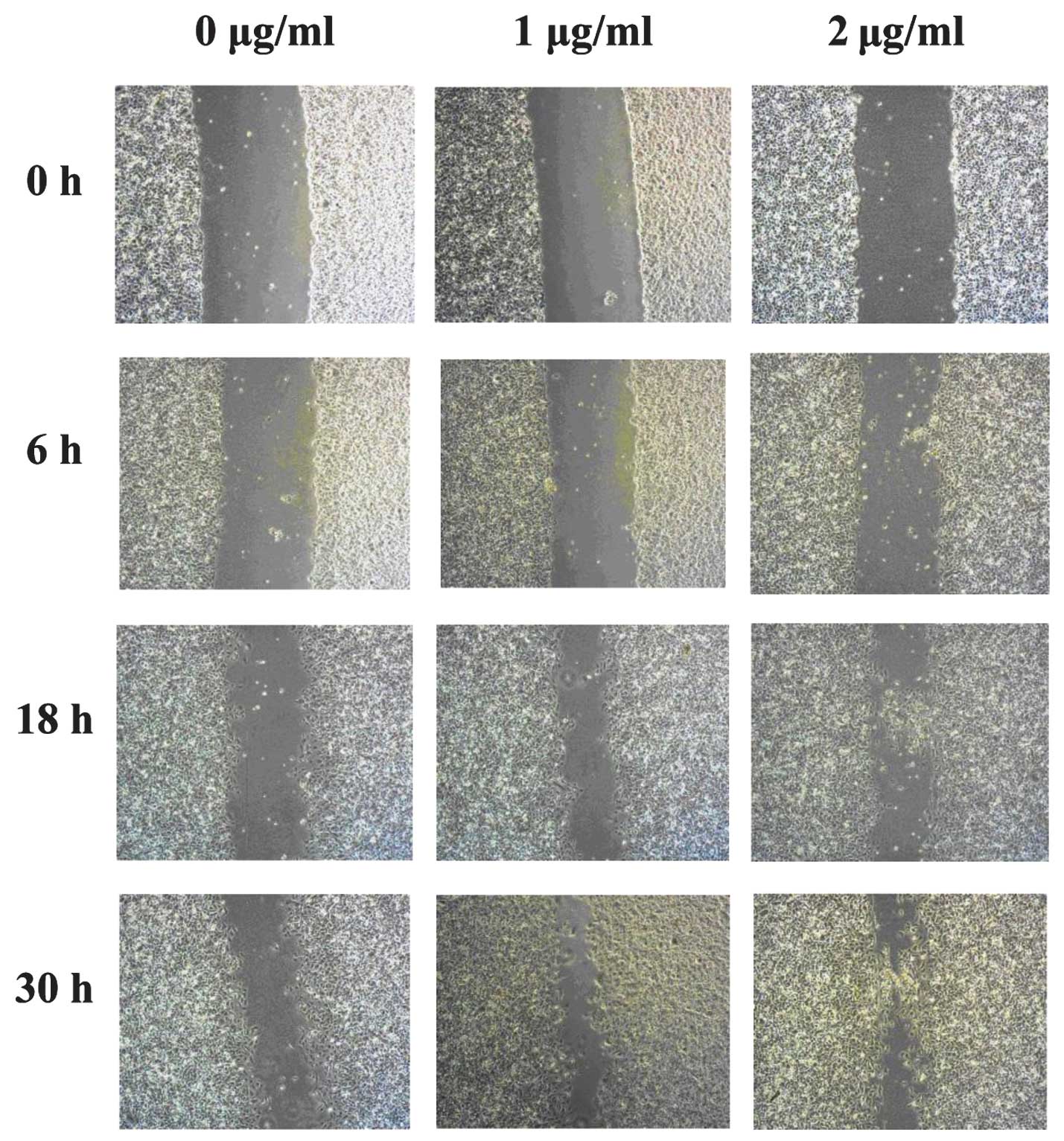
Pam3CSK4 induced migration of
HUVECs
The effect of Pam3CSK4 on the migration of HUVECs
was assessed using wound repair assays. HUVECs migrated more
rapidly in response to Pam3CSK4 stimulation after 18 h (Fig. 1), while after 30 h, HUVECs without
Pam3CSK4 stimulation showed a clear wound, where minimal cell
migration across the wound margin was observed. By contrast,
Pam3CSK4 induced cell migration across the wound margins resulting
in almost complete closure of the wound.
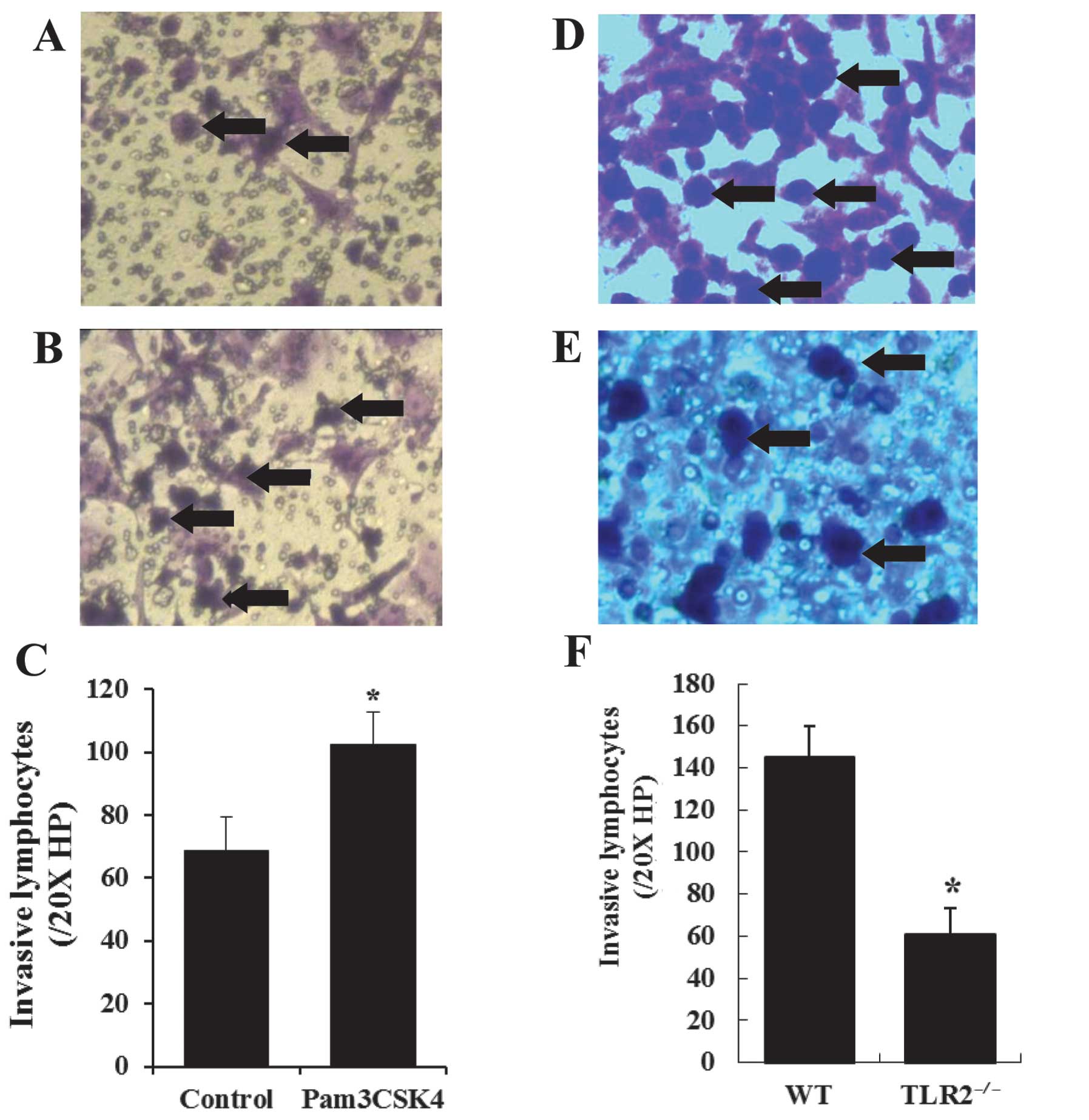
Permeability of HUVECs and lymphocyte
invasion
To assess the potential role of TLR2 activation in
endothelial cell permeability, HUVECs were pretreated with Pam3CSK4
(1 μg/ml), and then seeded in Transwell membranes for lymphocyte
invasion. Fig. 2A shows
lymphocyte invasion under basal conditions compared to those of
Pam3CSK4 pretreatment (Fig. 2B).
Quantification of lymphocyte invasion was performed in six
different fields/chamber under 20X high-power lens (HP), and the
mean values were used to evaluate the invasive lymphocytes for each
chamber. The lymphocytes were significantly increased by Pam3CSK4
pretreatment compared to the control (102.29±10.60/20X HP vs.
68.69±10.57/20X HP, P<0.001) (Fig.
2C).
In the lymphocyte invasion assay, lymphocytes
isolated from the peripheral blood of WT and TLR2−/−
mice were placed in the upper well of the Transwell chamber seeded
with HUVECs for lymphocyte migration. The lymphocytes invasion
(Fig. 2D and E) was quantified in
the same manner, and the results showed that the amount of invasive
lymphocytes from the WT mice was significantly more than that from
the TLR2−/− mice (145.07±14.49/20X HP vs.
60.92±12.27/20X HP, P<0.001) (Fig.
2F).
TLR2−/− delayed recovery
from ischemic injury
Following hindlimb ligation surgery, the operated
limb showed significant reduction in blood perfusion compared to
the sham-operated limb. On the 1st and 3rd day after ligation, the
operated limbs showed no differences in blood perfusion between
TLR2−/− and WT mice (n=6/time-point/group, 24.05±6.69
vs. 26.50±9.16, P>0.05; 38.14±8.82 vs. 39.61±9.45, P>0.05).
However, blood perfusion of operated limbs in TLR2−/−
mice was significantly lower than that in WT mice on the 7th and
14th day after ligation (39.15±3.71 vs. 52.40±2.93, P=0.001;
33.47±1.69 vs. 47.43±4.27, P=0.013) (Fig. 3A).
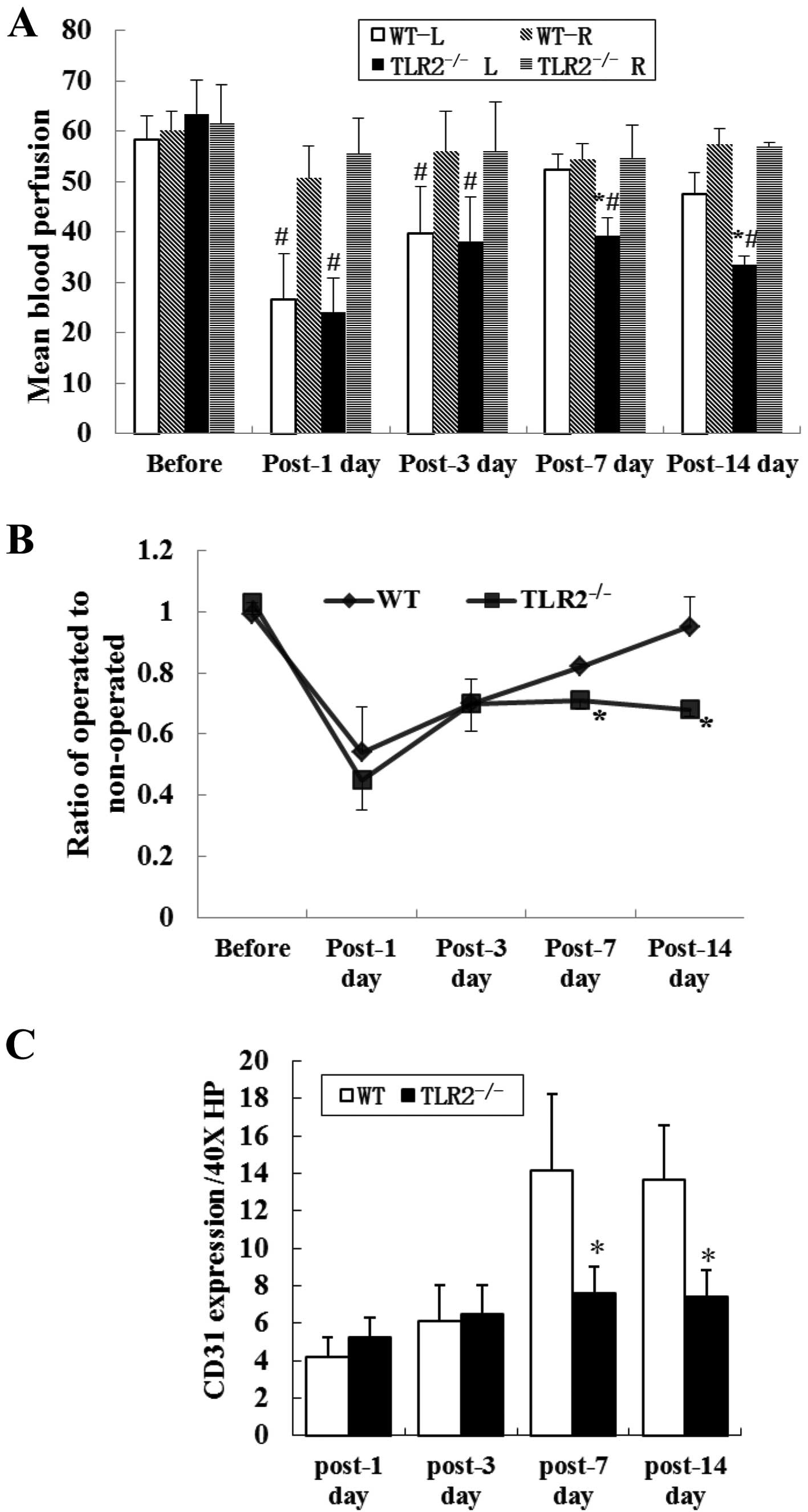 | Figure 3Blood perfusion and ischemic
gastrocnemius CD31 expression in WT and TLR2−/− mice
limbs. (A) L and R are the operated and non-operated limbs,
respectively. Blood perfusion of operated limbs in
TLR2−/− mice was significantly lower than that in WT
mice, on the 7th and 14th day after ligation. When compared to
those of non-operated limbs, blood perfusion was significantly
reduced in the ischemic limbs in the two groups on the 1st and 3rd
day after ligation. On the 7th and 14th day after ligation, blood
perfusion was still decreased in TLR2−/− mice but no
difference was observed in WT mice. *P<0.05,
TLR2−/−L vs. WT-L, #P<0.05, WT-L vs. WT-R,
or TLR2−/−L vs. TLR2−/−R. (B) Blood perfusion
ratio of operated to non-operated leg in each mouse reflected
recovery of the ischemic limbs. The ratio in TLR2−/−
mice was significantly lower than that in WT mice on the 7th and
14th day after ligation, but no significant difference was found on
the 1st and 3rd day after ligation. *P<0.05,
TLR2−/− vs. WT (C) CD31 expression in ischemic
gastrocnemiuses: TLR2−/− mice showed a reduced CD31
expression compared to WT mice on the 7th and 14th day after
ligation. No significant difference was detected between the two
groups of mice on the 1st and 3rd day after ligation. *P<0.05,
TLR2−/− vs. WT mice. Representative photomicrograph
shows the CD31 expression in (D) WT and (E) TLR2−/−
mice. |
To reduce the effect of individual diversities on
data, the results were plotted as a ratio of operated to
non-operated leg for each mouse, which reflected the recovery of
ischemic leg. The recovered blood perfusion of operated limbs in
TLR2−/− mice was significantly lower than that in WT
mice, on the 7th and 14th day after ligation, however, no
difference was detected on the 1st and 3rd day after ligation
(Fig. 3B).
TLR2−/− inhibited
angiogenesis following ischemic injury
To identify angiogenesis in the ischemic muscles,
ischemic gastrocnemius muscles were collected on the 1st, 3rd, 7th
and 14th day after ligation in order to detect CD31+
endothelial monolayer lining the lumen of vascular structures.
TLR2−/− mice showed a reduced CD31 expression compared
to WT mice on the 7th and 14th day after ligation (n=6/time-point/
group, 7.63±1.41/40X HP vs. 14.17±4.07/40X HP, P=0.001;
7.44±1.42/40X HP vs. 13.67±2.92/40X HP, P<0.001). CD31
expression was also detected on the 1st and 3rd day after ligation,
although there was no significant difference between the two groups
of mice (Fig. 3C).
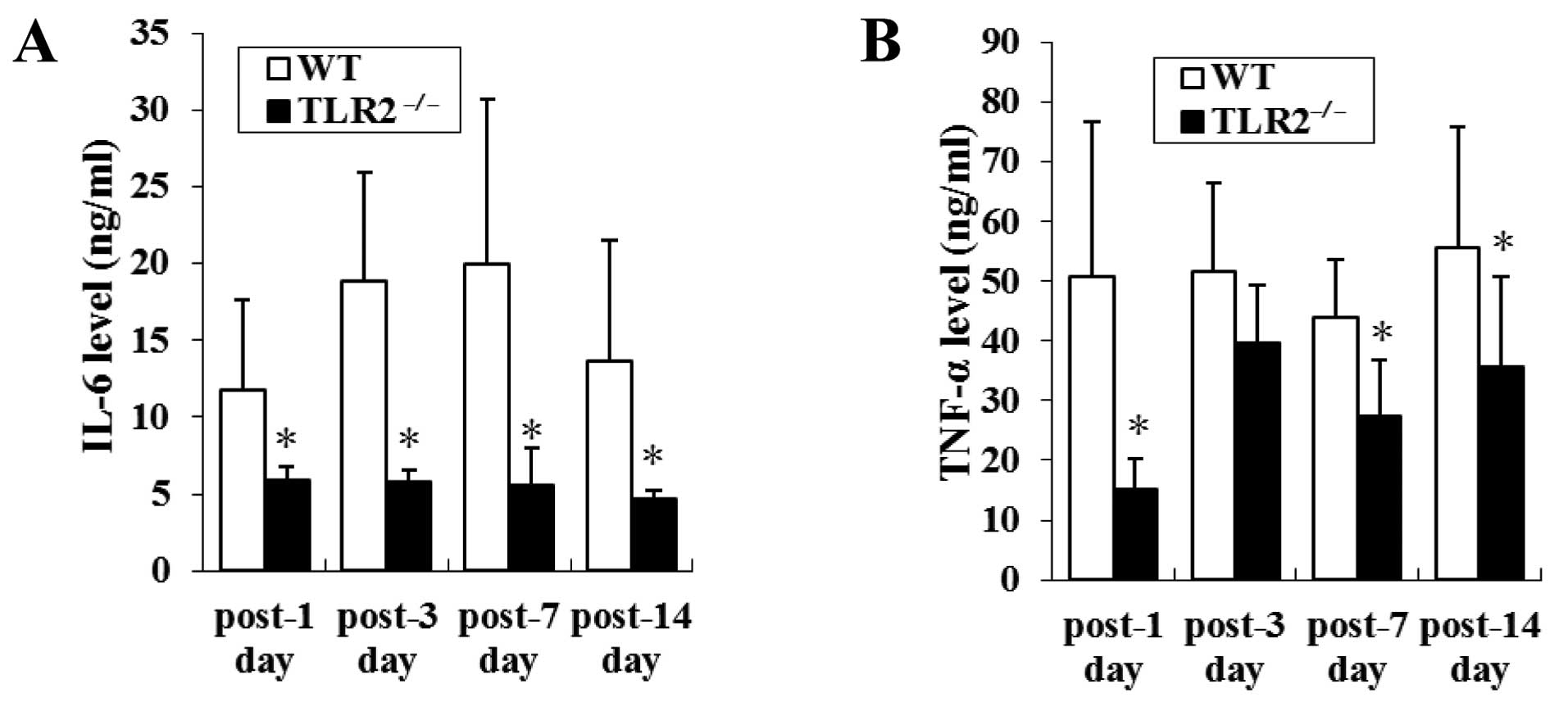
TLR2−/− reduced TNF-α
levels and IL-6 release following ischemic injury
To examine the effect of TLR2 deficiency on cytokine
production in ischemic injury, the sera of TLR2−/− and
WT mice were collected to measure the levels of TNF-α and IL-6 1,
3, 7 and 14 days after ligation. Elevated levels of IL-6 were
identified on 1, 3, 7 and 14 days after ligation in the serum of WT
mice, whereas little IL-6 was found in the serum of
TLR2−/− mice (11.79±5.80 vs. 5.85±0.94 ng/ml, P=0.043;
18.91±7.03 vs. 5.80±0.80 ng/ml, P=0.021; 19.95±10.81 vs. 5.60±2.39
ng/ml, P=0.003; 13.68±7.80 vs. 4.68±0.57 ng/ml, P=0.002) (Fig. 4A).
Similarly, TNF-α levels in TLR2−/− mice
were increased after ligation but were still significantly lower
than those in WT mice on the 1st, 7th and 14th day after ligation
(15.18±5.09 vs. 50.80±25.97 ng/ml, P=0.021; 27.35±9.44 vs.
44.04±9.68 ng/ml, P=0.005; 35.69±15.16 vs. 55.74±20.08 ng/ml,
P=0.046) (Fig. 4B).
Discussion
Angiogenesis is an important mechanism that protects
organs against imminent danger and tissue necrosis following acute
ischemic injury in various diseases. Inflammatory signaling
pathways are activated even in the absence of infection and are
believed to play an important role in angiogenesis during wound
healing. In the ischemic tissue, damage-associated molecular
patterns (DAMPs), released from necrosis cells, activates TLR2 and
sterile inflammation (17). The
aim of this study, was to clarify whether the TLR2 signaling
pathway induced angiogenesis to protect tissues that experienced
ischemia.
Lymphocyte invasion and recruitment in the ischemic
lesion are essential in inflammation and angiogenesis during
ischemic injury healing. Endothelial cell adhesion, migration and
permeability are also involved in this procedure. In this study, we
demonstrated that TLR2 activation induced HUVEC cell migration, and
increased its permeability to lymphocyte. TLR2 activation was able
to promote tube formation, as well as endothelial cell invasion and
migration in vitro (18).
However, lymphocytes in peripheral blood are required to cross
endothelial cells to assemble in ischemic tissue in vivo.
Therefore, lymphocyte invasion and endothelial cell permeability
are important, but have not been extensively studied. Thus, we
isolated the lymphocytes from TLR2−/− and WT mice for
invasion assay without Pam3CSK4 stimulation. TLR2−/−
lymphocytes showed a significantly reduced invasive ability
compared to WT lymphocytes. The results suggest that the TLR2
signaling pathway was involved in lymphocyte invasion and
endothelial cell permeability, promoting lymphocyte recruitment and
serving as important factors of angiogenesis.
Having established the role of TLR2 in cell
migration, invasion and endothelial cell permeability in
vitro, we assessed the association between TLR2 and
angiogenesis in vivo. An acute hindlimb ischemic model was
produced, and all the mice showed hindlimb disability following
ligation. However, TLR2−/− and WT mice showed different
responses after ligation. On the 7th and 14th day after ligation,
blood perfusion of ischemic legs in WT mice had almost recovered
and showed no difference compared with non-operated legs. However,
blood perfusion of ischemic legs in TLR2−/− mice
remained lower than that in non-operated and ischemic legs in WT
mice. Our results suggest that TLR2 deficiency induced the delayed
recovery of ischemic injury to leg muscles. Since the femoral
artery was ligated and excised, the blood perfusion recovery
resulted from neovascularization in ischemic muscles. To confirm
our hypothesis, CD31 was used to evaluate the extent of
revascularization. CD31, also known as platelet-endothelial-cell
adhesion molecule-1 (PECAM-1), is expressed at high density at
lateral borders of endothelial cells (19–21), and is associated with
angiogenesis. The revascularization labeled by CD31 in ischemic
gastrocnemius muscles showed similar results with the blood
perfusion results. The TLR2−/− mice also showed a
significantly decreased expression of CD31 on the 7th and 14th day
after ligation, but no difference was found on the 1st and 3rd day.
These data suggested that the TLR2 signaling pathway is important
in revascularization after ischemic injury, and promotes long-term
recovery. After ischemic injury, inflammatory cytokine and
proangiogenic factors were upregulated, and inflammatory cells
including lymphocytes were recruited in ischemic tissue. In
TLR2−/− mice, the ability of cytokine recognition and
arteriogenesis activation was apparently impaired, and the reduced
blood flow eventually induced the delayed recovery of the ischemic
legs. The importance of the TLR2 signaling pathway was not detected
at the earlier period of ischemic injury, but was strongly verified
in long-term revascularization.
A number of studies have clarified the relationship
between inflammatory cytokines and angiogenesis (22–27). Additionally, TLR2 signaling
contributed to the production of TNF-α and IL-6 (28–30). In the present study, we
investigated whether TNF-α and IL-6 were associated with
differences in angiogenesis between TLR2−/− and WT mice.
Expression of TNF-α and IL-6 in TLR2−/− mice was reduced
in both the early and long-term period of ischemic injury. These
findings suggest that the production of inflammatory cytokines in
ischemic injury depend on TLR2 expression in the host. After
TLR2−/−, the upregulated expression of NF-κB and AP-1 in
the nucleus after zero-flow ischemia may apparently be reduced
(31,32), causing the transcription and
expression of cytokine proteins (33) to be lower in TLR2−/−
mice compared to those of WT mice. In the earlier period of
ischemic injury, levels of TNF-α and IL-6 in WT mice were higher
than those in TLR2−/− mice. However, the formation of
new vessels was complicated and slow; therefore, detectable blood
reperfusion and new vessels in ischemic legs showed no differences.
The ischemic injury in WT mice had already recovered by the 7th day
after ligation. However, the neovascularization process in
TLR2−/− mice was significantly slower due to lack of
cytokines. Furthermore, lymphocyte invasion was reduced in
TLR2−/− mice. Our data suggest that TLR2 is important in
recovery of the ischemic injury, and that it is closely associated
with cytokine production and angiogenesis promotion of TLR2.
In conclusion, we have demonstrated that TLR2
activation promoted endothelial cell migration, cell permeability
and lymphocyte invasion. TLR2 activation promoted angiogenesis
in vivo, which was connected to the serum of TNF-α and IL-6
release, and lymphocyte invasion. These findings provide evidence
that the TLR2 signaling pathway is potentially a new target for
treating ischemic disease. Future studies should be performed to
clarify the intracytoplasm mechanism.
Abbreviations:
|
TLR2
|
Toll-like receptor 2;
|
|
HUVEC
|
human umbilical vein endothelial
cell;
|
|
TLR2−/−
|
TLR2 knockout;
|
|
WT
|
wild-type;
|
|
CD31
|
cluster of differentiation 31;
|
|
IL-6
|
interleukin-6;
|
|
TNF-α
|
tumor necrosis factor-α;
|
|
NF-κB
|
nuclear factor-κB;
|
|
AP-1
|
activator protein-1;
|
|
I/R
|
ischemia/reperfusion;
|
|
MAPK
|
mitogen-activated protein kinase;
|
|
OCT
|
optimal cutting temperature;
|
|
PBS
|
phosphate-buffered saline;
|
|
ELISA
|
enzyme-linked immunosorbent assay;
|
|
HP
|
high power lens;
|
|
DAMPs
|
damage-associated molecular
patterns
|
Acknowledgements
This study was funded by the Science
Technology Department of Zhejiang Province under grant no.
2012C33028.
References
|
1
|
Faxon DP, Fuster V, Libby P, et al:
Atherosclerotic Vascular Disease Conference: Writing Group III:
pathophysiology. Circulation. 109:2617–2625. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Dormandy J, Heeck L and Vig S: Acute limb
ischemia. Semin Vasc Surg. 12:148–153. 1999.
|
|
3
|
Costantini V and Lenti M: Treatment of
acute occlusion of peripheral arteries. Thromb Res. 106:V285–V294.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Isner JM: Therapeutic angiogenesis: a new
frontier for vascular therapy. Vasc Med. 1:79–87. 1996.PubMed/NCBI
|
|
5
|
Chen YC, Hsiao CC, Chen CJ, et al:
Toll-like receptor 2 gene polymorphisms, pulmonary tuberculosis,
and natural killer cell counts. BMC Med Genet. 11:172010.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Akira S, Uematsu S and Takeuchi O:
Pathogen recognition and innate immunity. Cell. 124:783–801. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Medzhitov R, Preston-Hurlburt P and
Janeway CA Jr: A human homologue of the Drosophila Toll
protein signals activation of adaptive immunity. Nature.
388:394–397. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carmeliet P: Angiogenesis in life, disease
and medicine. Nature. 438:932–936. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shigeoka AA, Holscher TD, King AJ, et al:
TLR2 is constitutively expressed within the kidney and participates
in ischemic renal injury through both MyD88-dependent and
-independent pathways. J Immunol. 178:6252–6258. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Leemans JC, Stokman G, Claessen N, et al:
Renal-associated TLR2 mediates ischemia/reperfusion injury in the
kidney. J Clin Invest. 115:2894–2903. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Favre J, Musette P, Douin-Echinard V, et
al: Toll-like receptors 2-deficient mice are protected against
postischemic coronary endothelial dysfunction. Arterioscler Thromb
Vasc Biol. 27:1064–1071. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mersmann J, Berkels R, Zacharowski P, et
al: Preconditioning by Toll-like receptor 2 agonist Pam3CSK4
reduces CXCL1-dependent leukocyte recruitment in murine myocardial
ischemia/ reperfusion injury. Crit Care Med. 38:903–909. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hua F, Ma J, Ha T, et al: Preconditioning
with a TLR2 specific ligand increases resistance to cerebral
ischemia/reperfusion injury. J Neuroimmunol. 199:75–82. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ha T, Lu C, Liu L, et al: TLR2 ligands
attenuate cardiac dysfunction in polymicrobial sepsis via a
phosphoinositide 3-kinase-dependent mechanism. Am J Physiol Heart
Circ Physiol. 298:H984–H991. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chang YJ, Wu MS, Lin JT and Chen CC:
Helicobacter pylori-induced invasion and angiogenesis of
gastric cells is mediated by cyclooxygenase-2 induction through
TLR2/TLR9 and promoter regulation. J Immunol. 175:8242–8252. 2005.
View Article : Google Scholar
|
|
16
|
Scholz D, Ziegelhoeffer T, Helisch A, et
al: Contribution of arteriogenesis and angiogenesis to
postocclusive hindlimb perfusion in mice. J Mol Cell Cardiol.
34:775–787. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Martin P, D’Souza D, Martin J, et al:
Wound healing in the PU.1 null mouse - tissue repair is not
dependent on inflammatory cells. Curr Biol. 13:1122–1128. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Saber T, Veale DJ, Balogh E, et al:
Toll-like receptor 2 induced angiogenesis and invasion is mediated
through the Tie2 signalling pathway in rheumatoid arthritis. PLoS
One. 6:e235402011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Muller WA, Ratti CM, McDonnell SL and Cohn
ZA: A human endothelial cell-restricted, externally disposed
plasmalemmal protein enriched in intercellular junctions. J Exp
Med. 170:399–414. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Newman PJ, Berndt MC, Gorski J, et al:
PECAM-1 (CD31) cloning and relation to adhesion molecules of the
immunoglobulin gene superfamily. Science. 247:1219–1222. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Muller WA: Leukocyte-endothelial-cell
interactions in leukocyte transmigration and the inflammatory
response. Trends Immunol. 24:327–334. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Balkwill F: Tumour necrosis factor and
cancer. Nat Rev Cancer. 9:361–371. 2009. View Article : Google Scholar
|
|
23
|
Balkwill F: TNF-alpha in promotion and
progression of cancer. Cancer Metastasis Rev. 25:409–416. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lin WW and Karin M: A cytokine-mediated
link between innate immunity, inflammation, and cancer. J Clin
Invest. 117:1175–1183. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ancrile B, Lim KH and Counter CM:
Oncogenic Ras-induced secretion of IL6 is required for
tumorigenesis. Genes Dev. 21:1714–1719. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang SP, Wu MS, Shun CT, et al:
Interleukin-6 increases vascular endothelial growth factor and
angiogenesis in gastric carcinoma. J Biomed Sci. 11:517–527. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fan Y, Ye J, Shen F, et al: Interleukin-6
stimulates circulating blood-derived endothelial progenitor cell
angiogenesis in vitro. J Cereb Blood Flow Metab. 28:90–98. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lin H, Yan J, Wang Z, et al: Loss of
immunity-supported senescence enhances susceptibility to
hepatocellular carcinogenesis and progression in TLR2-deficient
mouse. Hepatology. Aug 1–2012.(Epub ahead of print). View Article : Google Scholar
|
|
29
|
Amura CR, Renner B, Lyubchenko T, Faubel
S, Simonian PL and Thurman JM: Complement activation and Toll-like
receptor-2 signaling contribute to cytokine production after renal
ischemia/reperfusion. Mol Immunol. 52:249–257. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Correa-Costa M, Braga TT, Semedo P, et al:
Pivotal role of Toll-like receptors 2 and 4, its adaptor molecule
MyD88, and inflammasome complex in experimental tubule-interstitial
nephritis. PLoS One. 6:e290042011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Xie W, Wang Y, Huang Y, Yang H, Wang J and
Hu Z: Toll-like receptor 2 mediates invasion via activating
NF-kappaB in MDA-MB-231 breast cancer cells. Biochem Biophys Res
Commun. 379:1027–1032. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Shuto T, Ono T, Ohira Y, et al: Curcumin
decreases Toll-like receptor-2 gene expression and function in
human monocytes and neutrophils. Biochem Biophys Res Commun.
398:647–652. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jang JH, Yang ES, Min KJ and Kwon TK:
Inhibitory effect of butein on tumor necrosis factor-α-induced
expression of cell adhesion molecules in human lung epithelial
cells via inhibition of reactive oxygen species generation, NF-κB
activation and Akt phosphorylation. Int J Mol Med. 30:1357–1364.
2012.
|